 Fang Xu1,2†
Fang Xu1,2† Yangyang Wu
Yangyang Wu Yunguo Zhou
Yunguo Zhou Hong Li
Hong Li- 1Cardiology Treatment Center, Jiangxi Provincial Children's Hospital, Nanchang, China
- 2JXHC Key Laboratory of Children's Cardiovascular Diseases, Jiangxi Provincial Children's Hospital, Nanchang, China
- 3Pediatric Medical Department, Nanchang University, Nanchang, China
The subject of the study is an 11-month old IVF baby girl with the typical clinical manifestation of malonyl coenzyme A decarboxylase deficiency, including developmental delay, limb weakness, cardiomyopathy, and excessive excretion of malonic acid and methylmalonic acid. Whole genome sequencing (WGS) revealed a novel heterozygous nonsense mutation (c.672delG, p.Trp224Ter) in the MLYCD gene of the proband and her father and a novel heterozygous deletion in 5'-UTR-exon1-intron1 of the MLYCD gene of the proband and her mother. The patient's cardiac function and limb weakness improved considerably after 3 months of a low-fat diet supplemented with L-carnitine. Furthermore, mapping of gene mutations and clinical manifestations was done by case collection.
Introduction
Malonyl-CoA decarboxylase (MCD, E.C.4.1.1.9) activity deficiency caused by MLYCD gene mutation contributes to Malonic aciduria (OMIM 248360) (1). When MCD activity is affected, malonyl coenzyme A accumulates in the cytoplasm and strongly and effectively inhibits carnitine palmitoyltransferase (CPT1) in the outer mitochondrial membrane, resulting in the inability of long-chain fatty acids to enter the mitochondrial matrix for further β-oxidation and energy supply (2). Elevation of plasma malonylcarnitine (C3DC) level and urinary malonate is unique to this autosomal recessive disorder. After the report of the first case in 1984 (3), 55 cases have been reported worldwide, among which three were Chinese. In this study, we reported a case of MCD deficiency caused by novel compound heterozygous mutations at MLYCD gene loci, expanding the spectrum of genetic variants in this disease.
Methods
All cases in the literature review, except for one new case reported in this article, were searched in PubMed and CNKI database by the search term “(Malonyl Coenzyme A Decarboxylase Deficiency) OR (Malonyl-CoA Decarboxylase Deficiency)”, and 55 cases from 30 papers were collected. Information on clinical presentation as well as gene mutation was counted from the 30 retrieved papers and one case was excluded from the study for lacking clinical information.
Ethical compliance
The study was approved by the Institutional Review Ethics Board of Jiangxi Provincial Children's Hospital (JXSETYY-YXKY−20220277). Written informed consent for molecular tests and publication was obtained from the patient's parents.
Result
Clinical report
The 11-month-old patient is the second child of a healthy non-consanguineous Chinese couple, with a birth weight of 2.9 kg. Both the proband and her healthy older sister are IVF babies. No obvious abnormality could be observed in the patient. Biochemical metabolites test performed on postnatal Day 3 disclosed that C3DC+C4OH in whole blood and glutaric acid, 3-hydroxyglutaric acid, and methylmalonic acid in urine were all upregulated. This subject was suspected of having MCD deficiency according to the marked elevation of malonic acid. Whole-exome sequencing (WES) revealed a novel paternally inherited heterozygous mutation (p. Trp224Ter) in the MLYCD gene (NM_012213.2). No further treatment was given to this child. During the following months, symptoms including developmental delay, weakness of limbs, and inability to stand with support gradually arose in the baby, and she was hospitalized in the Cardiac Center owing to developmental delay, weakness of limbs, and poor cardiac function.
Physical examination showed that the patient experienced a relatively delayed physical development, weakened muscle strength, decreased muscle tone in the four extremities, and weakened knee and Achilles' tendon reflexes.
Laboratory tests
Urinary gas chromatography-mass spectrometry (GC/MS) showed significantly elevated levels of malonic acid (195.54 mmol/mol creatinine; reference 0–0.1 mmol/mol creatinine) and methylmalonic acid (66.25 mmol/mol creatinine; reference 0.2–3.6 mmol/mol creatinine). Slightly increased blood malonylcarnitine (0.54 uM; reference 0.03–0.31uM) was detected by tandem mass spectrometry (MS/MS). Biochemical test results indicated increased levels of B-type brain natriuretic peptide (530 pg/ml; reference 0–125 pg/ml) and ultrasensitive troponin (0.026 ng/ml, reference 0–0.012 ng/ml). Additional tests, including complete blood count, urinalysis, blood ammonia, and other blood biochemical indexes were all in the normal range.
Magnetic resonance imaging of the brain
Magnetic resonance imaging of the brain showed slightly broadened bilateral frontal space and widening and deepening of the sulcus.
Echocardiogram
Echocardiogram showed significant enlargement of the left ventricle accompanied by the low systolic function of the left heart with an ejection fraction of 41% and fractional shortening of 20.2% (Supplementary Figure 1 in Supporting Information).
Computerized tomography
Computerized tomography revealed an enlarged left ventricle and slight thickening of the left ventricular septal bundle and wall bundle.
No abnormality in 16-lead EEG and EMG was observed.
Denver developmental screening
Denver developmental screening implies the patient experiences delayed development in five developmental domains, including personal-social, gross-motor, fine-motor, language, and perceptive-cognitive.
Genetic testing
Peripheral blood samples were collected from the patient and the patient's parents and sister, followed by genomic DNA extraction. Whole genome sequencing (WGS) was conducted to examine the MLYCD gene, and a novel heterozygous mutation 5'-UTR-exon1-intron1 deletion was identified which her mother and sister also preserved. In addition, the copy number of MLYCD was estimated by quantitative PCR (qPCR), and the normal reference value is between 0.7 and 1.3. The test values for the patient, her mother, her sister, and her father were 0.47712, 0.32836, 0.51949, and 0.82228, respectively, indicating that the first three lost one copy of the MLYCD gene (Figure 1A). QPCR products of the three individuals were examined by agarose electrophoresis and all were found to have a truncated fragment of 250 bp (Figure 1B). Another mutation c.672delG in the patient and father, previously identified by WES, was also detected by WGS and confirmed by Sanger sequencing (Figure 2). Thus, the observed MCD deficiency was proved to be driven by a compound heterozygous pathogenic mutation comprised of the c.672delG mutation and the 5'-UTR-exon1-intron1 deletion in MLYCD. Both sites are not in the HGMD and gnomAD databases.
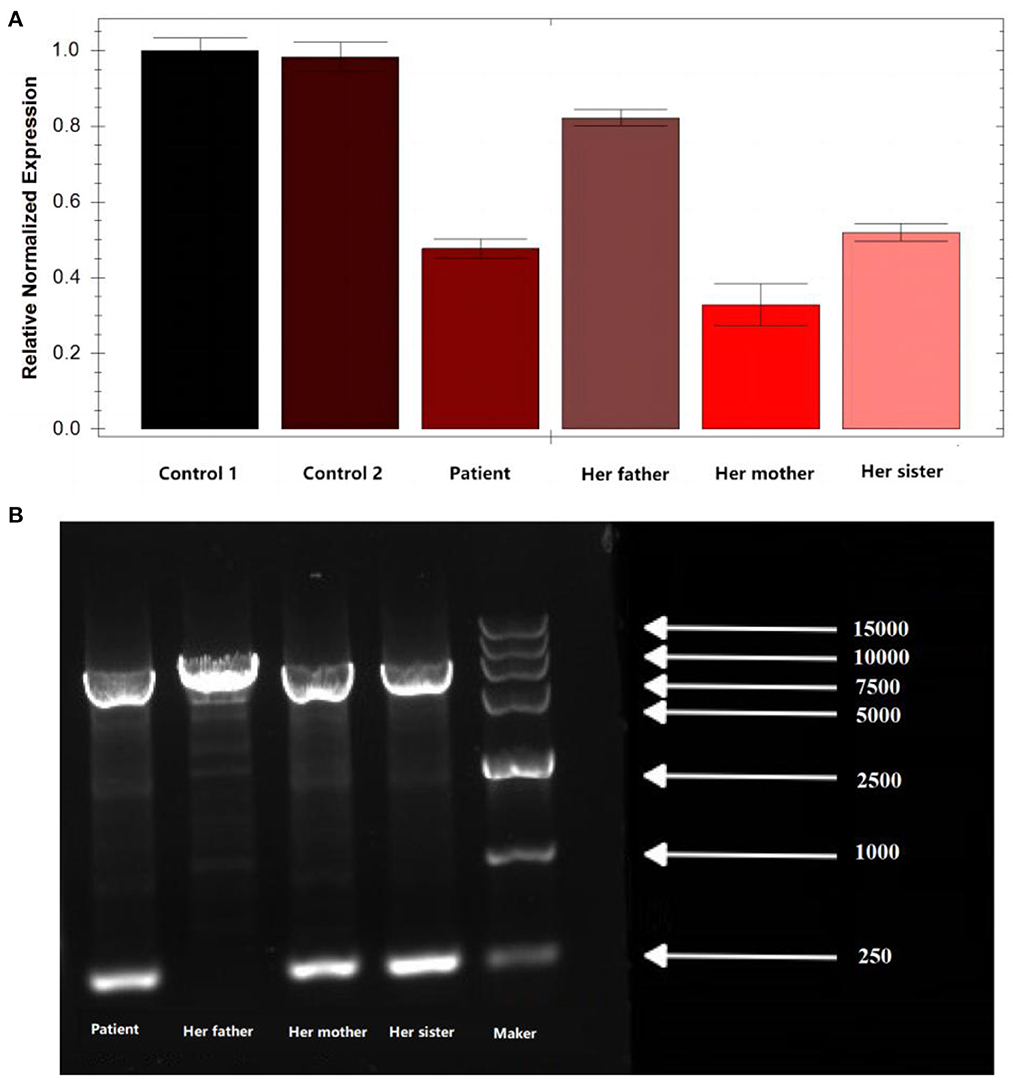
Figure 1. qPCR and agarose electrophoresis results from the patient's family. (A) qPCR results from the patient's family. (B) Results of agarose electrophoresis of the qPCR product from the patient's family. Her father has only one chromosome band with a fragment length of 7,181, which is the full length of the amplification product. In addition to this, the patient, her mother, and her sister have an additional chromosome band with a fragment length of 250 bp, which is the length of the truncated fragment.
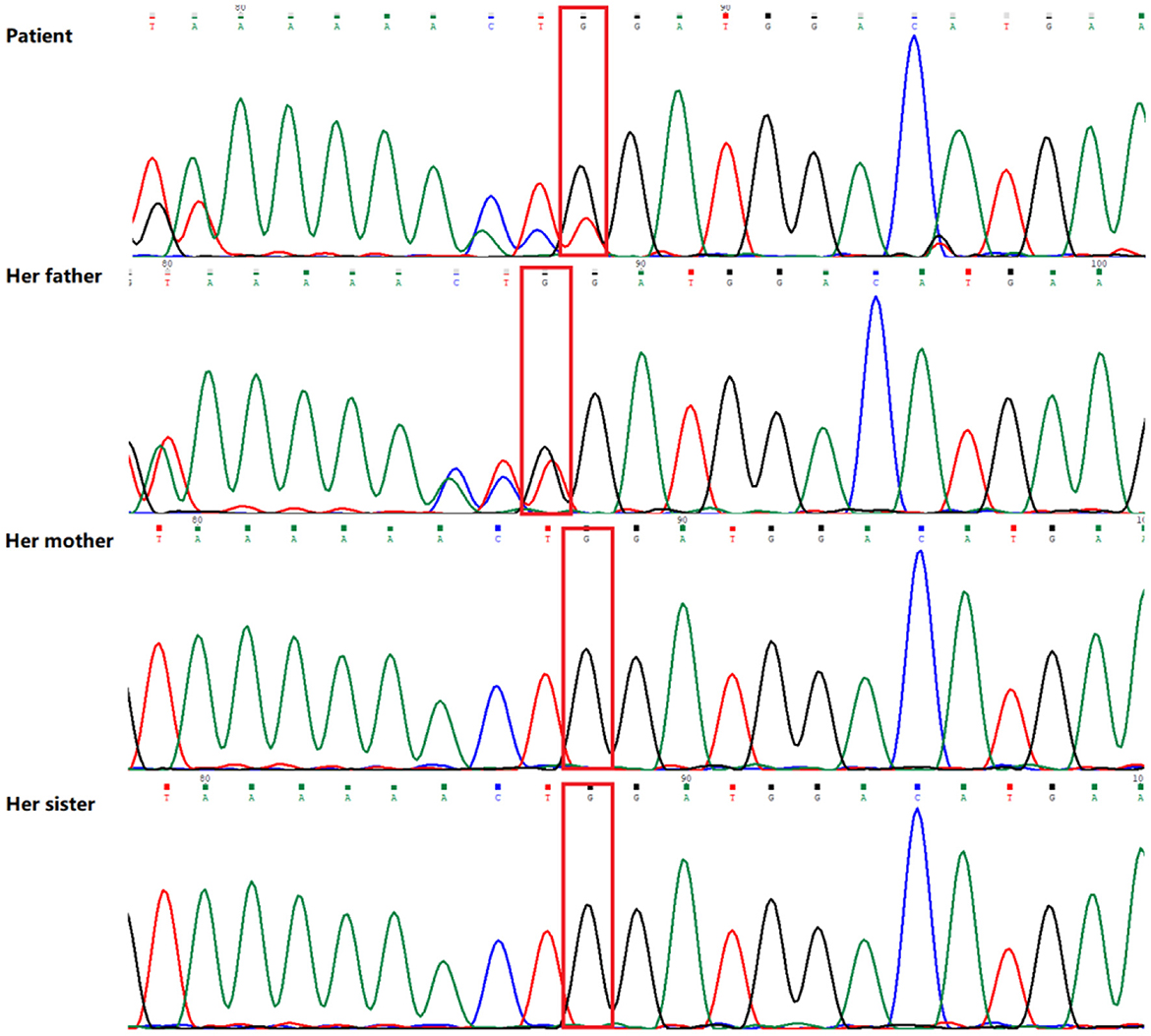
Figure 2. Sequence analysis results of the MLYCD gene of the patient and her family. The patient and her father have a novel heterozygous mutation (red box) in exon 3 (c.672del/p.Trp224*).
Treatment
After admission, the child was given a low-fat-high-carbohydrate, high-MCT formula diet. Levocarnitine was commenced at a dose of 100 mg/kg/day. Anti-cardiac failure therapy comprised of digoxin, diuretics, and enalapril maleate was initiated. Anti-symptomatic measures such as taking sodium creatine phosphate and vitamin C to nourish the myocardium were also applied. Eight days after treatment, muscle tension and tone showed improvement. Additional echocardiography showed the heart size recovered slightly and left ventricular systolic function becomes slightly better than at admission. On day 9 after admission, the child was discharged and the original treatment protocol was continued. After 3 months of follow-up, her heart function has improved to a normal level. At the end of the 6-month treatment, digoxin was discontinued. The subject's growth and development were close to a normal level. She is now able to stand and walk with support. The results of the follow-up cardiac ultrasound are presented in Supplementary Table 1 in Supporting Information.
Literature review
According to the review, 54 patients have been described in the literature so far (3–32). Development retardation, cardiomyopathy, hypotonia, and hypothermia were the most common symptoms. Among all the cases reviewed in this study, there were twice as many male patients as female patients, and nearly 42% of families were consanguineous. Of these, developmental delay was the most common clinical sign (75%, 41/55), cardiomyopathy was seen in about half of the patients, and central nervous system disorders were also common; only one patient was found being described with renal hypoplasia. Urinary malonic acid is a key diagnostic marker for MCD deficiency and 84% of patients demonstrated a high level of malonic acid. Thirteen cases were diagnosed through newborn screening programs; the way of diagnosis for other cases was not mentioned (Figure 3).
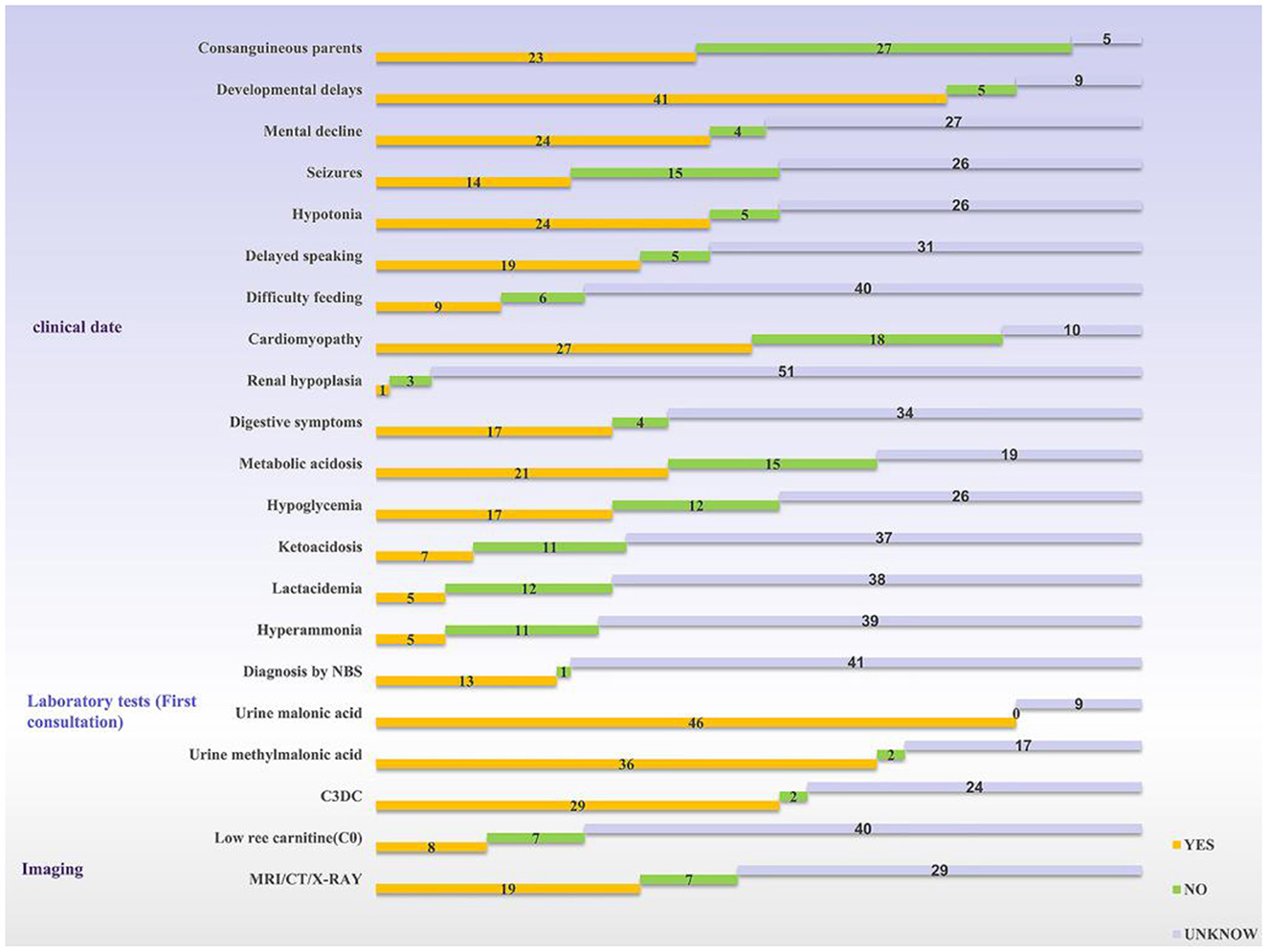
Figure 3. Statistics of clinical information reported in the literature. The yellow bar means the logo is present, the green bar means it is not, and the silver bar means it is not mentioned in the article. NBS, newborn screening; C3DC, malonylcarnitine.
To date, including the two mutant loci in this newly reported case, a total of 51 mutant loci have been reported to be associated with this disease. The mutation profile includes 10 CNV, 34 SNP, and 13 INDEL. Most variants are localized to coding regions (n = 52) and seldom to regulatory regions (n = 3) (Figure 4).
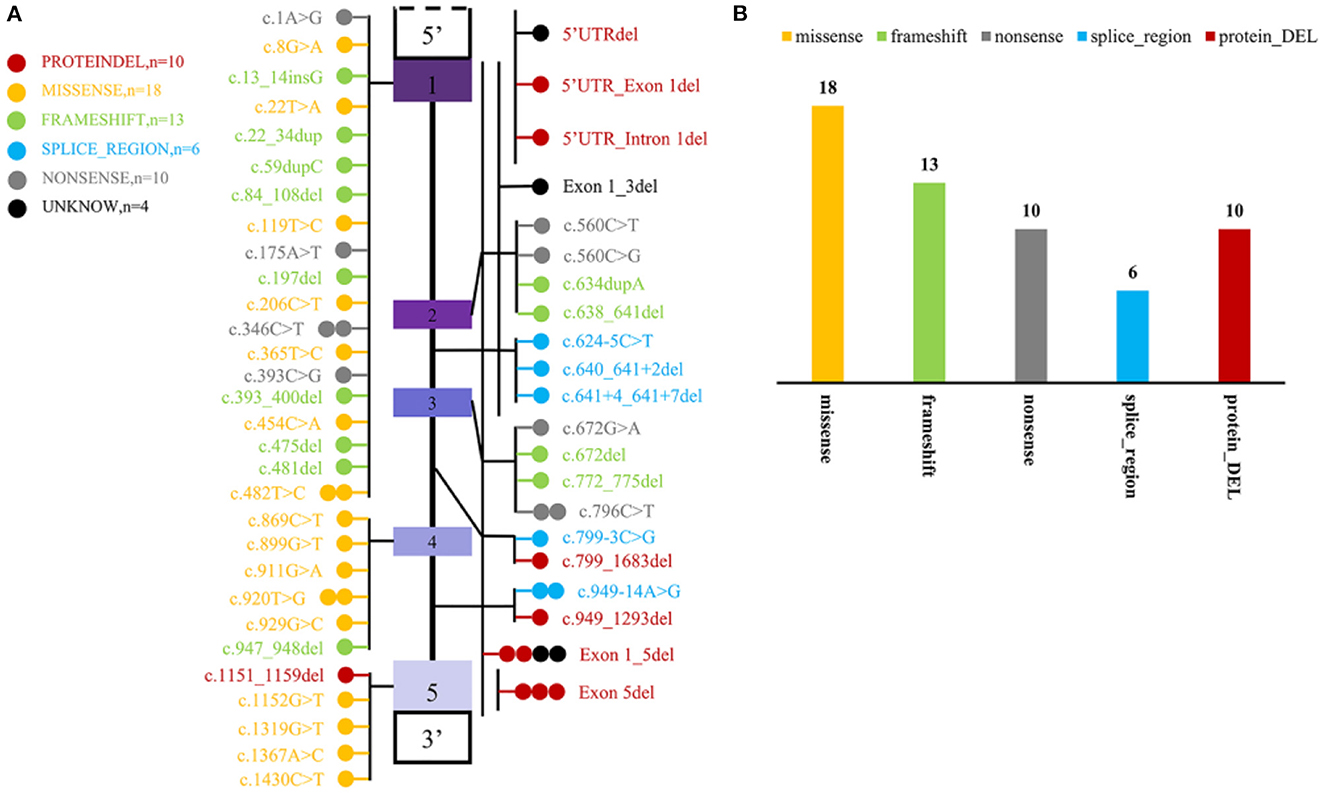
Figure 4. Statistics of mutation information of MYLCD gene reported in the literature. (A) Summary of MYLCD gene mutation loci described in the literature. The upper and lower blank boxes represent the regulatory regions of the MLYCD gene, and the colored boxes represent each of the five exons, with the region between them being introns. Each of these colored circles represents a different variant, and one circle means that there is one case of the mutant form represented by that circle. Twenty-nine variations are truncated variants, including non-sense, frameshift indels, and splicing variants, and 28 are missense variants or deletions without frameshift. The other four remaining variants are tentatively unclear and not found explicitly in the article; these four are indicated by black circles. (B) Statistical plot of protein effects produced by mutant loci. The specific colors representing the variants are described in the figure notes and are the same as in (A).
Discussion
The clinical manifestations of malonic aciduria involve multiple phenotypes, but the majority include impaired heart and skeletal muscles and central nervous system (3). Here, we reported a girl with typical clinical features of malonic aciduria, including developmental retardation, hypotonia, and cardiomyopathy. The patient harbored novel compound heterozygous mutations in MLYCD which consisted of a non-sense mutation in the coding region, c.672del (p.Trp224*), and a large fragment deletion mutation in the promoter region from 5'UTR to intron1. After receiving an adaptive diet and symptomatic treatment, the patient recovered to a near-normal level of development.
The impaired catalytic activity of MCD caused by loss-of-function mutation of the MLYCD gene contributes to disorders of malonic acid metabolism. An extensive review of published case reports suggested that MLYCD mutations are comprised of point mutations, indels, and copy number alterations, which were detected by molecular techniques such as DNA-NGS, MLPA, array, and PCR. While most of the mutations occurred in the coding regions, there were still a few exceptions. In our case, one private mutation located in the regulatory region far beyond the detection limit of whole exome sequencing led to the failure of the first round of WES sequencing. Therefore, we used WGS which allows the detection of exons, introns, and regulatory sequences simultaneously.
We summarized the genotypes and phenotypes of previous cases in Supplementary Table 2 in Supporting Information and found that the correlation between the genotypes and phenotypes of MCD deficiency is still a puzzle. The degree of severity and onset time of the disease varied even among the patients who had the same mutation (15, 24). The level of C3DC, an indicator of enzymatic activity of MCD, was less related to the severity of the disorders. There are some explanations for this discrepancy. First, besides the major gene, modifier genes may also exert their influence on monogenic inherited diseases as conveyed by genomic analysis. Second, epigenomic factors may play a role in the pathogenesis of inherited disorders. Thus, DNA-seq combined with RNA-seq could be a solution that can better discover the correlation between the genotypes and phenotypes of MCD deficiency. Newborn screening may be a powerful tool for rapid diagnosis of MCD deficiency, enabling a pre-clinical treatment before more prominent symptoms appeared. According to our summary of the literatures, only a small fraction of patients were screened neonatally, partly due to the rarity of MCD deficiency. Training clinicians to recognize the high-risk factor of inherited diseases is urgently needed.
Currently, the etiological treatment for MCD deficiency is unavailable, while diet is the mainstay of treatment. In this reported case, the patient was treated with a special diet, L-carnitine supplementation, digitalis, and ACEI drugs after diagnosis. Three months after treatment, the cardiomyopathy improved significantly, the heart size and cardiac function recovered gradually, and the motor development improved to almost normal.
In conclusion, novel heterozygous MLYCD mutations NM_012213.2: c.672delG and 5'-UTR-exon1-intron1 can reduce the catalytic activity of MCD, leading to MCD deficiency. MCD deficiency is extremely rare, with diverse clinical manifestations. Neonatal metabolic screenings detect the disease at an early stage and genetic tests confirm it. After the diagnosis of the disease, follow-up monitoring should be performed even in the absence of clinical symptoms, for the major organs (heart, brain, etc.) are more susceptible to the disease. Dietary modification and L-carnitine supplementation can significantly improve the prognosis of cardiomyopathy.
Data availability statement
To protect the privacy of the patients, all data related to the patients cannot be made available for public access, but all sequencing results from this manuscript are secured at Jiangxi Provincial Children's Hospital and are available from the corresponding author (eWVkdWFuamtAMTYzLmNvbQ==; aWNlbWFkZUBob3RtYWlsLmNvbQ==) upon reasonable request and approval by the Ethics Review Committee.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Ethics Board of Jiangxi Provincial Children's Hospital. Written informed consent for molecular test and publication of this case report were obtained from the patient's parents.
Author contributions
FaX and JH collected the clinical information and peripheral blood samples. YZ helped to collect the clinical samples. YW collected literature. FaX and YW wrote the original draft preparation. HL contributed to the genetic counseling. JD, FeX, and HL supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No. 81860065).
Acknowledgments
We are grateful to the patients and their families.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1160879/full#supplementary-material
References
1. FitzPatrick DR, Hill A, Tolmie JL, Thorburn DR, Christodoulou J. The molecular basis of malonyl-CoA decarboxylase deficiency. Am J Hum Genet. (1999) 65:318–26. doi: 10.1086/302492
2. Cheng ML, Yang CH, Wu PT, Li YC, Sun HW, Lin G, et al. Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation. Int J Mol Sci. (2023) 24:4418. doi: 10.3390/ijms24054418
3. Brown GK, Scholem RD, Bankier A, Danks DM. Malonyl coenzyme A decarboxylase deficiency. J Inherit Metab Dis. (1984) 7:21–6. doi: 10.1007/BF01805615
4. Haan EA, Scholem RD, Croll HB, Brown GK. Malonyl coenzyme A decarboxylase deficiency. Clinical and biochemical findings in a second child with a more severe enzyme defect. Eur J Pediatr. (1986) 144:567–70. doi: 10.1007/BF00496037
5. MacPhee GB, Logan RW, Mitchell JS, Howells DW, Tsotsis E, Thorburn DR. Malonyl coenzyme A decarboxylase deficiency. Arch Dis Child. (1993) 69:433–6. doi: 10.1136/adc.69.4.433
6. Matalon R, Michaels K, Kaul R, Whitman V, Rodriguez-Novo J, Goodman S, et al. Malonic aciduria and cardiomyopathy. J Inherit Metab Dis. (1993) 16:571–3. doi: 10.1007/BF00711684
7. Ozand PT, Nyhan WL, al Aqeel A, Christodoulou J. Malonic aciduria. Brain Dev. (1994) 16: 7–11. doi: 10.1016/0387-7604(94)90091-4
8. Krawinkel MB, Oldigs HD, Santer R, Lehnert W, Wendel U, Schaub J. Association of malonyl-CoA decarboxylase deficiency and heterozygote state for haemoglobin C disease. J Inherit Metab Dis. (1994) 17:636–7. doi: 10.1007/BF00711609
9. Yano S, Sweetman L, Thorburn DR, Mofidi S, Williams JC, A. new case of malonyl coenzyme A decarboxylase deficiency presenting with cardiomyopathy. Eur J Pediatr. (1997) 156:382–3. doi: 10.1007/s004310050619
10. Gregg AR, Warman AW, Thorburn DR, O'Brien WE. Combined malonic and methylmalonic aciduria with normal malonyl-coenzyme A decarboxylase activity: a case supporting multiple aetiologies. J Inherit Metab Dis. (1998) 21:382–90. doi: 10.1023/A:1005302607897
11. Buyukgebiz B, Jakobs C, Scholte HR, Huijmans JG, Kleijer WJ. Fatal neonatal malonic aciduria. J Inherit Metab Dis. (1998) 21:76–7. doi: 10.1023/A:1005371616609
12. Sacksteder KA, Morrell JC, Wanders RJ, Matalon R, Gould SJ, MCD. encodes peroxisomal and cytoplasmic forms of malonyl-CoA decarboxylase and is mutated in malonyl-CoA decarboxylase deficiency. J Biol Chem. (1999) 274:24461–8. doi: 10.1074/jbc.274.35.24461
13. Gao J, Waber L, Bennett MJ, Gibson KM, Cohen JC. Cloning and mutational analysis of human malonyl-coenzyme A decarboxylase. J Lipid Res. (1999) 40:178–82. doi: 10.1016/S0022-2275(20)33354-X
14. Surendran S, Sacksteder KA, Gould SJ, Coldwell JG, Rady PL, Tyring SK, et al. Malonyl CoA decarboxylase deficiency: C to T transition in intron 2 of the MCD gene. J Neurosci Res. (2001) 65:591–4. doi: 10.1002/jnr.1189
15. Wightman PJ, Santer R, Ribes A, Dougherty F, McGill N, Thorburn DR, et al. MLYCD mutation analysis: evidence for protein mistargeting as a cause of MLYCD deficiency. Hum Mutat. (2003) 22:288–300. doi: 10.1002/humu.10264
16. Ficicioglu C, Chrisant MR, Payan I, Chace DH. Cardiomyopathy and hypotonia in a 5-month-old infant with malonyl-coa decarboxylase deficiency: potential for preclinical diagnosis with expanded newborn screening. Pediatr Cardiol. (2005) 26:881–3. doi: 10.1007/s00246-005-1045-x
17. de Wit MC, de Coo IF, Verbeek E, Schot R, Schoonderwoerd GC, Duran M, et al. Brain abnormalities in a case of malonyl-CoA decarboxylase deficiency. Mol Genet Metab. (2006) 87:102–6. doi: 10.1016/j.ymgme.2005.09.009
18. Salomons GS, Jakobs C, Pope LL, Errami A, Potter M, Nowaczyk M, et al. Clinical, enzymatic and molecular characterization of nine new patients with malonyl-coenzyme A decarboxylase deficiency. J Inherit Metab Dis. (2007) 30:23–8. doi: 10.1007/s10545-006-0514-6
19. Malvagia S, Papi L, Morrone A, Donati MA, Ciani F, Pasquini E, et al. Fatal malonyl CoA decarboxylase deficiency due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Ann Hum Genet. (2007) 71:705–12. doi: 10.1111/j.1469-1809.2007.00373.x
20. Footitt EJ, Stafford J, Dixon M, Burch M, Jakobs C, Salomons GS, et al. Use of a long-chain triglyceride-restricted/medium-chain triglyceride-supplemented diet in a case of malonyl-CoA decarboxylase deficiency with cardiomyopathy. J Inherit Metab Dis. (2010) 33 Suppl 3:S253–256. doi: 10.1007/s10545-010-9137-z
21. Xue J, Peng J, Zhou M, Zhong L, Yin F, Liang D, et al. Novel compound heterozygous mutation of MLYCD in a Chinese patient with malonic aciduria. Mol Genet Metab. (2012) 105:79–83. doi: 10.1016/j.ymgme.2011.09.007
22. Prada CE, Jefferies JL, Grenier MA, Huth CM, Page KI, Spicer RL, et al. Malonyl coenzyme A decarboxylase deficiency: early dietary restriction and time course of cardiomyopathy. Pediatrics. (2012) 130:e456–460. doi: 10.1542/peds.2011-2927
23. Celato A, Mitola C, Tolve M, Giannini MT, De Leo S, Carducci C, et al. A new case of malonic aciduria with a presymptomatic diagnosis and an early treatment. Brain Dev. (2013) 35:675–80. doi: 10.1016/j.braindev.2012.10.014
24. Baertling F, Mayatepek E, Thimm E, Schlune A, Kovacevic A, Distelmaier F, et al. Malonic aciduria: long-term follow-up of new patients detected by newborn screening. Eur J Pediatr. (2014) 173:1719–22. doi: 10.1007/s00431-014-2421-4
25. Polinati PP, Valanne L, Tyni T. Malonyl-CoA decarboxylase deficiency: long-term follow-up of a patient new clinical features and novel mutations. Brain Dev. (2015) 37:107–13. doi: 10.1016/j.braindev.2014.02.001
26. Liu H, Tan D, Han L, Ye J, Qiu W, Gu X, et al. A new case of malonyl-CoA decarboxylase deficiency with mild clinical features. Am J Med Genet A. (2016) 170A:1347–51. doi: 10.1002/ajmg.a.37590
27. Ersoy M, Akyol MB, Ceylaner S, Cakir Bicer N, A. novel frameshift mutation of malonyl-CoA decarboxylase deficiency: clinical signs and therapy response of a late-diagnosed case. Clin Case Rep. (2017) 5:1284–8. doi: 10.1002/ccr3.1013
28. Chapel-Crespo C, Gavrilov D, Sowa M, Myers J, Day-Salvatore DL, Lynn H, et al. Clinical, biochemical and molecular characteristics of malonyl-CoA decarboxylase deficiency and long-term follow-up of nine patients. Mol Genet Metab. (2019) 128:113–21. doi: 10.1016/j.ymgme.2019.07.015
29. Lee SH, Ko JM, Song MK, Song J, Park KS, A. Korean child diagnosed with malonic aciduria harboring a novel start codon mutation following presentation with dilated cardiomyopathy. Mol Genet Genomic Med. (2020) 8:e1379. doi: 10.1002/mgg3.1379
30. Kasapkara CS, Civelek Urey B, Ceylan AC, Unal Uzun O, Cetin II. Malonyl coenzyme A decarboxylase deficiency with a novel mutation. Cardiol Young. (2021) 31:1535–7. doi: 10.1017/S104795112100113X
31. Snanoudj S, Torre S, Sudrie-Arnaud B, Abily-Donval L, Goldenberg A, Salomons GS, et al. Heterogenous clinical landscape in a consanguineous malonic aciduria family. Int J Mol Sci. (2021) 22:12633. doi: 10.3390/ijms222312633
Keywords: MLYCD, malonyl coenzyme A decarboxylase deficiency, cardiomyopathy, novel mutation, metabolic disease
Citation: Xu F, Wu Y, Huang J, Zhou Y, Xu F, Duan J and Li H (2023) Case report: A novel 5'-UTR-exon1-intron1 deletion in MLYCD in an IVF child with malonyl coenzyme A decarboxylase deficiency and literature review. Front. Med. 10:1160879. doi: 10.3389/fmed.2023.1160879
Received: 10 March 2023; Accepted: 07 April 2023;
Published: 03 May 2023.
Edited by:
Fu Lijun, Shanghai Children's Medical Center, ChinaReviewed by:
Shiwei Yang, Children's Hospital of Nanjing Medical University, ChinaWenhao Zhou, Fudan University, China
Copyright © 2023 Xu, Wu, Huang, Zhou, Xu, Duan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junkai Duan, eWVkdWFuamtAMTYzLmNvbQ==; Hong Li, aWNlbWFkZUBob3RtYWlsLmNvbQ==
†These authors share first authorship