 Nirupama Ramadas
Nirupama Ramadas Erica M. Sparkenbaugh
Erica M. Sparkenbaugh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 11 July 2023
Sec. Hematology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1141020
This article is part of the Research TopicRising Stars in Hematology: 2022View all 12 articles
Sickle Cell Disease (SCD) is a group of inherited hemoglobinopathies. Sickle cell anemia (SCA) is caused by a homozygous mutation in the β-globin generating sickle hemoglobin (HbS). Deoxygenation leads to pathologic polymerization of HbS and sickling of erythrocytes. The two predominant pathologies of SCD are hemolytic anemia and vaso-occlusive episodes (VOE), along with sequelae of complications including acute chest syndrome, hepatopathy, nephropathy, pulmonary hypertension, venous thromboembolism, and stroke. SCD is associated with endothelial activation due to the release of danger-associated molecular patterns (DAMPs) such as heme, recurrent ischemia–reperfusion injury, and chronic thrombin generation and inflammation. Endothelial cell activation is mediated, in part, by thrombin-dependent activation of protease-activated receptor 1 (PAR1), a G protein coupled receptor that plays a role in platelet activation, endothelial permeability, inflammation, and cytotoxicity. PAR1 can also be activated by activated protein C (APC), which promotes endothelial barrier protection and cytoprotective signaling. Notably, the APC system is dysregulated in SCD. This mini-review will discuss activation of PAR1 by APC and thrombin, the APC-EPCR-PAR1 axis, and their potential roles in SCD.
Sickle cell disease (SCD) is the most common inherited hemoglobinopathy worldwide. More than 300,000 babies are born with SCD annually and this rate is expected to increase over the next 30 years (1). The majority of cases are concentrated in sub-Saharan Africa and southern Asia, where sickle cell trait provides protection from malaria (2). Sickle Cell Anemia (SCA) is caused by a single nucleotide mutation in the gene for beta (β) globin. Normal hemoglobin is a tetramer of two α and two β subunits, each of which contain a heme molecule, and is the critical oxygen carrying protein in red blood cells (RBCs). In SCA, an A to T transversion in the sixth codon results in the substitution of a valine (Val) for a glutamine (Glu). This hydrophobic valine residue confers an adhesive property to HbS (comprised of two α and two βS subunits); thus, when it becomes deoxygenated it forms rigid polymers in RBCs (Figure 1). This results in the sickling of RBCs, causing hemolysis and anemia. Hemolysis releases HbS and free heme into the circulation, acting as danger associated molecular patterns (DAMPs) to activate the endothelium and leukocytes. The activated endothelium upregulates adhesion molecules E-selectin, P-selectin, vascular cell adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), and von Willebrand Factor (VWF). Activated neutrophils, platelets, and sickled RBCs form multicellular aggregates in the circulation, which adhere to the circulating endothelium, leading to vaso-occlusive episodes (VOE). These two primary pathologies, hemolytic anemia and VOE, are accompanied by sequelae of acute and chronic complications such as acute painful crises (3), chronic pain, stroke (4–8), venous thrombosis and pulmonary embolism (9, 10), pulmonary hypertension (11), acute chest syndrome (12), sickle nephropathy (13, 14), among others. More comprehensive reviews of SCD and its complications can be found elsewhere (3, 15).
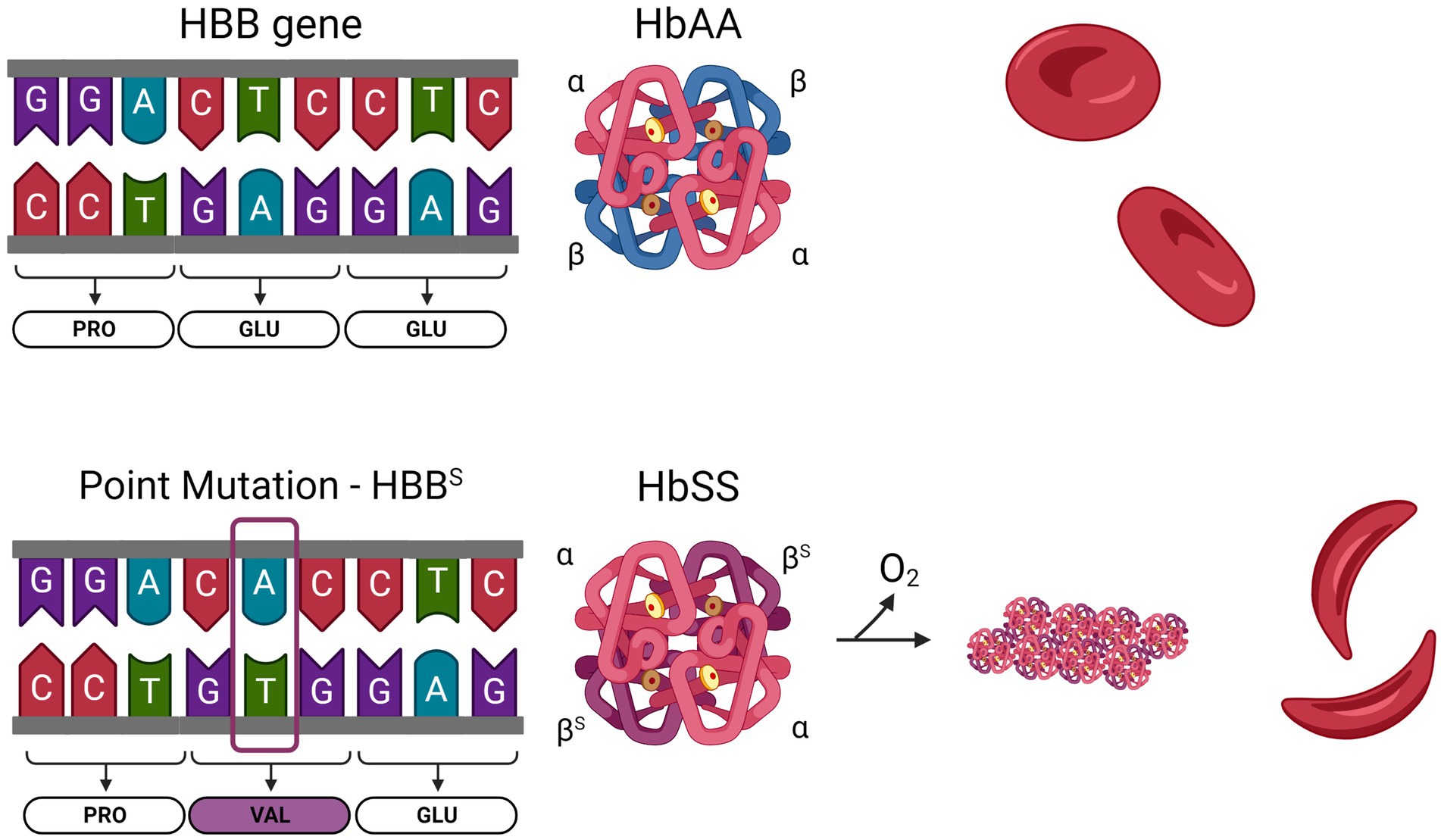
Figure 1. Sickle cell anemia is caused by a single nucleotide mutation in the HBB gene. Normal hemoglobin (HbAA) is formed from two α-globin and two β-globin subunits. The HBB gene encodes β-globin. In SCD, an adenine to thymine substitution changes the 6th codon of the mature protein from a glutamine to a valine. Sickle hemoglobin (HbSS) contains two α-globin and two βS-globin. Upon deoxygenation, the presence of a hydrophobic valine residue in HbSS causes the molecules to polymerize, leading to stiffening of the RBC.
Despite the significant global burden of SCA, there are only four FDA-approved drugs currently available to patients: hydroxyurea, L-glutamine, crizanlizumab, and Voxelotor (16). Hydroxyurea inhibits HbS polymerization and sickling by increasing production of fetal Hb (HbF) (17). L-glutamine reduces oxidative stress (18). Crizanlizumab inhibits P-selectin-dependent sRBC-endothelial interactions (19). Voxelotor changes the affinity of HbS for oxygen and inhibits hemoglobin polymerization (20). These therapies modestly limit the severity and frequency of VOC (17–19). Many SCA patients also routinely undergo whole blood transfusions and red blood cell exchange therapy (3, 21). Curative options such as hematopoietic stem cell transplantation (HSCT) and gene therapy (GT) are also being investigated (22). Allogeneic HSCT has been performed in approximately 2,000 patients in the past 30 years. A recent meta-analysis revealed that HSCT reduces the incidence of VOE, but it also identified risks including graft-versus host disease, graft failure, mortality, and secondary malignancies (23). The GT strategies currently being evaluated are correction of the HbS mutation, gene transfer to overexpress HbA in hematopoietic stem cells, and knockdown of BCL11a, the negative regulator of HbF, to increase HbF production and prevent sickling (3, 22–24). Although these therapies are promising, the FDA may require long-term follow up of 10–15 years after gene therapy to evaluate safety risks before they will be approved for clinical use (25). HSCT and GT carry significant costs and medical resources, and may not be feasible in low-resource countries where SCA is most prevalent (22). Recent GT trials have also been paused due to unexpected toxicity (26). The limited range of approved drugs for SCA, combined with an ageing population facing severe clinical complications, highlights the need to investigate new treatment options that are accessible and effective. Several drugs targeting downstream events are currently being evaluated in Phase II and III clinical trials, including anti-sickling agents, anti-inflammatory agents, and anticoagulants (3, 15).
A hallmark of SCA is activation of coagulation (27–33). Tissue factor (TF) is the primary initiator of extrinsic coagulation and is not normally expressed on intravascular cells. In SCA, TF expression is upregulated on leukocytes and endothelial cells (34–37). TF is a transmembrane protein and obligate cofactor for coagulation factor VIIa (FVIIa), activating factor X (FX) to FXa, which converts prothrombin to thrombin. Thrombin cleaves fibrinogen into fibrin, leading to clot formation. We and others have shown that TF (4, 5, 37, 38), FXa (6, 39), thrombin (39, 40), and fibrin(ogen) (41, 42) contribute to inflammation, cardiovascular dysfunction, vascular congestion, nephropathy, and microvascular stasis (43) in mouse models of SCA. In addition to its prothrombotic role, thrombin can induce signaling through Protease Activated Receptor-1 (PAR1).
Protease activated receptors (PARs) are a family of G-protein coupled receptors (GPCR) consisting of PAR1, PAR2, PAR3, and PAR4. PARs share a conserved mechanism of irreversible activation by proteolytic cleavage of specific amino acid residues on the extracellular N-terminus. This results in the exposure of a novel N-terminal peptide, or tethered ligand, which binds to extracellular loop 2 and induces a conformational change in the GPCR to signal through intracellular G proteins (44). PARs are subject to proteolysis by multiple proteases at different amino acid residues on the N-terminus, resulting in activation of different signaling pathways and outputs (45). The focus of this review is PAR1, which was first identified as the main thrombin receptor on platelets (46–48), triggering activation and aggregation of platelets that is critical for both hemostasis and thrombosis (49). Importantly, PAR1 is also expressed on leukocytes and endothelial cells. Thrombin cleaves PAR1 at arginine 41 (R41) (50), generating a tethered ligand that binds to a conserved sequence in extracellular loop 2 (51). This enables the C-terminus to engage with Gαq and Gα12/13 which leads to inflammation, endothelial barrier permeability, and cytotoxicity (52, 53) (Figure 2). Thrombin/PAR1 activation of Gαq and Gα12/13 upregulates inflammatory cytokines [interleukin-1 (IL-1), IL-6, and tumor necrosis factor α (TNFα)] and endothelial adhesion molecules [E-selectin, P-selectin, intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1)] via activation of MAPK and NFκB. G-protein mediated Ca2+ signaling promotes the release of Weibel Palade bodies to the endothelial surface, increasing P-selectin and von Willebrand Factor (VWF) release, thus increasing adhesion. These signaling pathways also cause apoptosis, through caspase activation, and endothelial barrier permeability, by modulating the cytoskeleton and disrupting tight junctions (54). Thrombin/PAR1 signaling is rapid, transient, and irreversible, due to the proteolytic cleavage of the protein. Signal termination is mediated by endocytosis of the receptor in clathrin-coated pits, and lysosomal degradation (55). Matrix metalloproteases MMP-1 and MMP-13 also cleave PAR1 at aspartate 39 (D39) and serine 42 (S42), respectively, with signaling outputs similar to thrombin (53).
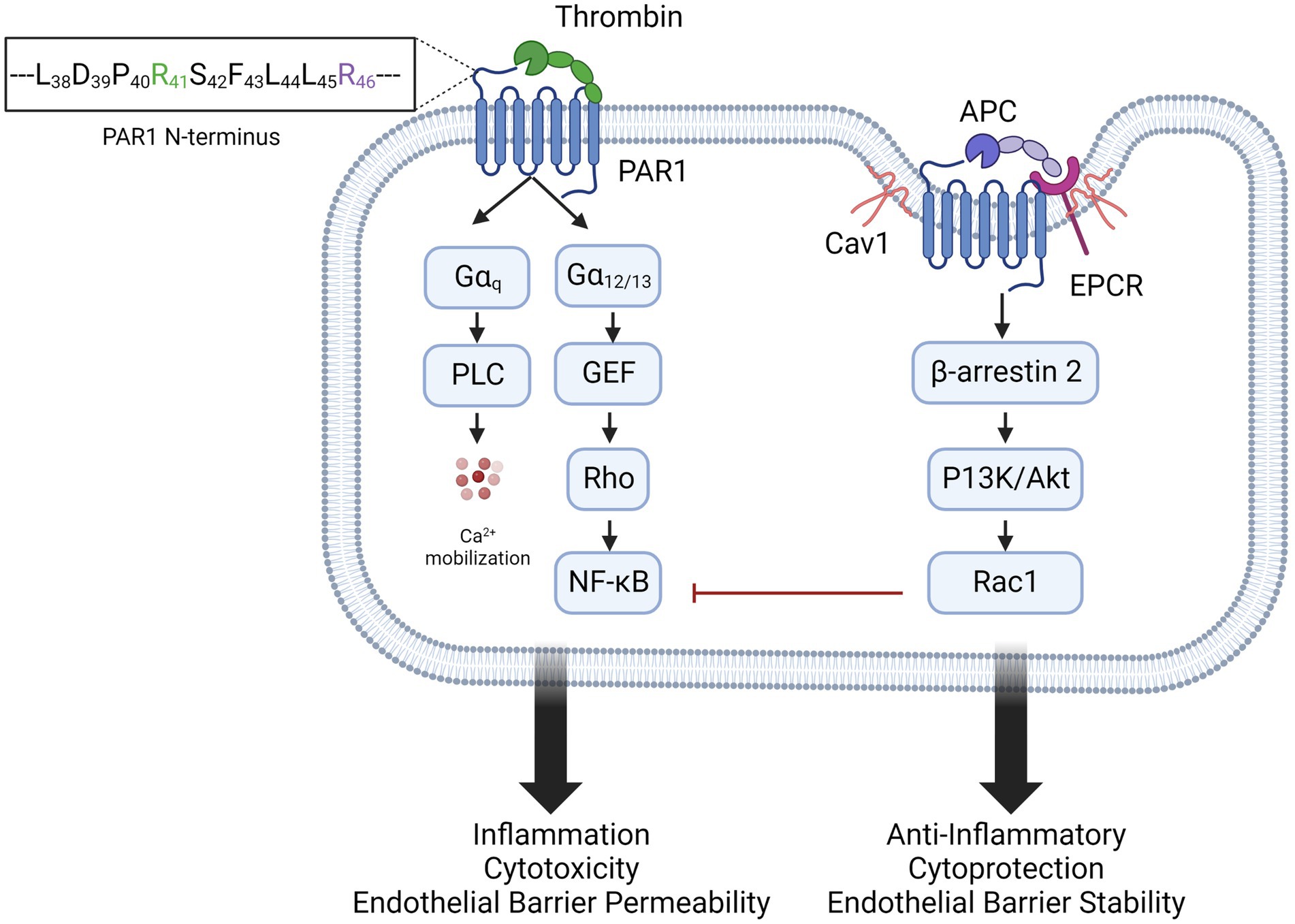
Figure 2. Biased agonism of PAR1 by thrombin and APC. Thrombin activation of PAR1, by cleavage at arginine 41 (R41) favors activation of Gαq and Gα12/13 signaling pathways. This results in inflammation, endothelial activation and barrier permeability, and cytotoxicity. In caveolin 1 (Cav1)-positive lipid rafts, PAR1 colocalizes with endothelial protein C receptor (EPCR). Activated protein C (APC) binds EPCR and activates PAR1 by cleavage of arginine 46 (R46), recruiting β-arrestin and inducing anti-inflammatory and endothelial stabilizing signaling.
Interestingly, PAR1 is also cleaved by activated protein C (APC), although with lower affinity than thrombin (56). The zymogen protein C binds to endothelial protein C receptor (EPCR), a type I transmembrane protein that binds the Gla domain of protein C and APC (57). On endothelial cells, the majority of EPCR is found in caveolin-1 (Cav1)-positive lipid rafts, where it colocalizes with PAR1 (58), which is required for cytoprotective signaling (59, 60). When bound to EPCR, protein C is cleaved by thrombomodulin-bound thrombin to generate the active serine protease APC, which cleaves PAR1 at Arg46 and activates multiple signaling pathways. APC/PAR1 signaling recruits and phosphorylates β arrestin-2, which activates Rac1, inhibits NFκB, and increases the barrier integrity of the endothelium (59). β arrestin-2 also activates sphingosine kinase 1 (SphK1), which converts the lipid messenger sphingosine into sphingosine-1-phosphate (S1P). In turn, S1P activates Sphingosine-1-phosphate receptor-1 (S1PR1), which signals through Gi/Akt and disheveled 2 (Dvl2) for anti-apoptotic and anti-inflammatory signaling (61, 62). aPC/PAR1-induced β-arrestin 2 also phosphorylates ERK1/2 (61). APC thus promotes anti-inflammatory and cytoprotective signaling on ECs and preserves endothelial barrier integrity (60, 63–65) (Figure 2). APC’s cytoprotective and anti-inflammatory activity is not limited to ECs, as it can also modulate activation of monocytes, macrophages, and neutrophils, but has no effect on platelets (66). It is expressed on leukocytes (67–69), keratinocytes (70), vascular smooth muscle cells (71), cardiomyocytes (72), and neurons (73, 74), The opposing effects of PAR1 activation by thrombin and APC is a classic example of biased agonist signaling (65, 75), well described for other G-protein-coupled receptors including the neurokinin 1, angiotensin II type 1A, parathyroid hormone 1, μ opioid, and D2 dopamine receptors (76).
APC is an important natural anticoagulant (77), in addition to its critical anti-inflammatory and cytoprotective role on the endothelium (78). Zymogen protein C is a glycoprotein produced by the liver. When activated by thrombin to its serine protease form APC, it irreversibly inactivates FVa and FVIIIa by proteolysis at arginine residues (79). Anticoagulant APC activity requires its cofactor protein S, a glycoprotein that binds negatively charged phospholipid membranes via its Gla domain (78). Dysfunction or deficiencies in the protein C—protein S system are associated with venous thrombosis (80–82). It is well-documented that individuals with SCD have deficiencies in both the antigen and activity levels of protein C and protein S (29, 83–89), and that they are further decreased during crisis (90). Protein C and protein S levels negatively correlate with markers of coagulation activation (88). Although deficiencies in this system have not been linked to VOC (29), lower levels of protein C and S are associated with a higher incidence of stroke in children and adolescents with SCA (87, 89, 91, 92). The low levels of protein C and S are likely caused by multiple factors common in SCA, including decreased synthesis due to liver disease (93), consumption due to chronic activation of coagulation, and binding to phosphatidylserine-positive sickled RBCs (94).
Decreased EPCR expression and EPCR shedding occurs in inflammatory bowel disease (95), malaria (96), diabetes (97), lupus, cardiovascular ischemia–reperfusion injury (98), and endotoxemia (99). EPCR shedding is mediated by pro-inflammatory cytokines and proteases such as TNFα converting enzyme (TACE), A Disintegrin and Metalloproteinase-10 (ADAM-10) and ADAM-17. Interestingly, EPCR shedding has been observed in individuals and mice with SCD (100, 101), and EPCR-positive microparticles are found in the circulation of individuals with SCD (90). A recent abstract described loss of EPCR expression in the kidney vasculature and presence of soluble EPCR in the urine of aged sickle mice, a phenomenon that could also be triggered in young sickle mice by infusion of a low dose of heme to mimic an acute sickling event (102).
Together, these observations describe dysfunction in regulation of the vascular endothelium. In SCD, the decreased availability of the natural anticoagulant APC and its cofactor protein S, along with diminished presence of endothelial EPCR could result in reduced cytoprotective APC/PAR1 signaling. Moreover, in a disease setting characterized by chronic thrombin generation, this imbalance might favor detrimental thrombin/PAR1 signaling. This imbalance could contribute to endothelial barrier dysfunction and vascular inflammation in SCD.
The endothelium is chronically activated in SCA due to hemolysis (103), which contributes to the pathogenesis of vaso-occlusive events (VOE). Increased expression of adhesion molecules P-selectin and E-selectin on the endothelial surface promotes interaction with P-selectin glycoprotein ligand-1 (PSGL-1) and Cd11b/Cd18 (Mac1) on leukocytes, respectively (104). This event recruits sRBCs and platelets to form multicellular aggregates that drive vascular stasis and ultimately occlusion (105, 106). In vitro studies have demonstrated that activation of PAR1 with either thrombin or PAR1 agonist peptide drives the interactions between sickle RBCs and endothelial cells. This was found to be dependent on the release of P-selectin and von Willebrand Factor (VWF) from endothelial Weibel-Palade bodies (107, 108). Infusion of PAR1 agonist peptide caused rolling adhesion of sRBCs to the vascular endothelium that was p-selectin dependent in sickle mice, suggesting a role of PAR1 in vascular stasis (109). We also investigated the thrombin/PAR1 axis in sickle mice at steady state. To determine the role of PAR1, we transplanted sickle bone marrow (BMSS) in PAR1−/− mice. Endothelial PAR1 deficiency did not affect the increased levels of thrombin generation (thrombin anti-thrombin complexes, TAT), systemic inflammation (IL-6), endothelial activation (sVCAM) or neutrophil recruitment in the lung vasculature. Interestingly thrombin inhibition with dabigatran reduced TAT, IL-6 and neutrophil recruitment to the organs (39). Agreeing with these results, Arumugam and colleagues found that decreasing expression of prothrombin reduces inflammation, vascular congestion, and improves survival of sickle mice (40). One possible interpretation of these data is that thrombin plays a role in endothelial activation and inflammation in SCD independent of PAR1, at least at steady state. An alternative hypothesis is that in BMSS PAR1−/− mice, the lack of PAR1 also prevents beneficial APC/PAR1 signaling. Indeed, it has been shown that administration of APC to sickle mice can attenuate thrombus formation in the cerebral microvasculature (101), indicating that APC is beneficial in SCD.
Since it is known that thrombin/PAR1 signaling contributes to endothelial P-selectin expression and sickle RBC adhesion, we also evaluated the role of this pathway in microvascular stasis. Using a dorsal skinfold chamber to evaluate blood flow in the skin microvasculature, we found that inhibition of PAR1 with the irreversible orthosteric antagonist vorapaxar protected sickle mice from heme-induced microvascular stasis (43). Similar results were obtained in BMSS PAR1−/− mice, which also had significantly less endothelial P-selectin and VWF expression in lung tissue after heme treatment (43). These data suggest that thrombin/PAR1 activation might play a role in the cell–cell interactions that lead to VOC (43, 109), and that APC/PAR1 signaling can be beneficial (101), in SCD. Future studies should be aimed at determining the role of PAR1 in other acute and chronic complications of SCD, including stroke, thrombosis, and acute chest syndrome.
Most PAR1 antagonists were designed to attenuate thrombin-mediated platelet activation and reduce thrombosis. Vorapaxar is an orally available antagonist that binds the extracellular pocket of PAR1 irreversibly and with high affinity. It effectively blocks PAR1 activation by both thrombin and APC. In clinical trials, administration of vorapaxar in combination with dual antiplatelet therapy improved cardiovascular outcomes but increased the risk of bleeding, especially in patients with a history of stroke (110). Thus, its use is counter-indicated in SCD patients. Recombinant APC (Xigris) was tested in pre-clinical models of sepsis, but had limited success and also increased the risk of bleeding in larger clinical trials (45). A signaling-selective variant of APC with limited anti-coagulant activity, 3K3A-APC, is also being tested for the treatment of stroke and amyotrophic lateral sclerosis (NCT02222714 and NCT05039268).
Another option for targeting PAR1 are small molecules. Q94 is an allosteric modulator that is thought to act at the intracellular face of PAR1. Although it inhibits PAR1-dependent platelet activation, it has limited efficacy on endothelial PAR1 signaling (111). Pepducins are a family of PAR1 modulators; they are biomimetic lipidated peptides that can enter the cell and target the intracellular loops of a receptor (112–114). One pepducin, PZ-128, is currently being tested in clinical trials for coronary artery disease (114) and has a promising safety profile. Parmodulins are small molecule allosteric modulators of PAR1, which bind the intracellular C-terminus and recruit β arrestin (115). They not only block thrombin-dependent PAR1 signaling; they can actually induce APC-like cytoprotective and anti-inflammatory signaling. Parmodulins have been shown to have significant anti-thrombotic and anti-inflammatory effects in mouse models of venous thrombosis (116, 117), neurologic diseases (115), virus (118), and diabetes (119).
The APC-EPCR-PAR1 axis is plays an important role in maintaining vascular endothelial homeostasis, and several studies described herein suggest that this pathway is dysfunctional in SCA. We speculate that chronic thrombin generation which can activate detrimental PAR1 signaling, paired with decreased APC/PAR1 signaling due to APC consumption and EPCR shedding, might play a role in the activated vascular endothelium in SCD.
NR and ES wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by NHLBI R01HL155193.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Piel, FB , Patil, AP , Howes, RE , Nyangiri, OA , Gething, PW , Dewi, M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. (2013) 381:142–51. doi: 10.1016/S0140-6736(12)61229-X
2. Cyrklaff, M , Sanchez, CP , Kilian, N , Bisseye, C , Simpore, J , Frischknecht, F, et al. Hemoglobins S and C interfere with actin remodeling in plasmodium falciparum-infected erythrocytes. Science. (2011) 334:1283–6. doi: 10.1126/science.1213775
3. Kavanagh, PL , Fasipe, TA , and Wun, T . Sickle cell disease: a review. JAMA. (2022) 328:57–68. doi: 10.1001/jama.2022.10233
4. Francis, RB . Large-vessel occlusion in sickle cell disease: pathogenesis, clinical consequences, and therapeutic implications. Med Hypotheses. (1991) 35:88–95. doi: 10.1016/0306-9877(91)90029-X
5. Prengler, M , Pavlakis, SG , Prohovnik, I , and Adams, RJ . Sickle cell disease: the neurological complications. Ann Neurol. (2002) 51:543–52. doi: 10.1002/ana.10192
6. Adams, RJ . Big strokes in small persons. Arch Neurol. (2007) 64:1567–74. doi: 10.1001/archneur.64.11.1567
7. Strouse, JJ , Lanzkron, S , and Urrutia, V . The epidemiology, evaluation and treatment of stroke in adults with sickle cell disease. Expert Rev Hematol. (2011) 4:597–606. doi: 10.1586/ehm.11.61
8. DeBaun, MR , Jordan, LC , King, AA , Schatz, J , Vichinsky, E , Fox, CK, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. (2020) 4:1554–88. doi: 10.1182/bloodadvances.2019001142
9. Shet, AS , and Wun, T . How I diagnose and treat venous thromboembolism in sickle cell disease. Blood. (2018) 132:1761–9. doi: 10.1182/blood-2018-03-822593
10. Lizarralde-Iragorri, MA , and Shet, AS . Sickle cell disease: a paradigm for venous thrombosis pathophysiology. Int J Mol Sci. (2020) 21:5279. doi: 10.3390/ijms21155279
11. Vichinsky, EP . Pulmonary hypertension in sickle cell disease. N Engl J Med. (2004) 350:857–9. doi: 10.1056/NEJMp038250
12. Vichinsky, EP , Styles, LA , Colangelo, LH , Wright, EC , Castro, O , and Nickerson, B . Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood. (1997) 89:1787–92. doi: 10.1182/blood.V89.5.1787
13. Ataga, KI , and Orringer, EP . Renal abnormalities in sickle cell disease. Am J Hematol. (2000) 63:205–11. doi: 10.1002/(SICI)1096-8652(200004)63:4<205::AID-AJH8>3.0.CO;2-8
14. Nath, KA , and Vercellotti, GM . Renal functional decline in sickle cell disease and trait. J Am Soc Nephrol. (2020) 31:236–8. doi: 10.1681/ASN.2019121291
15. Kato, GJ , Piel, FB , Reid, CD , Gaston, MH , Ohene-Frempong, K , Krishnamurti, L, et al. Sickle cell disease. Nat Rev Dis Primers. (2018) 4:18010. doi: 10.1038/nrdp.2018.10
16. Ballas, SK . The evolving Pharmacotherapeutic landscape for the treatment of sickle cell disease. Mediterr J Hematol Infect Dis. (2020) 12:e2020010. doi: 10.4084/mjhid.2020.010
17. Sethy, S , Panda, T , and Jena, RK . Beneficial effect of low fixed dose of hydroxyurea in vaso-occlusive crisis and transfusion requirements in adult HbSS patients: a prospective study in a tertiary care center. Indian J Hematol Blood Transfus. (2018) 34:294–8. doi: 10.1007/s12288-017-0869-x
18. Niihara, Y , Miller, ST , Kanter, J , Lanzkron, S , Smith, WR , Hsu, LL, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. (2018) 379:226–35. doi: 10.1056/NEJMoa1715971
19. Ataga, KI , Kutlar, A , Kanter, J , Liles, D , Cancado, R , Friedrisch, J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2016) 376:429–39. doi: 10.1056/NEJMoa1611770
20. Vichinsky, E , Hoppe, CC , Ataga, KI , Ware, RE , Nduba, V , El-Beshlawy, A, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. (2019) 381:509–19. doi: 10.1056/NEJMoa1903212
21. Han, H , Hensch, L , and Tubman, VN . Indications for transfusion in the management of sickle cell disease. Hematol Am Soc Hematol Educ Program. (2021) 2021:696–703. doi: 10.1182/hematology.2021000307
22. Tanhehco, YC , Nathu, G , and Vasovic, LV . Development of curative therapies for sickle cell disease. Front Med Lausanne. (2022) 9:1055540. doi: 10.3389/fmed.2022.1055540
23. Rotin, LE , Viswabandya, A , Kumar, R , Patriquin, CJ , and Kuo, KHM . A systematic review comparing allogeneic hematopoietic stem cell transplant to gene therapy in sickle cell disease. Hematology. (2023) 28:2163357. doi: 10.1080/16078454.2022.2163357
24. Ghiaccio, V , Chappell, M , Rivella, S , and Breda, L . Gene therapy for beta-hemoglobinopathies: milestones, new therapies and challenges. Mol Diagn Ther. (2019) 23:173–86. doi: 10.1007/s40291-019-00383-4
25. Bailey, AM , Arcidiacono, J , Benton, KA , Taraporewala, Z , and Winitsky, S . United States Food and Drug Administration regulation of gene and cell therapies In: M Galli and M Serabian, editors. Regulatory aspects of gene therapy and cell therapy products. Advances in experimental medicine and biology. Cham: Springer (2015)
26. Quach, D , Jiao, B , Basu, A , Bender, MA , Hankins, J , Ramsey, S, et al. A landscape analysis and discussion of value of gene therapies for sickle cell disease. Expert Rev Pharmacoecon Outcomes Res. (2022) 22:891–911. doi: 10.1080/14737167.2022.2060823
27. Ataga, KI , and Key, NS . Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematol Am Soc Hematol Educ Program. (2007) 2007:91–6. doi: 10.1182/asheducation-2007.1.91
28. Leslie, J , Langler, D , Serjeant, GR , Serjeant, BE , Desai, P , and Gordon, YB . Coagulation changes during the steady state in homozygous sickle-cell disease in Jamaica. Br J Haematol. (1975) 30:159–66. doi: 10.1111/j.1365-2141.1975.tb00530.x
29. Green, D , and Scott, JP . Is sickle cell crisis a thrombotic event? Am J Hematol. (1986) 23:317–21. doi: 10.1002/ajh.2830230403
30. Stuart, MJ , and Setty, BN . Hemostatic alterations in sickle cell disease: relationships to disease pathophysiology. Pediatr Pathol Mol Med. (2001) 20:27–46. doi: 10.1080/15513810109168816
31. Shah, N , Thornburg, C , Telen, MJ , and Ortel, TL . Characterization of the hypercoagulable state in patients with sickle cell disease. Thromb Res. (2012) 130:e241–5. doi: 10.1016/j.thromres.2012.08.307
32. Sparkenbaugh, E , and Pawlinski, R . Prothrombotic aspects of sickle cell disease. J Thromb Haemost. (2017) 15:1307–16. doi: 10.1111/jth.13717
33. Faes, C , Sparkenbaugh, EM , and Pawlinski, R . Hypercoagulable state in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68:301–18. doi: 10.3233/CH-189013
34. Solovey, A , Gui, L , Key, NS , and Hebbel, RP . Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. (1998) 101:1899–904. doi: 10.1172/JCI1932
35. Belcher, JD , Marker, PH , Weber, JP , Hebbel, RP , and Vercellotti, GM . Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. (2000) 96:2451–9. doi: 10.1182/blood.V96.7.2451
36. Shet, AS , Aras, O , Gupta, K , Hass, MJ , Rausch, DJ , Saba, N, et al. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. (2003) 102:2678–83. doi: 10.1182/blood-2003-03-0693
37. Chantrathammachart, P , Mackman, N , Sparkenbaugh, E , Wang, JG , Parise, LV , Kirchhofer, D, et al. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood. (2012) 120:636–46. doi: 10.1182/blood-2012-04-424143
38. Sparkenbaugh, E , Faes, C , Noubouossie, D , Kirchhofer, D , Gruber, A , Key, N, et al. FXIIa differentially regulates thrombin generation during steady state and vaso-occlusive crisis in sickle cell mice. Blood. (2016) 128:162. doi: 10.1182/blood.V128.22.162.162
39. Sparkenbaugh, EM , Chantrathammachart, P , Mickelson, J , van Ryn, J , Hebbel, RP , Monroe, DM, et al. Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood. (2014) 123:1747–56. doi: 10.1182/blood-2013-08-523936
40. Arumugam, PI , Mullins, ES , Shanmukhappa, SK , Monia, BP , Loberg, A , Shaw, MA, et al. Genetic diminution of circulating prothrombin ameliorates multiorgan pathologies in sickle cell disease mice. Blood. (2015) 126:1844–55. doi: 10.1182/blood-2015-01-625707
41. Nasimuzzaman, M , Arumugam, PI , Mullins, ES , James, JM , Vanden Heuvel, K , Narciso, MG, et al. Elimination of the fibrinogen integrin alphaMbeta2-binding motif improves renal pathology in mice with sickle cell anemia. Blood Adv. (2019) 3:1519–32. doi: 10.1182/bloodadvances.2019032342
42. Narciso, MG , Hoeting, B , James, JM , Vanden Heuvel, K , and Nasimuzzaman, M . A mutant fibrinogen that is unable to form fibrin can improve renal phenotype in mice with sickle cell anemia. eJHaem. (2021) 2:462–5. doi: 10.1002/jha2.204
43. Sparkenbaugh, EM , Chen, C , Brzoska, T , Nguyen, J , Wang, S , Vercellotti, GM, et al. Thrombin activation of PAR-1 contributes to microvascular stasis in mouse models of sickle cell disease. Blood. (2020) 135:1783–7. doi: 10.1182/blood.2019003543
44. Coughlin, SR . Thrombin signalling and protease-activated receptors. Nature. (2000) 407:258–64. doi: 10.1038/35025229
45. Coughlin, SR . Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. (2005) 3:1800–14. doi: 10.1111/j.1538-7836.2005.01377.x
46. Vu, TK , Hung, DT , Wheaton, VI , and Coughlin, SR . Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cells. (1991) 64:1057–68. doi: 10.1016/0092-8674(91)90261-V
47. Vu, TK , Wheaton, VI , Hung, DT , Charo, I , and Coughlin, SR . Domains specifying thrombin-receptor interaction. Nature. (1991) 353:674–7. doi: 10.1038/353674a0
48. Brass, LF , Vassallo, RR , Belmonte, E , Ahuja, M , Cichowski, K , and Hoxie, JA . Structure and function of the human platelet thrombin receptor. Studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J Biol Chem. (1992) 267:13795–8. doi: 10.1016/S0021-9258(19)49635-X
49. Brass, LF . Thrombin and platelet activation. Chest. (2003) 124:18S–25S. doi: 10.1378/chest.124.3_suppl.18S
50. Schuepbach, RA , Madon, J , Ender, M , Galli, P , and Riewald, M . Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost. (2012) 10:1675–84. doi: 10.1111/j.1538-7836.2012.04825.x
51. Nanevicz, T , Ishii, M , Wang, L , Chen, M , Chen, J , Turck, CW, et al. Mechanisms of thrombin receptor agonist specificity: chimeric receptors and complementary mutations identify an agonist recognition site (∗). J Biol Chem. (1995) 270:21619–25. doi: 10.1074/jbc.270.37.21619
52. Han, X , and Nieman, MT . The domino effect triggered by the tethered ligand of the protease activated receptors. Thromb Res. (2020) 196:87–98. doi: 10.1016/j.thromres.2020.08.004
53. Willis Fox, O , and Preston, RJS . Molecular basis of protease-activated receptor 1 signaling diversity. J Thromb Haemost. (2020) 18:6–16. doi: 10.1111/jth.14643
54. Rezaie, AR . Protease-activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost. (2014) 112:876–82. doi: 10.1160/TH14-02-0167
55. Grimsey, N , Lin, H , and Trejo, J . Chapter twenty-two-endosomal signaling by protease-activated receptors In: PM Conn , editor. Methods in enzymology. Cambridge, MA: Academic Press (2014). 389–401.
56. Ludeman, MJ , Kataoka, H , Srinivasan, Y , Esmon, NL , Esmon, CT , and Coughlin, SR . PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. (2005) 280:13122–8. doi: 10.1074/jbc.M410381200
57. Fukudome, K , and Esmon, CT . Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. (1994) 269:26486–91. doi: 10.1016/S0021-9258(18)47220-1
58. Mohan Rao, LV , Esmon, CT , and Pendurthi, UR . Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. (2014) 124:1553–62. doi: 10.1182/blood-2014-05-578328
59. Roy, RV , Ardeshirylajimi, A , Dinarvand, P , Yang, L , and Rezaie, AR . Occupancy of human EPCR by protein C induces beta-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood. (2016) 128:1884–93. doi: 10.1182/blood-2016-06-720581
60. Bae, JS , Yang, L , and Rezaie, AR . Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci U S A. (2007) 104:2867–72. doi: 10.1073/pnas.0611493104
61. Molinar-Inglis, O , Birch, CA , Nicholas, D , Orduña-Castillo, L , Cisneros-Aguirre, M , Patwardhan, A, et al. aPC/PAR1 confers endothelial anti-apoptotic activity via a discrete, β-arrestin-2-mediated SphK1-S1PR1-Akt signaling axis. Proc Natl Acad Sci U S A. (2021) 118:e2106623118. doi: 10.1073/pnas.2106623118
62. Soh, UJK , and Trejo, J . Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. (2011) 108:E1372–80. doi: 10.1073/pnas.1112482108
63. Bae, JS , Yang, L , Manithody, C , and Rezaie, AR . The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. (2007) 110:3909–16. doi: 10.1182/blood-2007-06-096651
64. Mosnier, LO , Sinha, RK , Burnier, L , Bouwens, EA , and Griffin, JH . Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. (2012) 120:5237–46. doi: 10.1182/blood-2012-08-452169
65. Griffin, JH , Zlokovic, BV , and Mosnier, LO . Activated protein C: biased for translation. Blood. (2015) 125:2898–907. doi: 10.1182/blood-2015-02-355974
66. Healy, LD , Rigg, RA , Griffin, JH , and McCarty, OJT . Regulation of immune cell signaling by activated protein C. J Leukoc Biol. (2018) 103:1197–203. doi: 10.1002/JLB.3MIR0817-338R
67. Sturn, DH , Kaneider, NC , Feistritzer, C , Djanani, A , Fukudome, K , and Wiedermann, CJ . Expression and function of the endothelial protein C receptor in human neutrophils. Blood. (2003) 102:1499–505. doi: 10.1182/blood-2002-12-3880
68. Feistritzer, C , Sturn, DH , Kaneider, NC , Djanani, A , and Wiedermann, CJ . Endothelial protein C receptor-dependent inhibition of human eosinophil chemotaxis by protein C. J Allergy Clin Immunol. (2003) 112:375–81. doi: 10.1067/mai.2003.1609
69. Galligan, L , Livingstone, W , Volkov, Y , Hokamp, K , Murphy, C , Lawler, M, et al. Characterization of protein C receptor expression in monocytes. Br J Haematol. (2001) 115:408–14. doi: 10.1046/j.1365-2141.2001.03187.x
70. Xue, M , Campbell, D , Sambrook, PN , Fukudome, K , and Jackson, CJ . Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J Invest Dermatol. (2005) 125:1279–85. doi: 10.1111/j.0022-202X.2005.23952.x
71. Bretschneider, E , Uzonyi, B , Weber, AA , Fischer, JW , Pape, R , Lotzer, K, et al. Human vascular smooth muscle cells express functionally active endothelial cell protein C receptor. Circ Res. (2007) 100:255–62. doi: 10.1161/01.RES.0000255685.06922.c7
72. Wang, J , Yang, L , Rezaie, AR , and Li, J . Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost. (2011) 9:1308–17. doi: 10.1111/j.1538-7836.2011.04331.x
73. Gleeson, EM , O’Donnell, JS , and Preston, RJ . The endothelial cell protein C receptor: cell surface conductor of cytoprotective coagulation factor signaling. Cell Mol Life Sci. (2012) 69:717–26. doi: 10.1007/s00018-011-0825-0
74. Gorbacheva, L , Davidova, O , Sokolova, E , Ishiwata, S , Pinelis, V , Strukova, S, et al. Endothelial protein C receptor is expressed in rat cortical and hippocampal neurons and is necessary for protective effect of activated protein C at glutamate excitotoxicity. J Neurochem. (2009) 111:967–75. doi: 10.1111/j.1471-4159.2009.06380.x
75. Zhao, P , Metcalf, M , and Bunnett, NW . Biased signaling of protease-activated receptors. Front Endocrinol Lausanne. (2014) 5:67. doi: 10.3389/fendo.2014.00067
76. Rajagopal, S , Rajagopal, K , and Lefkowitz, RJ . Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. (2010) 9:373–86. doi: 10.1038/nrd3024
77. Walker, FJ , Sexton, PW , and Esmon, CT . The inhibition of blood coagulation by activated protein C through the selective inactivation of activated factor V. Biochim Biophys Acta. (1979) 571:333–42. doi: 10.1016/0005-2744(79)90103-7
78. Leon, G , Rehill, AM , and Preston, RJS . The protein C pathways. Curr Opin Hematol. (2022) 29:251–8. doi: 10.1097/MOH.0000000000000726
79. Mosnier, LO , Gale, AJ , Yegneswaran, S , and Griffin, JH . Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. (2004) 104:1740–4. doi: 10.1182/blood-2004-01-0110
80. Schwarz, HP , Fischer, M , Hopmeier, P , Batard, MA , and Griffin, JH . Plasma protein S deficiency in familial thrombotic disease. Blood. (1984) 64:1297–300. doi: 10.1182/blood.V64.6.1297.1297
81. Griffin, JH . Clinical studies of protein C. Semin Thromb Hemost. (1984) 10:162–6. doi: 10.1055/s-2007-1004419
82. Gladson, CL , Scharrer, I , Hach, V , Beck, KH , and Griffin, JH . The frequency of type I heterozygous protein S and protein C deficiency in 141 unrelated young patients with venous thrombosis. Thromb Haemost. (1988) 59:18–22. doi: 10.1055/s-0038-1642558
84. Karayalcin, G , and Lanzkowsky, P . Plasma protein C levels in children with sickle cell disease. Am J Pediatr Hematol Oncol. (1989) 11:320–3.
85. Marfaing-Koka, A , Boyer-Neumann, C , Wolf, M , Leroy-Matheron, C , Cynober, T , and Tchernia, G . Decreased protein S activity in sickle cell disease. Nouv Rev Fr Hematol 1978. (1993) 35:425–30.
86. Peters, M , Plaat, BE , ten Cate, H , Wolters, HJ , Weening, RS , and Brandjes, DP . Enhanced thrombin generation in children with sickle cell disease. Thromb Haemost. (1994) 71:169–72.
87. Tam, DA . Protein C and protein S activity in sickle cell disease and stroke. J Child Neurol. (1997) 12:19–21. doi: 10.1177/088307389701200103
88. Westerman, MP , Green, D , Gilman-Sachs, A , Beaman, K , Freels, S , Boggio, L, et al. Antiphospholipid antibodies, proteins C and S, and coagulation changes in sickle cell disease. J Lab Clin Med. (1999) 134:352–62. doi: 10.1016/S0022-2143(99)90149-X
89. Schnog, JB , Mac Gillavry, MR , van Zanten, AP , Meijers, JC , Rojer, RA , Duits, AJ, et al. Protein C and S and inflammation in sickle cell disease. Am J Hematol. (2004) 76:26–32. doi: 10.1002/ajh.20052
90. Piccin, A , Eakins, E , Murphy, CV , Mod, WGM , Mcmahon, C , and Smith, O . Circulating microparticles, plasma protein C and free protein S levels in children with sickle cell Anemia. Blood. (2008) 112:1431. doi: 10.1182/blood.V112.11.1431.1431
91. Khanduri, U , Gravell, D , Christie, BS , Al Lamki, Z , Zachariah, M , and Cherian, E . Reduced protein C levels--a contributory factor for stroke in sickle cell disease. Thromb Haemost. (1998) 79:879–80. doi: 10.1055/s-0037-1615083
92. el-Hazmi, MAF , Warsy, AS , and Bahakim, H . Blood proteins C and S in sickle cell disease. Acta Haematol. (1993) 90:114–9. doi: 10.1159/000204390
93. Wright, JG , Malia, R , Cooper, P , Thomas, P , Preston, FE , and Serjeant, GR . Protein C and protein S in homozygous sickle cell disease: does hepatic dysfunction contribute to low levels? Br J Haematol. (1997) 98:627–31. doi: 10.1046/j.1365-2141.1997.2663083.x
94. Whelihan, MF , Lim, MY , Mooberry, MJ , Piegore, MG , Ilich, A , Wogu, A, et al. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost. (2016) 14:1941–52. doi: 10.1111/jth.13416
95. Kondreddy, V , Keshava, S , Esmon, CT , Pendurthi, UR , and Rao, LVM . A critical role of endothelial cell protein C receptor in the intestinal homeostasis in experimental colitis. Sci Rep. (2020) 10:20569. doi: 10.1038/s41598-020-77502-3
96. Maknitikul, S , Luplertlop, N , Grau, GER , and Ampawong, S . Dysregulation of pulmonary endothelial protein C receptor and thrombomodulin in severe falciparum malaria-associated ARDS relevant to hemozoin. PLoS One. (2017) 12:e0181674. doi: 10.1371/journal.pone.0181674
97. Lattenist, L , Ochodnicky, P , Ahdi, M , Claessen, N , Leemans, JC , Satchell, SC, et al. Renal endothelial protein C receptor expression and shedding during diabetic nephropathy. J Thromb Haemost. (2016) 14:1171–82. doi: 10.1111/jth.13315
98. Ren, D , Fedorova, J , Davitt, K , Van Le, TN , Griffin, JH , Liaw, PC, et al. Activated protein C strengthens cardiac tolerance to ischemic insults in aging. Circ Res. (2022) 130:252–72. doi: 10.1161/CIRCRESAHA.121.319044
99. Gu, JM , Katsuura, Y , Ferrell, GL , Grammas, P , and Esmon, CT . Endotoxin and thrombin elevate rodent endothelial cell protein C receptor mRNA levels and increase receptor shedding in vivo. Blood. (2000) 95:1687–93. doi: 10.1182/blood.V95.5.1687.005k08_1687_1693
100. Guo, Y , Uy, T , Wandersee, N , Scott, JP , Weiler, H , Holzhauer, S, et al. The protein C pathway in human and murine sickle cell disease: alterations in protein C, Thrombomodulin (TM), and endothelial protein C receptor (EPCR) at baseline and during acute vaso-occlusion. Blood. (2008) 112:538–202. doi: 10.1182/blood.V112.11.538.538
101. Gavins, FN , Russell, J , Senchenkova, EL , De Almeida, PL , Damazo, AS , Esmon, CT, et al. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood. (2011) 117:4125–33. doi: 10.1182/blood-2010-08-301366
102. Hazra, R , Chen, Q , Crosby, D , Lenhart, D , Tan, R , Kim, K, et al. Heme induced progressive loss of endothelial protein C receptor promotes development of chronic kidney disease in sickle cell disorders. Blood. (2021) 138:188. doi: 10.1182/blood-2021-154027
103. Hebbel, RP , Osarogiagbon, R , and Kaul, D . The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. (2004) 11:129–51. doi: 10.1080/mic.11.2.129.151
104. Zhang, D , Xu, C , Manwani, D , and Frenette, PS . Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. (2016) 127:801–9. doi: 10.1182/blood-2015-09-618538
105. Chen, G , Chang, J , Zhang, D , Pinho, S , Jang, JE , and Frenette, PS . Targeting Mac-1-mediated leukocyte-RBC interactions uncouples the benefits for acute vaso-occlusion and chronic organ damage. Exp Hematol. (2016) 44:940–6. doi: 10.1016/j.exphem.2016.06.252
106. Belcher, JD , Chen, C , Nguyen, J , Milbauer, L , Abdulla, F , Alayash, AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
107. Cleator, JH , Zhu, WQ , Vaughan, DE , and Hamm, HE . Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood. (2006) 107:2736–44. doi: 10.1182/blood-2004-07-2698
108. Matsui, NM , Borsig, L , Rosen, SD , Yaghmai, M , Varki, A , and Embury, SH . P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. (2001) 98:1955–62. doi: 10.1182/blood.V98.6.1955
109. Embury, SH , Matsui, NM , Ramanujam, S , Mayadas, TN , Noguchi, CT , Diwan, BA, et al. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood. (2004) 104:3378–85. doi: 10.1182/blood-2004-02-0713
110. Franchi, F , Rollini, F , Faz, G , Rivas, JR , Rivas, A , Agarwal, M, et al. Pharmacodynamic effects of Vorapaxar in prior myocardial infarction patients treated with potent Oral P2Y(12) receptor inhibitors with and without aspirin: results of the VORA-PRATIC study. J Am Heart Assoc. (2020) 9:e015865. doi: 10.1161/JAHA.120.015865
111. Francis, LRA , Millington-Burgess, SL , Rahman, T , and Harper, MT . Q94 is not a selective modulator of proteinase-activated receptor 1 (PAR1) in platelets. Platelets. (2022) 33:1090–5. doi: 10.1080/09537104.2022.2026911
112. Kuliopulos, A , and Covic, L . Blocking receptors on the inside: pepducin-based intervention of PAR signaling and thrombosis. Life Sci. (2003) 74:255–62. doi: 10.1016/j.lfs.2003.09.012
113. Covic, L , and Kuliopulos, A . Protease-activated receptor 1 as therapeutic target in breast, lung, and ovarian cancer: Pepducin approach. Int J Mol Sci. (2018) 19:2237. doi: 10.3390/ijms19082237
114. Kuliopulos, A , Gurbel, PA , Rade, JJ , Kimmelstiel, CD , Turner, SE , Bliden, KP, et al. PAR1 (protease-activated receptor 1) Pepducin therapy targeting myocardial necrosis in coronary artery disease and acute coronary syndrome patients undergoing cardiac catheterzation: a randomized, placebo-controlled, phase 2 study. Arter Thromb Vasc Biol. (2020) 40:2990–3003. doi: 10.1161/ATVBAHA.120.315168
115. Festoff, BW , and Dockendorff, C . The evolving concept of neuro-Thromboinflammation for neurodegenerative disorders and Neurotrauma: a rationale for PAR1-targeting therapies. Biomol Ther. (2021) 11:11. doi: 10.3390/biom11111558
116. Aisiku, O , Peters, CG , De Ceunynck, K , Ghosh, CC , Dilks, JR , Fustolo-Gunnink, SF, et al. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood. (2015) 125:1976–85. doi: 10.1182/blood-2014-09-599910
117. De Ceunynck, K , Peters, CG , Jain, A , Higgins, SJ , Aisiku, O , Fitch-Tewfik, JL, et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A. (2018) 115:E982–91. doi: 10.1073/pnas.1718600115
118. Subramaniam, S , Ogoti, Y , Hernandez, I , Zogg, M , Botros, F , Burns, R, et al. A thrombin-PAR1/2 feedback loop amplifies thromboinflammatory endothelial responses to the viral RNA analogue poly(I:C). Blood Adv. (2021) 5:2760–74. doi: 10.1182/bloodadvances.2021004360
Keywords: sickle cell disease, vaso-occlusion, inflammation, sickle cell anemia, protease activated receptor 1 (PAR1), endothelial protein C receptor (EPCR), activated protein C (APC), thrombin
Citation: Ramadas N and Sparkenbaugh EM (2023) The APC-EPCR-PAR1 axis in sickle cell disease. Front. Med. 10:1141020. doi: 10.3389/fmed.2023.1141020
Edited by:
Marcos De Lima, The Ohio State University, United StatesReviewed by:
Prasenjit Guchhait, Regional Centre for Biotechnology (RCB), IndiaCopyright © 2023 Ramadas and Sparkenbaugh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erica M. Sparkenbaugh, RXJpY2Ffc3BhcmtlbmJhdWdoQG1lZC51bmMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.