 Neza Molk1
Neza Molk1 Mojca Bitenc1Darja Urlep2,3
Mojca Bitenc1Darja Urlep2,3 Mojca Zerjav Tansek1,2Sara Bertok1,2
Mojca Zerjav Tansek1,2Sara Bertok1,2 Katarina Trebusak Podkrajsek3,4
Katarina Trebusak Podkrajsek3,4 Ursa Sustar1,4
Ursa Sustar1,4 Jernej Kovac2,4
Jernej Kovac2,4 Tadej Battelino1,2
Tadej Battelino1,2 Marusa Debeljak2,4
Marusa Debeljak2,4 Urh Groselj1,2*
Urh Groselj1,2*- 1Department of Pediatric Endocrinology, Diabetes and Metabolism, University Medical Center-University Children's Hospital, Ljubljana, Slovenia
- 2Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 3Department of Gastroenterology, Hepatology and Nutrition, University Children's Hospital Ljubljana, University Medical Center, Ljubljana, Slovenia
- 4Clinical Institute for Special Laboratory Diagnostics, University Children's Hospital, University Medical Center Ljubljana, Ljubljana, Slovenia
Background: Familial hypobetalipoproteinemia (FHBL) is an autosomal semi-dominant disorder usually caused by variants in the APOB gene that frequently interferes with protein length. Clinical manifestations include malabsorption, non-alcoholic fatty liver disease, low levels of lipid-soluble vitamins, and neurological, endocrine, and hematological dysfunction.
Methods: Genomic DNA was isolated from the blood samples of the pediatric patient with hypocholesterolemia and his parents and brother. Next-generation sequencing (NGS) was performed, and an expanded dyslipidemia panel was employed for genetic analysis. In addition, a systematic review of the literature on FHBL heterozygous patients was performed.
Case report: Genetic investigation revealed the presence of a heterozygous variant in the APOB (NM_000384.3) gene c.6624dup[=], which changes the open reading frame and leads to early termination of translation into the p.Leu2209IlefsTer5 protein (NP_000375.3). The identified variant was not previously reported. Familial segregation analysis confirmed the variant in the mother of the subject, who also has a low level of low-density lipoprotein and non-alcoholic fatty liver disease. We have introduced therapy that includes limiting fats in the diet and adding lipid-soluble vitamins E, A, K, and D and calcium carbonate. We reported 35 individuals with APOB gene variations linked to FHBL in the systematic review.
Conclusion: We have identified a novel pathogenic variant in the APOB gene causing FHBL in pediatric patients with hypocholesterolemia and fatty liver disease. This case illustrates the importance of genetic testing for dyslipidemias in patients with significant decreases in plasma cholesterol as we can avoid damaging neurological and ophthalmological effects by sufficient vitamin supplementation and regular follow-ups.
1. Introduction
Hypobetalipoproteinemias (HBLs) encompass a diverse range of disorders that are characterized by reduced levels of total cholesterol (TC), low-density lipoprotein-cholesterol (LDL-C), and apolipoprotein B (apoB) in the blood, which fall below the 5th percentile of the population distribution (1).
It may develop due to primary and secondary causes (1). Primary hypobetalipoproteinemia comprises various genetic conditions, such as abetalipoproteinemia (ABL) (genetic defect in MTP gene), chylomicron retention disease (genetic defect in SAR1B gene), and combined familial hypolipidemia (genetic defect in ANGPTL3 gene). These disorders share an autosomal recessive mode of inheritance. Another condition falling under primary hypobetalipoproteinemias is familial hypobetalipoproteinemia (FHBL) (2). The main secondary causes are malnutrition, intestinal fat malabsorption, severe liver disease, and hyperthyroidism (3).
Familial hypobetalipoproteinemia has an autosomal semi-dominant pattern of inheritance (4). Variants in the APOB gene are the main cause of FHBL. Additionally, less frequent variants in the PCSK9 gene have also been associated with the disease (5, 6). Most variants in the APOB gene cause the formation of truncated forms of apoB, which depending on the length may or may not be secreted into the plasma (7, 8). LDL-C levels in heterozygous FHBL are low but not absent (9). Due to the lower production rate, predicted apoB levels in FHBL heterozygotes would be 50% of normal; however, they are ~24% (10). Homozygous FHBL individuals commonly exhibit significantly low levels of total cholesterol (TC) in their bloodstream, with the presence of plasma apoB either completely absent or found only in minimal quantities (2).
We report a new heterozygous variant in the APOB gene identified in a patient whose symptoms were characteristic of FHBL. This case prompted us to carry out a systematic review to better define the genetic causes, already identified variants, and the clinical course in heterozygous FHBL.
2. Methods
2.1. Study design and family description
The patient has been followed regularly at the University Medical Centre Ljubljana. The medical records were used to acquire his clinical information. This report is a part of the research project approved by the National Medical Ethics Committee of Slovenia (#0120-273/2019/9). The principles of the Declaration of Helsinki were followed. Written informed consent using local consent forms was obtained from the parents of the patient for the publication of any data included in this article.
2.2. Liver enzymes, lipid profile, and fat-soluble vitamins analysis
Laboratory measurements were analyzed for lipids including TC, LDL-C, high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) using the direct enzymatic colorimetric method. Additionally, liver enzymes, including aspartate transaminase (AST) and alanine transaminase (ALT), and fat-soluble vitamins E, A, and D were measured.
2.3. Genetic analysis
Genetic analysis was performed at the University Children's Hospital Ljubljana in Slovenia. Using the FlexiGene DNA Kit 250, genomic DNA was extracted from peripheral blood samples (Qiagen, Hilden, Germany). Next-generation sequencing (NGS) was performed. Using TruSight One, Illumina, FC-14111006 (Illumina, San Diego, CA, United States), the regions of interest were enriched. Sequencing was performed on the MiSeq desktop sequencer together with MiSwq reagent kit v3 (Illumina, San Diego, CA, United States). Coding regions of 4,813 genes were analyzed. A panel of seven genes associated with hypocholesterolemia, including the APOB gene, was used for filtering variants. VarAFT tool was applied for annotation and filtration (11). The detected variants were classified according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG AMP) (12) classification criteria as (likely) benign, variants of uncertain significance (VUS), and (likely) pathogenic. The variant identified in the APOB gene was subsequently confirmed by Sanger sequencing.
2.4. Systematic literature review
We have collected accessible scientific report publications for the systemic review. A systematic literature review was performed on 20 June 2022, following PRISMA reporting guidelines. We searched the PubMed database for available case report articles on heterozygous patients with pathogenic and likely pathogenic variants in the APOB gene related to FHBL. The following search terms were used: “hypocholesterolemia” and “APOB”. In addition to that, we also searched Human Gene Variant Database Professional (13) and the Franklin by Genoox tool (14) based on the variant confirmed in our patient and using scope “Gene.” We found 63 articles. We read the abstracts and included all articles that (1) were published in English, (2) were fully accessible, (3) contained human data and clinical data on patients, and (4) described heterozygous cases. We excluded all articles that did not meet the criteria. All articles that met the criteria were read and analyzed in full-text form. In the end, 19 articles describing 35 cases were included in our systematic literature review. All data were collected in Microsoft Office 365 (Microsoft Corporation, Redmond, WA, USA). Systematic literature workflow is presented in Figure 4.
3. Case report
The proband was a male in his early twenties who was referred to our department for HBL at the age of 10 years. He was hospitalized in another hospital for gastroenterocolitis at the age of 7 years, where laboratory tests revealed severe hypobetalipoproteinemia. The family history revealed that his two siblings as well as his mother and his maternal grandmother all have low serum levels of LDL-C. His mother and sister have been diagnosed with celiac disease.
The patient was born after a fourth pregnancy, the second pregnancy ended with a miscarriage. He has had atopic dermatitis since birth and in the last years, allergic rhinitis has developed. The patient also reported getting tired very quickly, with unpleasant breath in the morning. On physical examination, he was quite high for his age with a body mass index of 17.5 kg/m2. Respiratory and cardiovascular examinations were normal. His abdomen was a little bit tender with a palpable liver of 2 cm under the rib cage and impalpable other organs or masses. Other physical findings included an asymmetric chest and irregular growth of teeth. After examination, several blood tests were performed, revealing mildly elevated levels of AST 54 IU/L (0.9 μkat/L) (<36.6 IU/L, <0.61 μkat/L) and ALT 100.8 IU/L (1.68 μkat/L) (<28.8 IU/L, <0.48 μkat/L) with normal gamma-glutamyl transferase (gGT) and bilirubin concentrations. Low levels of LDL-C of 0.4 mmol/L (2.0–3.5 mmol/L) and apoB of 0.2 (0.55–1.40 mmol/L) were found. Hormones and antibodies regarding any thyroid gland disorder were in a normal range. Coeliac disease was excluded. The proband was afterward regularly followed by our endocrinology department and by a gastroenterologist. Laboratory tests are chronologically presented in Figures 1–4.
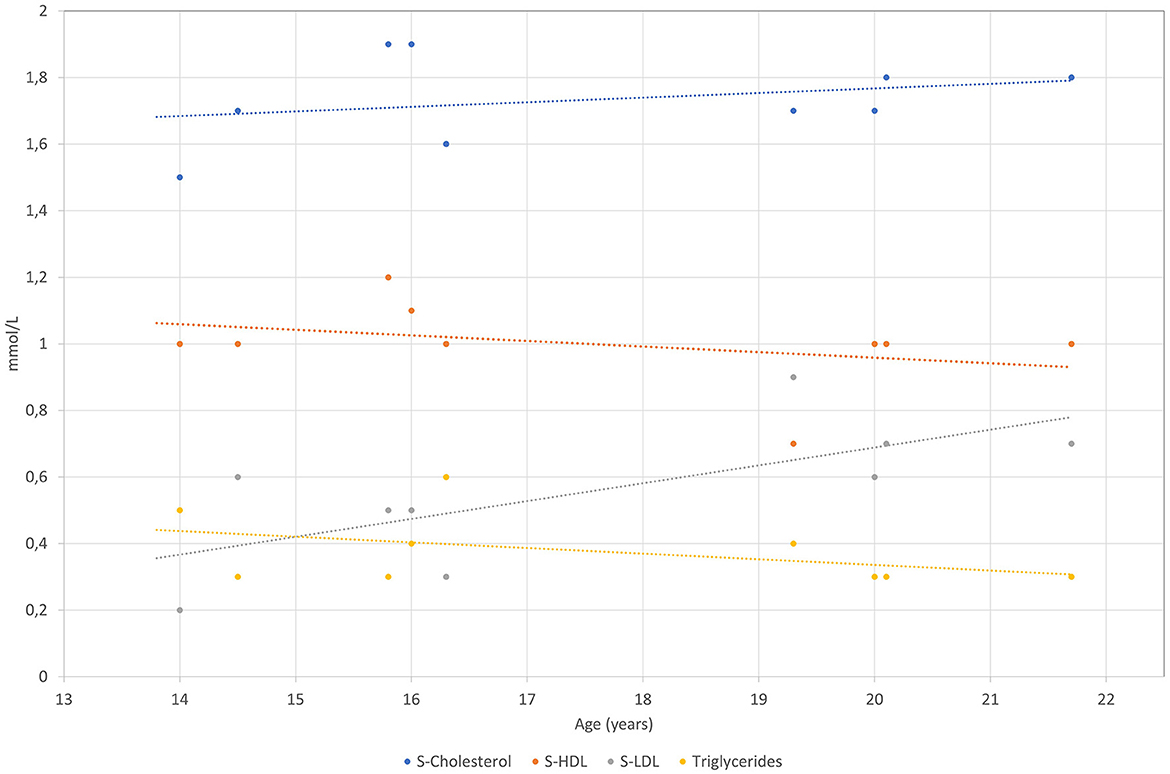
Figure 1. S-Cholesterol, S-HDL-C, S-LDL-C and S-TAG levels are presented over time for the patient measured in mmol/L.
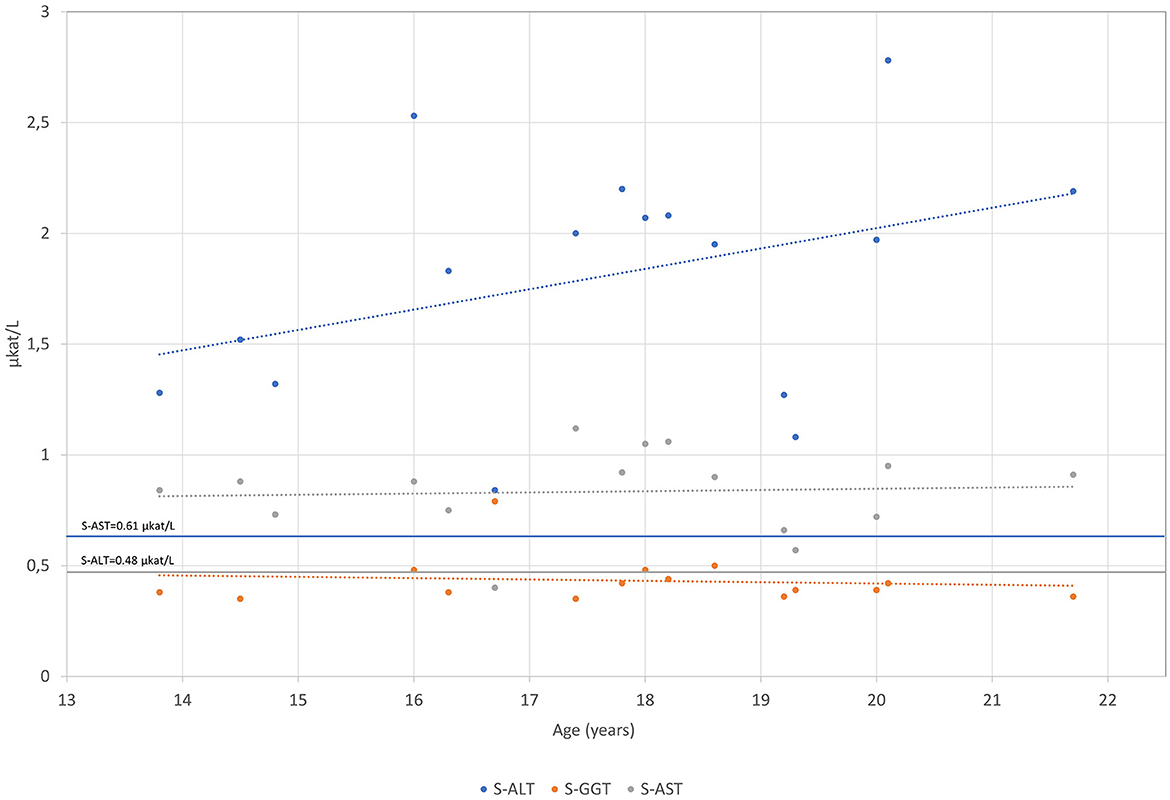
Figure 2. S-AST, S-ALT and S-GGT levels are presented over time for the patient measured in μkat/L.
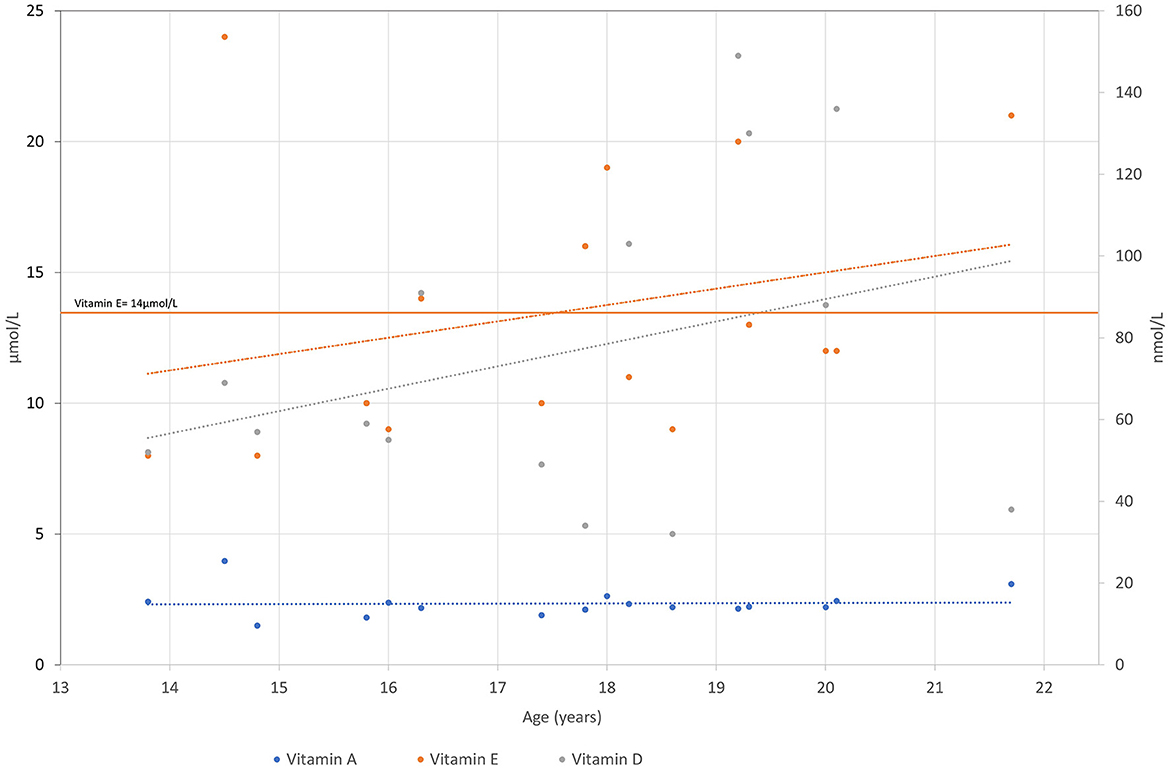
Figure 3. Vitamin A, E and D levels are presented over time for the patient. Vitamin A and E are measured in μkat/L and vitamin D in nmol/L.
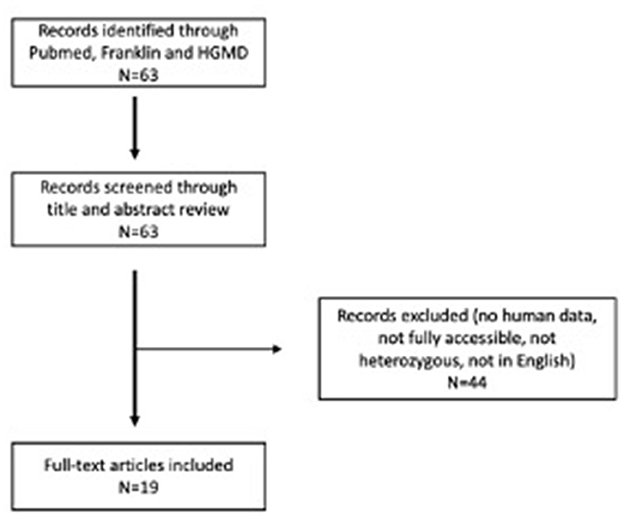
Figure 4. PRISMA flow diagram for systematic literature review.
The mildly elevated ALT and AST persisted until the end of the follow-up at our tertiary center. The concentration of ALT varied from 63.5 to 163.5 IU/L (1.08–2.78 μkat/L) and AST from 33.5 to 55.9 IU/L (0.57 to 0.95 μkat/L) (Figure 2). The concentrations of gGT and bilirubin were within normal limits. Abdominal ultrasound revealed a marginally enlarged liver and spleen (at the upper limit for age and gender). The gallbladder and bile ducts appeared normal. Other abdominal organs were without evident pathology. Magnetic resonance of the upper abdomen showed changes characteristic of hepatic steatosis. A liver biopsy was performed at the age of 16 years, which confirmed liver steatosis (steatosis was present in 50% of hepatocytes). There were no signs of liver cell injury (e.g., ballooning degeneration) or inflammation; however, there was evidence of mild fibrosis along the central veins and mild sinusoidal-pericellular fibrosis. Other causes of chronic liver diseases were excluded (e.g., viral liver disease, Wilson's disease, alpha-1-antitrypsin deficiency, autoimmune liver disease, hemochromatosis, drug-induced liver steatosis, and others). During the follow-up, the synthetic liver function was normal. There were no signs of portal hypertension or hypersplenism. The patient was without gastrointestinal symptoms throughout the follow-up, and he passed normally formed stools once or twice a day. Serological tests for coeliac disease were repeated several times and were always negative. He was not nourished, and his body mass index (BMI) ranged between 17.5 kg/m2 at the diagnosis of hypobetalipoproteinemia and 24.2 kg/m2 at the last follow-up examination at the age of 18 years. At that time, the values of ALT and AST were 74.7 IU/L (1.27 μkat/L) and 38.8 IU/L (0.66 μkat/L) respectively, with normal gGT and bilirubin concentrations, and there were no signs of portal hypertension on Doppler ultrasound of the portal veins.
Low levels of vitamin E were one of the major concerns regarding our proband. The blood test of proband aged 10 years revealed vitamin E levels of 13.6 μmol/L (14–23 μmol/L). He was prescribed a treatment of 10 mg of vitamin E per day and 1,000 IE of vitamin D per day. He was also treated with lecithin, complex B, and omega-3 fatty acids. As the measured vitamin E level did not improve, the dose was initially increased to 100 mg per day and later gradually to 1,000 mg per day to achieve adequate blood concentrations.
The first bone densitometry performed in 2011 was unremarkable (L1–L4 Z-score +0.3; whole body Z-score −0.3). He was prescribed calcium carbonate 1,000 mg per day and 1,000 IE of vitamin D per day due to osteopenia that was revealed on the second densitometry (L1–L4: Z-score −0.5; whole body Z-score −1.6).
During the follow-up, he was regularly followed by a clinical dietician. At the start of the follow-up, an analysis of the dietary diary revealed that the proband's intake was 2,000 kcal/day (recommended 3,000 kcal/day), fat intake was as recommended (<30% of total calories), but with long-chain fatty acids around 30 g/day, whereas 15 g/day are recommended for the patients with the HBL.
According to the following US of the abdomen in 2017, improvement was noticed. A liver biopsy revealed histologic changes in line with metabolic liver defects. It showed microvesicular steatosis in 50% of hepatocytes without steatohepatitis, fibrosis along the central veins, and mild sinusoidal-pericellular fibrosis. The first genetic analysis was performed in 2012. Exon 26 of the APOB gene was amplified by a polymerase chain reaction and sequenced directly to analyze the presence of variants NM_000384.3:c.10707C>T and NM_000384.3:c.10708G>A. Analyzed variants were not confirmed, thus FHBL could not be confirmed as the cause for the clinical manifestation of the disease. The diagnosis was confirmed after a second genetic analysis was performed using NGS. In the APOB (OMIM: +107730) gene, heterozygous variant c.6624dup has been identified, which we then confirmed with Sanger sequencing. The variant is not found in dbSNP, HGMD, or gnomAD databases. The in silico prediction tools report variants as pathogenic (CADD (https://cadd.gs.washington.edu/), Variant Taster). The variant is classified as pathogenic (PVS1 very strong, PM2 moderate, and PP1 supporting) according to the ACMG-AMP criteria (12). Thus, this variant was considered causal for the clinical manifestations. Later, Sanger sequencing-based segregation analysis confirmed the presence of this variant in proband's mother, but it was not confirmed in proband's brother and father. A family peedigree with c.6624dup p.Leu2209IlefsTer5 variant in the APOB gene is presented in Figure 5. We were unable to perform a genetic analysis on his sister who has normal cholesterol levels. In 2019, we expended genetic testing which revealed the variant in the COL2A1 gene (NM_001844.5) c.375+1G>A[=], confirming the Stickler syndrome.
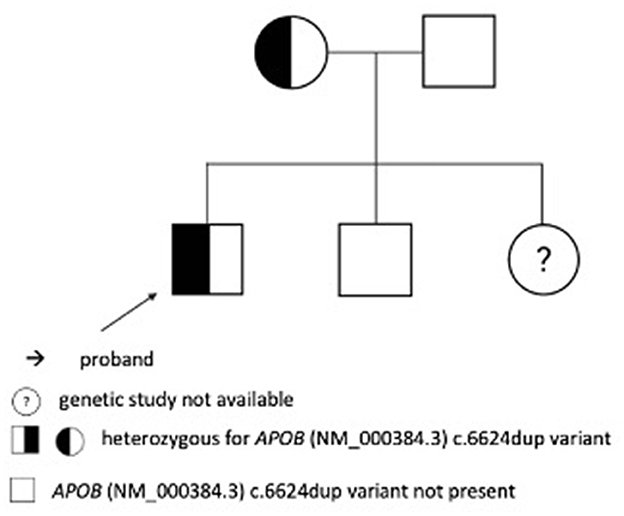
Figure 5. A family pedigree with c.6624dup p.Leu2209IlefsTer5 variant in the APOB gene.
In the next follow-ups in 2018 and 2019, treatment and diet remained the same. The US of the abdomen in 2019 showed regression of the steatohepatitis compared with the previous US of the abdomen at the beginning of the same year, which showed moderate hepatosplenomegaly and hepatopathy. Vitamin E level was still at the lower limit of the normal range, and the supplementation dose was increased to 2,000 mg/day. The last follow-up visit was in 2022. Apart from elevated liver enzymes with AST 53.5 IU/L (0.91 μkat/L) (<35.9 IU/L, <0.61 μkat/L) and ALT 128.8 IU/L (2.19 μkat/L) (<28.2 IU/L, <0.48 μkat/L), everything else remained stable and treatment remained unchanged. All biochemical measurements are collected and presented in Supplementary Table 1 and on the Mendeley Data Repository https://doi.org/10.17632/sm84p6vp26.1.
4. Discussion
Our study presented the results of a follow-up conducted over an extended period for a proband carrying a new variant in the APOB gene, which is associated with FHBL.
FHBL type 1 (OMIM: #615558) is a semi-dominant disorder mainly caused by protein-truncating variants (PTVs) in the APOB gene (15). In 1979, Steinberg and his colleagues were the first to identify a distinct condition characterized by hypobetalipoproteinemia and normal triglyceridemia. Young et al. described the first kindred with two distinct abnormal APOB alleles associated with FHBL (16). Multiple following kindreds have been documented since then, which has allowed the identification of numerous gene deletions, single nucleotide substitutions, and splicing variants.
The APOB gene, positioned on chromosome 2p24.1, is composed of 29 exons. The same gene produces two forms of protein, apoB-48 and apoB-100. ApoB-48 is produced by tissue-specific mRNA C → U RNA editing at nucleotide position 6666 which causes premature stop codon. The modification results in the production of apoB-48, representing 48% of the amino terminus of apoB-100 (17). ApoB-48 is produced by the small intestine and lacks the LDL-receptor binding domain, but it plays a crucial role in chylomicron synthesis (18, 19). Full-length apoB-100 is produced in the liver. It plays a role as a structural component of very low-density lipoprotein (VLDL) and a ligand for receptor-mediated endocytosis of low-density lipoprotein (LDL).
Due to their enormous size and hydrophobicity, apoB-100 and apoB-48's 3D structures have not yet been fully characterized at the anatomic level. However, attempts have been made to determine the structure of the apoB-100 domains using different algorithms (20). The modeled apoB-100 protein is composed of five domains NH3-βα1-β1-α2-β2-α3-COOH. The initial assembly of triglyceride-rich lipoproteins in enterocytes and hepatocytes depends on the N-terminal βα1 superdomain (N-terminal amino acids 1–827), which has both α-helical and β-sheet secondary structures. β1 domain, which is located roughly between amino residues 827 and 2000, is involved in irreversible lipid-binding, while the β2 provides LDL receptor-binding abilities (21). The α2 domain, located roughly between amino residues 2,075 and 2,575, and the α3 domain, located roughly between amino residues 4,100 and 4,550, form amphipathic α -helices with both hydrophilic and hydrophobic characteristics. They provide a flexible area that gives the molecule elasticity and enables the recruitment of varying amounts of core lipids (22). ApoB-48 is composed of the domains βα1, β1, and a portion of the domain α2 (20).
The majority of the FHBL-causing variants identified interfere with the development of a complete apoB-100 molecule (19). The size and density of lipoprotein particles depend on the length of apoB truncation. Patients with variants causing truncated proteins have lower plasma levels of apoB possibly because of lower apoB production and higher clearance rate (23). The ability of apoB to create plasma lipoproteins in the liver or intestine and export lipids from these organs is lost when apoB-48 is in an inadequate form (24). The severity of symptoms is related to truncation lengths; long-truncated apoBs continue to have some lipid-binding ability, whereas truncations that are shorter than apoB-29/30 are primarily degraded within cells and are not released as part of lipoprotein particles. Truncations shorter than apoB-29/30 lead to a phenotype similar to ABL, characterized by neurologic dysfunctions, fatty liver, acanthocytosis, a lack of fat-soluble vitamins, and fat malabsorption (1, 25). Carriers with longer truncations are less affected (e.g., longer than apoB-75) as they still have some capacity to bind lipids and form lipoprotein particles that are then released into the bloodstream. However, in the presence of certain factors, such as a high-fat diet, alcohol consumption, or obesity, individuals with longer truncated apoBs may still develop fatty liver. Most people with heterozygous FHBL do not experience any symptoms but may have mild liver dysfunction and hepatic steatosis. However, around 5–10% of them may develop more severe non-alcoholic steatohepatitis that requires medical treatment and, in rare cases, may progress to cirrhosis (26). Other symptoms observed in heterozygous patients include increased stool frequency, chronic steatorrhea, mild vitamin deficiencies, and malabsorption, particularly after consuming high-fat meals (27).
The clinical severity of FHBL in individuals who are homozygous or compound heterozygous for apoB truncations is determined by the ability of truncated apoBs to bind lipids and form lipoprotein particles. FHBL homozygotes or compound heterozygotes carrying apoB truncations, that result in both alleles encoding truncated apoBs shorter than ApoB-29/30, do not have detectable apoB in their bloodstream. For individuals who are homozygous or compound heterozygous for truncations longer than apoB-50, the clinical phenotype can vary widely. In severe cases, it may lead to neurological complications due to the malabsorption of vitamin E (28). The malabsorption of fat can be managed through dietary modifications, such as limiting the consumption of long-chain fatty acids, and supplementing fat-soluble vitamins (1).
In Supplementary Table 2, we conducted a literature review on the variants in the APOB gene that have been observed in heterozygous FHBL patients so far. Our review includes 19 articles describing 35 cases. The most common type of variants observed are substitutions in exons (43%). Truncation lengths vary from 6.46–83% of the full apoB-100 length. Among the reported symptoms, fatty liver was described in 57% of cases, and it was also exhibited in our proband.
We report a novel single nucleotide duplication [NM_000384.3:c.6624dup[=]] in exon 26 of the APOB gene, which was found in a family with HBL. This variant causes a frameshift in translation p.Leu2209IlefsTer5 protein (NP_000375.3), early termination, and results in a truncated protein. In our case, frameshift causes termination codon TGA at the 2213th amino acid. This represents 48.5% of full apoB length or intermediate size truncation which is longer than the wild-type counterpart. This variant is located in α2 based on protein structure. Previous studies have shown that individuals who are FHBL heterozygotes and carry truncated apoBs have lower-than-expected levels of plasma LDL-C and apoB, which are approximately one-third of normal levels. This is due to a combination of reduced hepatic secretion of apoB-100, increased catabolism of VLDL, and decreased secretion of the truncated apoBs (29). Martín-Morales et al. presented a heterozygous case with the Ser2184fsVal2193X variant that produces a protein apoB-48.32 (a truncated apoB comparable in size to that found in our kindred) (2013). Like our proband, their case also showed symptoms of steatorrhea, fatty liver, and low concentrations of TC, LDL-C, and apoB. In addition, we also discovered the variant in the COL2A1 gene (NM_001844.5) c.375+1G>A[=], confirming the Stickler syndrome. Stickler syndrome affects connective tissue and can involve multiple systems in the body, including the inner ear, eyes, joints, and skeleton. The disorder is characterized by a range of symptoms, including myopia (nearsightedness), vitreoretinal degeneration, cataracts, and retinal detachment at a young age (30). These eye manifestations were also observed in the case being discussed. Since some of FHBL and Stickler syndrome symptoms overlap, we cannot solely attribute all the characteristics observed in our proband to the variant discovered in the APOB gene.
According to previous studies, patients with FHBL have been shown to have lower levels of lipid-soluble vitamins E, A, K, and D in their cellular membranes. Vitamin A plays a crucial role in phototransduction and vitamin E is important for normal retinal function. Vitamin E transport is hindered in FHBL due to the fact that it heavily relies on transport through chylomicrons and other particles that contain apo B, which are impaired in FHBL (8). Supplementation of vitamin E can prevent the onset of retinopathy, or if it has already developed, it can halt its progression (31). Homozygous FHBL patients may present neurological and ophthalmological abnormalities, which include progressive spinocerebellar degeneration, areflexia, ataxia, and retinitis pigmentosa due to lipid-soluble vitamins deficiency (4). They usually experience neurological symptoms in their first or second decade of life. Early identification of these conditions through an effective screening program is crucial, as proper treatment can prevent adverse neurological and ophthalmological outcomes (2).
The current standard treatment involves dietary restrictions and vitamin supplementation. It is recommended to consider supplementation when low levels of lipid-soluble vitamins are detected. Vitamin E doses should be close to the recommended daily intake (15 mg/day), with tolerable upper intake levels of 1,000 mg/day. In our case, our patient was prescribed up to 2,000 mg/day due to irregular intake. To prevent ophthalmologic complications, high doses of vitamin A (100–400 IU/kg/day) should be given if a deficiency is detected. Additionally, it is recommended to supplement vitamin D (800–1,200 IU/day) in all patients. Due to malabsorption, it is recommended to reduce long-chain fatty acids in the diet. Regular clinical judgment is recommended for all homozygous patients and heterozygous patients with vitamin deficiencies and fatty liver disease. Periodic transaminase tests and abdominal ultrasound can be used to monitor hepatic function. Follow-up evaluations for older patients include ophthalmologic assessments to detect atypical retinitis pigmentosa and tests to measure bone mineral density to check for osteopenia (2). Our patient was managed with vitamin E, vitamin D3, calcium carbonate, lecithin, complex B, and omega-3 fatty acids. Doses were adjusted based on the deficiency.
In conclusion, early detection and treatment of patients with FHBL are of great importance since damaging neurological and ophthalmological effects may be avoided by following up with sufficient vitamin supplementation. Early detection is possible through genetic testing of subjects showing symptoms of malabsorption and a decrease in plasma cholesterol or, ideally, through a screening program for familial hypercholesterolemia (32–35). Our proband had a novel heterozygous APOB variant causing significant liver steatosis, malabsorption of fat-soluble vitamins, and osteopenia. Treatment with sufficient vitamins and an adequate diet helped at limiting disease symptoms progression (36).
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the National Medical Ethics Committee (#0120-273/2019/9). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
NM and MB collected the data and prepared the first draft of the manuscript. MZ, DU, and UG followed and treated the patient, performed follow-up tests, and collected the data. MD, SB, US, and JK performed the molecular genetic analysis and data analysis with interpretation. UG supervised the work and helped write the manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Funding
The study was partly funded by the Slovenian Research Agency Program P3-0343 and Project J3-2536.
Acknowledgments
We would like to thank the patient and his family, and all the other physicians and nurses involved in the management and treatment of the patient.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1106441/full#supplementary-material
References
1. Tarugi P, Averna M. Hypobetalipoproteinemia: genetics, biochemistry, and clinical spectrum. Adv Clin Chem. (2011) 54:81–107. doi: 10.1016/B978-0-12-387025-4.00004-2
2. Moutzouri E, Elisaf M, Liberopoulos EN. Hypocholesterolemia. Curr Vasc Pharmacol. (2011) 9:200–12. doi: 10.2174/157016111794519354
3. Yalouris A. Hypocholesterolemia: a blessing or a problem? Hosp Chronicles. (2020) 15:31. doi: 10.2015/hc.v15i1.891
4. Bredefeld C, Hussain MM, Averna M, Black DD, Brin MF, Burnett JR, et al. Guidance for the diagnosis and treatment of hypolipidemia disorders. J Clin Lipidol. (2022) 16:797–812. doi: 10.1016/j.jacl.2022.08.009
5. Cefalù AB, Pirruccello JP, Noto D, Gabriel S, Valenti V, Gupta N, et al. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscl Thromb Vasc Biol. (2013) 33:2021–5. doi: 10.1161/ATVBAHA.112.301101
6. Tarugi P, Averna M. Genetics of familial hypobetalipoproteinemia. Fut Lipidol. (2007) 2:615–624. doi: 10.2217/17460875.2.6.615
7. Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol. (2014) 61:891–902. doi: 10.1016/j.jhep.2014.05.023
8. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol. (2014) 25:161–8. doi: 10.1097/MOL.0000000000000072
9. Bredefeld C, Peretti N, Hussain MM, Di Filippo M, Granot E, Cuerq C, et al. New classification and management of abetalipoproteinemia and related disorders. Gastroenterology. (2021) 160:1912–6. doi: 10.1053/j.gastro.2020.11.040
10. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia: liver disease and cardiovascular disease. Curr Opin Lipidol. (2020) 31:49–55. doi: 10.1097/MOL.0000000000000663
11. Desvignes JP, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, et al. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. (2018) 46:W545–53. doi: 10.1093/nar/gky471
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
13. Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. (2020) 139:1197–207. doi: 10.1007/s00439-020-02199-3
14. Genoox F. The Future of Genomic Medicine. (2022). Available online at: https://franklin.genoox.com/clinical-db/home (accessed June 20, 2022).
15. Tada H, Okada H, Nomura A, Nohara A, Takamura M, Kawashiri M-A. A healthy family of familial hypobetalipoproteinemia caused by a protein-truncating variant in the PCSK9 gene. Intern Med. (2020) 59:783–7. doi: 10.2169/internalmedicine.3737-19
16. Young SG, Bertics SJ, Curtiss LK, Dubois BW, Witztum JL. Genetic analysis of a kindred with familial hypobetalipoproteinemia. Evidence for two separate gene defects: one associated with an abnormal apolipoprotein B species, apolipoprotein B-37; and a second associated with low plasma concentrations of apolipop. J Clin Invest. (1987) 79:1842–51. doi: 10.1172/JCI113026
17. Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, Scott J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell. (1987) 50:831–40. doi: 10.1016/0092-8674(87)90510-1
18. Fazio S, Linton MF. Clinical Lipidology: A Companion to Braunwald's Heart Disease. Elsevier (2009).
19. Whitfield AJ, Barrett PHR, Van Bockxmeer FM, Burnett JR. Lipid disorders and mutations in the APOB gene. Clin Chem. (2004) 50:1725–32. doi: 10.1373/clinchem.2004.038026
20. Jeiran K, Gordon SM, Sviridov DO, Aponte AM, Haymond A, Piszczek G, et al. A new structural model of apolipoprotein b100 based on computational modeling and cross linking. Int J Mol Sci. (2022) 23:11480. doi: 10.3390/ijms231911480
21. Wang L, Walsh MT, Small DM. Apolipoprotein B is conformationally flexible but anchored at a triolein/water interface: a possible model for lipoprotein surfaces. Proc Natl Acad Sci USA. (2006) 103:6871–6. doi: 10.1073/pnas.0602213103
22. Salter JD, Bennett RP, Smith HC. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci. (2016) 41:578–94. doi: 10.1016/j.tibs.2016.05.001
23. Davidson NO. Genetic regulation of intestinal lipid transport and metabolism. In:Said HM, , editor. Physiology of the Gastrointestinal Tract (Sixth Edition). Academic Press (2018). p. 1109–31. doi: 10.1016/B978-0-12-809954-4.00049-9
24. Di Costanzo A, Di Leo E, Noto D, Cefalù AB, Minicocci I, Polito L, et al. Clinical and biochemical characteristics of individuals with low cholesterol syndromes: a comparison between familial hypobetalipoproteinemia and familial combined hypolipidemia. J Clin Lipidol. (2017) 11:1234–42. doi: 10.1016/j.jacl.2017.06.013
25. Hooper AJ, van Bockxmeer FM, Burnett JR. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit Rev Clin Lab Sci. (2005) 42:515–45. doi: 10.1080/10408360500295113
26. Vilar-Gomez E, Gawrieh S, Liang T, McIntyre AD, Hegele RA, Chalasani N. Interrogation of selected genes influencing serum LDL-Cholesterol levels in patients with well characterized NAFLD. J Clin Lipidol. (2021) 15:275–91. doi: 10.1016/j.jacl.2020.12.010
27. Martín-Morales R, García-Díaz JD, Tarugi P, González-Santos P, Saavedra-Vallejo P, Magnolo L, et al. Familial hypobetalipoproteinemia: analysis of three Spanish cases with two new mutations in the APOB gene. Gene. (2013) 531:92–6. doi: 10.1016/j.gene.2013.08.049
28. Schonfeld G, Lin X, Yue P. Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci. (2005) 62:1372–8. doi: 10.1007/s00018-005-4473-0
29. Tarugi P, Averna M, Di Leo E, Cefalu AB, Noto D, Magnolo L, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. (2007) 195:e19–27. doi: 10.1016/j.atherosclerosis.2007.05.003
30. Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. Seattle, Seattle, WA: University of Washington (1993). Available online at: http://europepmc.org/abstract/MED/20301479 (accessed January 15, 2023).
31. Burnett JR, Hooper AJ, Hegele RA. APOB-Related Familial hypobetalipoproteinemia. In: GeneReviews®. Seattle, WA: University of Washington (1993).
32. Gidding SS, Wiegman A, Groselj U, Freiberger T, Peretti N, Dharmayat KI, et al. Paediatric familial hypercholesterolaemia screening in Europe: public policy background and recommendations. Eur J Prev Cardiol. (2022) 29:2301–2311. doi: 10.1093/eurjpc/zwac200
33. Groselj U, Wiegman A, Gidding SS. Screening in children for familial hypercholesterolaemia: start now. Eur Heart J. (2022) 43:3209–12. doi: 10.1093/eurheartj/ehac224
34. Sustar U, Groselj U, Trebusak Podkrajsek K, Mlinaric M, Kovac J, Thaler M, et al. Early discovery of children with lysosomal acid lipase deficiency with the universal familial hypercholesterolemia screening program. Front Genet. (2022) 13:936121. doi: 10.3389/fgene.2022.936121
35. Sustar U, Kordonouri O, Mlinaric M, Kovac J, Arens S, Sedej K, et al. Universal screening for familial hypercholesterolemia in 2 populations. Genet Med. (2022) 24:2103–11. doi: 10.1016/j.gim.2022.06.010
Keywords: APOB, APOB gene, familial hypobetalipoproteinemia, nonalcoholic steatohepatitis, hypocholesterolemia, pediatric, fatty liver, systematic review
Citation: Molk N, Bitenc M, Urlep D, Zerjav Tansek M, Bertok S, Trebusak Podkrajsek K, Sustar U, Kovac J, Battelino T, Debeljak M and Groselj U (2023) Non-alcoholic fatty liver disease in a pediatric patient with heterozygous familial hypobetalipoproteinemia due to a novel APOB variant: a case report and systematic literature review. Front. Med. 10:1106441. doi: 10.3389/fmed.2023.1106441
Received: 23 November 2022; Accepted: 09 May 2023;
Published: 13 June 2023.
Edited by:
Julia Wattacheril, Columbia University, United StatesReviewed by:
Antonina Giammanco, University of Palermo, ItalyAlbert Wiegman, Amsterdam University Medical Center, Netherlands
Copyright © 2023 Molk, Bitenc, Urlep, Zerjav Tansek, Bertok, Trebusak Podkrajsek, Sustar, Kovac, Battelino, Debeljak and Groselj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Urh Groselj, dXJoLmdyb3NlbGpAa2Nsai5zaQ==