 Gaetano La Rocca1
Gaetano La Rocca1 Francesco Ferro1
Francesco Ferro1 Chiara Baldini1
Chiara Baldini1 Alessandro Libra2Domenico Sambataro3
Alessandro Libra2Domenico Sambataro3 Michele Colaci4
Michele Colaci4 Lorenzo Malatino4Stefano Palmucci5
Lorenzo Malatino4Stefano Palmucci5 Carlo Vancheri2
Carlo Vancheri2 Gianluca Sambataro2,3*
Gianluca Sambataro2,3*- 1Rheumatology Unit, Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
- 2Regional Referral Centre for Rare Lung Disease, Azienda Ospedaliero Universitaria Policlinico “G. Rodolico-San Marco”, University of Catania, Catania, Italy
- 3Artroreuma S.R.L., Rheumatology Outpatient Clinic, Catania, Italy
- 4Internal Medicine Unit, Rheumatology Clinic, Azienda Ospedaliera per l’Emergenza Cannizzaro, University of Catania, Catania, Italy
- 5Department of Medical Surgical Sciences and Advanced Technologies “GF Ingrassia”, University Hospital Policlinico “G. Rodolico-San Marco”, Catania, Italy
In recent decades, several pieces of evidence have drawn greater attention to the topic of innate immunity, in particular, interferon (IFN) and Interleukin 6 in the pathogenesis of idiopathic inflammatory myopathies (IIM). Both of these molecules transduce their signal through a receptor coupled with Janus kinases (JAK)/signal transducer and activator of transcription proteins (STAT). In this review, we discuss the role of the JAK/STAT pathway in IIM, evaluate a possible therapeutic role for JAK inhibitors in this group of diseases, focusing on those with the strongest IFN signature (dermatomyositis and antisynthetase syndrome).
Introduction
Idiopathic inflammatory myopathies (IIM) represent a group of systemic autoimmune disorders sharing striated muscles as their preferred target, but potentially involving various organ systems including the skin, lungs, joints and gastrointestinal tract (1). Specifically, interstitial lung disease (ILD) is a common manifestation, representing one of the main causes of mortality (2).
In recent decades it has become progressively more evident that IIM cannot be bundled into a single entity. In fact, IIM patients exhibit heterogeneous clinical phenotypes, histologic findings and peripheral autoantibody repertoires, pointing to the existence of different underlying pathogenetic pathways (3).
In 1975, Bohan and Peter distinguished between polymyositis and dermatomyositis relying on the presence or absence of a typical skin rash, while the most recent 2017 EULAR/ACR classification criteria allow for the stratification of IIM patients into 6 major subgroups based on clinical and pathological features: polymyositis (PM), inclusion body myositis (IBM), amyopathic dermatomyositis (ADM), dermatomyositis (DM), Juvenile dermatomyositis (JDM) and Juvenile myositis other than JDM (4–6).
Moreover, the discovery of new myositis specific antibodies (MSA), strongly associated with the clinical features of IIM patients (7), will probably lead to new classification criteria discriminating distinct entities such as antisynthetase syndrome (ASS) and immune-mediated necrotizing myopathies (IMNM), in the spirit of a modern phenotype-based approach. Indeed, various authors reported an overdiagnosis of PM according to the classical Bohan and Peter criteria, and a recent cluster analysis on a large French cohort showed that PM patients were more correctly classified in other subgroups, mainly IMNM and ASS (8, 9). Accordingly, nowadays the existence of a pure PM entity is debated.
Therapeutic options for IIM patients are limited, and mortality rates are still especially high for some IIM subsets, such as anti-MDA5+ IIM (10). Steroids continue to be a therapeutic cornerstone, with concerns about their chronic use causing potential side effects, especially considering that steroid sparing drugs are currently less efficacious compared with other connective tissue diseases (11).
In view of this, in this era of precision medicine, dissecting the exact pathogenetic mechanisms underlying IIM is a fundamental pre-requisite in order to forge a new path toward effective, patient- and phenotype-tailored target therapies.
Given recent evidence from the successful employment of Janus kinase inhibitors (JAKi) in the treatment of diverse interferon (IFN)-mediated autoinflammatory processes, the present review will discuss the involvement of Janus kinases (JAK) mediated pathways in IIM and their potential as a therapeutic target.
We specifically focus on IIM subsets with a strong IFN signature, namely DM, ASS and anti-MDA5+ ADM.
DM, ASS, and ADM
Dermatomyositis and ASS are distinguished clinically from PM mainly by the association of myositis with a typical inflammatory skin involvement. DM patients usually present serum autoantibodies such as anti-Mi2, anti-TIF1gamma, anti-NXP2, and anti-SAE which are correlated with specific clinical features, risk for associated cancer and prognosis (12). In contrast, the hallmark of ASS is the association of serum anti-aminoacyl tRNA synthetase autoantibodies with the triad of myositis, arthritis and ILD (13). Indeed, at disease onset, a significant proportion of ASS patients show only one item of the classic triad: the risk of developing other signs seems to be associated with the presence of a specific antibody (14). Finally, ADM is defined by the presence of typical vasculitic skin lesions, with little (hypomyopathic DM) or no clinical or histologic muscle involvement. Anti-MDA5 autoantibodies are detected in at least 50% of patients in ADM cohorts, a prevalence which is probably influenced by the laboratory techniques employed, and are strongly associated with a high risk of rapidly progressing ILD (RP-ILD) (15). A clusterisation of ADM patients into 3 main phenotypic groups with different RP-ILD risk and prognosis has been proposed, with some differences between European and Asiatic cohorts (10, 16).
The JAK/STAT pathway
The term JAK/signal transducer and activator of transcription proteins (STAT) refers to molecules mediating a widespread intracellular signaling pathway, physiologically involved in haematopoiesis, adipogenesis, apoptosis and immune response, influencing both innate and adaptive immunity (17). However, the aberrant activation of this pathway is associated with the development of autoimmune diseases and carcinogenesis (18). The discovery of this pathway has led to great advances in knowledge of the pathogenesis of several diseases, and more importantly, has highlighted a promising therapeutic target.
Janus kinases-coupled cytokine receptors undergo dimerisation after binding by their extracellular ligands. Dimerisation induces the phosphorylation of JAKs, which in turn phosphorylate STAT. This latter activated form of STAT translocates into the nucleus regulating gene transcription. This represents the canonical process of JAK/STAT activation. The pathway can also be activated in a non-canonical way, by oxidative stress-induced tyrosine kinases, 7-elix membrane receptors or cellular hypertonicity (19).
There are currently 4 known forms of JAK: JAK1, JAK2, JAK3, and TYK2. JAK1, JAK2, and TYK2 are ubiquitous. JAK1 phosphorylation is induced by both IFN-I (mainly α/β), IFN-II (γ) and cytokines belonging to the interleukin (IL) 2, IL6 and IL10 families. JAK2 is activated by similar ligands, but also by hormones, while TYK2 mainly mediates the signaling of IFNs, IL6 and IL10. Finally, JAK3 is mostly involved in the negative selection and production of lymphocytes, and is therefore only present in bone marrow and lymphoid tissue and is activated by cytokines belonging to the IL2 family (20).
The STAT family includes 7 proteins: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STAT1 is activated by all of the IFN, IL2, and IL6 families, tumour necrosis factor (TNF) and other chemokines. Its role is to favor apoptosis, inhibit cell growth, and regulate cell differentiation. STAT1 also plays a role in the regulation of the immune system, transducing the signal of major histocompatibility complexes (MHC) after antigen presentation and allowing the development of B cells. STAT2, STAT3, and STAT4 are activated by IFN-I, exerting different actions.
Signal transducer and activator of transcription proteins (STAT2) regulates the immune response of macrophages and T cells, with an antiviral effect. STAT3 also mediates IL6 and IL10 signaling, resulting in activation of the Th17 response and inhibition of apoptosis. STAT4 is also phosphorylated by receptors recognizing IL12 family cytokines, favoring a Th1 response. STAT5a and STAT5b are activated by IL2 family cytokines and prolactin, with a role in apoptosis, lactation and production of immune cells. Finally, STAT6 is mainly activated by IL4 and IL13. It is crucial for Th2 differentiation, and the proliferation and maturation of B cells, as well as for the expression of MHC II and IgE (20).
Numerous cytokines and hormones transduce signals through the JAK-STAT pathway, but with different strengths, and with the ability to activate in multiple ways. For example, IFN-I is classically associated with a strong activation of STAT1 and a weaker activation of STAT3 and 4. Other cytokines with opposite functions are able to act in the same way (for example IL6 and IL10 on STAT3). The final effect of an immune stimulus depends on the composition and relative quantities of the cytokine milieu released, the duration and intensity of JAK/STAT signaling, the types of STAT proteins coupled with JAKs and the cell types involved in the process (21).
The idea of interfering with JAK/STAT mediated immune processes led to the synthesis of small molecules acting as JAKi, initially approved for the treatment of rheumatoid arthritis (RA). First generation JAKi are non-selective. The first two molecules approved were Tofacitinib, a pan-inhibitor of JAKs, with strong activity against JAK1 and JAK3, and minor activity against JAK2 and TYK2, and Baricitinib, exerting a significant inhibitory action on JAK1 and JAK2, moderate activity against TYK2 and minimal activity against JAK3 (22). New generation JAKis display greater selectivity for JAK1, thus potentially limiting the hematological side effects related to interference with JAK3 (23). Up to the present moment, the only JAK1 inhibitors approved for RA treatment, Upadacitinib and Filgotinib, have shown great efficacy coupled with an acceptable safety profile (24, 25).
In light of the ability of JAKis to inhibit production of both Th1 and Th2 cytokines, particularly IL6, thus also impairing the polarization of Th17 lymphocytes, they are currently being tested for the treatment of a broad range of inflammatory disorders (26).
Current pathogenetic models in DM, ADM, and ASS
JAK-STAT pathway in IIM
The JAK1-STAT1/STAT3 axis is pivotal in the physiology of skeletal muscles.
It promotes myogenesis by exerting a potent antidifferentiation action on myoblasts’ premature differentiation, and premature formation of myotubes (27). In contrast, activation of the JAK2-STAT2/STAT3 axis leads to the opposite effects.
Therefore, STAT3 is located downstream on a physiological axis potentially able to cause both muscle growth and wasting. IL6 plays a pivotal role in both pathways, as its receptor is associated with the two axes (28). IFN-γ can interact with JAK1 and JAK2, but is also able to activate STAT3 inducing muscle wasting via NFκb, independently of IL6 (29). This data highlights the “double-edged sword” role of STAT3 in skeletal muscle physiology.
Signal transducer and activator of transcription proteins (STAT3) is also crucial in skin homeostasis. It is phosphorylated by JAK1, JAK2, and TYK2, regulating the migration of keratinocytes (30). Physiologically, STAT3 is necessary in wound healing and protection from ultraviolet rays, but if constantly activated is associated with the development of cutaneous rashes in lupus erythematosus, psoriasis, and atopic dermatitis, as well as with carcinogenesis (30, 31).
Finally, in normal lung tissue, STAT1, STAT5a, and STAT5b promote inflammation, while STAT2 and STAT6, respectively, have a pro and an anti-apoptotic effect. STAT4 is involved in the response to IFN-γ for the regulation of immune response, and STAT3 in the transduction of the IL6 pathway, favoring cell proliferation (32).
The principal role in ILD patients seems to be played by the JAK2/STAT3 axis. STAT 3 is overexpressed in lung macrophages, endothelial cells, myofibroblasts and neutrophils in idiopathic pulmonary fibrosis (IPF) and systemic sclerosis patients, allowing the deposition of extracellular matrix leading to fibrosis (33). Finally, transforming growth factor β (TGFβ) is recognized by JAK2-coupled receptors that are highly expressed in fibroblasts, hyperplastic alveolar epithelial type II cells, and in the small pulmonary arteries of IPF patients (34).
The JAK2/STAT3 pathway therefore mediates TGFβ and IL6 induced pro-fibrotic and pro-inflammatory effects leading to ILD in connective tissue diseases (33). In a recent in vitro assay, Baricitinib was able to inhibit JAK2/STAT3 and therefore prevent IL6 induced epithelial-mesenchymal transition (35). Moreover, Ruxolitinib (another JAK1/JAK2 inhibitor) was successfully employed in the treatment of ILD secondary to a STAT3 gain of function mutation (36).
The prominent role of IFNs and IL-6
On a pathologic level, DM is characterized by mononuclear cells infiltrates with predominant T CD4+ lymphocytes and prevalent perivascular distribution, leading to perifascicular myofiber atrophy in muscle samples and vasculitic changes in skin biopsies. This key contribution of adaptive immunity to the pathogenesis of the disease is further supported by known genetic associations with human leukocyte antigen (HLA) alleles, the presence of specific serum autoantibodies, and the proven efficacy of immunosuppressive strategies targeting cellular (calcineurin inhibitors) and humoral (anti-CD20 monoclonal antibodies) autoimmunity (37, 38).
However, the main focus of translational research has progressively shifted toward the role of innate immunity players in initiating the autoimmune process (39).
Perimysial atrophic fibers in DM biopsies typically show upregulation of MHC-I molecules, whose expression is known to be induced by IFN-I (40). Actually, IFN-α/β induced genes were found to be highly expressed in muscle biopsies of DM patients compared to controls, PM, and IBM, together with the presence of MxA, an IFN-I induced protein (41). The IFN-I signature was also found in peripheral blood and skin biopsies, longitudinally correlating with disease activity in DM and ADM patients. Since then, the presence of an IFN-I signature was also identified in peripheral blood and skin samples from DM and ADM patients (42, 43).
In contrast to the predominant type 1 IFN axis activation observed in DM, gene expression analysis of ASS muscle biopsies identified a type 2 IFN signature (44, 45). This is in line with the expression of IFN-II induced MHC-II molecules on the membranes of necrotic perifascicular myofibers in ASS, a rare occurrence in DM muscle samples (46).
Both IFN-I and IFN-II are widely produced by innate immunity phagocytes: mainly plasmacytoid dendritic cells (pDC) for IFN-I, monocytes-macrophages and NK lymphocytes for IFN-II. IFN production is stimulated by the non-specific binding of molecules shared between phylogenetically correlated pathogens to pattern recognition receptors (PRRs) expressed by innate immunity cells. However, activation of PRRs, including membrane, cytoplasmic and endosomal toll-like receptors (TLRs) can also be triggered by endogenous DNA or RNA particles released by apoptotic cells (or neutrophils undergoing NETosis) either alone or complexed with autoantibodies (47). In this regard, it is notable that most autoantigens targeted by DM associated autoantibodies are DNA- or RNA-complexed proteins (48).
Immature muscle precursors are an alternative source of IFN-I. They also have an enhanced expression of autoantigens, possibly providing a further local stimulus for the production of IFN through PRRs (49, 50). Finally, the presence of pDC was reported in DM and JDM patients’ muscles and may be related to in loco production of IFN-I in muscle tissue (51).
The role of IFN-I in IIM was confirmed in the trial involving Sifalimumab, an anti-IFN-α antibody. Treated patients showed suppression of IFN signature associated with improved strength, however, a subsequent trial emphasized excessive side effects (NCT00979654) (52).
Along with IFNs, recent studies have highlighted the involvement of IL-6 in the physiopathology of IIM related organ damage. IL-6 is a cytokine with pleiotropic effects, physiologically involved in immune response and the hematopoietic, endocrine and nervous systems (53). IL-6 knockout mice were less prone to develop muscular inflammation in a murine model of myositis (54). More importantly, in vivo studies demonstrated that adult and JDM patients exhibit higher IL-6 levels compared to healthy controls, and IL-6 levels correlate with disease activity and an IFN-I signature (55–57).
Interestingly, DM, ASS, and anti-MDA5+ DM patients with ILD display higher IL-6 serum levels than patients without ILD (56, 58).
Indeed, IL-6 has been shown to exert pro-inflammatory and pro-fibrotic effects, by stimulating fibroblasts, both in preclinical and in vivo studies of IPF and non-IPF ILD patients (59).
Various case reports have highlighted the efficacy of Tocilizumab (an anti-IL6R) in the treatment of refractory DM, ASS and anti-MDA5+ ADM, both with chronic fibrosing- and RP-ILD (60–63). However, a recent phase IIb trial comparing Tocilizumab to a placebo in the treatment of DM and PM patients failed to meet its primary endpoint (64).
In light of the aforementioned evidence pointing to the relevance of IFNs and IL-6 in the pathogenetic mechanisms underlying skin, muscle and lung damage in IIM, blocking their signaling with JAKi represents a promising therapeutic strategy that is currently being explored intensively (Figure 1).
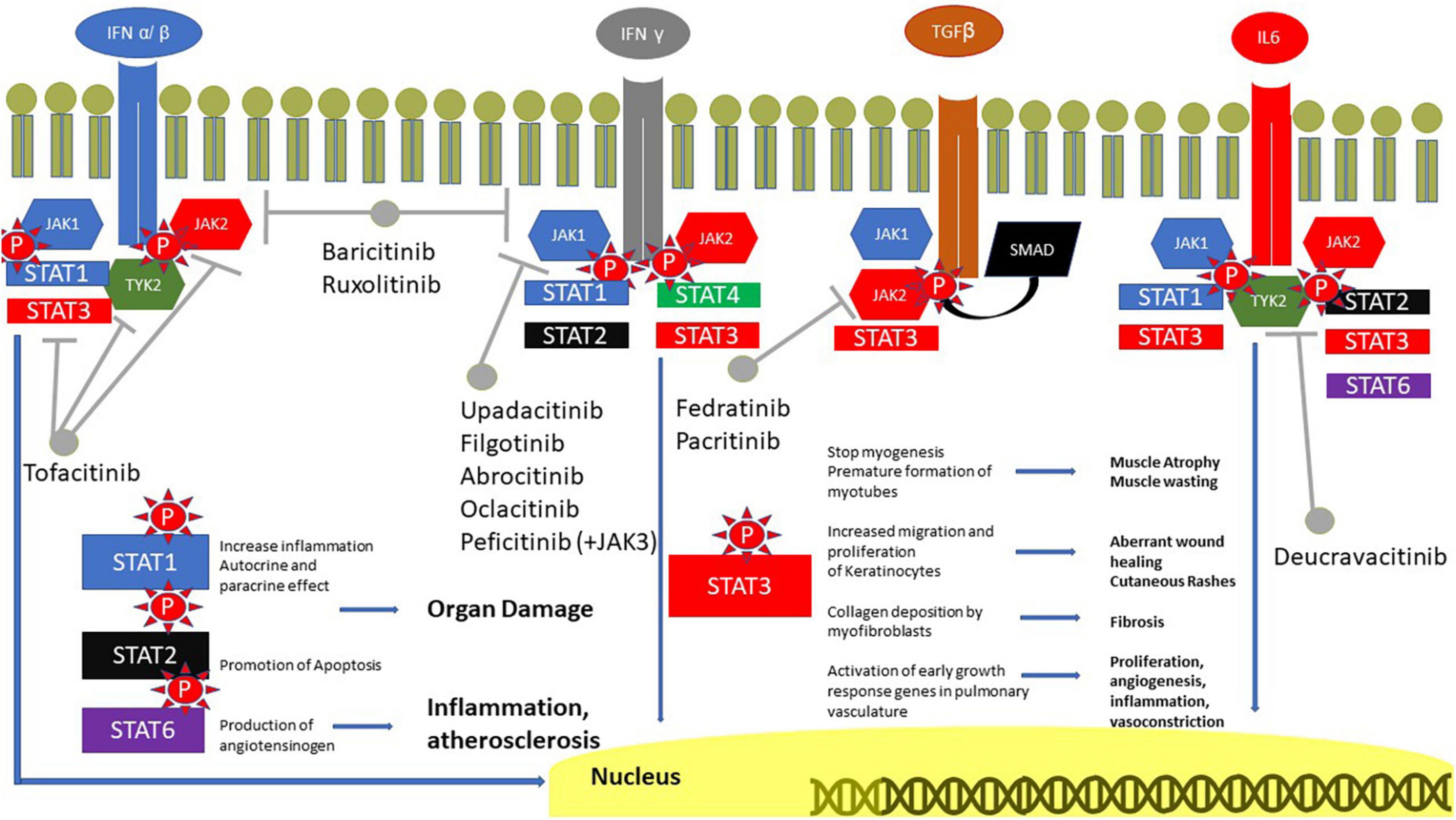
Figure 1. Role of JAK in IIM. IFN receptors are coupled with JAK1 and 2, IFN α/β and IL6 also with TYK2. TGFβ is coupled with SMAD, however, it can also phosphorylate JAK and JAK2. The signal is transmitted to the nucleus through STAT, with a great role of STAT3. STAT1 is involved in inflammation, STAT2 in the promotion of apoptosis and STAT3 in the production of angiotensinogen. JAK, Janus kinases; IFN, interferon; IL, interleukin; SMAD, suppressor of mothers against decapentaplegic; STAT, signal transducer and activator of transcription proteins; TGF, transforming growth factor.
Targeting intracellular pathways in IIM with JAKi: Where are we now?
In the last few years, an impressive number of reports have highlighted Tofacitinib efficacy in diverse domains of DM patients with heterogeneous phenotypes (65).
In a small case series, 3 patients with refractory cutaneous DM were treated with Tofacitinib (either 5 mg bid or 10 mg bid, in monotherapy or combined with Hydroxychloroquine), improving their lesions according to the cutaneous dermatomyositis disease area and severity index (CDASI) activity score (66). In a subsequent open-label trial (STIR), 10 patients with mainly cutaneous refractory DM were treated with Tofacitinib. The primary endpoint of the international myositis assessment and clinical studies group (IMACS) definition of improvement was met for all patients and the CDASI showed a mean decrease of 66% from baseline (67). Last year, the results of the long-term extension of this study were published: notably 7/10 patients retained Tofacitinib therapy for a mean 1.2 years, demonstrating a sustained response without the reintroduction of steroids (68).
Regarding the muscular domain, according to a systematic review published last year, 15/16 (93.8%) DM patients with muscular involvement treated with JAKi experienced clinical and/or imaging improvement (69).
Moreover, numerous studies have reported on the beneficial effects of Tofacitinib in the management of anti-MDA5+ ADM related ILD. Particularly, 18 anti-MDA5+ patients with early stage ILD were treated with combined steroids and Tofacitinib in an open label trial, showing higher 6-month survival rates compared to historical controls treated conventionally (70, 71). Another retrospective study compared in a small cohort of anti-MDA5+ ADM the efficacy of Tofacitinib and Tacrolimus, proving a better outcome in patients exposed to JAKi (72). Survival rate was low in both groups, notably, however, a great proportion of the enrolled patients were classifiable as RP-ILD, a condition known to portend a very poor prognosis, especially in anti-MDA5+ ADM patients (73).
Baricitinib also showed rapid beneficial effects in several case series including refractory adult and JDM (74–76). In 2022, two open label trials met their primary endpoint consisting of a clinically significant improvement of the CDASI activity score in DM patients treated with Baricitinib (77, 78).
Tofacitinib and Baricitinib also seem to be effective in calcinosis, as described in numerous real-life experiences (76, 79, 80).
Additional trials are ongoing, with the aim of investigating the efficacy of Baricitinib in both adult and JDM patients (NCT04208464, NCT05524311, NCT04972760, NCT05361109).
Conversely, only two case reports are available on the use of JAKis in ASS: Tofacitinib improved two ASS patients with an RP-ILD resembling ADM, though positive for anti-Jo1 and anti-EJ, respectively (81, 82).
Table 1 summarizes the current experiences and available evidence on JAKi in IIM.
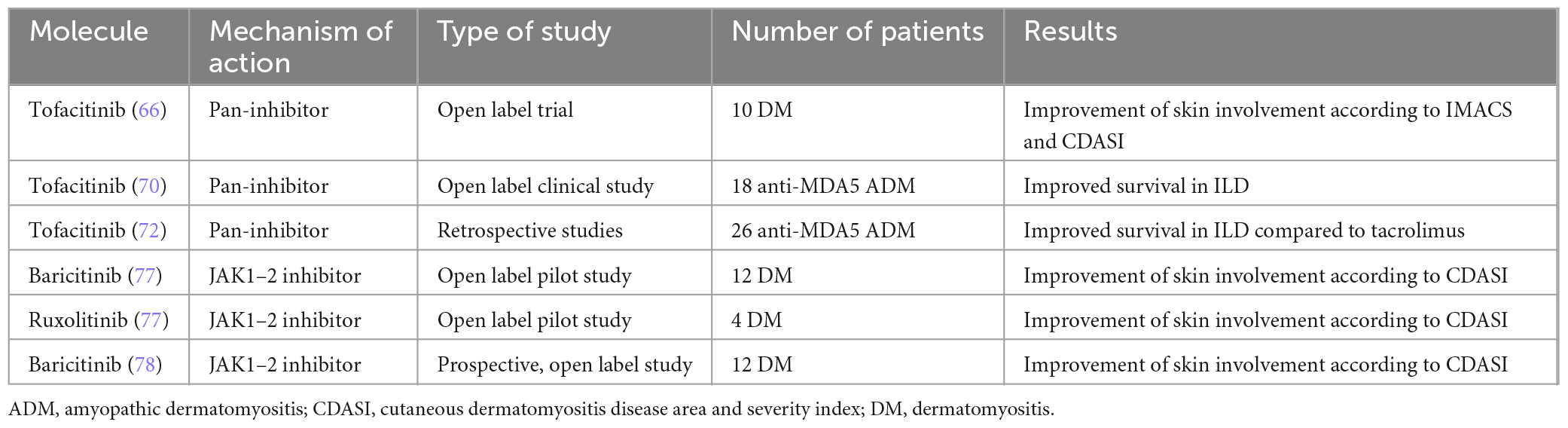
Table 1. Published clinical trials on the use of JAK inhibitors in idiopathic inflammatory myopathies.
Besides the aforementioned JAKi, alternative approaches to target intracellular pathways in IIM are being explored. Brepocitinib, a dual TYK2 and JAK1 inhibitor, showed efficacy in preliminary studies and is currently being investigated in a phase III RCT on DM patients (NCT05437263). Similarly, TLRs are located upstream in the cascade leading to IFN hyperproduction and 2 different anti-TLR agents will be the object of phase II RCTs (NCT05650567) (83). Table 2 resumes recent and ongoing RCT investigating new promising molecules in IIM patients.
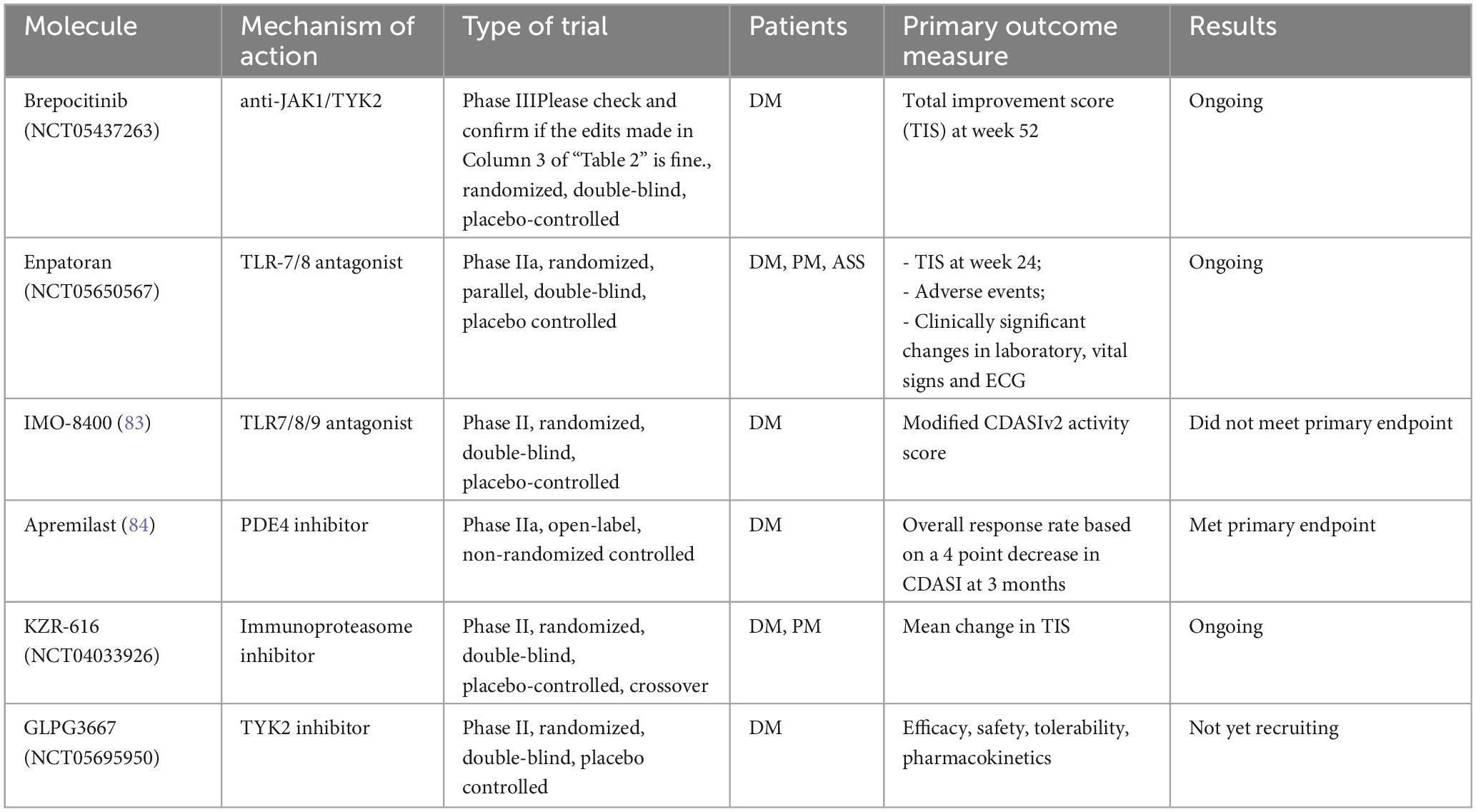
Table 2. Recent and ongoing trials investigating new molecules interfering with intracellular pathways in IIM.
RP-ILD in IIM: Lesson learned from COVID-19 pandemic and future perspectives
An increasing amount of evidence suggests that RP-ILD in ADM patients is probably driven by IFN-γ axis upregulation, in line with the presence in their serum of biohumoural markers typically reflecting macrophagic hyperactivation, namely hyperferritinemia, high C Reactive Protein (CRP), peripheral cytopenia (especially lymphopenia), and high Lactic Dehydrogenase (LDH) and IL1 levels. Moreover, higher levels of these biomarkers have been associated with adverse lung and global outcomes in ADM subjects (85–88).
In view of this, important clues regarding the pathophysiology of ADM related RP-ILD come from experience acquired in the management of SARS-CoV-2 related severe interstitial pneumonia. Indeed, the severe hyperinflammatory phenotype of COVID-19 pneumonia shares striking biohumoural, clinical and histopathologic similarities with ADM related RP-ILD.
Wang et al. (89) also demonstrated that the presence of serum anti-MDA5 autoantibodies in COVID-19 patients represents a marker of severe disease and anti-MDA5 titres in severe COVID-19 patients correlate with mortality.
Furthermore, MDA5 is physiologically involved in the IFN-mediated response to viral infections and anti-MDA5 autoantibodies are thought to be directly pathogenic in ADM (90). Accordingly, plasma exchange and intravenous immunoglobulins showed efficacy in the treatment of both COVID-19 and anti-MDA5 ILD (91–93).
Of note, IFN-γ axis seems to be the main driver of inflammation and fibrosis also in SARS-CoV-2 pneumonia (94, 95).
As Baricitinib is the most effective available JAKi acting on JAK2 (31), it has been widely employed in clinical trials and real-life experience, proving to be efficacious in reducing the need for supplemental oxygen, as well as reducing mortality and the duration of hospitalization in intensive care units (96–101).
Based on these physiopathological considerations, we believe that in the near future, treatment with Baricitinib of both DM-related fibrosing ILD and ADM related RP-ILD may provide important advantages compared with Tofacitinib. Moreover, anti-MDA5+ RP-ILD patients still present dramatically high mortality rates due to progression of respiratory failure, despite exposure to aggressive combined conventional immunosuppressants. Therefore, current evidences probably justify early administration of JAKi as a first line treatment in these subset of patients, possibly combined with therapies targeting the humoral immune response (namely IVIg, PEX, and Rituximab) (102).
Serum biomarkers reflecting the hyperactivation of IFN-γ may not only serve as prognostic factors but also represent important clues when choosing the most appropriate JAKi agent.
Similarly, despite lack of studies exploring the potentialities of Upadacitinib and Filgotinib in IIM, selective JAK1 inhibition may show considerable efficacy in the management of IFN-I mediated manifestations, such as cutaneous and muscular involvement in DM patients. However, in light of the complexity of intracellular signaling pathways these considerations are purely speculative and still await confirmation by solid evidences coming from clinical experiences and trials.
Author contributions
GS and GL conceived the manuscript. GS, GL, DS, AL, and FF wrote the manuscript. CV, CB, SP, MC, and LM critically revised the manuscript. All authors provided approval for publication of this content and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved and participated in the selection of articles and their interpretation.
Conflict of interest
CV is part of the F. Hoffmann-La Roche Ltd., Scientific Board. He has received consulting fees and/or speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, F. Hoffmann-La Roche Ltd., and Menarini. GS and SP received honoraria from Boehringer Ingelheim outside of the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. (2021) 7:86. doi: 10.1038/s41572-021-00321-x
2. Ashton C, Paramalingam S, Stevenson B, Brusch A, Needham M. Idiopathic inflammatory myopathies: a review. Intern Med J. (2021) 51:845–52. doi: 10.1111/imj.15358
3. Lundberg IE, de Visser M, Werth VP. Classification of myositis. Nat Rev Rheumatol. (2018) 14:269–78. doi: 10.1038/nrrheum.2018.41
4. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. (1975) 292:344–7. doi: 10.1056/NEJM197502132920706
5. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. (1975) 292:403–7. doi: 10.1056/NEJM197502202920807
6. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. (2017) 69:2271–82.
7. McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol. (2018) 14:290–302. doi: 10.1038/nrrheum.2018.56
8. van der Meulen MF, Bronner IM, Hoogendijk JE, Burger H, van Venrooij WJ, Voskuyl AE, et al. Polymyositis: an overdiagnosed entity. Neurology. (2003) 61:316–21. doi: 10.1212/WNL.61.3.316
9. Mariampillai K, Granger B, Amelin D, Guiguet M, Hachulla E, Maurier F, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol. (2018) 75:1528–37. doi: 10.1001/jamaneurol.2018.2598
10. Yang Q, Lyu K, Li J, Zhang P, Guan W, Zhang L, et al. Anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis exhibit three clinical phenotypes with different prognoses. Clin Exp Rheumatol. (2022) 40:304–8.
11. Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. (2018) 14:279–89. doi: 10.1038/nrrheum.2018.42
12. Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. (2016) 280:8–23. doi: 10.1111/joim.12451
13. Opinc AH, Makowska JS. Antisynthetase syndrome - much more than just a myopathy. Semin Arthritis Rheum. (2021) 51:72–83. doi: 10.1016/j.semarthrit.2020.09.020
14. Cavagna L, Trallero-Araguás E, Meloni F, Cavazzana I, Rojas-Serrano J, Feist E, et al. Influence of antisynthetase antibodies specificities on antisynthetase syndrome clinical spectrum time course. J Clin Med. (2019) 8:2013. doi: 10.3390/jcm8112013
15. Nombel A, Fabien N, Coutant F. Dermatomyositis with anti-MDA5 antibodies: bioclinical features, pathogenesis and emerging therapies. Front Immunol. (2021) 12:773352. doi: 10.3389/fimmu.2021.773352
16. Allenbach Y, Uzunhan Y, Toquet S, Leroux G, Gallay L, Marquet A, et al. Different phenotypes in dermatomyositis associated with anti-MDA5 antibody: study of 121 cases. Neurology. (2020) 95:e70–8. doi: 10.1212/WNL.0000000000009727
17. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers (Basel). (2019) 11:2002. doi: 10.3390/cancers11122002
18. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
19. Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front Immunol. (2017) 8:29. doi: 10.3389/fimmu.2017.00029
20. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
21. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of JAK/STAT signaling in immunity and disease. J Immunol. (2015) 194:21–7. doi: 10.4049/jimmunol.1401867
22. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. (2014) 57:5023–38. doi: 10.1021/jm401490p
23. Lin CM, Cooles FA, Isaacs JD. Basic mechanisms of JAK inhibition. Mediterr J Rheumatol. (2020) 31(Suppl. 1):100–4. doi: 10.31138/mjr.31.1.100
24. Kim ES, Keam SJ. Filgotinib in rheumatoid arthritis: a profile of its use. Clin Drug Investig. (2021) 41:741–9. doi: 10.1007/s40261-021-01055-0
25. Tanaka Y. A review of upadacitinib in rheumatoid arthritis. Mod Rheumatol. (2020) 30:779–87. doi: 10.1080/14397595.2020.1782049
26. Harigai M, Honda S. Selectivity of janus kinase inhibitors in rheumatoid arthritis and other immune-mediated inflammatory diseases: is expectation the root of all headache? Drugs. (2020) 80:1183–201. doi: 10.1007/s40265-020-01349-1
27. Sun L, Ma K, Wang H, Xiao F, Gao Y, Zhang W, et al. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J Cell Biol. (2007) 179:129–38. doi: 10.1083/jcb.200703184
28. Moresi V, Adamo S, Berghella L. The JAK/STAT pathway in skeletal muscle pathophysiology. Front Physiol. (2019) 10:500. doi: 10.3389/fphys.2019.00500
29. Ma JF, Sanchez BJ, Hall DT, Tremblay AK, Di Marco S, Gallouzi IE. STAT3 promotes IFNγ/TNFα-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol Med. (2017) 9:622–37. doi: 10.15252/emmm.201607052
30. Sano S, Chan KS, DiGiovanni J. Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J Dermatol Sci. (2008) 50:1–14. doi: 10.1016/j.jdermsci.2007.05.016
31. Welsch K, Holstein J, Laurence A, Ghoreschi K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol. (2017) 47:1096–107. doi: 10.1002/eji.201646680
32. Bousoik E, Montazeri Aliabadi H. “Do We Know Jack” About JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. (2018) 8:287.
33. Huo R, Guo Q, Hu J, Li N, Gao R, Mi L, et al. Therapeutic potential of janus kinase inhibitors for the management of interstitial lung disease. Drug Des Devel Ther. (2022) 16:991–8. doi: 10.2147/DDDT.S353494
34. Milara J, Hernandez G, Ballester B, Morell A, Roger I, Montero P, et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir Res. (2018) 19:24. doi: 10.1186/s12931-018-0728-9
35. Liu Y, Hu M, Fan G, Xing N, Zhang R. Effect of baricitinib on the epithelial-mesenchymal transition of alveolar epithelial cells induced by IL-6. Int Immunopharmacol. (2022) 110:109044. doi: 10.1016/j.intimp.2022.109044
36. Silva-Carmona M, Vogel TP, Marchal S, Guesmi M, Dubus JC, Leroy S, et al. Successful treatment of interstitial lung disease in STAT3 gain-of-function using JAK inhibitors. Am J Respir Crit Care Med. (2020) 202:893–7. doi: 10.1164/rccm.201906-1204LE
37. Ceribelli A, De Santis M, Isailovic N, Gershwin ME, Selmi C. The immune response and the pathogenesis of idiopathic inflammatory myositis: a critical review. Clin Rev Allergy Immunol. (2017) 52:58–70. doi: 10.1007/s12016-016-8527-x
38. Dalakas MC. Pathogenesis and therapies of immune-mediated myopathies. Autoimmun Rev. (2012) 11:203–6. doi: 10.1016/j.autrev.2011.05.013
39. Cardelli C, Zanframundo G, Cometi L, Marcucci E, Biglia A, Cavagna L, et al. Idiopathic inflammatory myopathies: one year in review 2021. Clin Exp Rheumatol. (2022) 40:199–209. doi: 10.55563/clinexprheumatol/vskjxi
40. Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature muscle precursors are a source of interferon-β in myositis: role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum. (2012) 64:533–41. doi: 10.1002/art.33350
41. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. (2005) 57:664–78. doi: 10.1002/ana.20464
42. Bolko L, Jiang W, Tawara N, Landon-Cardinal O, Anquetil C, Benveniste O, et al. The role of interferons type I, II and III in myositis: a review. Brain Pathol. (2021) 31:e12955. doi: 10.1111/bpa.12955
43. Greenberg SA, Higgs BW, Morehouse C, Walsh RJ, Kong SW, Brohawn P, et al. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun. (2012) 13:207–13. doi: 10.1038/gene.2011.61
44. Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology. (2019) 93:e1193–204. doi: 10.1212/WNL.0000000000008128
45. Rigolet M, Hou C, Baba Amer Y, Aouizerate J, Periou B, Gherardi RK, et al. Distinct interferon signatures stratify inflammatory and dysimmune myopathies. RMD Open. (2019) 5:e000811. doi: 10.1136/rmdopen-2018-000811
46. Uruha A, Goebel HH, Stenzel W. Updates on the immunopathology in idiopathic inflammatory myopathies. Curr Rheumatol Rep. (2021) 23:56. doi: 10.1007/s11926-021-01017-7
47. Chasset F, Dayer JM, Chizzolini C. Type I interferons in systemic autoimmune diseases: distinguishing between afferent and efferent functions for precision medicine and individualized treatment. Front Pharmacol. (2021) 12:633821. doi: 10.3389/fphar.2021.633821
48. Baechler EC, Bilgic H, Reed AM. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. (2011) 13:249. doi: 10.1186/ar3531
49. Tournadre A, Miossec P. A critical role for immature muscle precursors in myositis. Nat Rev Rheumatol. (2013) 9:438–42. doi: 10.1038/nrrheum.2013.26
50. Suber TL, Casciola-Rosen L, Rosen A. Mechanisms of disease: autoantigens as clues to the pathogenesis of myositis. Nat Clin Pract Rheumatol. (2008) 4:201–9. doi: 10.1038/ncprheum0760
51. de Padilla CM, Reed AM. Dendritic cells and the immunopathogenesis of idiopathic inflammatory myopathies. Curr Opin Rheumatol. (2008) 20:669–74. doi: 10.1097/BOR.0b013e3283157538
52. Higgs BW, Zhu W, Morehouse C, White WI, Brohawn P, Guo X, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-α monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis. (2014) 73:256–62. doi: 10.1136/annrheumdis-2012-202794
53. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. (2019) 50:812–31. doi: 10.1016/j.immuni.2019.03.027
54. Okiyama N, Sugihara T, Iwakura Y, Yokozeki H, Miyasaka N, Kohsaka H. Therapeutic effects of interleukin-6 blockade in a murine model of polymyositis that does not require interleukin-17A. Arthritis Rheum. (2009) 60:2505–12. doi: 10.1002/art.24689
55. Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. (2009) 60:3436–46. doi: 10.1002/art.24936
56. Gono T, Kaneko H, Kawaguchi Y, Hanaoka M, Kataoka S, Kuwana M, et al. Cytokine profiles in polymyositis and dermatomyositis complicated by rapidly progressive or chronic interstitial lung disease. Rheumatology (Oxford). (2014) 53:2196–203. doi: 10.1093/rheumatology/keu258
57. Wilkinson MGL, Radziszewska A, Wincup C, Ioannou Y, Isenberg DA, Manson JJ, et al. Using peripheral blood immune signatures to stratify patients with adult and juvenile inflammatory myopathies. Rheumatology (Oxford). (2020) 59:194–204. doi: 10.1093/rheumatology/kez252
58. Lou Y, Zheng Y, Fan B, Zhang L, Zhu F, Wang X, et al. Serum levels of interleukins and S100A8/A9 correlate with clinical severity in patients with dermatomyositis-associated interstitial lung disease. BMC Pulm Med. (2020) 20:196. doi: 10.1186/s12890-020-01226-3
59. Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci. (2021) 22:6211. doi: 10.3390/ijms22126211
60. Lu Z, Chen Y, Xue J, Liu L. NXP2-positive dermatomyositis complicated with refractory skin edema: successful treatment with tocilizumab. Dermatol Ther. (2021) 34:e14712. doi: 10.1111/dth.14712
61. Su CF, Liao HT, Tsai CY. Tocilizumab and rituximab for anti-MDA-5 positive amyopathic dermatomyositis complicated with macrophage activation syndrome and progressive fibrosing interstitial lung disease. Scand J Rheumatol. (2022) 51:166–8. doi: 10.1080/03009742.2021.1972519
62. Beaumel A, Muis-Pistor O, Tebib JG, Coury F. Antisynthetase syndrome treated with tocilizumab. Joint Bone Spine. (2016) 83:361–2. doi: 10.1016/j.jbspin.2015.03.016
63. Zhang X, Zhou S, Wu C, Li M, Wang Q, Zhao Y, et al. Tocilizumab for refractory rapidly progressive interstitial lung disease related to anti-MDA5-positive dermatomyositis. Rheumatology (Oxford). (2021) 60:e227–8. doi: 10.1093/rheumatology/keaa906
64. Oddis CV, Rockette HE, Zhu L, Koontz DC, Lacomis D, Venturupalli S, et al. Randomized trial of tocilizumab in the treatment of refractory adult polymyositis and dermatomyositis. ACR Open Rheumatol. (2022) 4:983–90. doi: 10.1002/acr2.11493
65. Paudyal A, Zheng M, Lyu L, Thapa C, Gong S, Yang Y, et al. JAK-inhibitors for dermatomyositis: a concise literature review. Dermatol Ther. (2021) 34:e14939. doi: 10.1111/dth.14939
66. Kurtzman DJ, Wright NA, Lin J, Femia AN, Merola JF, Patel M, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. (2016) 152:944–5. doi: 10.1001/jamadermatol.2016.0866
67. Paik JJ, Casciola-Rosen L, Shin JY, Albayda J, Tiniakou E, Leung DG, et al. Study of tofacitinib in refractory dermatomyositis: an open-label pilot study of ten patients. Arthritis Rheumatol. (2021) 73:858–65. doi: 10.1002/art.41602
68. Paik JJ, Shneyderman M, Gutierrez-Alamillo L, Albayda J, Tiniakou E, Perin J, et al. Long-term extension study of tofacitinib in refractory dermatomyositis. Arthritis Rheumatol. (2022) 74:371–2. doi: 10.1002/art.41944
69. Paik JJ, Lubin G, Gromatzky A, Mudd PN, Ponda MP, Christopher-Stine L. Use of Janus kinase inhibitors in dermatomyositis: a systematic literature review. Clin Exp Rheumatol. (2022): doi: 10.55563/clinexprheumatol/hxin6o [Epub ahead of print].
70. Chen Z, Wang X, Ye S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N Engl J Med. (2019) 381:291–3. doi: 10.1056/NEJMc1900045
71. McPherson M, Economidou S, Liampas A, Zis P, Parperis K. Management of MDA-5 antibody positive clinically amyopathic dermatomyositis associated interstitial lung disease: a systematic review. Semin Arthritis Rheum. (2022) 53:151959. doi: 10.1016/j.semarthrit.2022.151959
72. Fan L, Lyu W, Liu H, Jiang H, Chen L, Liu Y, et al. A retrospective analysis of outcome in melanoma differentiation-associated gene 5-related interstitial lung disease treated with tofacitinib or tacrolimus. J Rheumatol. (2022) 49:1356–64. doi: 10.3899/jrheum.220367
73. Li Y, Wang Y, Shi L, Lin F, Zhang Z, Zhang J, et al. A clinical risk model to predict rapidly progressive interstitial lung disease incidence in dermatomyositis. Front Med (Lausanne). (2021) 8:733599. doi: 10.3389/fmed.2021.733599
74. Fischer K, Aringer M, Steininger J, Heil J, Beissert S, Abraham S, et al. Improvement of cutaneous inflammation and panniculitis in patients with dermatomyositis by the Janus kinase inhibitor baricitinib. Br J Dermatol. (2022) 187:432–5. doi: 10.1111/bjd.21252
75. Delvino P, Bartoletti A, Monti S, Biglia A, Montecucco C, Carducci M, et al. Successful treatment with baricitinib in a patient with refractory cutaneous dermatomyositis. Rheumatology (Oxford). (2020) 59:e125–7. doi: 10.1093/rheumatology/keaa184
76. Wang Z, Zheng Q, Xuan W, Xu X, Lu M, Wu J, et al. Short-term effectiveness of baricitinib in children with refractory and/or severe juvenile dermatomyositis. Front Pediatr. (2022) 10:962585. doi: 10.3389/fped.2022.962585
77. Landon-Cardinal O, Guillaume-Jugnot P, Toquet S, Sbeih N, Rigolet A, Champtiaux N, et al. JAK inhibitors for the treatment of adult dermatomyositis: a pilot study. J Am Acad Dermatol. (2022): doi: 10.1016/j.jaad.2022.10.055 [Epub ahead of print].
78. Zhao Q, Zhu Z, Fu Q, Shih Y, Wu D, Chen L, et al. Baricitinib for the treatment of cutaneous dermatomyositis: a prospective, open-label study. J Am Acad Dermatol. (2022) 87:1374–6. doi: 10.1016/j.jaad.2022.08.025
79. Wendel S, Venhoff N, Frye BC, May AM, Agarwal P, Rizzi M, et al. Successful treatment of extensive calcifications and acute pulmonary involvement in dermatomyositis with the Janus-Kinase inhibitor tofacitinib - A report of two cases. J Autoimmun. (2019) 100:131–6. doi: 10.1016/j.jaut.2019.03.003
80. Shneyderman M, Ahlawat S, Christopher-Stine L, Paik JJ. Calcinosis in refractory dermatomyositis improves with tofacitinib monotherapy: a case series. Rheumatology (Oxford). (2021) 60:e387–8. doi: 10.1093/rheumatology/keab421
81. Conca W, Weheba I, Abouzied ME, Abdelsayed A, Aleyouni Y, Al-Mutairy E, et al. Iacta Alea Est: the inexorable advance of tofacitinib in the treatment of dermatomyositis-associated rapidly progressive interstitial lung disease. A case report. Front Pharmacol. (2020) 11:585761. doi: 10.3389/fphar.2020.585761
82. Tseng CW. Tofacitinib treatment in anti-glycyl-tRNA synthetase antibody interstitial lung disease - A case report. Int J Rheum Dis. (2022): [Epub ahead of print].
83. Kim YJ, Schiopu E, Dankó K, Mozaffar T, Chunduru S, Lees K, et al. A phase 2, double-blinded, placebo-controlled trial of toll-like receptor 7/8/9 antagonist, IMO-8400, in dermatomyositis. J Am Acad Dermatol. (2021) 84:1160–2. doi: 10.1016/j.jaad.2020.07.122
84. Bitar C, Ninh T, Brag K, Foutouhi S, Radosta S, Meyers J, et al. Apremilast in recalcitrant cutaneous dermatomyositis: a nonrandomized controlled trial. JAMA Dermatol. (2022) 158:1357–66. doi: 10.1001/jamadermatol.2022.3917
85. Ishikawa Y, Iwata S, Hanami K, Nawata A, Zhang M, Yamagata K, et al. Relevance of interferon-gamma in pathogenesis of life-threatening rapidly progressive interstitial lung disease in patients with dermatomyositis. Arthritis Res Ther. (2018) 20:240. doi: 10.1186/s13075-018-1737-2
86. Coutant F, Bachet R, Pin JJ, Alonzo M, Miossec P. Monoclonal antibodies from B cells of patients with anti-MDA5 antibody-positive dermatomyositis directly stimulate interferon gamma production. J Autoimmun. (2022) 130:102831. doi: 10.1016/j.jaut.2022.102831
87. Peng QL, Zhang YM, Liang L, Liu X, Ye LF, Yang HB, et al. A high level of serum neopterin is associated with rapidly progressive interstitial lung disease and reduced survival in dermatomyositis. Clin Exp Immunol. (2020) 199:314–25.
88. Zhou J, Huang W, Ren F, Luo L, Huang D, Tang L. Evaluation of prognostic factors in anti-MDA5 antibody-positive patients in Chongqing, China: a retrospective study. Int J Gen Med. (2021) 14:4775–81. doi: 10.2147/IJGM.S327751
89. Wang G, Wang Q, Wang Y, Liu C, Wang L, Chen H, et al. Presence of anti-MDA5 antibody and its value for the clinical assessment in patients with COVID-19: a retrospective cohort study. Front Immunol. (2021) 12:791348. doi: 10.3389/fimmu.2021.791348
90. Hu H, Yang H, Liu Y, Yan B. Pathogenesis of anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis: a concise review with an emphasis on Type I interferon system. Front Med (Lausanne). (2021) 8:833114. doi: 10.3389/fmed.2021.833114
91. Yaqinuddin A, Ambia AR, Elgazzar TA, AlSaud MBM, Kashir J. Application of intravenous immunoglobulin (IVIG) to modulate inflammation in critical COVID-19 - A theoretical perspective. Med Hypotheses. (2021) 151:110592. doi: 10.1016/j.mehy.2021.110592
92. Wang LM, Yang QH, Zhang L, Liu SY, Zhang PP, Zhang X, et al. Intravenous immunoglobulin for interstitial lung diseases of anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Rheumatology (Oxford). (2022) 61:3704–10. doi: 10.1093/rheumatology/keab928
93. Wilfong EM, Matthay MA. Intravenous immunoglobulin therapy for COVID-19 ARDS. Lancet Respir Med. (2022) 10:123–5. doi: 10.1016/S2213-2600(21)00450-1
94. Sposito B, Broggi A, Pandolfi L, Crotta S, Clementi N, Ferrarese R, et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell. (2021) 184:4953–68.e16. doi: 10.1016/j.cell.2021.08.016
95. Yang L, Xie X, Tu Z, Fu J, Xu D, Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. (2021) 6:255. doi: 10.1038/s41392-021-00679-0
96. Group RC. Baricitinib in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial and updated meta-analysis. Lancet. (2022) 400:359–68.
97. Marconi VC, Ramanan AV, de Bono S, Kartman CE, Krishnan V, Liao R, et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. (2021) 9:1407–18.
98. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med. (2021) 384:795–807.
99. Ely EW, Ramanan AV, Kartman CE, de Bono S, Liao R, Piruzeli MLB, et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir Med. (2022) 10:327–36. doi: 10.1101/2021.10.11.21263897
100. Ferro F, Elefante E, Italiano N, Moretti M, La Rocca G, Mozzo R, et al. Baricitinib and pulse steroids combined treatment in severe COVID-19 pneumonia: preliminary data from a rheumatologic experience in intensive care unit. Ann Rheum Dis. (2022) 81:955. doi: 10.1136/annrheumdis-2022-eular.3231
101. Ferro F, Elefante E, Italiano N, La Rocca G, Schilirò D, Moretti M, et al. Combination treatment with baricitinib and pulse steroids in severe COVID-19. Arthritis Rheumatol. (2022) 74(suppl 9). Available online at: https://acrabstracts.org/abstract/combination-treatment-with-baricitinib-and-pulse-steroids-in-severe-covid19-a/ (accessed March 3, 2023).
102. Shirai T, Machiyama T, Sato H, Ishii T, Fujii H. Intensive induction therapy combining tofacitinib, rituximab and plasma exchange in severe anti-melanoma differentiation-associatedprotein-5 antibody-positive dermatomyositis. Clin Exp Rheumatol. (2023) 41:291–300 doi: 10.55563/clinexprheumatol/8kulbf
Keywords: JAK/STAT 1, 2, 3, dermatomyositis, antisynthetase syndrome, Baricitinib, Tofacitinib
Citation: La Rocca G, Ferro F, Baldini C, Libra A, Sambataro D, Colaci M, Malatino L, Palmucci S, Vancheri C and Sambataro G (2023) Targeting intracellular pathways in idiopathic inflammatory myopathies: A narrative review. Front. Med. 10:1158768. doi: 10.3389/fmed.2023.1158768
Received: 04 February 2023; Accepted: 27 February 2023;
Published: 13 March 2023.
Edited by:
João Eurico Fonseca, University of Lisbon, PortugalReviewed by:
Lorenzo Cavagna, San Matteo Hospital Foundation (IRCCS), ItalySofia Carvalho Barreira, Santa Maria Hospital, Portugal
Gonçalo Boleto, Hôpital Cochin, France
Copyright © 2023 La Rocca, Ferro, Baldini, Libra, Sambataro, Colaci, Malatino, Palmucci, Vancheri and Sambataro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluca Sambataro, ZG90dG9yc2FtYmF0YXJvQGdtYWlsLmNvbQ==