
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 18 April 2023
Sec. Hematology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1101501
This article is part of the Research Topic Developments in Sickle Cell Disease Therapy and Potentials for Gene Therapy View all 17 articles
 John D. Belcher1*
John D. Belcher1* Selvaraj Nataraja2
Selvaraj Nataraja2 Fuad Abdulla1
Fuad Abdulla1 Ping Zhang1
Ping Zhang1 Chunsheng Chen1
Chunsheng Chen1 Julia Nguyen1
Julia Nguyen1 Conglin Ruan1
Conglin Ruan1 Maneet Singh2†
Maneet Singh2† Shilpa Demes3
Shilpa Demes3 Lyndsay Olson2
Lyndsay Olson2 Domi Stickens2†Jeff Stanwix2Emer Clarke4Yongzhao Huang4Margaret Biddle2†
Domi Stickens2†Jeff Stanwix2Emer Clarke4Yongzhao Huang4Margaret Biddle2† Gregory M. Vercellotti1
Gregory M. Vercellotti1In sickle cell disease (SCD), heme released during intravascular hemolysis promotes oxidative stress, inflammation, and vaso-occlusion. Conversely, free heme can also activate expression of antioxidant and globin genes. Heme binds to the transcription factor BACH1, which represses NRF2-mediated gene transcription. ASP8731, is a selective small molecule inhibitor of BACH1. We investigated the ability of ASP8731 to modulate pathways involved in SCD pathophysiology. In HepG2 liver cells, ASP8731 increased HMOX1 and FTH1 mRNA. In pulmonary endothelial cells, ASP8731 decreased VCAM1 mRNA in response to TNF-α and blocked a decrease in glutathione in response to hemin. Townes-SS mice were gavaged once per day for 4 weeks with ASP8731, hydroxyurea (HU) or vehicle. Both ASP8731 and HU inhibited heme-mediated microvascular stasis and in combination, ASP8731 significantly reduced microvascular stasis compared to HU alone. In Townes-SS mice, ASP8731 and HU markedly increased heme oxygenase-1 and decreased hepatic ICAM-1, NF-kB phospho-p65 protein expression in the liver, and white blood cell counts. In addition, ASP8731 increased gamma-globin expression and HbF+ cells (F-cells) as compared to vehicle-treated mice. In human erythroid differentiated CD34+ cells, ASP8731 increased HGB mRNA and increased the percentage of F-cells 2-fold in manner similar to HU. ASP8731 and HU when given together induced more HbF+ cells compared to either drug alone. In CD34+ cells from one donor that was non-responsive to HU, ASP8731 induced HbF+ cells ~2-fold. ASP8731 and HU also increased HBG and HBA, but not HBB mRNA in erythroid differentiated CD34+ cells derived from SCD patients. These data indicate that BACH1 may offer a new therapeutic target to treat SCD.
Sickle cell disease (SCD) is caused by a point mutation in the β-globin chain of hemoglobin, resulting in a valine substitution for glutamate at position 6 resulting in the mutation of hemoglobin A (HbA, α2/β2) to hemoglobin S (HbS, α2/βS2). When deoxygenated during transit through the venous circulation, HbS polymerizes, which leads to the formation of red blood cells (RBCs) that are dehydrated, stiffer, more adhesive, and abnormally shaped (1). Repeated bouts of HbS polymerization and depolymerization as RBCs circulate through the venous and arterial vasculature shortens their lifespan and promotes intravascular and extravascular hemolysis that releases toxic free heme. Free heme promotes oxidative stress, inflammation, and vaso-occlusion (2, 3).
Despite its toxicity, free heme also activates antioxidant and globin gene expression (4–9). Heme binds to BTB and CNC homolog 1 (BACH1), which functions as a transcriptional repressor of nuclear factor erythroid 2-related factor 2 (NRF2) (10). The release of heme-BACH1 from antioxidant response elements (ARE) permits the binding of NRF2 to ARE and the cell-specific transcription of antioxidant genes such as HMOX1 (heme oxygenase 1, HO-1), GR (glutathione reductase), and NQO1 (NAD(P)H dehydrogenase [quinone] 1). Increasing HO-1 expression has been shown to reduce inflammation, adhesion molecules, and stasis in a mouse model of SCD (11, 12). Additionally, polymorphisms in the HMOX1 promoter that correspond to increased HO-1 levels have been correlated with reduced incidents of acute chest syndrome in pediatric SCD patients and overall reductions in vaso-occlusive pain crises and hospitalization rates (13, 14).
Previous studies demonstrated increases in nuclear Nrf2 expression and downstream expression of Nrf2-responsive genes including Hmox1, Nqo1, and Hbg (gamma globin) after administration of dimethyl fumarate (DMF) to murine SCD models (15, 16). Importantly, heme-induced microvascular stasis was inhibited in an HO-1 dependent manner (15). In addition, DMF increased the expression of other NRF2-responsive genes including proteins involved in hemoglobin, heme, and iron clearance as well as a decrease in markers of inflammation such as nuclear factor kappa B (NF-ĸB) phospho-p65, toll-like receptor 4 (TLR4), adhesion molecules, and pro-inflammatory cytokines (3, 15, 16). A decrease in hepatic lesions and increased circulating hemoglobin F (HbF)-containing RBCs (F-cells) was also observed in these studies (3, 15, 16). Conversely, a loss of NRF2 function exacerbates SCD pathophysiology and inhibits HbF expression (17, 18).
ASP8731 (previously known as ML-0207) was identified as a selective small molecule BACH1 inhibitor that relieves BACH1 repression of NRF2 pathways in human and murine cells (19). We investigated the capability of ASP8731 to increase antioxidant and anti-inflammatory gene expression in cell culture, decrease microvascular stasis (vaso-occlusion) and white blood cell (WBC) counts, and induce gamma globin and F-cells in a preclinical murine model of SCD and human CD34 cells during erythroid differentiation.
Animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Minnesota. These studies used male and female knock-in Townes-sickle (HbSS) mice (hα/hα, hγA/hγA, hβS/hβS) on a 129/B6 mixed genetic background (20). All mice were genotyped, housed in specific pathogen-free rooms to limit infections, and kept on a 12-h (h) light/dark cycle at 21°C. All animals were monitored daily for health problems, food and water levels, and cage conditions. All animals were included in each analysis and there were no adverse events that required changes to the protocol. Mice were 12–16 weeks of age.
ASP8731 is a selective small molecule BACH1 inhibitor, which activates antioxidant NRF2-responsive genes (19). We examined the cytoprotective effects of ASP8731 on human cells and in Townes HbSS mice. Mice were gavaged (10 ml/kg) once daily for 14 days consecutively or 6 days per week for 4 weeks with the indicated dose of ASP8731 or hydroxyurea (HU) suspended in 0.05% w/v Tween 80 + 0.45% hydroxypropyl methylcellulose (HPMC).
At the end of each experiment, mice were euthanized in a CO2 atmosphere and blood was collected from the inferior vena cava into EDTA tubes and processed at 4°C. Livers were excised, flash frozen in liquid N2, and stored at -85°C until use.
Human hepatoma cells (HepG2, ATCC HB-8065) were grown in Minimum Essential Media (EMEM Corning #MT-10-010-CV) supplemented with 1X MEM Non-essential Amino Acids Solution (Gibco 100X Solution #11140–50), 1 mM sodium pyruvate (Gibco 100 mM solution, #MT-25-000-CI) and 10% heat-inactivated fetal bovine serum (Gibco #16140–071). Cells were spilt 2–3 times weekly at 1:4 or 1:6. HepG2 cells were plated at a density of 250 k cells in 12-well plates coated with collagen (ENZO Life Sciences, Cat#ALX-522-440-0050). Approximately 24 h post-seeding, cells were treated with a dose titration of ASP8731 or DMF (Fisher Scientific Cat#50–144-5120) as a positive control via media change at three biological replicates per treatment. Compounds were dissolved in 0.1% DMSO, untreated wells had the same concentration of DMSO to account for any effect of the vehicle. At 24 h post-treatment, cells were washed in PBS and frozen (-80°C) before RNA isolation and analysis as described below. Further, in independent experiment, cells were checked for treatment toxicity by MTT assay (no toxicity observed- data not shown).
Human primary pulmonary arterial endothelial cells (PAEC, Lonza CC-2530) were grown in EGM-2 media (Lonza CC-3162). Cells were sub-cultured when they reached 70–85% confluence. Cells were seeded at 5 K viable cells/cm2. HPAEC were plated at 200 K/cells per well on 12-well plates (TrueLine #TR5001) in growth media and cultured at 37°C in 5% CO2. Approximately 24 h post-seeding, cells were treated with dose titration of ASP8731 in 0.1% DMSO via media change, control cells had the same amount of DMSO (n = 3 replicates per treatment). At 24 h post-treatment with ASP8731, TNF-α (10 ng/ml, Invitrogen) or hemin (50 μM, Sigma-Aldrich) was added to the media and the cells were incubated an additional 4 h (TNF-α) or 30 min (hemin). Treatment of 50 uM hemin for 30 min was determined to be the minimal dose that elicits a decrease in GSH, and is within the physiological levels reported in SCD and thalassemia patients. After treatment was complete, media was aspirated off, cells were washed with PBS and aspirated dry. For the TNF-α experiments, RNA was isolated and VCAM1 mRNA was measured by Nanostring gene expression analysis as described below. For the hemin experiments, cellular glutathione (GSH) was measured using a GSH-Glo™ glutathione assay kit (Promega) following the manufacturer’s protocol. Cell viability was observed by MTT assay as described by the manufacturer (Sigma-Aldrich, Cat# CT02).
Six normal human bone marrow (BM) samples from 2 African Americans, 3 Hispanics, and 1 Caucasian were obtained from three commercial sources (Lonza, NorCal Biologics, and iSpecimen). SCD subject whole blood (~50 ml/subject) was obtained from Sanguine Biosciences (n = 3 subjects). The whole blood was diluted 2-fold with PBS containing 2% FBS and then layered on ficoll for peripheral blood mononuclear blood cell (PBMC) isolation using density gradient centrifugation. The whole blood was centrifuged at 1200 rpm for 25 min and PBMC were harvested and counted. PBMC and BM derived CD34+ cells were enriched using Miltenyi’s CD34+ microbeads (positive selection method). BM derived CD34+ cells were stored in liquid nitrogen (−152°C) until required for HbF induction. On the day of the experiment, CD34+ cells were thawed in Iscove’s modified Dulbecco’s medium (IMDM, Hyclone) containing 10% heat-inactivated fetal bovine serum (FBS) and then cells were pelleted by centrifugation. The cell pellets were re-suspended in X-Vivo medium (Lonza) and viability was assessed with trypan blue. CD34+ cells from SCD whole blood were cultured soon after isolation. CD34+ cells were expanded in X-Vivo media containing rhIL-3 (10 ng/ml), rhSCF (100 ng/mL) and rhFlt-3 ligand (100 ng/mL). Approximately 10,000–12,500 cells in 250 μL volume were plated per well in round-bottom 96-well plates and culture for 7 days in a humidified incubator at 37°C/5% CO2. After 7 days, the cells were washed twice with X-Vivo media and resuspended in freshly prepared erythroid differentiation media containing X-Vivo-15, rhIL-3 (10 ng/mL), rhSCF (100 ng/mL), and Epo (3 U/mL). BACH1 inhibitor ASP8731 (1, 3, 10 μM) and/or HU (3, 10 μM) positive control for HbF induction were added either alone or in combination to the media. Standard cultures (solvent and compound free) and solvent control cultures (containing DMSO) were also initiated. Each test condition was set up in triplicate. Following culture in differentiation media for 7 days, the induction of HbF was assessed by flow cytometry. In addition, cells were collected on days 3 or 7 of differentiation for gene expression analysis. RNA from erythroid differentiated CD34 cells was isolated and analyzed for gene expression as described below.
RNA was isolated using Machery-Nagel isolation kit (NucleoSpin RNA, Cat#740955.250) and quantified using a NanoDrop spectrophotometer (ThermoFisher Scientific). Isolated RNA was analyzed on a NanoString SPRINT profiler and nSolver version 4.0 software. A custom Tag-24 CodeSet and exploratory gene expression biomarker probes, were hybridized with a total mRNA input of 200 ng. To hybridize the probes to the target genes of interest, a thermocycler block was set to 67C (with the lid set to 72C) and samples were incubated overnight for a total of 16 h. The samples were cooled to 4C at the end of 16 h and remained at 4C until samples were pooled and added to wells of a SPRINT cartridge. Positive and negative controls were assessed for run QC, ensuring linearity and ample assay limit of detection, respectively. Raw data from the reference genes in study samples were assessed for stability across the experiment utilizing the GeNorm protocol for assessment of expression. Reference genes that exhibited differential expression ie oscillated randomly or excessively above/below the experiment geometric mean were excluded from content normalization calculations. The limit of detection for each sample was determined as the average of negative controls in each sample plus two times the standard deviation of the negative controls within the sample; the experimental limit of detection was calculated as the geometric mean of all individual limit of detection calculations across the experiment. CLTC, POLR2A, RPL27, and TBP were used as reference genes for normalization. Data was graphed as fold-change compared to DMSO treatment. Probe Sequences are listed in Supplementary Table 1.
After 4 weeks treatment with ASP8731 or HU, HbSS mice were anesthetized with ketamine and xylazine and dorsal skinfold chambers were implanted (21). Flowing subcutaneous venules (20–23 venules/mouse) in the chamber window were selected and mapped using intravital microscopy as previously described (21). After selection of flowing venules, mice were infused with a bolus infusion via tail vein with hemin (3.2 μmol/kg). One hour after hemin infusion, the venules were visually re-examined for stasis (no flow). The static venules in each mouse were counted and percent stasis at 1 h was calculated by dividing the number of static venules by the total (static + flowing) number of venules.
Hepatic microsomes and nuclear extracts were isolated from frozen organs as previously described (12). Immunoblots of cellular subfractions (30–50 μg protein) were run on 4–20% SDS PAGE gels (Bio-Rad #3450033), transferred to PVDF membranes, and immunostained with primary antibodies to NF-ĸB phospho-p65 (Ser536, Cell Signaling #3031), total p65 (Cell Signaling #3034), ICAM-1 (Abcam #ab124759), HO-1 (Enzo #ADI-111), BACH1 (US Biological Life Sciences #220980), or GAPDH (Sigma-Aldrich #G9545). Primary antibodies were detected with suitable secondary antibodies conjugated to alkaline phosphatase and visualized with ECF substrate (GE Healthcare) and a Typhoon FLA 9500 imager (GE Healthcare).
EDTA blood was diluted 1:20 or 1:40 in 3% glacial acetic acid to lyse RBCs. WBCs were counted manually using a hemocytometer and a light microscope.
Differentiated CD34+ cells collected at day 7 of erythroid differentiation were washed with PBS. For flow cytometry analysis, non-specific binding of antibodies was blocked with 10% FBS with human IgG (20 μg/mL) at 4°C for 20 min. Following blocking, the surface markers of the cells were stained with an antibody cocktail containing CD235a-PE/Cy7, and CD71-APC at 4°C for 30 min. The cells were washed with PBS, re-suspended in 1x Cytofix/Cytoperm solution (BD Biosciences), and fixed for 20 min at 4°C. Following fixation, cells were washed with Perm/Wash Buffer (BD Biosciences) and stained with anti-HbF-PE for 30 min at room temperature. For isotype control, IgG-PE was used in place of HbF-PE. Finally, the cells were washed twice with Perm/Wash Buffer, resuspended in PBS containing 2% FBS, and then analyzed by flow cytometry using a Beckman Coulter CytoFlex cytometer. Both percentages of various cell populations as well as events (cells) per μL were collected. The HbF+ cells were found within the CD71+ bright cell population. HU was used as a positive control throughout the experiment.
Comparisons of multiple treatment groups were made using one-way ANOVA with Tukey’s or Dunnett’s multiple comparison test (GraphPad Prism version 9.4). Normality of data was assessed using the Shapiro–Wilk test. Non-normally distributed data was analyzed for differences between treatment groups using the Kruskal-Wallis test with Dunn’s multiple comparisons test.
Since BACH1 represses NRF2-responsive pathways in cells, we investigated the ability of ASP8731 to modulate antioxidant and anti-inflammatory genes in cell culture. HEPG2 cells were treated with increasing concentrations of the BACH1 inhibitor ASP8731 (0.1–50 μM) or DMF (1–250 μM), a positive control for NRF2 gene transcription, for 24 h. ASP8731 potently induced HO-1 (HMOX1) and ferritin heavy chain (FTH) mRNA in HEPG2 cells, even more potently than DMF (Figures 1A,B). Human primary pulmonary arterial endothelial cells (PAEC) were incubated with ASP8731 (0, 1, 3, 10 μM) or DMSO vehicle for 24 h and then TNF-α (10 ng/ml), was added for an additional 4 h followed by RNA isolation and measurement of VCAM1 mRNA. VCAM1 mRNA was maximally increased in TNF-α-treated cells in the absence of BACH1 inhibitor ASP8731 and decreased as the concentration of ASP8731 increased (Figure 1C). PAEC were incubated with DMSO vehicle or ASP8731 (0, 1, or 3 μM) for 24 h and then cells were exposed to heme-mediated oxidative stress. Hemin (50 μM) was added for an additional 30 min followed by measurement of glutathione (GSH) levels as a marker of oxidative stress. GSH levels were decreased in hemin-treated cells in the absence of BACH1 inhibitor ASP8731 and increased as the concentration of ASP8731 increased (Figure 1D). The effects of treatments on HepG2 and PAEC viability were measured by MTT assay. ASP8731 at the doses tested did not affect the viability of HepG2 cells or HPAEC cells in the presence of DMSO or hemin.
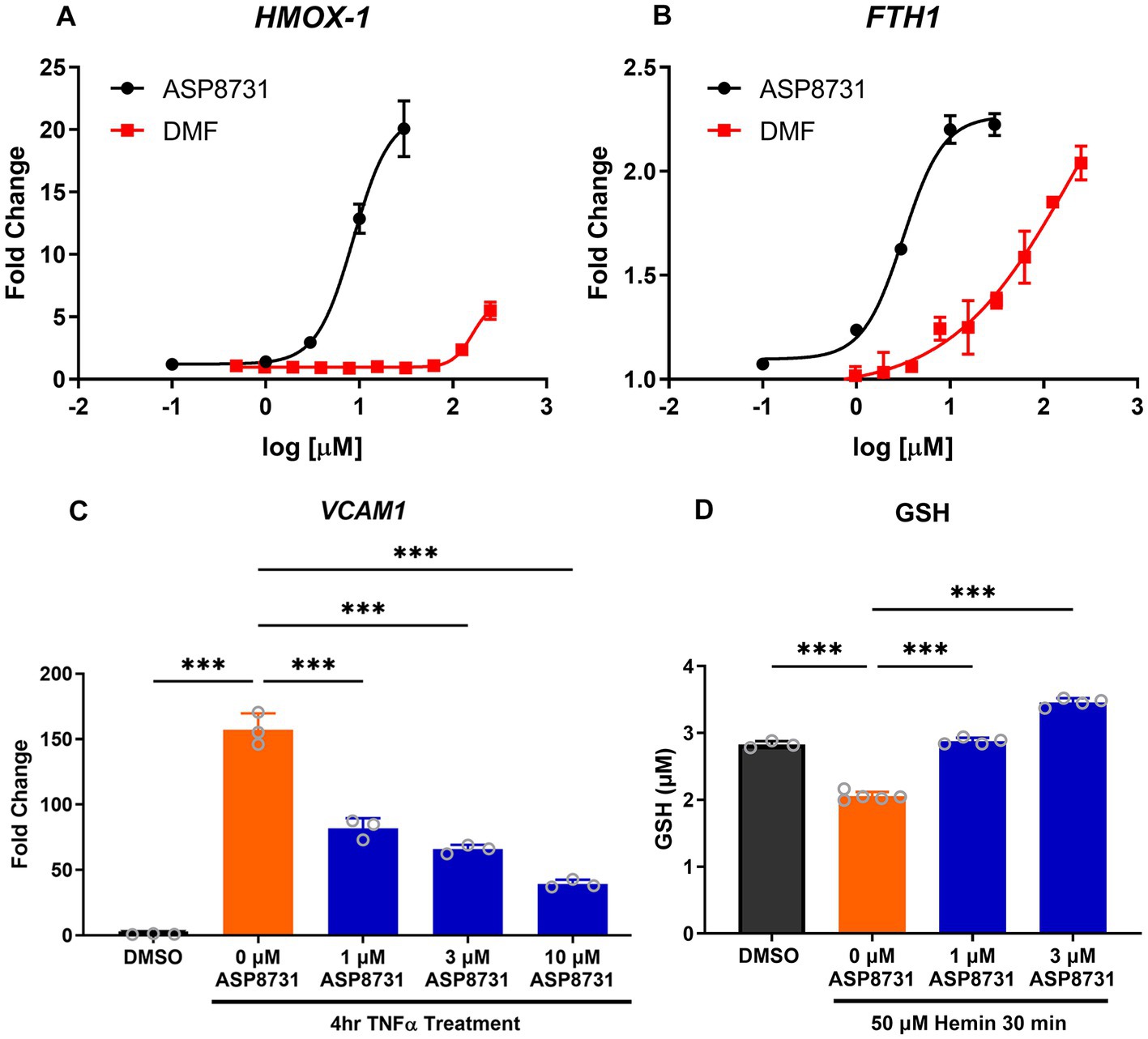
Figure 1. BACH1 inhibitor ASP8731 modulated multiple pathways in vitro that are involved in SCD pathophysiology. ASP8731, dimethyl fumarate (DMF), or DMSO vehicle were incubated with HepG2 cells for 24 h and (A) HMOX1 (HO-1) and (B) FTH (ferritin heavy chain) mRNA were measured. (C) Human primary pulmonary arterial endothelial cells (PAEC) were incubated with ASP8731 (1, 3, 10 μM) or DMSO vehicle for 24 h and then TNF-α (10 ng/ml), was added for an additional 4 h followed by RNA isolation and measurement of VCAM1 mRNA. (D) PAEC were incubated with DMSO vehicle or ASP8731 (0, 1, or 3 μM) for 24 h and then hemin (50 μM) was added for an additional 30 min followed by measurement of glutathione (GTH) levels. Values are means ± SEM. ***p < 0.001, one-way ANOVA with Tukey’s multiple comparison test.
Since activation of NRF2-responsive genes has been shown to be beneficial in SCD mice, BACH1 inhibition was also evaluated. Townes HbSS mice were gavaged once daily, 6 days per week for 4 weeks with vehicle (VEH), ASP8731 (1, 3, 25 mg/kg body weight), hydroxyurea (HU, 100 mg/kg), or ASP8731 (25 mg/kg) + HU (100 mg/kg). On the last day of treatment, dorsal skinfold chambers were implanted on the mice. Flowing venules (20–23 venules/mouse) in the chamber window were selected and mapped using intravital microscopy. Mice were then infused with hemin (3.2 μmol/kg body weight) via the tail vein. One hour after hemin infusion, the venules were visually re-examined for stasis (no flow). The static venules in each mouse were counted and percent stasis at 1 h was calculated by dividing the number of static venules by the total (static + flowing) number of venules (Figure 2A). ASP8731 inhibited hemin-induced stasis in a dose responsive manner with the highest stasis occurring in HbSS mice pretreated with vehicle (33%) and decreasing to 26, 21, and 7% stasis in mice treated with 1, 3, and 25 mg/kg ASP8731, respectively. Treatment with HU (100 mg/kg) decreased stasis to 13%, with the lowest stasis (3.5%) seen in mice treated with ASP8731 (25 mg/kg) + HU (100 mg/kg).
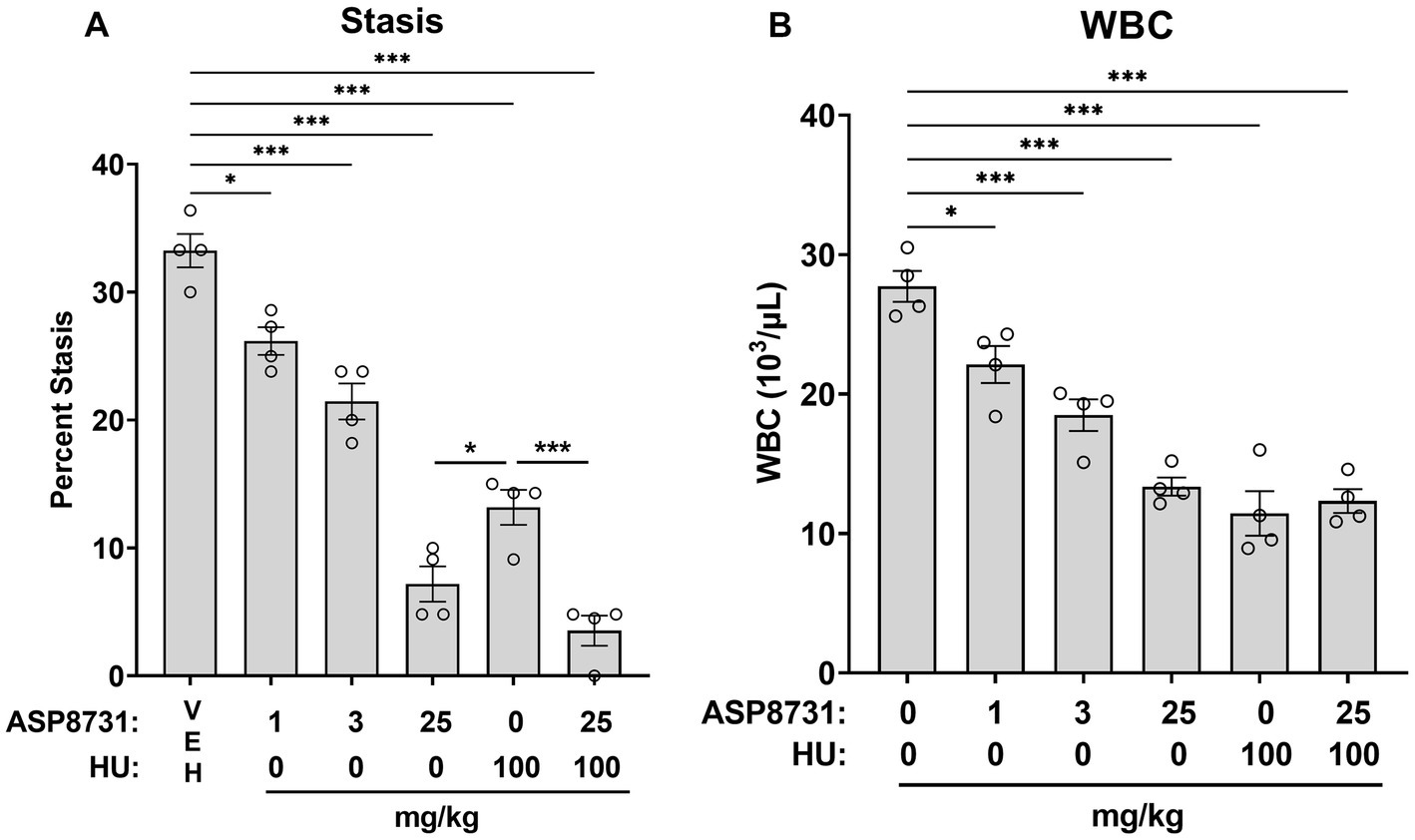
Figure 2. Bach1 inhibitor ASP8731 decreases microvascular stasis and WBC counts in Townes HbSS mice. Townes HbSS mice were gavaged once daily, 6 days per week for 4 weeks with vehicle (VEH), ASP8731, and/or HU at the indicated doses (n = 4 mice/group, 2 male and 2 female). On the last day of treatment, mice were implanted with dorsal skinfold chambers and infused with hemin (3.2 μmol/kg body weight). Flowing venules (20–24 venules/mouse) in the chamber window were selected and mapped using intravital microscopy prior to hemin infusion. (A) Microvascular stasis was measured in the same venules 1 h after hemin infusion. (B) WBC counts were measured in blood 4 h after hemin infusion. Individual values are presented as circles with bars representing the means ± SEM; *p < 0.05 and ***p < 0.001, one-way ANOVA with Tukey’s multiple comparison test.
After stasis measurements, blood samples were collected 4 h after the infusion of hemin. White blood cells (WBC) were counted manually in whole blood. WBC counts decreased in a manner similar to stasis (Figure 2B). ASP8731 significantly inhibited WBC counts in a dose-responsive manner with the highest WBC counts occurring in HbSS mice pretreated with vehicle (28 × 103/μL) and decreasing to 22 × 103/μL, 18 × 103/μL, and 13 × 103/μL in mice treated with 1, 3, and 25 mg/kg ASP8731, respectively. Treatment with HU (100 mg/kg) decreased the WBC count to 11 × 103/μL. Treatment with ASP8731 (25 mg/kg) + HU (100 mg/kg) decreased the WBC count to 12 × 103/μL.
There were no significant differences in any of the red blood cell indices, including red blood cell counts, hemoglobin levels, hematocrits, and reticulocyte counts in any of the treatment groups as compared to vehicle treated HbSS mice (Supplementary Figure 1). In addition, the spleen weights as a percent of body weight were also not significantly different between treatment groups suggesting that BACH1 inhibition did not significantly inhibit hemolysis in HbSS Townes mice.
In a separate cohort of HbSS mice, we examined the effects of ASP8731 and HU on HO-1, NF-ĸB phospho-p65 and ICAM-1 expression in the liver. HbSS mice (n = 3 mice/treatment, 2 males and 1 female) were gavaged once daily for 14 consecutive days with vehicle (VEH), ASP8731, or HU at the indicated doses. On the last day of treatment, mice were infused with hemin (3.2 μmol/kg) via the tail vein. Livers were collected 4 h after the infusion of hemin. Hepatic HO-1, NF-ĸB phospho-p65, and ICAM-1 proteins were analyzed on Western blots (Figures 3A–C) Relative expression of the bands on the Western blots were quantified using densitometry (Supplementary Figure 2). Microsomal HO-1 was significantly increased in the livers of HbSS mice treated with ASP8731 (25 mg/kg), HU (100 mg/kg), and ASP8731 (25 mg/kg) + HU (100 mg/kg). Nuclear NF-ĸB phospho-p65, a marker of NF-ĸB activation, was significantly decreased in the livers of HbSS mice at all treatment doses of ASP8731 and HU. Microsomal ICAM-1 was also significantly decreased in livers at all doses of ASP8731 and HU. Treatment of HbSS mice with ASP8731 increased nuclear BACH1 expression in the liver (Supplementary Figure 3). Similarly, HU also increased nuclear BACH1 protein expression.
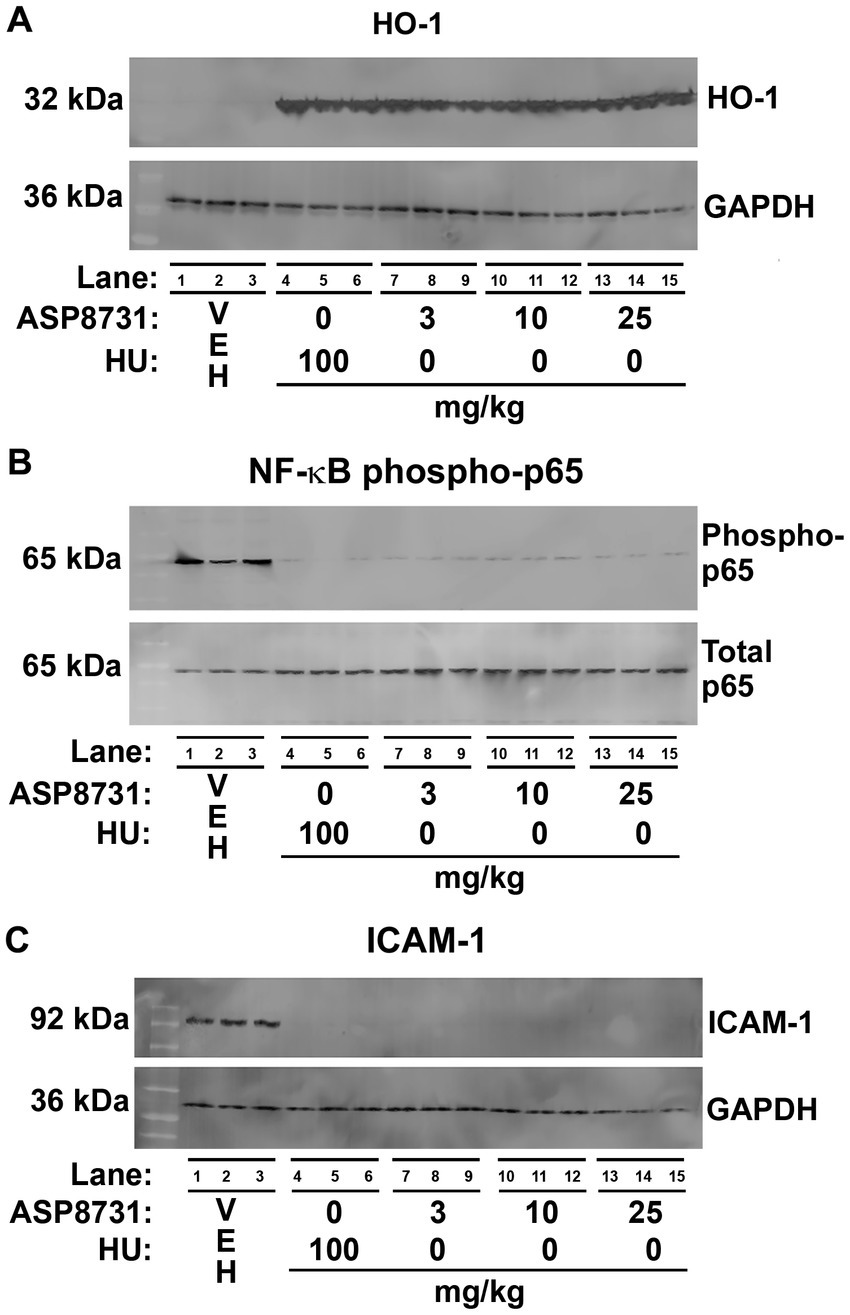
Figure 3. Bach1 inhibitor ASP8731 had anti-inflammatory properties in Townes HbSS mice. Townes HbSS mice (n = 3 mice/treatment, 2 males and 1 female) were gavaged once daily for 14 consecutive days with vehicle (VEH), ASP8731, or HU at the indicated doses. On the last day of treatment, mice were infused with hemin (3.2 μmol/kg). Livers were excised 4 h after the infusion of hemin. Proteins in liver were analyzed by Western blots. (A) HO-1 and GAPDH. (B) NF-ĸB phospho-and total p65. (C) ICAM-1 and GAPDH using liver microsomes (A,C) and nuclear extracts (B).
In a separate set of studies, ASP8731 dose responsively increased heme oxygenase (HO) activity in the livers of HbSS mice (Supplementary Figure 4). HU also increased HO activity to a somewhat lesser degree than ASP8731, but the highest HO activity was measured in mice treated with ASP8731 + HU.
BACH1 inhibitor ASP87311 was examined for its ability to increase expression of gamma globins and HbF-containing F-cells. Gamma and beta-S globin levels were measured by reverse phase HPLC in the red blood cells of Townes HbSS mice after treatment with ASP8731 (3 or 25 mg/kg) or vehicle once daily for 14 days. The ratio of gamma globin to gamma globin + beta-S globin increased from a mean of 5.8% in vehicle treated HbSS mice to 7.8 and 8.3% in HbSS mice treated with 3 and 25 mg/kg ASP8731, respectively (Figure 4A). Similarly, HbF containing F-cells measured by FACS significantly increased from 1.0% of red blood cells in vehicle-treated HbSS mice to 1.8 and 2.1% in HbSS mice treated with 3 and 25 mg/kg ASP8731, respectively (Figure 4B).
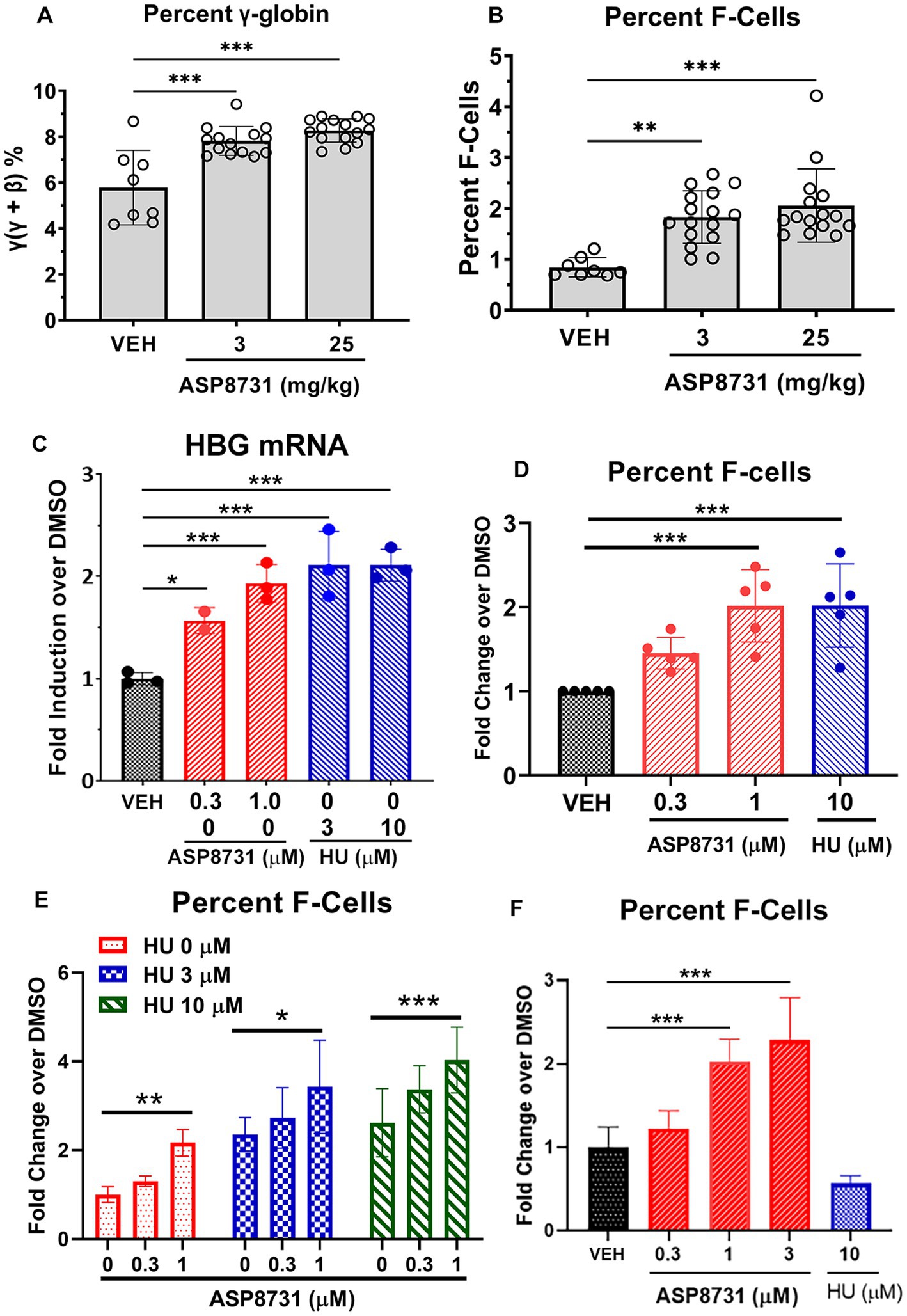
Figure 4. BACH1 inhibitor ASP8731 induces gamma globin and F-cells in HbSS mice and human erythroid differentiated CD34 cells. Townes HbSS mice (n = 8–16 mice/treatment, equal males and females) were gavaged once daily for 14 consecutive days with vehicle (VEH, n = 8 mice) or ASP8731 at 3 mg/kg (n = 16 mice) or 25 mg/kg (n = 16 mice). Whole blood was collected on the last day of treatment and the red blood cells (RBC) were lysed in water and membrane debris was moved by centrifugation and filtration. (A) Gamma (ɣ)-A and beta (β)-S globins in the lysed RBC supernatants were measured by reverse phase HPLC and expressed as percent gamma-globin (ɣ/ɣ + β). (B) HbF containing F-cells in whole blood were measured by FACS and expressed as percent F-cells [(HbF+ and Ter119+)/total RBC (Ter119+)]. (C–F) Human BM derived CD34+ cells were expanded (days 1–7) and differentiated (days 7–14) to erythroid cells in cell culture. Cells were differentiated in the presence of the indicated concentrations of ASP8731, HU, or DMSO (VEH) on days 7–14. Cells were collected for HBG mRNA or percent F-cell FACS analysis on day 14. (C) HBG mRNA was measured by Nanostring analysis (n = 3 subjects). (D–F) Percent F-cells were measured by FACS. Percent F-cells [(CD71+ and HbF+)/(CD71+)] was calculated and expressed as fold change over DMSO (VEH)-treated cells. (D) Values are mean ± SD of 5 subjects. (E) Values are the mean of 3 subjects measured in triplicate. (F) In one subject’s CD34 cells, HbF was not increased in response to HU, but these cells were responsive to ASP8731. Mean ± SD of triplicate measurements from one donor. *p < 0.05, **p < 0.01, and ***p < 0.001, one-way ANOVA with Tukey’s multiple comparison test.
The effects of BACH1 inhibitor ASP8731 and HU on gamma globin gene (HBG) mRNA levels and F-cell production were measured in human BM-derived CD34 cells during erythroid differentiation. HGB mRNA was significantly increased in CD34 cells differentiated in the presence of ASP8731 at 0.3 and 1 μM or in the presence of HU at 3 and 10 μM (Figure 4C) as compared to the DMSO vehicle (VEH). The percent F-cells was significantly increased in CD34 cells differentiated in 1 μM ASP8731 or 10 μM HU (Figure 4D; Supplementary Table 2). Differentiation of cells to CD71+ cells (reticulocyte marker) was not affected by treatment with HU or ASP8731 (Supplementary Table 2). In preliminary time course study, incubation of compounds for different time (days, 3, 7, and 10) did not change the % CD71+ cells (data not shown). F-cells were increased by both ASP8731 and HU individually, but the combination of both compounds increased F-cells higher than either drug alone (Figure 4E). In one healthy CD34 donor’s cells that were non-responsive to HU, ASP8731 was able to induce F-cells at 1 and 3 μM (Figure 4F).
The effects of BACH1 inhibitor ASP8731 and HU were examined in erythroid differentiated CD34 cells derived from the peripheral blood of SCD subjects (Figure 5). Levels of (A) gamma globin (HBG), (B) alpha globin (HBA), (C) beta globin (HBB), (D) HO-1 (HMOX1), (E) NAD(P)H quinone dehydrogenase 1 (NQO1), (F) glutamate-cysteine ligase modifier subunit (GCLM), (G) solute carrier family 7 member 11 (SLC7A11), and (H) solute carrier family 48 member 1 (SLC48A1) mRNA levels were measured using Nanostring. ASP8731 (10 μM) and the combination of ASP8731 (10 μM) + HU (10 μM) significantly increased the mRNA of every gene tested except HBB and HMOX1. The lack of statistical difference in HMOX1 gene was primarily due to very high level of expression in 1 patient sample, thus fold change at 3, 1, and 0.3 μM ASP8731 and 10 μM HU were 38.16 ± 41.5; 3.96 ± 1.28, 1.55 ± 0.8, and 1.51 ± 0.3, respectively. In contrast HU alone at 10 μM concentration did not significantly increase the mRNA of any of the genes tested. These data suggest that the BACH1 inhibitor ASP8731 is a potent inducer of HbF and antioxidant response genes in SCD patients.
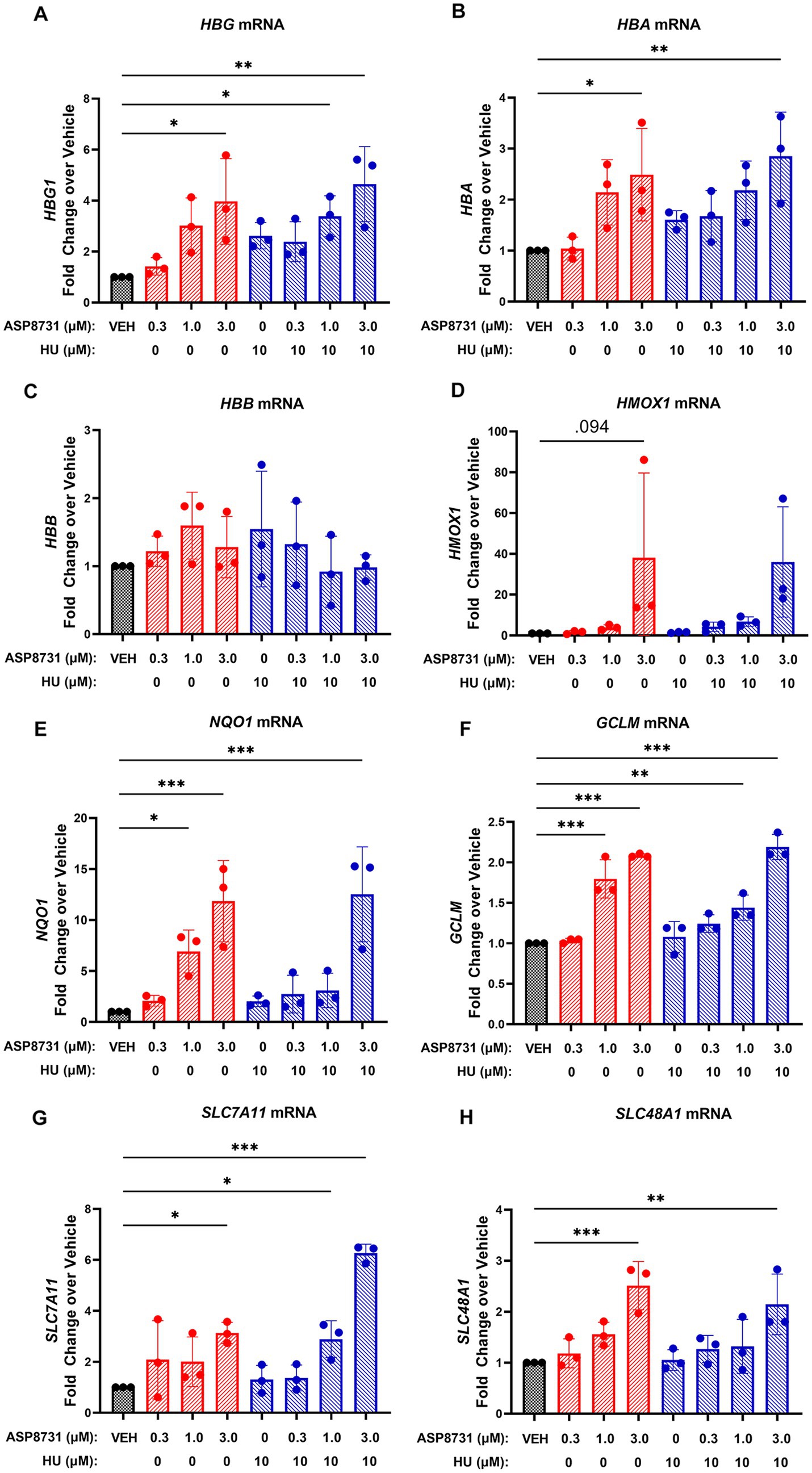
Figure 5. BACH1 inhibitor ASP8731 induces gamma-and alpha-globin, but not beta-globin in erythroid differentiated CD34 cells derived from SCD subjects. Peripheral blood CD34+ cells isolated from SCD subjects (n = 3) were expanded (days 1–7) and differentiated (days 7–10) to erythroid cells in cell culture. Cells were differentiated in the presence of the indicated concentrations of ASP8731, HU, or DMSO (VEH) on days 7–10. Cells were collected for mRNA analysis by Nanostring on day 10. (A) HBG mRNA; (B) HBA mRNA; (C) HBB mRNA – the probes used for Nanostring for HBB do not distinguish between normal and sickle; (D) HMOX1 mRNA; (E) NQO1 mRNA; (F) GCLM mRNA; (G) SLC7A11 mRNA; and (H) SLC48A11 mRNA. Values are expressed as fold change over DMSO vehicle and bars are mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to vehicle, one-way ANOVA followed by Dunnett’s multiple comparison test.
BACH1 inhibitor ASP8731 potently modulated multiple pathways that affect SCD pathophysiology. In HepG2 liver cells, ASP8731 increased HMOX1 and FTH1 mRNA. In pulmonary endothelial cells, ASP8731 decreased VCAM1 mRNA in response to TNF-α and prevented a decrease in glutathione in response to hemin. BACH1 has been reported to repress transcription of genes that reduce labile iron, oxidative stress, and ferroptosis (22).
Both ASP8731 and HU markedly inhibited heme-mediated microvascular stasis in a dorsal skin-fold chamber model in HbSS mice while increasing hepatic HO-1 expression. HO-1 induction might have had a large impact on the inhibition of microvascular stasis in HbSS mice in part due to the many anti-inflammatory effects of HO-1 (11, 12). Markers of inflammation including ICAM-1 and NF-kB phospho-p65 expression in the liver and blood WBC counts were significantly decreased by ASP8731 treatment as compared to vehicle controls. Inhibition of HO-1 activity with tin protoporphyrin has been previously shown to abrogate the beneficial effects of NRF2 activation on microvascular stasis (15). The potent anti-inflammatory effects of HO-1 are related in part to the release of carbon monoxide and biliverdin and the post-transcriptional induction of ferritin heavy chain (FTH1) by iron generated by HO-1-mediated heme degradation (11, 23, 24).
Paradoxically, treatment of HbSS mice with ASP8731 increased nuclear BACH1 expression in the liver, suggesting a there is a feedback loop that increases nuclear BACH1 protein expression upon inhibition of BACH1. Similarly, HU also increased nuclear BACH1 protein expression. HU is a known nitric oxide (NO) donor (25). The ability of NO alone or in combination with heme to induce Nrf2/HO-1 has been reported (26).
Microvascular stasis was measured in the subcutaneous venules in the skin, but we did not directly measure inflammatory markers in the skin. However, it has previously been shown that inflammation in HbSS mice is occurring in blood vessels throughout the vasculature including the skin, liver, kidneys, and lungs (3, 11, 15, 27). We measured hepatic HO-1 expression and activity in HbSS mice, but not in other organs. We previously showed that genetic overexpression of hepatic HO-1 has systemic effects on microvascular stasis in the distal skin (12). It is likely that ASP8731 had anti-inflammatory effects in all tissues as evidenced by the significant reduction in the WBC counts.
HU induced HO-1 to a similar degree as ASP8731 in HbSS mice. To our knowledge, this is the first time HU has been reported to increase HO-1 expression. Lanaro and colleagues (28) reported increased HMOX1 mRNA in the mononuclear cells and neutrophils of SCD patients. In that study, SCD patients taking HU had a trend toward higher HMOX1 mRNA than SCD patients not on HU, but this difference did not appear to be significant. HU increases nitric oxide (NO) production, acting as both a NO donor and a stimulator of NO synthase (25, 29). Bioactivation of NO is a plausible mechanism for HMOX1 induction by HU as NO can activate KEAP1/NRF2 signaling (30).
Clinical studies have shown that increasing HbF levels with HU inhibits the polymerization of HbS, decreases the severity of many clinical features of SCD, and improves survival (31–33). SCD patients with hereditary persistence of F-cells have a more benign clinical course (34). Given the modest increase in F-cells in HbSS mice treated with ASP8731 (0.8% F-cells in vehicle treated mice compared to 1.8 and 2.1% in mice treated with 3 mg/kg and 25 mg/kg of ASP8731, respectively), F-cell induction is probably not a major factor in the anti-vaso-occlusion protection seen in HbSS mice. Humans at birth express 2 gamma-globin genes (HBG1 and HBG2). HBG1 codes for A-gamma (Aɣ)-globin and HBG2 codes for G-gamma (Gɣ)-globin. The 2 globin chains differ by one amino acid, with (Aɣ) having an alanine and (Gɣ) having a glycine at position 136. Two gamma globin chains with two alpha (α)-globin chains form HbF (α2/ɣ2), which is normally replaced by HbA shortly after birth. Townes HbSS mice have the human HBG1 gene, but not the human HBG2 gene, and thus they can only express Aɣ-globin. Murine HBB genes in the Townes model were replaced by human HBG1 and HBBS genes and some proximal but not some distal gene-regulatory elements. The HbSS Townes humanized knock-in mouse model recapitulates human globin gene switching (35). HU suppresses erythropoiesis in Townes HbSS mice to create an early persistent F-cell phenotype without re-activating γ-globin transcription (35). Townes mouse HSCs, in contrast to human HSCs, fail to induce HbF to therapeutic levels after genetic disruption of the BCL11A binding site in the HBG1 promoter (36). This is likely due to missing distal regulatory elements of the globin genes in the Townes model (36). Thus, interpretation of HbF induction in Townes HbSS mice has limitations.
BACH1 and BACH2 support erythropoiesis by regulating heme metabolism in committed erythroid cells, driving cells toward erythroid differentiation and away from myeloid differentiation (37) by repressing C/EBPβ, a transcription factor regulating myelopoiesis and inflammation. Heme-BACH1 interaction also plays an important role in globin gene expression. The binding of heme to CP motifs in BACH1 inhibits BACH1 repressor activity. The human globin gene cluster spans ~70 kb and contains 5 globin genes including ϵ, γG, γA, δ, and β that are controlled by the microlocus control region (μLCR) (38). BACH1 forms heterodimers with small Maf proteins that repress globin expression by binding to Maf recognition elements (MARE) in the μLCR (9). Heme increases globin gene expression by binding to BACH1 and blocking the interaction of BACH1 with MARE regions in the μLCR (38, 39). A role for BACH1 in the control of gamma globin expression has been suggested, but not shown directly (40, 41). Our studies indicate that BACH1 inhibition by ASP8731 induces gamma-globin and HbF expression in human erythroid cells.
In BM derived CD34+ cells from normal subjects, BACH1 inhibitor induced HBG mRNA and percent F-cells. ASP8731 and heme alleviate BACH1 repression, leading to activation of NRF2-responsive genes. Activation of NRF2 promotes Aɣ- and Gɣ-globin expression in erythroid cells (42–46), which increase HbF expression in red blood cells of SCD patients. Interestingly, ASP8731 + HU potentiated the induction of F-cells over HU alone. In one donor’s CD34 cells that were non-responsive to HU, ASP8731 was able to induce F-cells. The effectiveness of HU to induce HbF is variable in SCD patients and there are concerns over the long-term efficacy of HU (31, 47–49). Induction of F-cells in an HU non-responder by BACH1 inhibition, albeit a single subject, suggests this therapeutic might be an option for patients who are not responsive to HU. During differentiation of CD34+ cells from SCD patients, BACH1 inhibition increased the expression of HBG, HBA and other NRF2 responsive genes.
In conclusion, BACH1 inhibitors are promising therapeutics for SCD that enhance the cytoprotective oxidative stress responses and thereby reduce inflammation and vaso-occlusion. BACH1 inhibitors have the added benefit of inducing HbF expression and thereby potentially reducing HbS polymerization and hemolysis. The present data support evaluation of BACH1 inhibitors as therapeutics for SCD patients.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by University of Minnesota’s Institutional Animal Care and Use Committee.
JB wrote the manuscript, analyzed data, oversaw the studies in mice, and prepared figures. FA, CC, PZ, JN, and CR conducted mouse experiments and data collection, processing, and analysis. GV analyzed data and edited the manuscript. JS, LO, EC, and YH planned and conducted cell culture experiments, data collection, processing, and analysis, and prepared figures. SD, MS, MB, DS, and SN: conception, study design and data interpretation. All authors contributed to the article and approved the submitted version.
This study was funded by Astellas Pharma Inc. and investigators received funding to conduct this study from the study sponsor.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Astellas-Mitobridge. The funder had the following involvement: study design, data collection, analysis, interpretation of data, and the writing of this article.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1101501/full#supplementary-material
1. Telen, MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood. (2016) 127:810–9. doi: 10.1182/blood-2015-09-618553
2. Charache, S, Dover, GJ, Moore, RD, Eckert, S, Ballas, SK, Koshy, M, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. (1992) 79:2555–65. doi: 10.1182/blood.V79.10.2555.2555
3. Belcher, JD, Chen, C, Nguyen, J, Milbauer, L, Abdulla, F, Alayash, AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
4. Ogawa, K. Heme metabolism in stress response. Nihon Eiseigaku Zasshi. (2002) 56:615–21. doi: 10.1265/jjh.56.615
5. Sun, J, Hoshino, H, Takaku, K, Nakajima, O, Muto, A, Suzuki, H, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. (2002) 21:5216–24. doi: 10.1093/emboj/cdf516
6. Sun, J, Brand, M, Zenke, Y, Tashiro, S, Groudine, M, and Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A. (2004) 101:1461–6. doi: 10.1073/pnas.0308083100
7. Dhakshinamoorthy, S, Jain, AK, Bloom, DA, and Jaiswal, AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. (2005) 280:16891–900. doi: 10.1074/jbc.M500166200
8. Ishikawa, M, Numazawa, S, and Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic Biol Med. (2005) 38:1344–52. doi: 10.1016/j.freeradbiomed.2005.01.021
9. Igarashi, K, and Sun, J. The heme-Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal. (2006) 8:107–18. doi: 10.1089/ars.2006.8.107
10. Zhang, X, Guo, J, Wei, X, Niu, C, Jia, M, Li, Q, et al. Bach1: function, regulation, and involvement in disease. Oxidative Med Cell Longev. (2018) 2018:1347969. doi: 10.1155/2018/1347969
11. Belcher, JD, Mahaseth, H, Welch, TE, Otterbein, LE, Hebbel, RP, and Vercellotti, GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. (2006) 116:808–16. doi: 10.1172/JCI26857
12. Belcher, JD, Vineyard, JV, Bruzzone, CM, Chen, C, Beckman, JD, Nguyen, J, et al. Heme oxygenase-1 gene delivery by sleeping beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med (Berl). (2010) 88:665–75. doi: 10.1007/s00109-010-0613-6
13. Bean, CJ, Boulet, SL, Ellingsen, D, Pyle, ME, Barron-Casella, EA, Casella, JF, et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. (2012) 120:3822–8. doi: 10.1182/blood-2011-06-361642
14. Bakr, S, Khorshied, M, Talha, N, Jaffer, KY, Soliman, N, Eid, K, et al. Implication of HMOX1 and CCR5 genotypes on clinical phenotype of Egyptian patients with sickle cell anemia. Ann Hematol. (2019) 98:1805–12. doi: 10.1007/s00277-019-03697-9
15. Belcher, JD, Chen, C, Nguyen, J, Zhang, P, Abdulla, F, Nguyen, P, et al. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid Redox Signal. (2017) 26:748–62. doi: 10.1089/ars.2015.6571
16. Krishnamoorthy, S, Pace, B, Gupta, D, Sturtevant, S, Li, B, Makala, L, et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI. Insight. (2017) 2:e96409. doi: 10.1172/jci.insight.96409
17. Zhu, X, Xi, C, Thomas, B, and Pace, BS. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood. (2018) 131:558–62. doi: 10.1182/blood-2017-10-810531
18. Li, B, Zhu, X, Ward, CM, Starlard-Davenport, A, Takezaki, M, Berry, A, et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp Hematol. (2019) 70:85–96.e5. doi: 10.1016/j.exphem.2018.11.002
19. Nataraja, S, Singh, M, Demes, S, Olson, L, Stanwix, J, Biddle, M, et al. ML-0207/ASP8731: a novel BACH1 inhibitor that induces fetal hemoglobin in treatment of sickle cell disease. Blood. (2021) 138:854–4. doi: 10.1182/blood-2021-151606
20. Wu, LC, Sun, CW, Ryan, TM, Pawlik, KM, Ren, J, and Townes, TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. (2006) 108:1183–8. doi: 10.1182/blood-2006-02-004812
21. Kalambur, VS, Mahaseth, H, Bischof, JC, Kielbik, MC, Welch, TE, Vilbäck, Å, et al. Microvascular blood flow and stasis in transgenic sickle mice: utility of a dorsal skin fold chamber for intravital microscopy. Am J Hematol. (2004) 77:117–25. doi: 10.1002/ajh.20143
22. Nishizawa, H, Matsumoto, M, Shindo, T, Saigusa, D, Kato, H, Suzuki, K, et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J Biol Chem. (2020) 295:69–82. doi: 10.1074/jbc.RA119.009548
23. Belcher, JD, Gomperts, E, Nguyen, J, Chen, C, Abdulla, F, Kiser, ZM, et al. Oral carbon monoxide therapy in murine sickle cell disease: beneficial effects on vaso-occlusion, inflammation and anemia. PLoS One. (2018) 13:e0205194. doi: 10.1371/journal.pone.0205194
24. Vercellotti, GM, Khan, FB, Nguyen, J, Chen, C, Bruzzone, CM, Bechtel, H, et al. H-ferritin ferroxidase induces cytoprotective pathways and inhibits microvascular stasis in transgenic sickle mice. Front Pharmacol. (2014) 5:79. doi: 10.3389/fphar.2014.00079
25. Gladwin, MT, Shelhamer, JH, Ognibene, FP, Pease-Fye, ME, Nichols, JS, Link, B, et al. Nitric oxide donor properties of hydroxyurea in patients with sickle cell disease. Br J Haematol. (2002) 116:436–44. doi: 10.1046/j.1365-2141.2002.03274.x
26. Naughton, P, Hoque, M, Green, CJ, Foresti, R, and Motterlini, R. Interaction of heme with nitroxyl or nitric oxide amplifies heme oxygenase-1 induction: involvement of the transcription factor Nrf2. Cell Mol Biol (Noisy-le-Grand). (2002) 48:885–94.
27. Vercellotti, GM, Dalmasso, AP, Schaid, TR Jr, Nguyen, J, Chen, C, Ericson, ME, et al. Critical role of C5a in sickle cell disease. Am J Hematol. (2019) 94:327–37. doi: 10.1002/ajh.25384
28. Lanaro, C, Franco-Penteado, CF, Albuqueque, DM, Saad, STO, Conran, N, and Costa, FF. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol. (2009) 85:235–42. doi: 10.1189/jlb.0708445
29. Subotički, T, Ajtić, OM, Đikić, D, Santibanez, JF, Tošić, M, and Čokić, VP. Nitric oxide synthase dependency in hydroxyurea inhibition of Erythroid progenitor growth. Genes (Basel). (2021) 12:1145. doi: 10.3390/genes12081145
30. Um, HC, Jang, JH, Kim, DH, Lee, C, and Surh, YJ. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide. (2011) 25:161–8. doi: 10.1016/j.niox.2011.06.001
31. Charache, S, Terrin, ML, Moore, RD, Dover, GJ, Barton, FB, Eckert, SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. (1995) 332:1317–22. doi: 10.1056/NEJM199505183322001
32. Steinberg, MH, Barton, F, Castro, O, Pegelow, CH, Ballas, SK, Kutlar, A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. (2003) 289:1645–51. doi: 10.1001/jama.289.13.1645
33. Smith, WR, Ballas, SK, McCarthy, WF, Bauserman, RL, Swerdlow, PS, Steinberg, MH, et al. The association between hydroxyurea treatment and pain intensity, analgesic use, and utilization in ambulatory sickle cell anemia patients. Pain Med. (2011) 12:697–705. doi: 10.1111/j.1526-4637.2011.01096.x
34. Steinberg, MH. Fetal hemoglobin in sickle cell anemia. Blood. (2020) 136:2392–400. doi: 10.1182/blood.2020007645
35. Walker, AL, Crosby, D, Miller, V, Weidert, F, and Ofori-Acquah, S. Hydroxyurea decouples persistent F-cell elevation and induction of γ-globin. Exp Hematol. (2022) 112-113:15–23.e1. doi: 10.1016/j.exphem.2022.07.005
36. Woodard, KJ, Doerfler, PA, Mayberry, KD, Sharma, A, Levine, R, Yen, J, et al. Limitations of mouse models for sickle cell disease conferred by their human globin transgene configurations. Dis Model Mech. (2022) 15:dmm049463. doi: 10.1242/dmm.049463
37. Kato, H, Itoh-Nakadai, A, Matsumoto, M, Ishii, Y, Watanabe-Matsui, M, Ikeda, M, et al. Infection perturbs Bach2-and Bach1-dependent erythroid lineage ‘choice’ to cause anemia. Nat Immunol. (2018) 19:1059–70. doi: 10.1038/s41590-018-0202-3
38. Tahara, T, Sun, J, Nakanishi, K, Yamamoto, M, Mori, H, Saito, T, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. (2004) 279:5480–7. doi: 10.1074/jbc.M302733200
39. Tahara, T, Sun, J, Igarashi, K, and Taketani, S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. (2004) 324:77–85. doi: 10.1016/j.bbrc.2004.09.022
40. Lee, TY, Muniandy, L, Teh, LK, Abdullah, M, George, E, Sathar, J, et al. Correlation of BACH1 and hemoglobin E/Beta-thalassemia globin expression. Turk J Haematol. (2016) 33:15–20. doi: 10.4274/tjh.2014.0197
41. Zakaria, NA, Islam, MA, Abdullah, WZ, Bahar, R, Mohamed Yusoff, AA, Abdul Wahab, R, et al. Epigenetic insights and potential modifiers as therapeutic targets in β-thalassemia. Biomol Ther. (2021) 11:755. doi: 10.3390/biom11050755
42. Marini, MG, Chan, K, Casula, L, Kan, YW, Cao, A, and Moi, P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. J Biol Chem. (1997) 272:16490–7. doi: 10.1074/jbc.272.26.16490
43. Macari, ER, and Lowrey, CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood. (2011) 117:5987–97. doi: 10.1182/blood-2010-10-314096
44. Pullarkat, V, Meng, Z, Tahara, SM, Johnson, CS, and Kalra, VK. Proteasome inhibition induces both antioxidant and hb f responses in sickle cell disease via the nrf2 pathway. Hemoglobin. (2014) 38:188–95. doi: 10.3109/03630269.2014.898651
45. Promsote, W, Makala, L, Li, B, Smith, SB, Singh, N, Ganapathy, V, et al. Monomethyl fumarate induces gamma-globin expression and fetal hemoglobin production in cultured human retinal pigment epithelial (RPE) and erythroid cells, and in intact retina. Invest Ophthalmol Vis Sci. (2014) 55:5382–93. doi: 10.1167/iovs.14-14179
46. Zhu, X, Xi, C, Ward, A, Takezaki, M, Shi, H, Peterson, KR, et al. NRF2 mediates γ-globin gene regulation through epigenetic modifications in a β-YAC transgenic mouse model. Exp Biol Med (Maywood). (2020) 245:1308–18. doi: 10.1177/1535370220945305
47. Steinberg, MH, McCarthy, W, Castro, O, Ballas, SK, Armstrong, FD, Smith, W, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. (2010) 85:403–8. doi: 10.1002/ajh.21699
48. Steinberg, MH, Lu, ZH, Barton, FB, Terrin, ML, Charache, S, Dover, GJ, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Mult Study Hydroxyurea Blood. (1997) 89:1078–88. doi: 10.1182/blood.V89.3.1078
Keywords: BACH1, Nrf2, sickle cell disease, hemoglobin F, gamma globin, HMOX1, vaso-occlusion, NF-kappa B
Citation: Belcher JD, Nataraja S, Abdulla F, Zhang P, Chen C, Nguyen J, Ruan C, Singh M, Demes S, Olson L, Stickens D, Stanwix J, Clarke E, Huang Y, Biddle M and Vercellotti GM (2023) The BACH1 inhibitor ASP8731 inhibits inflammation and vaso-occlusion and induces fetal hemoglobin in sickle cell disease. Front. Med. 10:1101501. doi: 10.3389/fmed.2023.1101501
Edited by:
Magali J. Fontaine, University of Maryland, United StatesReviewed by:
Wassim El Nemer, French Blood Establishment (EFS), FranceCopyright © 2023 Belcher, Nataraja, Abdulla, Zhang, Chen, Nguyen, Ruan, Singh, Demes, Olson, Stickens, Stanwix, Clark, Huang, Biddle and Vercellotti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John D. Belcher, YmVsY2hlckB1bW4uZWR1
†Present addresses: Domi Stickens, Arrakis Therapeutics, Waltham, MA, United States
Maneet Singh, Wave Life Sciences, Cambridge, MA, United States
Margaret Biddle, Entact Bio, Watertown, MA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.