 Ganesh Yadagiri
Ganesh Yadagiri Aakriti Singh
Aakriti Singh Kanika Arora
Kanika Arora Shyam Lal Mudavath
Shyam Lal Mudavath- 1Infectious Disease Biology Laboratory, Chemical Biology Unit, Institute of Nano Science and Technology, Mohali, Punjab, India
- 2Department of Veterinary Biosciences, The Ohio State University, Columbus, OH, United States
Visceral leishmaniasis (VL), a vector-borne disease, is caused by an obligate intramacrophage, kinetoplastid protozoan parasite of the genus Leishmania. Globally, VL is construed of diversity and complexity concerned with high fatality in tropics, subtropics, and Mediterranean regions with ~50,000–90,000 new cases annually. Factors such as the unavailability of licensed vaccine(s), insubstantial measures to control vectors, and unrestrained surge of drug-resistant parasites and HIV-VL co-infections lead to difficulty in VL treatment and control. Furthermore, VL treatment, which encompasses several problems including limited efficacy, emanation of drug-resistant parasites, exorbitant therapy, and exigency of hospitalization until the completion of treatment, further exacerbates disease severity. Therefore, there is an urgent need for the development of safe and efficacious therapies to control and eliminate this devastating disease. In such a scenario, biotherapy/immunotherapy against VL can become an alternative strategy with limited side effects and no or nominal chance of drug resistance. An extensive understanding of pathogenesis and immunological events that ensue during VL infection is vital for the development of immunotherapeutic strategies against VL. Immunotherapy alone or in combination with standard anti-leishmanial chemotherapeutic agents (immunochemotherapy) has shown better therapeutic outcomes in preclinical studies. This review extensively addresses VL treatment with an emphasis on immunotherapy or immunochemotherapeutic strategies to improve therapeutic outcomes as an alternative to conventional chemotherapy.
Introduction
Leishmaniasis is one of the seven most important tropical diseases representing a serious world health problem on a broad spectrum of clinical manifestations with a potentially fatal outcome. It is a vector-borne chronic infectious disease attributable to an intracellular one-celled protozoan parasite of the genus Leishmania. It is systemically transmitted to humans by the bite of an infected female Phlebotomine sand fly (1). Its incidence has increased to more than 12 million people in 98 countries around the world, mostly afflicting developing and under developed countries (higher in rural areas than in urban areas) (2). The epidemiological burden also depends on the characteristics of the parasite, ecological characteristics of the transmission site, and the immune response of the host. In accordance with the World Health Organization (WHO), an estimated 70,000–100,000 new leishmaniasis cases are reported globally, and over 20,000–30,000 deaths occur annually (3). A hemoflagellate vector, a female sand fly of genus Phlebotomus in the Old World and Lutzomyia in the New World, is responsible for the transmission of this disease to humans (4). Leishmaniasis has gained more public consideration owing to the higher incidence of morbidity; “the London declaration community” on Neglected Tropical Diseases declared to eliminate leishmaniasis as a public health issue by 2020 (5). Discrete species of Leishmania cause diverse clinical manifestations ranging in severity from self-healing localized cutaneous ulcers to fatal multi-organ disease. It broadly manifests as cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis (MCL), and visceral leishmaniasis (VL). The outcome of the disease is unwavering by the interplay of parasite characteristics, vector biology, and host factors, with immune responses (6). VL or kala-azar, as it is called in its advanced stages, is a severe chronic systemic infection caused by Leishmania donovani (subtropics of South Asia and East Africa; anthroponotic mode of transmission) and Leishmania infantum (Europe, North Africa, and Latin America; zoo anthroponotic mode of transmission) (7). VL is the most severe clinical form of leishmaniasis characterized by hepatosplenomegaly, high fever, pancytopenia, and hypergammaglobulinemia, and if left untreated, the disease may worsen over time to severe progressive cachexia, multi-organ damage, secondary infections, and ultimately causes death of the patient (8). VL is ranked second in mortality and fourth in morbidity among tropical parasitic diseases, with 20,000 to 40,000 deaths annually and over 2,000,000 disability-adjusted life years (DALYs) lost (6). In conformity with the WHO, perennially 50,000–90,000 new VL cases arise globally, and >95% of new VL cases are reported in 10 countries: Brazil, China, Ethiopia, India, Iraq, Kenya, Nepal, Somalia, South Sudan, and Sudan (3). More characteristically, a conclusive 80% of the global burden of VL was reported in South Asia (e.g., in the year, 2007, 0.1–0.15 million VL cases were reported in India alone). The condition is particularly severe in eastern states of India, including Bihar, Jharkhand, Uttar Pradesh, and West Bengal (9). A total of 6,70,897 VL cases were reported officially from 1987 to 2011 from Bihar state exclusively. Some districts of Bihar state (Muzaffarpur, Purnea, Saharsa, Ararea, Vaishali, Madhepura, East Champaran, Samastipur, Saran, and Darbhanga) have confronted worst epidemic since the 1970s with 90% of the case reports (10). Collectively, an integrated elimination program was started by the government of Bangladesh, India, and Nepal started in 2005 with the aim to decline the VL case reports to less than one new case per 10,000 population per year at sub-district level (block level in India and Nepal and upazila level in Bangladesh) by 2015 (11).
The treatment and control of VL are limited and rely mainly on chemotherapy. Existing anti-leishmanial chemotherapeutic agents (meglumine antimoniate, sodium stibogluconate, pentamidine, amphotericin B, miltefosine, and liposomal amphotericin B) encompasses several problems with regard to safety and efficacy. Nephrotoxicity, pancreatitis, cardiotoxicity, teratogenicity, emergence of drug-resistant parasites, high cost, and exigency for hospitalization due to longevous intravenous treatment present challenges toward patient compliance (12). Hitherto, several clinical trials have been performed in India to improve the therapeutic regimens and to ameliorate the efficacy of the limited armamentarium of existing anti-leishmanials (11). The limited therapeutic efficacy of human vaccine(s) and inefficient vector control measures impose perplexity in the treatment of VL. Moreover, leishmania–HIV (human immunodeficiency virus) coinfected people have accelerated chances of developing a full-blown disease with high relapse and lethality. Such cases of co-infections are particularly reported in Western Europe, India, Brazil, Ethiopia, and Africa. These co-infections lead to diagnostic difficulty and therapeutic unresponsiveness to VL treatment (13). Post-kala-azar dermal leishmaniasis (PKDL) is a neglected complication of VL, playing a significant role in the inter-epidemic period as a potential reservoir for the Leishmania parasite. It usually appears as a macular, maculopapular, or nodular rash on the face, upper arms, trunks, and bared parts of the body. PKDL is a deadly, infectious disease that is reported in Africa particularly in Sudan (50%; L. infantum), and in Asia particularly in India (5–10%; L. donovani) (14). The emergence of drug-resistant strains of the Leishmania parasite, severe toxic effects of current anti-leishmanial therapy, advent of HIV-VL co-infections, PKDL, and absence of proper vector control measures and vaccine(s) against VL pose severe problems to VL treatment and control (15). Therefore, prevention and control of leishmaniasis require a combination of intervention strategies that would begin with the search for a novel system to actualize the treatment shorter, safer, efficacious, and more affordable (16). Drug discovery for such a poverty-ridden neglected tropical disease was never included as a high priority by pharmaceutical companies as it is unlikely to yield a good profit on research and drug development costs.
In this extant, we have extensively addressed the leading-edge and provocation in therapeutics and ministrations of VL with emphasis on immunotherapy or immunochemotherapeutic strategies to improve therapeutic outcomes as an alternative to prevalent chemotherapy.
Immunobiology of VL: cells and immune mediators correlated to susceptibility and resistance
The immunobiology and immunopathology of visceral infection for various species including humans, canines, and experimental small animal (rodent) models have been comprehensively studied, with many points exemplified but yet some to be explicated. General consent is that despite the uniqueness of each model, the infection extent and spread are basically influenced by many factors such as concomitant pathologies, pathogen–host interaction, geographic location, and host immune response (17). Other factors, for instance, genetic differences of the parasite between species and strains, host genetics, and environmental factors (18), also provocate the susceptibility or resistance to infection. Parasites have hence evolved to evade and evert host immune responses. Different types of immune cells such as macrophages, neutrophils, natural killer (NK) cells, and dendritic cells (DCs) play an essential role by sensing parasitic first occurrence via pattern recognition receptors (PRRs) and complement receptors present on host cells. Numerous toll-like receptors (TLRs) such as TLR2, TLR3, TLR4, TLR7, and TLR9 similarly contribute to the sensing and recognition of Leishmania parasites. Recognition and interaction stimulate intracellular signaling pathways that lead to the occurrence of inflammatory responses and control parasite multiplication via an innate immune response (19). The macrophages and DCs are known to play crucial roles in the initiation, expansion, and maintenance of an assertive immunity against Leishmania infection. Nonetheless, the intramacrophage amastigotes, once internalized, alter cell-signaling pathways in macrophages inhibiting cytokine responses, and this disrupts the protective capacity of cells. These work by inhibiting MAP kinases, transactivation of NF-κB by local stimulation of TGFβ32, and suppression of cytokine signaling (SOCS-3) (20). VL-infected patients evince an accretion of dysfunctional T cells due to their incapability to produce a cell-mediated immune response in defiance of inchoate antigens. This leads to immunocompromisation of VL patients and is prone to secondary pathogenic infections (21). Cellular immunity rather than humoral mediated immunity plays a crucial role in the control of Leishmania infection. The progression of type-I immune responses against leishmania is characterized by antigen-presenting cells (APCs) that stimulate the production of interleukin-12 (IL-12) which in turn facilitates interferon-γ (IFN-γ) via Th1 T cells. Consequently, it activates macrophage-mediated microbicidal mechanisms which function by producing nitric oxide (NO) and reactive oxygen species (ROS) which efficaciously kill intra-macrophage amastigotes. An immunosuppressive cytokine, IL-10, deactivates the detectable levels of Th1 cytokines which might result in the progression of VL regardless of immune functionality (22). The coaction of Th1 and Th2 cytokines seems to persist throughout the infectious phase which advocates significant roles in disease protection and pathogenesis by various cytokines (23). The resistance and susceptibility of infection depend on the host's immunological status. Enhanced levels of different cytokines of CD4 T-helper cells such as IFN-γ, IL-12, and IL-2 with a lack of IL-10 is requisite for the revivification of VL infection (24). Therefore, enhanced levels of Th2 cytokines result in disease progression while that of Th1 cytokines arbitrates disease clearance (1). Specifically, in BALB/c mice, the replication of amastigotes in the liver marks the first week of infection. Genetically resistant mice (C57BL/6) have natural resistance-associated macrophage protein 1 (NRAMP 1) gene (Slc11a1) mainly exhibited on macrophage surface and involved in macrophage activation that might function by killing the Leishmania parasite by nitric oxide (NO)-mediated mechanisms (25). It mainly elevates the expression of IFN-γ by CD4+ T cells and exacerbates the disease situation with the scarceness of IFN-γ levels in Leishmania-infected C57BL/6 (resistant) and BALB/c (susceptible) mouse strains, respectively (26). A balanced proportion of the cytokine release from the CD4+ and CD8+ T- ells is a critical factor of immune function against leishmaniasis infection. In active VL, both CD4 and CD8 cells function cooperatively for clearance of infection displaying diverse functions and expression of the cytokines. CD4+ cells are critical for the deterrence of primary infection, whereas CD8+ cells are role players during the secondary immune response (9). Over expression of chemokines, chemokine (C-X-C motif) ligand 9 [CXCL9], and CXCL10 in serum during active infection counseled that these chemokines accompanied by IFN-γ play a crucial role in the immunopathogenesis of VL (23) (Figure 1).
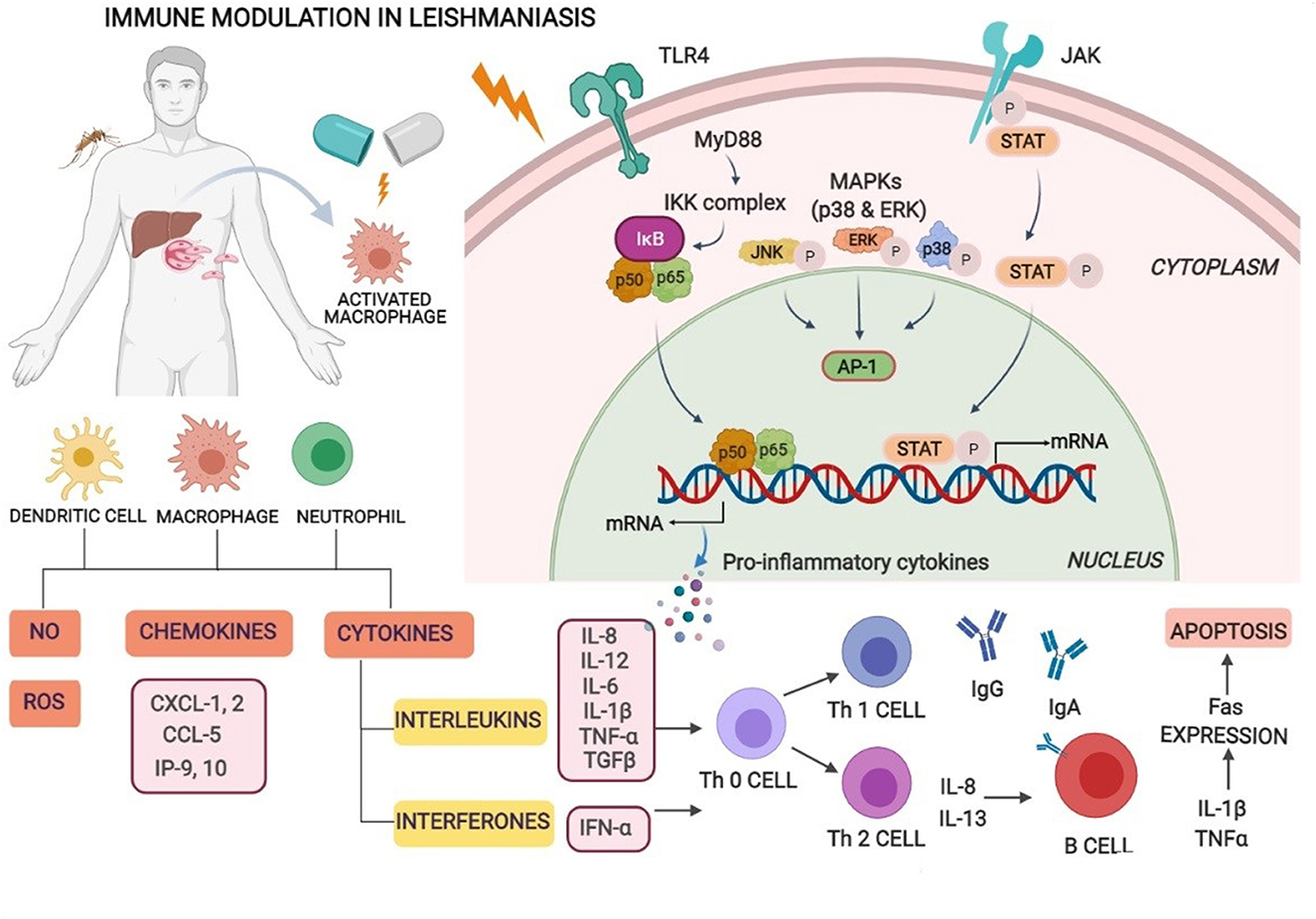
Figure 1. Immune modulation in visceral leishmaniasis. Macrophages, neutrophils, natural killer (NK) cells, and dendritic cells (DCs) play an essential role by sensing parasitic first occurrence via pattern recognition receptors (PRRs) and complement receptors present on host cells. Recognition and interaction stimulate intracellular signaling pathways that lead to the occurrence of inflammatory responses and control parasite multiplication. Leishmania-infected macrophages and dendritic cells activate Th1 cell differentiation and generate inflammatory cytokines especially IL-12, IFN-γ, reactive oxygen, and nitric oxide species supporting intra-cellular parasite clearance and enhanced levels of Th2 cytokines resulting in disease progression.
However, the unification of innate and acquired immunity together with the insufficiency of data on the human immune response is one of the crucial obstacles currently impeding vaccine development and application.
Current VL chemotherapy and its limitations
Certain factors accounted for the selection of specific drugs in the primary VL zones and therapeutic options including drug risk-benefit ratio, maintenance of health protection, and the accessibility of anti-leishmanial medication in the view of public wellbeing and the epidemiological aspects of vector, parasite, and domestic reservoir of VL (zooanthroponotic and anthroponotic) (27). In the Indian subcontinent, VL is anthroponotic and a major challenge is widespread resistance to antimonials, particularly in Bihar, which is the hyper endemic zone of VL. In Southern Europe and America, VL is zooanthroponotic, and dogs are reservoirs, and is a serious issue toward the control of the infection (28). Due to the non-availability of an effective vaccine against VL, control of VL exclusively relies on chemotherapy. The therapeutic options available for the treatment of VL are summarized in Table 1.
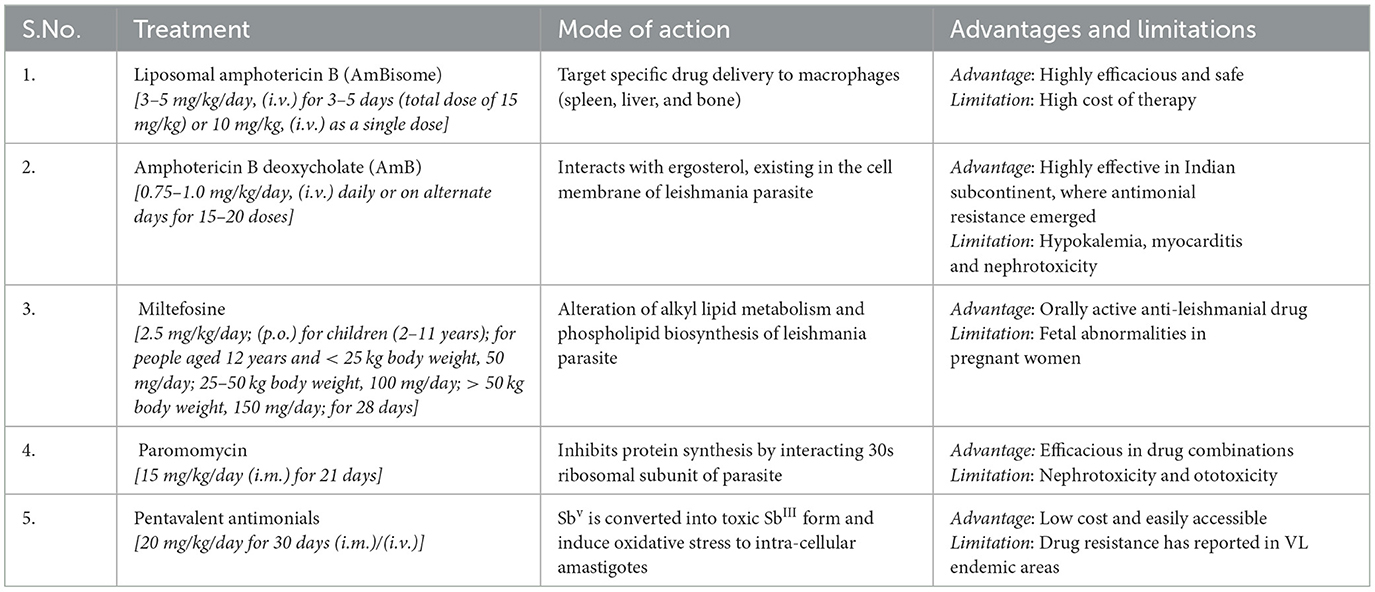
Table 1. WHO recommended treatment regimens for VL, in the Indian subcontinent and its mode of action, advantages, and limitations (11, 29).
Pentavalent antimonials
Antimony (Stibium) is a semi-metallic element having atomic number 51 that belongs to the vanadium family (Group VA) of the periodic table and has been used in therapeutics since antiquity. An Indian scientist named Dr. Upendranath Brahmachari for the first time in 1920 actualized an antimony-containing substance, that is, urea stibamine, for VL treatment. Later, sodium stibogluconate (Pentostam) and meglumine antimoniate (Glucantime) as synthetic injectable pentavalent antimonials were introduced as primary therapy for CL and VL treatments. It is commonly documented that pentavalent antimonials (SbV) work as a prodrug that is converted into toxic and active trivalent antimonials (SbIII) (reduced form) in the intracellular environment of macrophages, where amastigotes reside and multiply (30). Leishmania donovani DNA topoisomerase-I is inhibited by pentavalent antimonial drugs (31). Daily intramuscular injection (20 mg/kg for 28–30 days) has been used as the standard treatment for VL in most parts of the globe. The most frequent side effects of pentavalent antimonials are cardiovascular issues (QTc interval prolongation, ventricular tachycardia, ventricular fibrillation, and torsades de pointes) amongst which QTc interval prolongation (> 0.5 s) signals may lead to the onset of severe and fatal cardiac arrhythmias, arthralgia, pancreatitis, and nephrotoxicity. Pancreatitis is more common with antimonials in HIV-VL co-infected patients and further increases the mortality rate (32). Antimonials (Pentostam or Glucantime) are no longer recommended for clinical use for Indian VL patients due to the rampant rise of drug-resistant parasites. Initially, in 1980s, pentavalent antimonials were administered at a much lower dosage (10 mg/kg) for a short span of 6–10 days, which showed resistance in Indian KA patients. However, in the later years, as reported by Mohapatra (33), the high dosage of 20 mg/kg for a longer duration showed no success in treatment.
Amphotericin B
Amphotericin B deoxycholate (AmB), a polyene class of anti-fungal antibiotic has been recommended as a first-line drug for the treatment of VL in the Indian subcontinent for more than four decades. AmB has shown high therapeutic efficiency in Indian KA patients, especially in North Bihar, a VL hyper endemic region (34). Ergosterol, a component in the Leishmanial parasitic cell membrane, is the primary target of AmB. It can bind and sequester ergosterol, leading to the formation of pores in the cell membrane of the parasite (35). AmB has shown improved therapeutic efficiency (~100%) at the dose of 0.75–1 mg/kg, i.v., infusions for 15–20 doses every day or on alternate days; however, most of the KA-infected patients experience infusion-related problems such as fever, thrombophlebitis, chills, hypokalaemia, nephrotoxicity, and myocarditis. These adverse effects require continuous monitoring and hospitalization, thereby increasing the cost of therapy (36). Lipid-based carrier systems for AmB delivery have been developed to overcome the toxic adverse effects of conventional AmB. Liposomal AmB (AmBisome) was introduced with a unique safety profile. Single dose AmBisome (10 mg/kg) was found to be adequate to efficaciously treat VL with an improved safety profile and has now been recommended as a drug of choice to treat VL in the Indian subcontinent (37, 38). As per the recommendations of the WHO's expert committee on the control of leishmaniasis, AmBisome was declared as a first-line treatment drug for VL in India, Bangladesh, and Nepal (39).
Pentamidine
Pentamidine, chemically known as 4-[5-4-carbamimidoylphenoxy] pentoxyl benzenecarboximidamide, is an orphan drug approved for the treatment of Pneumocystis carinii, a serious fungal opportunistic infection in immunocompromised patients in the United States (40). The anti-protozoal effects of pentamidine isethionate, an aromatic diamidine, were reported against Trypanosoma cruzi in 1938. Soon after, reports of antimonial drug resistance, pentamidine, were recommended as a second-line anti-leishmanial drug in KA-infected patients (41). Pentamidine isethionate is recommended as secondary prophylaxis in addition to anti-retroviral drugs to inhibit relapse in HIV-VL co-infected immunocompromised patients (42). Pentamidine selectively binds to kinetoplastid DNA (kDNA) of Leishmania donovani resulting in the inhibition of parasite replication (43). Pain or formation of abscess at the site of injection, renal insufficiency, and allergic reactions, including Stevens–Johnson syndrome, cardiotoxicity, and metabolic disorders, are the common adverse effects of intravenous pentamidine isethionate injections (44).
Paromomycin
Paromomycin (formerly known as aminosidine) is an aminoglycoside antibiotic with a unique spectrum of anti-leishmanial activity. Paromomycin interacts with the ribosomes of mitochondria and induces respiratory dysfunction in Leishmania parasites. Paromomycin binds to the 30S subunit of ribosomes which causes the inhibition of protein synthesis (45). Paromomycin (11 mg/kg/day for 3 weeks, i.m. route) was recommended as a first-line treatment for VL in the Indian subcontinent (46–48). However, paromomycin was found to effect the renal (nephrotoxicity), vestibular, and auditory organs (ototoxicity) in VL-infected patients (49).
Miltefosine
Miltefosine (hexadecylphosphocholine, HePC), an alkyl phospholipid compound, is the first approved oral therapeutic agent registered for combating VL. Initially, it was used as an anti-neoplastic agent originally designed for breast cancer and other solid tumors. Miltefosine was licensed as an oral anti-leishmanial drug in the Indian subcontinent in 2002 (50). Miltefosine functions by disrupting lipid metabolism via inhibition of phosphatidylcholine synthesis, thereby affecting cell-signaling pathways and membrane synthesis of the Leishmania parasite. Miltefosine mainly interacts with the acidocalcisome and stimulates the sphingosine-dependent plasma membrane calcium channels of the Leishmania parasite (51). Miltefosine (2.5 mg/kg/day for 28 days, p.o.) has been recommended for CL- and VL-infected patients with good cure rates (52). Miltefosine is not recommended for pregnant women due to its teratogenicity (fetal abnormalities) (50).
Sitamaquine
Sitamaquine (WR-6026) is an orally active 8-aminoquinoline analog developed by Walter Reed Army Institute Research in collaboration with GlaxoSmithKline. Sitamaquine has shown improved efficacy against L. donovani-infected patients in Kenya and the Indian subcontinent in phase 2B clinical trials (53). Sitamaquine acts by entering the parasite through diffusion in a sterol-dependent and energy-independent electrical gradient manner (54). Renal toxicity and methemoglobinemia are very common side effects with higher doses of sitamaquine in VL-infected patients. Furthermore, the decision of its clinical development is hindered because of its safety concerns (55).
WHO recommended combination treatment regimens for VL, in the Indian subcontinent (29).
1. Combination of AmBisome (5 mg/kg (i.v.) as a single dose) + Miltefosine (2.5 mg/kg/day; (p.o.) for children (2–11 years); for people aged >12 years and <25 kg body weight, 50 mg/day; 25–50 kg body weight, 100 mg/day; > 50 kg body weight, 150 mg/day) for 7 days.
2. Combination of AmBisome (5 mg/kg (i.v.) as a single dose) + Paromomycin (15 mg/kg/day (i.m.)) for 10 days.
3. Combination of Miltefosine (2.5 mg/kg/day; (p.o.) for children (2–11 years); for people aged >12 years and <25 kg body weight, 50 mg/day; 25–50 kg body weight, 100 mg/day; > 50 kg body weight, 150 mg/day) for 10 days + Paromomycin 15 mg/kg/day (i.m.) for 10 days.
Immunotherapy and immunochemotherapy: promising strategies for VL treatment
Efficient chemotherapy is the alternative for the treatment of VL due to inefficacious vector preventive techniques available for humans so far. Nonetheless, the emergence of drug-resistant parasite strains, drug unresponsiveness, discrepancy in medical responses due to geographical distribution, and disease relapse to the conventional anti-leishmanial therapeutic strategies enforce the concerted efforts for the search of developing new therapies and treatment regimens which can be given orally (56). In such a situation, immunotherapy alone or in combination with conventional chemotherapy can become a viable alternative in the treatment and control of VL. The molecular pathways that lead to the development of parasites are inhibited using biological response modifiers to prevent leishmanial-associated immunosuppression. Thus, this immunotherapy approach has proven its novelty offering advantages of nominal toxicity and cost-effectiveness that could further ameliorate disease progression (9). To attain prophylactic and/or therapeutic success, a biological molecule that has a therapeutic potential to modulate the immunological status of the host is the need of the hour. Presently, immunotherapy strategies have been employed for the successful treatment of several diseases including cancers, allergy-mediated diseases, and some viral infections (hepatitis) (19). Immunotherapy in combination with conventional chemotherapy displays rapid parasite clearance, thereby augmenting the efficacy of treatment (57). Immunotherapy can clear intracellular amastigotes by modulating immunological mechanisms involving the activation of macrophages and cell-mediated immune responses in VL infection (58). For hindering VL infections, Th1-type cytokine responses are to be selectively induced. This approach could successfully augment molecular interactions of immune cells and released cytokines, such as Th1 cytokines, IFN-gamma, and IL-12 levels, and downregulate IL-10 and IL-14, thereby preventing the enhancement of disease severity leading to treatment and simultaneously offering a unique diagnostic approach for VL (59). VL developed as a substantial opportunistic infection in immunocompromised patients with HIV infection (HIV-VL co-infections) in African regions, the Indian subcontinent, and Western Europe and has also shown unresponsiveness to conventional chemotherapeutics. In HIV-VL co-infections, the treatment outcome with monotherapy is poor (60). The conventional approach of anti-leishmanial chemotherapy can be defeasible subject to a high degree of infection and therapeutic situation. To overcome such a stage, the incorporation of an immunomodulator with prevailing chemotherapies can be promising as this combination will stimulate cell-mediated immune response and can result in successful outcomes (Figure 2).
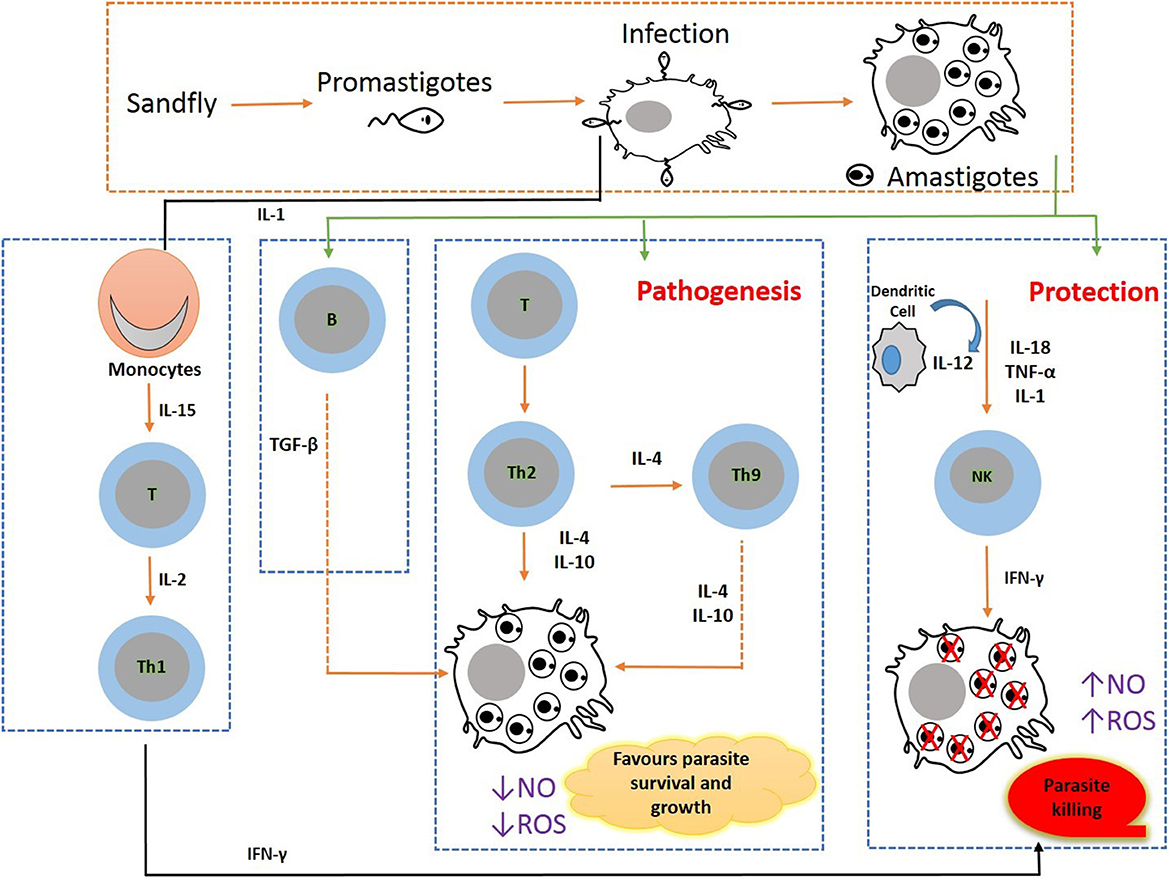
Figure 2. Immunobiology during pathogenesis in visceral leishmaniasis: Leishmania parasite invades macrophages and activates CD4 T cells to release Th1- (IL-12, IFN-γ, and nitric oxide) and Th2 (IL-10 and TGF-β)-type cytokine responses. VL pathogenesis is mediated through the augmented expression of Th2-type cytokines, and VL clearance is arbitrated through the increased Th1 cytokine responses.
The host immunological state in VL is a key factor that significantly affects infection susceptibility and resistance. The Th1-type cytokines in the CD4 T-helper cells are increased and activated by the microbicidal process mediated by macrophages. As a result, nitric oxide (NO) and reactive oxygen species (ROS) are produced, both of which are extremely efficient for eliminating intramacrophage amastigotes. The increased levels of Th2-type cytokines that can deactivate macrophages to prevent the generation of NO and ROS boost amastigotes' intracellular development.
The neutrophils, macrophages, dendritic cells, and natural killer cells work together to mediate the innate immune response in leishmaniasis. The female sand fly takes a blood meal to start the life cycle of the Leishmania parasite. The metacyclic promastigotes subsequently enter the bloodstream and are phagocytosed by neutrophils at the infection site. A greater number of neutrophils migrate to the infection site as a result of the neutrophils phagocytosing the parasite and secreting IL-8. Chemotaxis of macrophages to the infection site is caused by the apoptosis of infected neutrophils and the subsequent release of the macrophage chemokine MIP-1. The parasites' binding to C3b speeds up phagocytosis, and promastigotes are transformed into the amastigote form. The phagocytosed parasites can survive in apoptotic neutrophils and grow in macrophages, causing the spread of the disease. To induce the Th1 response necessary for parasite eradication in infected parasites, Leishmania parasites block the production of IL-12 in macrophages, which promotes parasite proliferation. Dendritic cells are prevented from presenting the T lymphocytes with the parasite-specific antigens by the suppression of interleukin (IL)-12 secretions. By enhancing the production of arginase, which inhibits NO generation, Leishmania parasites also promote the activation of ornithine from arginine. The conventionally activated macrophages produce pro-inflammatory microbicidal and tissue-damaging cytokines, while the alternatively activated macrophages are anti-inflammatory and regulate inflammation and tissue repair. The signals produced by the microenvironment are necessary for macrophage polarization into M1 and M2. LPS, IFN, TNF, and GM-CSF are the main causes of M1 macrophage polarization, which also activates the complement system and draws in immune cells. M1 macrophages can differentiate into M2 and vice versa, M2 macrophages repolarize more quickly than M1 macrophages, and M1 macrophages can develop into M2 and vice versa. By polarizing macrophages into the M2 subset and reducing dendritic cells, the Leishmania parasite impairs antigen presentation and establishes an environment that is immunosuppressive to sustain its survival (61). Furthermore, chemokines including MIP-1, cytokines, and complement proteins induce monocytes to the site of infection, facilitating parasite replication. IL-12, which is essential for the development of CD4+ T cells into protective Th1 cells for the expression of co-stimulatory molecules CD40, CD80, and CD86, is primarily produced by dendritic cells as the disease progresses. A higher parasite load results from blocking CD40–CD40L interaction because it reduces IL-12 and IFN-γ productions. In addition, NK cell suppression reduced IFN production and markedly increased the parasite load (13, 62). Numerous studies have shown that immunotherapy is a successful anti-leishmanial treatment. Activation of macrophages was increased by a single dose of an IL-27 or IL-10 monoclonal antibody, which improved parasite killing (56%). The synthesis of the anti-inflammatory IL-17 is adversely regulated by IL-27 inhibition, which worsens the condition (63). It has been discovered that IFN-induced macrophage killing of Leishmania is inhibited by the macrophage deactivation cytokines IL-4 and IL-10, which promote the development of VL. Additionally, TGF antibody neutralization increases IFN-γ production, which in turn increases IL-2-associated macrophage activation, nitric oxide generation, and proliferation of cytotoxic T lymphocytes, curing VL independently of TH2-type cytokines (IL-4). Additionally, IL-10 inhibition promotes VL regression (64). A pleiotropic cytokine called IL-6 regulates dendritic cell activity during infection with L. donovani. IFN-γ and tumor necrosis factor can be used to activate macrophages, but IL-6 directly impairs their ability to do so. It also promotes the repression of Th2-type responses and inhibits the development of Th1 cells. An achievable therapeutic target is the immunosuppressive, macrophage-deactivating nature. L. donovani infection in the liver has been linked to cytokine antagonist therapy with IL-6, IL-10, and IL-27. In contrast, IL-10, IL-6, and IL-27 receptor signaling directly affects L. donovani liver infection and, when absent, enhances Th1-type responses, speeding up parasite elimination (65). As inflammation reaches a particular level, the cytokine IL-22, a member of the IL-10 family, causes non-immune cells such as epithelial cells and fibroblasts to proliferate and migrate, aiding in tissue protection. Strong IL-17 and IL-22 induction by L. donovani leads to additional tissue healing that aids in immunosurveillance and suggests their complementary roles in host defense against the parasite (66). The immunotherapy and immunochemotherapeutic approaches against visceral leishmaniasis is highlighted in Table 2.
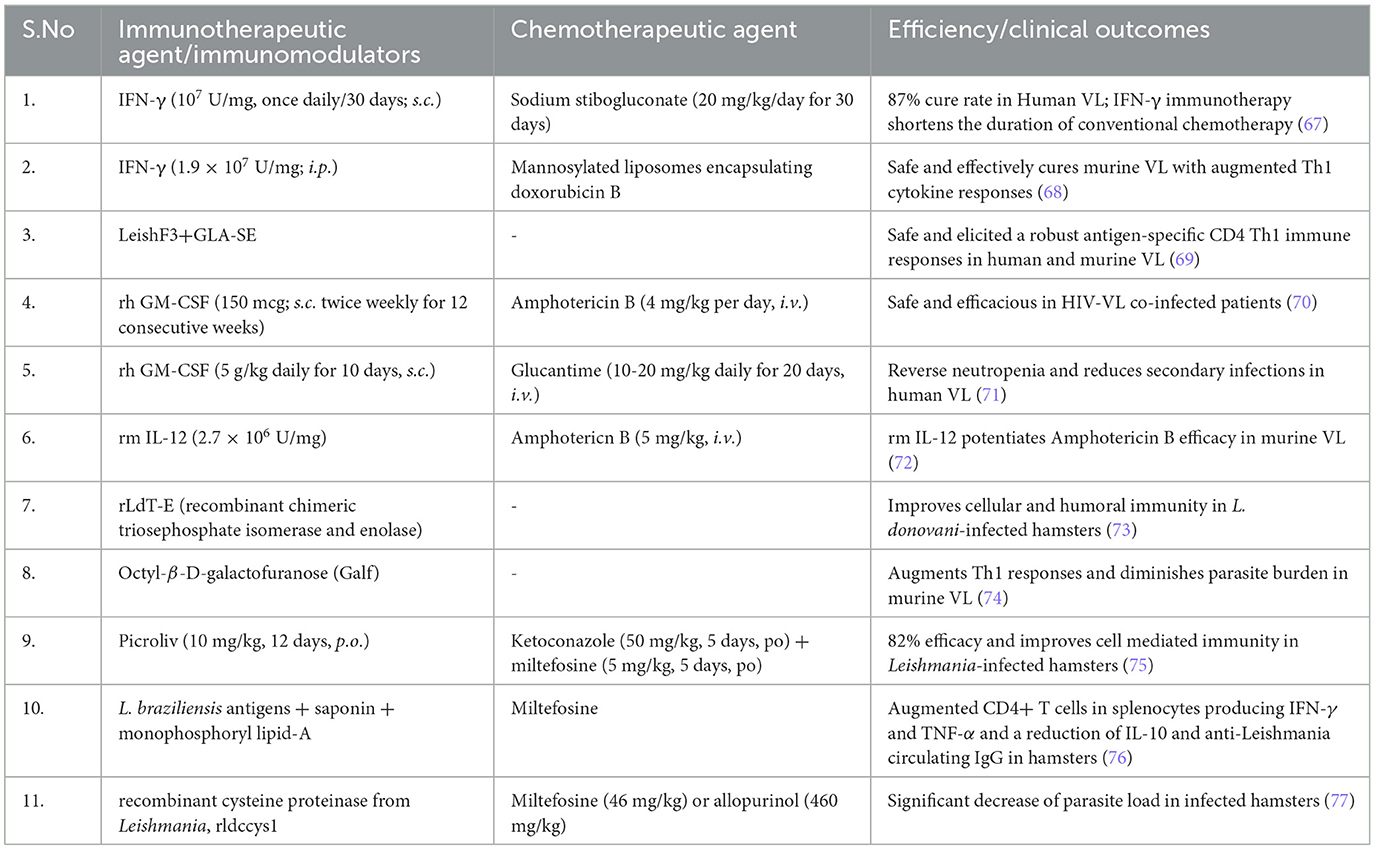
Table 2. Immunotherapy and immunochemotherapeutic approaches against visceral leishmaniasis.
Cytokine therapy for VL
Small, soluble proteins known as cytokines play a critical role in the control of innate immunity. They are primarily produced by mononuclear phagocytes such as macrophages and dendritic cells (78). Th2-type cytokines are expressed more frequently in VL, which regulates the course of the disease, and Th1 cytokines are produced at higher levels, which intervene to resolve the infection (1). The identification of cytokines that preferentially activate Th1-type cytokine responses may be useful in the treatment of VL. Anti-IL-10 antibodies are being investigated for use in monotherapy (immunotherapy) or in conjunction with conventional anti-leishmanials. Immunostimulatory cytokines (GM-CSF, IFN-γ, and IL-12) as well as antibodies that target suppressive or deactivating cytokines are also being investigated (immunochemotherapy).
Granulocyte-macrophage colony-stimulating factor
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is an immuno-regulatory glycoprotein cytokine that stimulates hematopoiesis by inducing the differentiation and proliferation of committed progenitor cells of the myeloid lineage in the bone marrow (79). In addition to playing a crucial role in the control of innate and adaptive immunity, GM-CSF activates macrophages to improve phagocytosis, antigen presentation, chemotaxis, and cell adhesion (80). By using an in vitro colony-stimulating factor activity assay, Burgess et al. for the first time extracted and purified GM-CSF from mouse lung conditioned medium, which stimulates the proliferation of granulocytes, macrophages, or both, in bone marrow progenitor cells (81). Infectious disorders such as leishmaniasis, malaria, and tuberculosis have all been successfully treated with immunotherapy using recombinant GM-CSF (1). In VL, GM-CSF causes granulocytopenia to improve as well as blood monocyte mobilization and macrophage activation, both of which may help maintain an infection-fighting immune system. Recombinant mouse GM-CSF (rm GM-CSF) therapy efficiently eliminated the infection in experimental VL, but anti-rm GM-CSF antibody therapy made the visceral infection worse (82). Recombinant human GM-CSF (rh GM-CSF) treatment effectively eliminated intracellular amastigotes in L. donovani-infected macrophages in an LPS-independent manner, and the amount of time required to stimulate macrophages to produce leishmanicidal effects is very short (36 h for rh GM-CSF and 48–72 h for rh IFN-γ to activate macrophages) (83). Combining rh GM-CSF and M-CSF with rh IFN-γ is a highly effective way to increase the control of intracellular parasites. In patients with VL infection, immunochemotherapy with rh GM-CSF (5 g/kg daily for 10 days) and Glucantime (10–20 mg/kg daily for 20 days) can reverse neutropenia and decrease subsequent infections (71). Patients with HIV and VL can successfully treat the visceral infection with increased safety using immunotherapy with rh GM-CSF (150 mcg, s.c. route, twice weekly for 12 consecutive weeks) and liposomal amphotericin B (4 mg/kg/day for 5 consecutive days, then on day 10, 17, 14, 31, and 38) (70).
Interferon-gamma
Interferon-gamma (IFN-γ) (molecular weight 20–25 kDa) is a sole type II IFN, IFN-γ is a pleiotropic soluble glycoprotein cytokine, crucial for persuading and modulating the innate and adaptive immune response. It induces the host defense mechanism and is hence used in immunotherapy against several pathogenic infectious diseases including malaria, tuberculosis, fungal diseases, leishmaniasis, and toxoplasmosis (84). The integral source for the formulation of IFN-γ is T-lymphocytes (helper T cells, cytotoxic T cells, and natural killer cells) which develop strength for parasitic infections by fortifying macrophages. IFN-γ augments the intra-cellular amastigote-killing impact to secrete Th1-type cytokines by aiding in the development of macrophages. Augmented expression of Th1-type cytokines is embroiled in eliminating the infection in the first place while Th2-type cytokines mediate VL pathology (85). For instance, treatment with L. donovani antigens (triosephosphate isomerase and enolase) clears the liver and splenic parasite burden with enhanced IFN-γ levels in Syrian golden hamsters (73). CGD4+ T cells were found to be critical for and the key basis of the IFNγ generation in Leishmania parasite-stimulated whole blood (WB) cultures. Endogenously generated IFN-γ in active VL patients helps in the control of intracellular parasite growth, and the inhibition of IFN-γ secretion in ex-vivo splenic aspirate cultures exacerbates the disease progression (86). CD4+ T cells stimulate the formation of augmented levels of IFN-γ which not only assists in controlling the Leishmania parasite but also deteriorates the disease condition by creating a deficiency of IFN-γ in L. major-infected resistant (C57BL/6) and susceptible (BALB/c) mouse strains, respectively (26). Immunotherapy with IFN-γ alone has shown anti-leishmanial protective effects in Indian VL-infected patients. In a pilot study, for 20 days, 9 VL-infected patients were treated with rh IFN-γ where patients showed diminished parasites in their splenic aspirates indicating the therapeutic efficiency of IFN-γ (87). Immunochemotherapy with rh IFN-γ in addition to pentavalent antimonials treated the formerly untreated Indian VL patients and proved to be beneficial in minimizing the length of standard chemotherapy (67). Immuno-nano-chemotherapy with rm IFN-γ and mannosylated liposome-incorporated doxorubicin effectively cures the experiment murine VL and this synergetic combined therapy clears the intracellular parasites by inducing nitric oxide synthase, modulates the T-cell expressions from a Th2 to Th1 pattern, and reveals long-lasting resistance (68).
Interleukin-12
Interleukin-12 (IL-12) is a heterodimeric pluripotent cytokine with a molecular weight of 70 kDa, comprising two subunits namely IL-12p35 (35 kDa) and IL-12p40 (40 kDa) linked by a covalent bond. Antigen-presenting cells (APCs) such as monocytes macrophages, neutrophils, and dendritic cells are the main source for the generation of IL-12. IFN-γ production, the generation of Th1-type cytokines, the activation of natural killer (NK) cells, and the differentiation of naive CD4+ cells from Th1 cells are all influenced by IL-12 (88). Immunotherapy of infectious diseases congruous with IL-12 can be considered where a Th1 response is necessary. IFN-γ and Th1-type cytokines with much more improvisation would help in controlling (bacterial, viral, or protozoan) pathogenic infectious diseases (89). Augmented levels of IL-12 p40 and IFN-γ were expressed, independently in peripheral blood mononuclear cells (PBMC) from treated VL patients in response to in vitro activation with Leishmania parasites, and treatment with exogenous rh IL-10 abrogates the IL-12 p40 levels. Consequently, treatment of active VL patient PBMC with IL-12 or anti-IL-10 modulates the response toward a Th1-type response with the release of IFN-γ and suggests that IL-12 may play a crucial role in the modulation of cellular immune responses in human VL (90). Immunotherapy with rm IL-12 efficiently reduced the established systemic intracellular infection in experimental murine VL in IFN-γ mediated mechanism (91). In experimental VL, IL-12 initiates control over Leishmania infection through the activation of Th1-type cytokine response, stimulation and release of IFN-γ, and formation of granuloma. L. donovani-challenged IL-12p35 gene KO mice allowed uncontrolled liver infection which failed to respond to pentavalent antimonials (which requires T cells and IFN-γ activation). Immunochemotherapy using rm IL-12 synergistically to pentavalent antimonial in normal mice was IFN-gamma dependent; nevertheless, IL-12 also augmented the responsiveness to antimonials in IFN-γ KO mice. Therefore, IL-12 plays a key role in regulating the host immune responses in both IFN-γ-dependent and independent manner that improvises the anti-leishmanial activity of antimonials (92). Co-administration of plasmid encoding for both p35 and p40 subunits of IL-12 and leishmanial recombinant open reading frame F (rORFF) protein provide immunity against experimental VL with an augmented proliferative response of splenocytes and ensuing secretion of Th1 cytokine IFN-γ. Interestingly, IL-12 DNA played a crucial role in regulating the humoral-mediated responses toward the IgG2a isotype signifying its use as a viable vaccine adjuvant against VL (93). Immunostimulatory compounds such as Octyl-β-D-galactofuranose (Galf) clear intracellular parasites in Leishmania-infected human monocyte-derived macrophages and murine models with augmented IL-12 expression (74).
IL-10 receptor blocker as an alternative approach against VL therapeutics
IL-10 is 18 kDa pleiotropic anti-inflammatory cytokine, predominantly generated by alternatively activated macrophages, DCs, natural killer cells, both Th1 and Th2 cells, B cells, CD4+CD25+ forkhead box protein 3 (Foxp3+) Treg cells, and keratinocytes. IFN-γ and IL-2 produced in Th2 cells are majorly inhibited by IL-10 which then effectively inhibits proliferation and cytokine responses in T cells. This also leads to the arbitration of both immunological insensitivity and the suppression of immune reactions (94). Moreover, IL-10 also functions to impede macrophage-facilitated stimulation of T cells which occurs by the decreased expression of class II major histocompatibility complex (MHC) and co-stimulatory molecules on the macrophage surface. This impedes both innate and T-cell-facilitated immunity (95). IL-10 levels markedly augmented in L. donovani-infected macrophages by inhibiting the release of nitric oxide, superoxide (O), and TNF-α. Treatment with anti-IL-10 monoclonal antibodies reduces intracellular amastigotes. IL-10 favors the intramacrophage survival of the Leishmania parasite through selective impairment of Ca2+-dependent protein kinase C-mediated signal transduction (96). A peculiar difference could be seen in symptomatic and asymptomatic VL patients where IL-10 levels show enhanced serum secretions in the former cases. IL-10 essentially functions to protect the tissues from a collateral impairment that occurs due to extreme inflammation. In active VL, CD8+ T cells could also play a significant function in the progression of the disease as these are associated with elevated IL-10 inception (97). Neutralization of IL-10 cytokine induces enhanced Leishmania parasite clearance in splenic aspirates of VL-infected patients with augmented levels of IFN-γ and TNF-α. This brings light toward another approach to target IL-10 in VL immunotherapy (98). Immunotherapy with anti-interleukin (IL)-10 receptor (IL-10R) monoclonal antibody (mAb) reduces the liver parasite burden in L. donovani-infected mice through the enhanced expression of IL-12 protein 40, IFN-γ inducible nitric oxide synthase (iNOS) (99). Human clinical trials have been perceived with the combination of AmBisome and anti-IL-10 mAb which anticipates to prompt synergistic effects in VL-infected patients. The threat of drug resistance and conceivably attaining a chemotherapeutic dose-sparing effect may be conquered through the outcomes in better therapeutic efficiency and treatment adherence (9).
Dendritic cell-based immunotherapy
Dendritic cells (DC), originally described by Steinman and Cohn, are professional antigen-presenting cells (APCs), that specifically stimulate naive T-cell activation and effector differentiation. These are mainly considered messengers between adaptive and innate immune systems. DC-based immunotherapies have been successfully employed in the treatment of cancers by manipulating the immune system to attain cancer control and, preferably, cure cancer. The flexibility of cancer immunotherapy was first revealed by Coley, who used a mixture of bacterial toxins to treat sarcomas (100). The traditional therapies being inefficient stipulates an alternative productive approach that can combat parasitic infections and can impart protection against immunity. DC manipulation serves the purpose. Earlier reports have shown that DC-based immunotherapy can prompt protection against different infectious pathogenic microbes, including bacteria, virus, and protozoan parasites. DCs sense microbes via TLR or C-type lectin receptors (101). Uptake of the Leishmania parasite by DCs requires parasite-reactive immunoglobulin (Ig) G and is mediated through FcγRI and FcγRIII, critical for the optimal development of protective immunity against infection (102). DC subsets contribute extensive polarizing effects on T helper cell differentiation and DC subset 1, exerting Th1 polarization by IL-12 secretion and activation of signal transducer and activator of transcription 4 (STAT4). DCs play a vital role in initial anti-leishmanial T-cell response and in promoting differentiation into memory T cells to accomplish long-lasting adaptive immunity (103). DCs are also the critical source of early IL-12 generation following Leishmania infection. IL-12 secretion by DC is transient, peaking at 24 h of post-infection and reaching the levels observed in uninfected mice by 72 h. DC-T-cell clusters offer a microenvironment for initial NK cell activation, which releases IFN-γ, through a pathway that is reliant on IL-2 and IL-12, crucial for the development of host-protective T-cell responses against the Leishmania parasite (104). DC-SIGN (DC-specific ICAM-3-grabbing non-integrin), a C-type lectin receptor expressed on tissue monocyte-derived DCs bind with distinctive Leishmania species, has been shown to encourage parasite survival. Therefore, DC-SIGN receptor can be considered a therapeutic target for VL (105). Dendritic cells (DCs) evolved from bone marrow (BM) or bone marrow-derived dendritic cells (BM-DCs) resulting in increased parasitic activity in association with L. infantum histone H1 in BALB/c mice. The decrease in the cells produced by 1L 10 and an increase in IFN-γ producing cells (parasite-specific) are noticed. Moreover, the increased value of the ratio IgG2a/IgG1 concludes the presence of immune responses of Th1. These immuno-stimulatory effects of Leishmania histone H1 can help in developing a vaccine against VL infections acting as a competitor protein (106). Immunochemotherapy with soluble L. donovani Ag (SLDA)-pulsed syngeneic BMDC and pentavalent antimonials successfully treated experimental murine VL (107).
Toll-like receptors
Toll-like receptors (TLRs) are trans-membrane proteins expressed as a membrane or cytosolic receptor on monocytes, neutrophils macrophages, dendritic cells, B lymphocytes, and T lymphocytes. The TLR signaling pathway is one of the first defense systems against invasive pathogenic microbes. TLR family consists of 11 members (TLR1–TLR11), with specificity to the innate immunity cells by pathogen-associated molecular patterns (PAMPs) and toll interleukin1 (IL-1) receptor (TIR) of numerous infectious pathogenic microorganisms (108). TLRs play a crucial role in the activation of macrophages and the control of intracellular parasitic infections. During Leishmania infection, TLR1, TLR2, TLR3, and TLR4 expressions were upregulated and stimulated tumor necrosis factor alpha (TNF-α) release by human primary macrophages which are vital for the elimination and control of intracellular parasites (109). Prevention of TLR-4 activation by Leishmania inhibitor of serine peptidase 2 in murine macrophages favors survival and growth of the Leishmania parasite (110).
During Leishmania infection, the involvement of TLR9, TLR4, and TLR2 is critical for causing a pro-inflammatory cytokine response. TLR-9 deficient mice have shown more susceptibility to Leishmania infection. Miltefosine treatment significantly induces both TLR4 and TLR9 in L. donovani-infected macrophages which might account for eliciting a potent antileishmanial pro-inflammatory response with augmented levels of IFN-γ, IL-12, and iNOS2 accompanied by a consequent reduction in IL-10 and TGF-β levels (111). Electrospray encapsulation of resiquimod, polymeric microparticles–TLR agonist, has shown protection in experimental VL (112). Release of IFN-γ, NO, and ROS via TLR7/8 agonists stimulated by macrophages may be proved to be helpful in the clearance of parasites. Liposomal formulation of resiquimod (a TLR 7/8 agonist) has been known to significantly reduce the parasitic burden in the liver, spleen, and bone marrow in addition to enhanced levels of IFN-γ in L. donovani-infected BALB/c mice (113). TLR-4 agonist (glucopyranosyl lipid A (GLA-SE) and LEISH-F3 showed protection in L. donovani and L. infantum infected murine models (69).
Vaccines
The conventional therapeutics for VL are accompanied by severe toxic adverse effects, including drug resistance against parasites and exorbitant cost. Vaccine-based therapies could provide a better alternative to the current therapeutics which may overcome the shortcomings of present-day anti-leishmanials and could be effectively efficacious in eliminating VL. Efforts are ongoing by researchers to find a suitable vaccine (s) to control VL (114). The development of a safe and effective vaccine mainly depends on the identification of antigens and their delivery platforms that could provoke T-cell responses (115). Generally, the vaccine against leishmaniasis can be categorized as first-generation vaccines (whole-killed parasites), second-generation vaccines (recombinant proteins), and third-generation vaccines. The currently available experimental vaccines for the control and elimination of VL are summarized in Table 3.
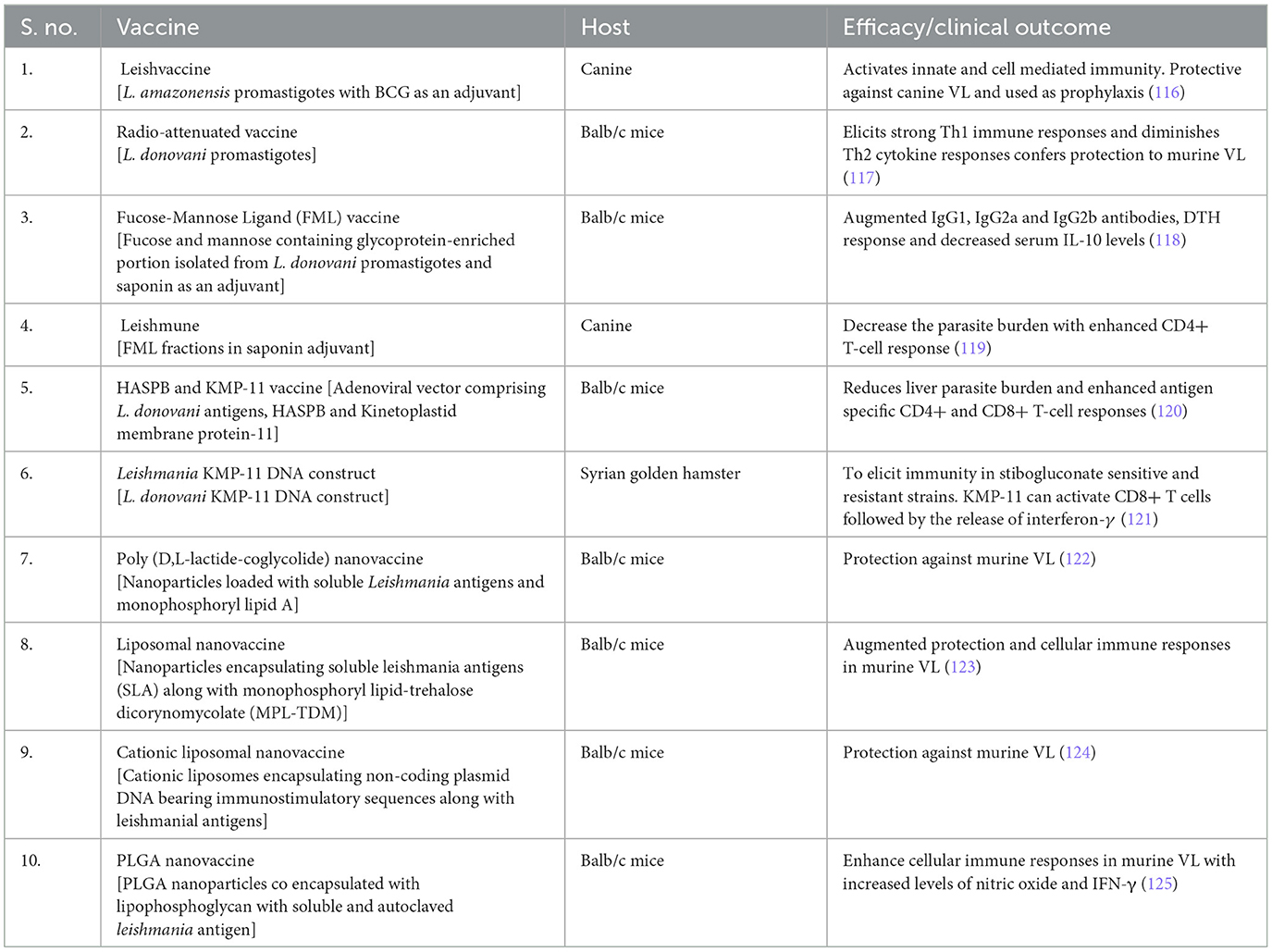
Table 3. Experimental vaccines for the control and elimination of VL.
First-generation vaccine candidates for visceral leishmaniasis
Whole-killed, fractionated antigens, and attenuated Leishmania parasites are the three main important components of first-generation vaccines. The first-generation vaccines are simple and easy to create in developing countries at low cost nevertheless, registration of vaccines fabricated from cultured parasites creates a major hurdle (126). For instance, the Leish vaccine comprises promastigotes of the L. amazonensis strain with BCG as an adjuvant that elicited excellent protective immunity in the canine VL model and was used for prophylactic treatment. This first-generation vaccine initially activates the innate immune system followed by cell-mediated immunity with the stimulation of a mixed cytokine profile with interferon-γ and IL-4 (116). Furthermore, γ-irradiated (radio-attenuated) L. donovani parasites were used as a vaccine candidate for VL. Intramuscular injection of radio-irradiated L. donovani parasites (2 doses once in 15 days) to Balb/c mice elicits strong Th1 immune responses and diminishes Th2 cytokine responses conferring protection to VL. This vaccine restores memory T cells which resulted in the clearing of intra-cellular amastigotes by phosphoinositide-dependent kinase 1 (PDK1), phosphoinositide 3 kinase (PI3K), and p38 mitogen-activated protein kinase (p38MAPK) signaling pathways leading to increased release of nitric oxide (117). The production of whole-killed vaccines is theoretically simple, less expensive, and helps in the prevention and control of VL endemic zones in middle- and low-income countries. Nevertheless, standardization of vaccine generated from in vitro cultured parasites is challenging, and killed parasites does not resemble a clinical infection and has created problems in commercial vaccine development efforts. Safety and stability are the main concerns for first-generation vaccines, which require further investigation (20).
Second-generation vaccine candidates for visceral leishmaniasis
Recombinant purified proteins, which are generated through genetic engineering, are named “second generation vaccines”. On contrary, recombinant proteins with adjuvants or expression in heterologous microbial vectors are used as second-generation vaccines against leishmaniasis. Scalable and cost-effective production approaches can be achieved by recombinant technology and implicit a more viable alternative for mass vaccination campaigns; however, stability is a major concern (127). Fucose-Mannose ligand (FML), the fucose and mannose containing glycoprotein-enriched portion isolated from L. donovani promastigotes, strongly inhibited both promastigote and amastigote forms of L. donovani. Furthermore, FML is a potent immunogen in rodents and rabbits, present on the surface of the parasite throughout their life cycle. Therefore, it is used in the serodiagnosis of human and canine VL (128). Recently, the immunotherapy with FML of L. donovani promastigotes formulated with saponin as an adjuvant has shown protection in L. donovani-infected BALB/c mice with enhanced IgG1, IgG2a, and IgG2b antibodies, a delayed-type hypersensitivity (DTH) response and decreased serum IL-10 levels (118). The first commercially licensed vaccine, Leishman, comprised FML fractions in saponin adjuvant against canine leishmaniasis. Administration of this vaccine has been shown to decrease the parasite burden and clinical disease of long-term L. donovani infection with enhanced CD4+ T-cell response and lowered serum antibody levels (119). A recombinant adenoviral vector comprising L. donovani antigens, hydrophilic acylated surface protein B (HASPB), and Kinetoplasmid membrane protein-11 (KMP11), administered in a single dose to the mice previously infected with L. donovani considerably decreased liver parasite burden. This novel vaccination strategy resulted in improved DTH with enhanced antigen-specific CD4+ and CD8+ T-cell responses (120). At present, numerous vaccine prospects, for instance, Leish-F1, F2, and F3, are in clinical trials developed by Infectious Disease Research Laboratory against leishmaniasis. Leish-F1, a human recombinant vaccine could confer a specific degree of protective immunity against Leishmania infection (116).
Third-generation vaccine candidates for visceral leishmaniasis
DNA vaccination is a novel technology, which uses genetically engineered DNA to elicit an immunological response. The immunogenic protein-expressing genes are cloned into a suitable vector and vaccinated to murine models through the i.d. (or) i.m. route causing strong Th1 immune responses, ensuing in robust cytotoxic T-cell immunity. Several molecules (A2, KMP-11, ORFF, P36LACK, and KMP-11 PPG) delivered through the DNA vaccine platform to preclinical animal models induce strong antibody and cellular immunity (129). Leishmania KMP-11 DNA construct has the potential to elicit immunity in stibogluconate-sensitive and resistant strains in the Syrian golden hamster model. KMP-11 can activate CD8+ T cells followed by the release of interferon-γ (121). A recombinant adenoviral vector comprising L. donovani antigens, hydrophilic acylated surface protein B (HASPB), and KMP11, administered in a single dose to the mice previously infected with L. donovani considerably decreased liver parasite burden. This novel vaccination strategy resulted in improved DTH with enhanced antigen specific CD4+ and CD8+ T-cell responses (120).
Nanovaccines for visceral leishmaniasis
Nanoparticle delivery of vaccines can boost humoral and cellular immune responses in the host. Nanoparticles facilitate the improved antigen uptake by immune cells such as dendritic cells macrophages, nasal-associated lymphoid tissue (NALT), and gut-associated lymphoid tissue (GALT), initiating an effective antigen processing and presentation. This also makes a vaccine more effective to target specific immune cell surface receptors to elicit strong protective immune responses. Nanomaterials can help co-deliver antigen and adjuvant in one cargo to target antigen-presenting cells (130, 131). Leishmania-infected BALB/c mice when treated with a combination of poly (D,L-lactide-coglycolide) nanoparticles loaded with soluble Leishmania antigens in addition to TNFα mimicking peptide or monophosphoryl lipid A actuate the protection against the disease (122). Vaccination of Balb/c mice with liposomal formulations of soluble Leishmania antigens (SLA) along with monophosphoryl lipid-trehalose dicorynomycolate (MPL-TDM) showed enhanced protection with strong cellular immune responses (123). Cationic liposomes encapsulating non-coding plasmid DNA bearing immunostimulatory sequences along with leishmanial antigens showed protection in experimental murine VL (124). PLGA nanoparticles co-encapsulated with lipophosphoglycan with soluble and autoclaved leishmania antigen were shown to enhance cellular immune responses in murine VL model with augmented levels of nitric oxide and IFN-γ (125).
Combination therapy: interplay of immunochemotherapy
Detailed understanding of host immune response aids in the implementation of a synergistic chemoimmunotherapeutic regime which could exacerbate inflammatory responses leading to direct parasiticidal effects. By inducing the factors that downregulate macrophage-derived nitric oxide function and deactivating macrophage-killing effector functions via enhancement of IL-10 Leishmania parasites, this therapy hijacks the host immunity by impairment of Th1 differentiation. A combinatorial host-directed chemoimmunotherapy where aiming immune responses in coalition with the chemotherapeutic agent could enhance Th1 response and the corresponding anti-leishmanial activity. An additive effect has been observed in combination with chemotherapy with significant improvement in the clinical condition of patients with severe side effects (132). Combinative therapy of IL-12 in association with an anti-CD40 antibody enhanced the effectuality of a substandard dose of AmB against L. donovani. Inhibition of cytotoxic T lymphocyte Ag-4 (CTLA-4) which acts as a negative regulator in the T-cell activation mechanism, enhanced the number of cells producing IFN-γ and IL-4 in both spleen as well as liver of L. donovani-infected mice. Co-consortium of anti-CD40 and anti-CTLA-4 with antimonials synergistically enhanced antileishmanial activity (133). Furthermore, IL-4 production stimulates dendritic cells to produce IL-12 mediating a beneficial Th1 response while IL-10 modulates dendritic cells' production of essential reactive nitrogen intermediates (134). Previous studies have reported the synergistic effect of anti-IL-10 with antimony substantiating its potential for immunochemotherapy (135). Antimony-based chemotherapy in coalition with dendritic cell-specific immunotherapy potentiated the antileishmanial activity by an additive effect of enhanced IFN-γ signaling and the ability to potentiate the production of reactive oxygen species leading to induction of programmed cell death in Leishmania parasite (107).
Currently, enhanced efficacy mainly relies on the action of type-1 and type-2 cytokines (IL-2, IL-4, IL-12, IFN-γ, and TNF-α), activation of reactive oxygen species and nitric oxide (NO) production in macrophages, and CD4+ and CD8+ T-cell subsets when infected macrophages are co-treated with pentavalent antimonials (136). Additionally, combinatorial therapy of AmB with IL-12 and anti-IL-10 receptor boosts Th1 implications and hence clearance of L. donovani (137). Miltefosine upsurges the production of IFN-γ, TNF-α, and IL-12, which increased phagocytosis in macrophages with augmented NO production favoring a protective immune response (134). Miltefosine along with immunomodulator picropliv and ketoconazole effectively treated the Leishmania donovani-infected hamsters with improved cell-mediated immune responses (75). Miltefosine in combination with L. braziliensis antigens, saponin, and monophosphoryl lipid-A enhances CD4+ T cells in splenocytes producing IFN-γ and TNF-α and a reduction of IL-10 and anti-Leishmania circulating IgG in hamsters (76). A combination of miltefosine and recombinant cysteine proteinase from Leishmania, rldccys1, significantly reduced the parasite load in infected hamsters (77). Notably, IL-2 reduced L. donovani parasite burdens by 50% while IL-12 treatment reduced parasitic burdens by 47% and IFN-γ decreased them by 40%. Combination therapy of IFN-γ in addition to IL-12 enhanced the efficacy of a suboptimal dose of AmB against L. donovani. IL-10 and IL-27 are both suitable antileishmanial targets for neutralization in L. donovani-infected liver. In conclusion, both immunotherapy and immunochemotherapy could boost the efficacy of drugs (62, 132).
Future prospective of immunotherapy and immunochemotherapy in VL elimination
Leishmaniasis is a severely neglected tropical disease with high mortality and morbidity rates especially in underdeveloped regions. The potential chemotherapeutic agents suffer from certain pitfalls such as parenteral administration, poor patient compliance, high dose, toxic side effects, and development of resistance (138). Regulation of certain co-stimulatory molecules, chemokines or chemokine receptor agonists, and costimulatory and cell signaling pathways has been known to reduce parasitic burden in Leishmania infection models. Therefore, focusing on the mechanism of host immune evasion by the parasite offers potential targets for immunotherapy. Such strategies aiming at enhancing the efficacy of chemotherapeutic agents with specific immunomodulators could serve as promising targets for immunochemotherapy (139). Moreover, immunochemotherapy can elicit an immune response clearing infection more efficaciously and providing more comeback trial possibility of recovery in patients. The current immuno-chemotherapies have shown a synergistic effect with antimonials which are known to show resistance in the Indian population. Therefore, future studies should be directed at the use of current first-line drugs such as AmB, and warrants further investigations as an immunochemotherapeutic. New research opportunities could advance the development of novel drug delivery systems encapsulating potential immunomodulators with and without drugs leading to the propagation of protective immunity (140). Nonetheless, the substantial cost of immunotherapy clearly necessitates the significance of new drug discovery platforms and pharmacological studies which are not only stable at tropical temperatures but can be administered in a patient-complaint manner. Although licensed vaccines are present, the scope for improvement remains undeterred which in future could lead to the development of safe and affordable vaccines (141).
Current limitations of immunotherapy
A single dose of IL-27 or IL-10 monoclonal antibody enhanced macrophage activation, leading to an enhanced parasitic killing (56%). However, combination treatment with both anti-IL-27 and anti-IL-10 did not augment the immunotherapeutic antileishmanial effects (63). Combinations of immunomodulators reduce the parasite burden and at the same time accelerate self-cure. Although, much of the area is still straggling due to the absence of robust clinical studies (64). Furthermore, antibody neutralization of TGF-β augments IFN-γ production and hence enhances Th1-associated (IL-2) macrophage activation, nitric oxide production, and cytotoxic T lymphocyte proliferation, leading to the cure of VL independently of Th2-type cytokines (IL-4) (142). Furthermore, this hypothesis needs to be explored further to be used as a distinct possibility for VL treatment. Inhibition of IL-27 imposes negative regulates on IL-17 production which leads to disease exacerbation. Additionally, the inhibition of IL-10 contributes to the regression of VL (62). Treatment with IFN alone was only marginally effective, and conversely, it was more effective in combination with chemotherapy in VL patients (142). The current immunochemotherapy has shown a synergistic effect with antimonials which are known to show resistance in the Indian population. Treatment with antimonials alone did not show any difference among Indian patients. However, few patients were cured when treated with the most efficient immunochemotherapy. Therefore, future studies should be directed at the use of first-line drugs such as AmB, and warrants further investigations as an immunochemotherapeutic agent (140). A rudimentary understanding of detailed pathways and precise mechanisms underlying pathogenesis is still unclear. Translating experimental results into treatment strategies serves as a major hurdle due to the difference in immunohistopathologies of experimental models from humans. Furthermore, even though immunotherapy has given promising antileishmanial results without any severe toxic side effects, the immunomodulators might exacerbate an impervious immune response and pose a higher risk of sensitization along with the development of allergic reactions. Finally, the prophylactic investigation into these treatment strategies remains unresolved (143).
Conclusion
The conventional and cost-effective therapeutic options for VL suffer from several limitations encompassing from being toxic to having adverse drug reactions (ADRs). Furthermore, the use of an effective lipid-based formulation (AmBisome) is confined mainly due to its high cost and instability in tropical conditions. To surmount these limitations, one best alternative approach could be an extensive understanding of pathogenesis and immunological events that ensue during VL infection, vital for the development of immunotherapeutic strategies against VL. Immunotherapy alone or its combination with conventional anti-leishmanial chemotherapeutic elixir (immunochemotherapy) could warrant an effective therapeutic option in future. However, such attempts need an explicit outlook toward the standardization procedures, and in vivo animal models could further help to understand the immunological footing and confer the possibility to translate it to clinical settings.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
GY is thankful to the Department of Science and Technology (DST) for providing financial support. This work was supported by the SERB (DST), Government of India for funding (EEQ/2020/000563).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yadagiri G, Singh PP. Chemotherapy and experimental models of visceral leishmaniasis. Infect Dis Your Health. (2018) 63–97. doi: 10.1007/978-981-13-1577-0_5
2. Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE. (2012) 7:e35671. doi: 10.1371/journal.pone.0035671
3. Leishmaniasis. (2020). Available online at: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed April 12, 2023).
4. Ready PD. Biology of phlebotomine sand flies as vectors of disease agents. Annu Rev Entomol. (2013) 58:227–50. doi: 10.1146/annurev-ento-120811-153557
5. de Vlas SJ, Stolk WA, le Rutte EA, Hontelez JA, Bakker R, Blok DJ, et al. Concerted efforts to control or eliminate neglected tropical diseases: how much health will be gained? PLoS Negl. Trop. Dis. (2016) 2:e0004386. doi: 10.1371/journal.pntd.0004386
6. McCall LI, Zhang WW, Matlashewski G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathogens. (2013) 9:e1003053. doi: 10.1371/journal.ppat.1003053
7. Yadagiri G, Mudavath SL. Enkephalins as a therapeutic intervention for visceral leishmaniasis. Med Hypotheses. (2020) 144:109956. doi: 10.1016/j.mehy.2020.109956
8. van Griensven J, Diro E. Visceral leishmaniasis. Infect Dis Clin North Am. (2012) 26:309–22. doi: 10.1016/j.idc.2012.03.005
9. Singh OP, Sundar S. Immunotherapy and targeted therapies in treatment of visceral leishmaniasis: current status and future prospects. Front Immunol. (2014) 5:1–9. doi: 10.3389/fimmu.2014.00296
10. Bhunia GS, Kesari S, Chatterjee N, Kumar V, Das P, Bhunia GS, et al. The burden of visceral leishmaniasis in India: challenges in using remote sensing and GIS to understand and control. ISRN Infect Dis. (2013) 2013:1–14. doi: 10.5402/2013/675846
11. Singh OP, Singh B, Chakravarty J, Sundar S. Current challenges in treatment options for visceral leishmaniasis in India: a public health perspective. Infect Dis Poverty. (2016) 5:19. doi: 10.1186/s40249-016-0112-2
12. Croft SL, Coombs GH. Leishmaniasis—current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. (2003) 19:502–8. doi: 10.1016/j.pt.2003.09.008
13. Didwania N, Shadab M, Sabur A, Ali N. Alternative to chemotherapy-the Unmet demand against leishmaniasis. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.01779
14. Zijlstra EE, Musa AM, Khalil EAG, El Hassan IM, El-Hassan AM. Post-kala-azar dermal leishmaniasis. BMC Infect Dis. (2003) 18:108. doi: 10.1016/S1473-3099(03)00517-6
15. Sundar S, Singh A, Singh OP. Strategies to overcome antileishmanial drugs unresponsiveness. J Trop Med. (2014). doi: 10.1155/2014/646932
16. Moore EM, Lockwood DN. Treatment of visceral leishmaniasis. J Glob Infect Dis. (2010) 2:151–8. doi: 10.4103/0974-777X.62883
17. Kaye PM, Aebischer T. Visceral leishmaniasis: Immunology and prospects for a vaccine. Clin Microbiol Infect. (2011) 17:1462–70. doi: 10.1111/j.1469-0691.2011.03610.x
18. Pérez-Cabezas B, Cecílio P, Gaspar TB, Gärtner F, Vasconcellos R, Cordeiro-Da-Silva A, et al. Understanding resistance vs. susceptibility in visceral leishmaniasis using mouse models of Leishmania infantumInfection. Front Cell Infect Microbiol. (2019) 9:1–14. doi: 10.3389/fcimb.2019.00030
19. Khadem F, Uzonna JE. Immunity to visceral leishmaniasis: implications for immunotherapy. Fut Microbiol. (2014) 9:901–15. doi: 10.2217/fmb.14.43
20. Srivastava S, Shankar P, Mishra J, Singh S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasites Vectors. (2016) 9:1–15. doi: 10.1186/s13071-016-1553-y
21. Faleiro RJ, Kumar R, Hafner LM, Engwerda CR. Immune regulation during chronic visceral leishmaniasis. PLoS Neglect Tropical Dis. (2014) 8:e2914. doi: 10.1371/journal.pntd.0002914
22. Rodrigues V, Cordeiro-Da-Silva A, Laforge M, Silvestre R, Estaquier J. Regulation of immunity during visceral Leishmania infection. Parasites Vectors. (2016) 9:1–13. doi: 10.1186/s13071-016-1412-x
23. Bhattacharya P, Ali N. Involvement and interactions of different immune cells and their cytokines in human visceral leishmaniasis. Rev Soc Bras Med Trop. (2013) 46:128–34. doi: 10.1590/0037-8682-0022-2012
24. Roberts MTM. Current understandings on the immunology of leishmaniasis and recent developments in prevention and treatment. Br Med Bull. (2005) 115–30. doi: 10.1093/bmb/ldl003
25. Loria-Cervera EN, Andrade-Narvaez FJ. Animal models for the study of leishmaniasis immunology. Rev Inst Med Trop. (2014) 56:1–11. doi: 10.1590/S0036-46652014000100001
26. Kima PE, Soong L. Interferon gamma in leishmaniasis. Front Immunol. (2013) 4:1–5. doi: 10.3389/fimmu.2013.00156
27. Roatt BM, Aguiar-Soares RDDO, Coura-Vital W, Ker HG, Moreira NDD, Vitoriano-Souza J, et al. Immunotherapy and immunochemotherapy in visceral leishmaniasis: promising treatments for this neglected disease. Front Immunol. (2014) 5:1–12. doi: 10.3389/fimmu.2014.00272
28. Werneck GL. Visceral leishmaniasis in Brazil: rationale and concerns related to reservoir control. Rev Saude Publica. (2014) 48:851–6. doi: 10.1590/S0034-8910.2014048005615
29. World Health Organization. (2010). Control of the Leishmaniases. Geneva: World Health Organization, p. 22–26.
30. Frezard F, Demicheli C, Ribeiro RR. Pentavalent antimonials: new perspectives for old drugs. Molecules. (2009) 14:2317–36. doi: 10.3390/molecules14072317
31. Walker J, Saravia NG. Inhibition of Leishmania donovani promastigote DNA topoisomerase I and human monocyte DNA topoisomerases I and II by antimonial drugs and classical antitopoisomerase agents. J Parasitol. (2004) 90:1155–62. doi: 10.1645/GE-3347
32. Sundar S, Chakravarty J. An update on pharmacotherapy for leishmaniasis. Expert Opin Pharmacother. (2015) 16:237–52. doi: 10.1517/14656566.2015.973850
33. Mohapatra S. Drug resistance in leishmaniasis : newer developments. Trop Parasitol. (2014) 4:4–9. doi: 10.4103/2229-5070.129142
34. Sundar S, Chakravarty J, Rai VK, Agrawal N, Singh SP, Chauhan V, et al. Amphotericin B treatment for indian visceral leishmaniasis: response to 15 daily versus alternate-day infusions. Clin Infect Dis. (2007) 45:556–61. doi: 10.1086/520665
35. Paila YD, Saha B, Chattopadhyay A. Amphotericin B inhibits entry of Leishmania donovani into primary macrophages. Biochem Biophys Res Commun. (2010) 399:429–33. doi: 10.1016/j.bbrc.2010.07.099
36. Sundar S, Jaya J. Liposomal amphotericin B and leishmaniasis: dose and response. J Glob Infect Dis. (2010) 2:159. doi: 10.4103/0974-777X.62886
37. Parvez S, Yadagiri G, Gedda MR, Singh A. Modified solid lipid nanoparticles encapsulated with Amphotericin B and Paromomycin : an effective oral combination against experimental murine visceral leishmaniasis. Sci Rep. (2020) 10:1–14. doi: 10.1038/s41598-020-69276-5
38. Singh A, Yadagiri G, Parvez S, Singh OP, Verma A, Sundar S, et al. Formulation, characterization and in vitro anti-leishmanial evaluation of amphotericin B loaded solid lipid nanoparticles coated with vitamin B12-stearic acid conjugate. Mater Sci Eng C. (2020) 117:111279. doi: 10.1016/j.msec.2020.111279
39. Balasegaram M, Ritmeijer K, Lima MA, Burza S, Ortiz Genovese G, Milani B, et al. Liposomal amphotericin B as a treatment for human leishmaniasis. Expert Opin Emerg Drugs. (2012) 17:493–510. doi: 10.1517/14728214.2012.748036
40. Goa KL, Campoli-Richards DM. Pentamidine isethionate: a review of its antiprotozoal activity, pharmacokinetic properties and therapeutic use in Pneumocystis carinii pneumonia. Drugs. (1987) 33:242–58. doi: 10.2165/00003495-198733030-00002
41. Pearson RD, Hewlett EL. Pentamidine for the treatment of Pneumocystis carinii pneumonia and other protozoal diseases. Ann Intern Med. (1985) 103:782–6. doi: 10.7326/0003-4819-103-5-782
42. Diro E, Ritmeijer K, Boelaert M, Alves F, Mohammed R, Abongomera C, et al. Use of pentamidine as secondary prophylaxis to prevent visceral leishmaniasis relapse in HIV infected patients, the first twelve months of a prospective cohort study. PLoS Negl Trop Dis. (2015) 9:1–15. doi: 10.1371/journal.pntd.0004087
43. Yang G, Choi G, No JH. Antileishmanial mechanism of diamidines involves targeting kinetoplasts. Antimicrob Agents Chemother. (2016) 60:6828–36. doi: 10.1128/AAC.01129-16
44. Yeung KT, Chan M, Chan CKN. The safety of IV pentamidine administered in an ambulatory setting. Chest. (1996) 110:136–40. doi: 10.1378/chest.110.1.136
45. Wiwanitkit V. Interest in paromomycin for the treatment of visceral leishmaniasis (Kala-azar). Ther Clin Risk Manag. (2012) 8:323–8. doi: 10.2147/TCRM.S30139
46. Parvez S, Yadagiri G, Karole A, Singh OP, Verma A, Sundar S, et al. Recuperating biopharmaceutical aspects of amphotericin B and paromomycin using a chitosan functionalized nanocarrier via oral route for enhanced anti-leishmanial activity. Front Cell Infect Microbiol. (2020) 10:1–12. doi: 10.3389/fcimb.2020.570573
47. Parvez S, Yadagiri G, Singh A, Karole A, Singh OP, Sundar S, et al. Improvising anti-leishmanial activity of amphotericin B and paromomycin using co-delivery in D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) tailored nano-lipid carrier system. Chem Phys Lipids. (2020) 231:104946. doi: 10.1016/j.chemphyslip.2020.104946
48. Sundar S, Agrawal N, Arora R, Agarwal D, Rai M, Chakravarty J, et al. Short-course paromomycin treatment of visceral leishmaniasis in India: 14-day vs. 21-day treatment. Clin Infect Dis. (2009) 49:914–8. doi: 10.1086/605438
49. Xie J, Talaska AE, Schacht J. New developments in aminoglycoside therapy and ototoxicity. Hear Res. (2011) 281:28–37. doi: 10.1016/j.heares.2011.05.008
50. Sundar S, Olliaro PL. Miltefosine in the treatment of leishmaniasis: clinical evidence for informed clinical risk management. Ther Clin Risk Manag. (2007) 3:733–40.
51. Pinto-Martinez AK, Rodriguez-Durán J, Serrano-Martin X, Hernandez-Rodriguez V, Benaim G. Mechanism of action of miltefosine on Leishmania donovani involves the impairment of acidocalcisome function and the activation of the sphingosine-dependent plasma membrane Ca 2+ channel. Antimicrob Agents Chemother. (2018) 62:1–10. doi: 10.1128/AAC.01614-17
52. Monge-Maillo B, López-Vélez R, Saravolatz LD. Miltefosine for visceral and cutaneous leishmaniasis: drug characteristics and evidence-based treatment recommendations. Clin Infect Dis. (2015) 60:1398–404. doi: 10.1093/cid/civ004
53. Carvalho L, Luque-Ortega JR, López-Martín C, Castanys S, Rivas L, Gamarro F, et al. The 8-aminoquinoline analogue sitamaquine causes oxidative stress in Leishmania donovani promastigotes by targeting succinate dehydrogenase. Antimicrob Agents Chemother. (2011) 55:4204–10. doi: 10.1128/AAC.00520-11
54. Coimbra ES, Libong D, Cojean S, Saint-Pierre-Chazalet M, Solgadi A, Le Moyec L, et al. Mechanism of interaction of sitamaquine with Leishmania donovani. J Antimicrob Chemother. (2010) 65:2548–55. doi: 10.1093/jac/dkq371
55. Loiseau PM, Cojean S, Schrevel J. Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite. (2011) 18:115–9. doi: 10.1051/parasite/2011182115
56. Clem A. A current perspective on leishmaniasis. J Glob Infect Dis. (2010) 2:124. doi: 10.4103/0974-777X.62863
57. Kumar R, Nylén S. Immunobiology of visceral leishmaniasis. Front Immunol. (2012) 3:1–10. doi: 10.3389/fimmu.2012.00251
58. Dasgupta S, Aghazadeh-Dibavar S, Bandyopadyay M. The role of toll-like receptor agonists in the immunotherapy of leishmaniosis. An update and proposal for a new form of anti-leishmanial therapy. Ann Parasitol. (2014) 60:75–82.
59. Scott P. Leishmaniasis: complexity at the host—pathogen interface. Nat Rev Microbiol. (2020) 9, 604–15. doi: 10.1038/nrmicro2608
60. Lindoso JAL, Moreira CHV, Cunha MA, Queiroz IT. Visceral leishmaniasis and HIV coinfection: current perspectives. HIV/AIDS Res Palliat Care. (2018) 10:193–201. doi: 10.2147/HIV.S143929
61. Tomiotto-Pellissier F, Bortoleti BT, Assolini JP, Gonçalves MD, Carloto AC, Miranda-Sapla MM, et al. Macrophage polarization in leishmaniasis: broadening horizons. Front Immunol. (2018) 9:2529. doi: 10.3389/fimmu.2018.02529
62. Ikeogu NM, Akaluka GN, Edechi CA, Salako ES, Onyilagha C, Barazandeh AF, et al. Leishmania immunity: advancing immunotherapy and vaccine development. Microorganisms. (2020) 8:1–21. doi: 10.3390/microorganisms8081201
63. Murray HW. Targeting IL-27 and/or IL-10 in experimental murine visceral leishmaniasis. Am J Trop Med Hyg. (2020) 103:1938–41. doi: 10.4269/ajtmh.20-0531
64. Kaye PM, Cruz I, Picado A, Van Bocxlaer K, Croft SL. Leishmaniasis immunopathology—impact on design and use of vaccines, diagnostics and drugs. Semin Immunopathol. (2020) 42:247–64. doi: 10.1007/s00281-020-00788-y
65. Stäger S, Maroof A, Zubairi S, Sanos SL, Kopf M, Kaye PM. Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur J Immunol. (2006) 36:1764–71. doi: 10.1002/eji.200635937
66. Gimblet C, Loesche MA, Carvalho L, Carvalho EM, Grice EA, Artis D, et al. IL-22 protects against tissue damage during cutaneous leishmaniasis. PLoS ONE. (2015) 10:e0134698. doi: 10.1371/journal.pone.0134698
67. Sundar S, Singh VP, Sharma S, Makharia MK, Murray HW. Response to interferon-γ plus pentavalent antimony in Indian visceral leishmaniasis. J Infect Dis. (1997) 176:1117–9. doi: 10.1086/516526
68. Kole L, Das L, Das PK. Synergistic effect of interferon-γ and mannosylated liposome- incorporated doxorubicin in the therapy of experimental visceral leishmaniasis. J Infect Dis. (1999) 180:811–20. doi: 10.1086/314929
69. Coler RN, Duthie MS, Hofmeyer KA, Guderian J, Jayashankar L, Vergara J, et al. From mouse to man: safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin Transl Immunol. (2015) 4:e35. doi: 10.1038/cti.2015.6
70. Mastroianni A. Liposomal amphotericin B and rHuGM-CSF for treatment of visceral leishmaniasis in AIDS. Infez med. (2004) 12:197–204.
71. Badaro R, Nascimento C, Carvalho JS, Badaro F, Russo D, Ho JL, et al. Granulocyte-macrophage colony-stimulating factor in combination with pentavalent antimony for the treatment of visceral leishmaniasis. Eur J Clin Microbiol Infect Dis. (1994) 13:23–8. doi: 10.1007/BF01973598
72. Murray HW, Brooks EB, DeVecchio JL, Heinzel FP. Immunoenhancement combined with amphotericin B as treatment for experimental visceral leishmaniasis. Antimicrob Agents Chemother. (2003) 47:2513–7. doi: 10.1128/AAC.47.8.2513-2517.2003
73. Ratnapriya S, Yadav NK, Dube A, Sahasrabuddhe AA. A Chimera of Th1 stimulatory proteins of leishmania donovani offers moderate immunotherapeutic efficacy with a Th1-inclined immune response against visceral leishmaniasis. Biomed Res. Int. (2021) 1–14. doi: 10.1155/2021/8845826
74. Guegan H, Ory K, Belaz S, Jan A, Dion S, Legentil L, et al. In vitro and in vivo immunomodulatory properties of octyl-β-d-galactofuranoside during Leishmania donovani infection. Parasites Vectors. (2019) 12:1–16. doi: 10.1186/s13071-019-3858-0
75. Shakya N, Sane SA, Vishwakarma P, Bajpai P, Gupta S. Improved treatment of visceral leishmaniasis (kala-azar) by using combination of ketoconazole, miltefosine with an immunomodulator—Picroliv. Acta Trop. (2011) 119:188–93. doi: 10.1016/j.actatropica.2011.05.017
76. Carvalho LM, Gusmão MR, Costa AFP, de Brito RCF, de Oliveira Aguiar-Soares RD, de Oliveira Cardoso JM, et al. Immunochemotherapy for visceral leishmaniasis: combinatorial action of Miltefosine plus LBSapMPL vaccine improves adaptative Th1 immune response with control of splenic parasitism in experimental hamster model. Parasitology. (2022) 149:371–9. doi: 10.1017/S0031182021001906
77. de Santana FR, da Silva DAM, Katz S, Orikaza CM, Oliveira KC, Barbiéri CL. A new immunochemotherapy schedule for visceral leishmaniasis in a hamster model. Parasitol Res. (2022) 121:2849–60. doi: 10.1007/s00436-022-07628-y
78. Gulati K, Guhathakurta S, Joshi J, Rai N, Ray A. Cytokines and their role in health and disease: A brief overview. MOJ Immunol. (2016) 4:1–10. doi: 10.15406/moji.2016.04.00121
79. Lotfi N, Thome R, Rezaei N, Zhang GX, Rezaei A, Rostami A, et al. Roles of GM-CSF in the pathogenesis of autoimmune diseases: an update. Front Immunol. (2019) 10:1–14. doi: 10.3389/fimmu.2019.01265
80. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. (2006) 16:126–33. doi: 10.1038/sj.cr.7310017
81. Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem. (1977) 252:1998–2003. doi: 10.1016/S0021-9258(18)71855-3
82. Murray HW, Cervia JS, Hariprashad J, Taylor AP, Stoeckle MY, Hockman H, et al. Effect of granulocyte-macrophage colony-stimulating factor in experimental visceral leishmaniasis. J Clin Invest. (1995) 95:1183–92. doi: 10.1172/JCI117767
83. Weiser WY, Van Niel A, Clark SC, David JR, Remold HG. Recombinant human granulocyte/macrophage colony-stimulating factor activates intracellular killing of Leishmania donovani by human monocyte-derived macrophages. J Exp Med. (1987) 166:1436–46. doi: 10.1084/jem.166.5.1436
84. Kak G, Raza M, Tiwari BK. Interferon-gamma (IFN-γ): Exploring its implications in infectious diseases. Biomol Concepts. (2018) 9:64–79. doi: 10.1515/bmc-2018-0007
85. Sundar S, Chatterjee M. Visceral leishmaniasis—current therapeutic modalities. Indian J Med Res. (2006) 123:345–52.
86. Kumar R, Singh N, Gautam S, Singh OP, Gidwani K, Rai M, et al. Leishmania specific CD4 T cells release IFN-gamma that limits parasite replication in patients with visceral leishmaniasis. PLoS Negl Trop Dis. (2014) 8:e3198. doi: 10.1371/journal.pntd.0003198
87. Sundar S, Rosenkaimer F, Lesser ML, Murray HW. Immunochemotherapy for a systemic intracellular infection: accelerated response using interferon-gamma in visceral leishmaniasis. J Infect Dis. (1995) 171:992–6. doi: 10.1093/infdis/171.4.992
88. Liu J, Cao S, Kim S, Chung E, Homma Y, Guan X, et al. Interleukin-12: an update on its immunological activities, signaling and regulation of gene expression. Curr Immunol Rev. (2005) 1:119–37. doi: 10.2174/1573395054065115
89. Hamza T, Barnett JB, Li B. Interleukin 12 a key immunoregulatory cytokine in infection applications. Int J Mol Sci. (2010) 11:789–806. doi: 10.3390/ijms11030789
90. Ghalib HW, Whittle JA, Kubin M, Hashim FA, El-Hassan AM, Grabstein KH, et al. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J Immunol. (1995) 154:4623–9. doi: 10.4049/jimmunol.154.9.4623
91. Murray HW, Hariprashad J. Interleukin 12 is effective treatment for an established systemic intracellular infection: experimental visceral leishmaniasis. J Exp Med. (1995) 181:387–91. doi: 10.1084/jem.181.1.387
92. Murray HW, Montelibano C, Peterson R, Sypek JP. Interleukin-12 regulates the response to chemotherapy in experimental visceral leishmaniasis. J Infect Dis. (2000) 182:1497–502. doi: 10.1086/315890
93. Tewary P, Saxena S, Madhubala R. Co-administration of IL-12 DNA with rORFF antigen confers long-term protective immunity against experimental visceral leishmaniaisis. Vaccine. (2006) 24:2409–16. doi: 10.1016/j.vaccine.2005.11.058
94. Schülke S. Induction of interleukin-10 producing dendritic cells as a tool to suppress allergen-specific T helper 2 responses. Front Immunol. (2018) 9. doi: 10.3389/fimmu.2018.00455
95. Mittal SK, Cho KJ, Ishido S, Roche PA. Interleukin 10 (IL-10)-mediated Immunosuppression March-i induction regulates antigen presentation by macrophages but not Dendritic cells. J Biol Chem. (2015) 290:27158–67. doi: 10.1074/jbc.M115.682708
96. Bhattacharyya S, Ghosh S, Jhonson PL, Bhattacharya SK, Majumdar S. Immunomodulatory role of interleukin-10 in visceral leishmaniasis: defective activation of protein kinase C-mediated signal transduction events. Infect Immun. (2001) 69:1499–507. doi: 10.1128/IAI.69.3.1499-1507.2001
97. Dayakar A, Chandrasekaran S, Kuchipudi SV, Kalangi SK. Cytokines: Key determinants of resistance or disease progression in visceral leishmaniasis: opportunities for novel diagnostics and immunotherapy. Front Immunol. (2019) 10:670. doi: 10.3389/fimmu.2019.00670
98. Gautam S, Kumar R, Maurya R, Nylén S, Ansari N, Rai M, et al. IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis. (2011) 204:1134–7. doi: 10.1093/infdis/jir461
99. Murray HW, Moreira AL, Lu CM, DeVecchio JL, Matsuhashi M, Ma X, et al. Determinants of response to interleukin-10 receptor blockade immunotherapy in experimental visceral leishmaniasis. J Infect Dis. (2003) 188:458–64. doi: 10.1086/376510
100. Patente TA, Pinho MP, Oliveira AA, Evangelista GCM, Bergami-Santos PC, Barbuto JAM, et al. Human dendritic cells: their heterogeneity and clinical application potential in cancer immunotherapy. Front Immunol. (2019) 10:1–18. doi: 10.3389/fimmu.2018.03176
101. Freitas-Silva R, Brelaz-de-Castro MC, Pereira VR. Dendritic cell-based approaches in the fight against diseases. Front. Immunol. (2014) 5:1–4. doi: 10.3389/fimmu.2014.00078
102. Woelbing F, Kostka SL, Moelle K, Belkaid Y, Sunderkoetter C, Verbeek S, et al. Uptake of Leishmania major by dendritic cells is mediated by Fcγ receptors and facilitates acquisition of protective immunity. J Exp Med. (2006) 203:177–88. doi: 10.1084/jem.20052288
103. Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat Immunol. (2000) 1:199–205. doi: 10.1038/79734
104. Gorak PMA, Engwerda CR, Kaye PM. Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol. (1998) 28:687–95.
105. Colmenares M, Corbí AL, Turco SJ, Rivas L. The dendritic cell receptor DC-SIGN discriminates among species and life cycle forms of leishmania. J Immunol. (2004) 172:1186–90. doi: 10.4049/jimmunol.172.2.1186
106. Agallou M, Smirlis D, Soteriadou KP, Karagouni E. Vaccination with Leishmania histone H1-pulsed dendritic cells confers protection in murine visceral leishmaniasis. Vaccine. (2012) 30:5086–93. doi: 10.1016/j.vaccine.2012.05.075
107. Ghosh M, Pal C, Ray M, Maitra S, Mandal L, Bandyopadhyay S, et al. Dendritic cell-based immunotherapy combined with antimony-based chemotherapy cures established murine visceral leishmaniasis. J Immunol. (2003) 170:5625–9. doi: 10.4049/jimmunol.170.11.5625
108. Fouzder C, Mukhuty A, Das S, Chattopadhyay D. “TLR signaling on protozoan and helminthic parasite infection,” in Toll-like Receptors. London, UK: IntechOpen (2020). doi: 10.5772/intechopen.84711
109. Gallego C, Golenbock D, Gomez MA, Saravia NG. Toll-like receptors participate in macrophage activation and intracellular control of Leishmania (Viannia) panamensis. Infect Immun. (2011) 79:2871–9. doi: 10.1128/IAI.01388-10
110. Faria MS, Reis FCG, Azevedo-Pereira RL, Morrison LS, Mottram JC, Lima APCA, et al. Leishmania inhibitor of serine peptidase 2 prevents TLR4 activation by neutrophil elastase promoting parasite survival in murine macrophages. J Immunol. (2011) 186:411–22. doi: 10.4049/jimmunol.1002175
111. Mukherjee AK, Gupta G, Adhikari A, Majumder S, Kar Mahapatra S, Bhattacharyya Majumdar S, et al. Miltefosine triggers a strong proinflammatory cytokine response during visceral leishmaniasis: role of TLR4 and TLR9. Int Immunopharmacol. (2012) 12:565–72. doi: 10.1016/j.intimp.2012.02.002
112. Duong AD, Sharma S, Peine KJ, Gupta G, Satoskar AR, Bachelder EM, et al. Electrospray encapsulation of toll-like receptor agonist resiquimod in polymer microparticles for the treatment of visceral leishmaniasis. Mol Pharm. (2013) 10:1045–55. doi: 10.1021/mp3005098
113. Peine KJ, Gupta G, Brackman DJ, Papenfuss TL, Ainslie KM, Satoskar AR, et al. Liposomal resiquimod for the treatment of leishmania donovani infection. J Antimicrob Chemother. (2014) 69:168–75. doi: 10.1093/jac/dkt320
114. Ghorbani M, Farhoudi R. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Develop Therapy. (2017) 25–40. doi: 10.2147/DDDT.S146521
115. Pollard AJ, Bijker EM. A guide to vaccinology: from basic principles to new developments. Nat Rev Immunol. (2021) 21:83–100. doi: 10.1038/s41577-020-00479-7
116. Moafi M, Sherkat R, Taleban R, Rezvan H. Leishmania vaccines entered in clinical trials: a review of literature. Int J Prev Med. (2019) 10:1–6. doi: 10.4103/ijpvm.IJPVM_116_18
117. Datta S, Roy S, Manna M. Therapy with radio-attenuated vaccine in experimental murine visceral leishmaniasis showed enhanced T cell and inducible nitric oxide synthase levels, suppressed tumor growth factor-beta production with higher expression of some signaling molecules. Brazilian J Infect Dis. (2015) 19:36–42. doi: 10.1016/j.bjid.2014.10.009
118. Santos, W. R., Santos WR, Aguiar IA, de Souza EP, de Lima VM, Palatnik M, Palatnik-de-Sousa CB. Immunotherapy against murine experimental visceral leishmaniasis with the FML-vaccine. Vaccine. (2003) 21:4668–76. doi: 10.1016/S0264-410X(03)00527-9
119. Saraiva EM, de Figueiredo Barbosa A, Santos FN, Borja-Cabrera GP, Nico D, Souza LOP. The FML-vaccine (Leishmune®) against canine visceral leishmaniasis: a transmission blocking vaccine. Vaccine. (2006) 24:2423–31. doi: 10.1016/j.vaccine.2005.11.061
120. Maroof A, Brown N, Smith B, Hodgkinson MR, Maxwell A, Losch FO, et al. Therapeutic vaccination with recombinant adenovirus reduces splenic parasite burden in experimental visceral leishmaniasis. J Infect Dis. (2012) 205:853–63. doi: 10.1093/infdis/jir842
121. Jain K, Jain NK. Vaccines for visceral leishmaniasis: a review. J Immunol Methods. (2015) 422:1–12. doi: 10.1016/j.jim.2015.03.017
122. Margaroni M, Agallou M, Athanasiou E, Kammona O, Kiparissides C, Gaitanaki C, et al. Vaccination with poly (D, L-lactide-co-glycolide) nanoparticles loaded with soluble Leishmania antigens and modified with a TNFα-mimicking peptide or monophosphoryl lipid A confers protection against experimental visceral leishmaniasis. Int J Nanomedicine. (2017) 12:6169. doi: 10.2147/IJN.S141069
123. Ravindran R, Maji M, Ali N. Vaccination with liposomal leishmanial antigens adjuvanted with MPL-TDM confers long-term protection against visceral leishmaniasis through human adminstrable route. Mol Pharm. (2012) 9:59–70. doi: 10.1021/mp2002494
124. Mazumder S, Ravindran R, Banerjee A, Ali N. Non-coding pDNA bearing immunostimulatory sequences co-entrapped with leishmanial antigens in cationic liposomes elicits almost complete protection against experimental visceral leishmaniasis in BALB/c mice. Vaccine. (2007) 25:8771–81. doi: 10.1016/j.vaccine.2007.10.028
125. Tosyali OA, Allahverdiyev A, Bagirova M, Abamor S, Aydogdu M, Dinparvar S, et al. Nano-Co-Delivery of Lipophosphoglycan With Soluble and Autoclaved Leishmania Antigens Into PLGA Nanoparticles: Evaluation of In Vitro and In Vivo. Amsterdam: Elsevier (2020). doi: 10.1016/j.msec.2020.111684
126. Sundar S, Singh B. Identifying vaccine targets for anti-leishmanial vaccine development. Expert Rev Vaccines. (2014) 13:489–505. doi: 10.1586/14760584.2014.894467
127. Duthie MS, Raman VS, Piazza FM, Reed SG. The development and clinical evaluation of second-generation leishmaniasis vaccines. Vaccine. (2012) 30:134–41. doi: 10.1016/j.vaccine.2011.11.005
128. Sousa CB, Gomes EM, Souza EPD, Santos WRD, Macedo SRD, Medeiros LVD, et al. The FML (Fucose Mannose Ligand) of Leishmania donovani: a new tool in diagnosis, prognosis, transfusional control and vaccination against human kala-azar. Rev Soc Bras Med Trop. (1996) 29:153–63. doi: 10.1590/S0037-86821996000200008
129. Joshi S, Rawat K, Yadav NK, Kumar V, Siddiqi MI, Dube A, et al. Visceral leishmaniasis: advancements in vaccine development via classical and molecular approaches. Front Immunol. (2014) 5:380. doi: 10.3389/fimmu.2014.00380
130. Gujjari L, Kalani H, Pindiprolu SK, Arakareddy BP, Yadagiri G. Current challenges and nanotechnology-based pharmaceutical strategies for the treatment and control of malaria. Parasite Epidemiol Control. (2022) 16:e00244. doi: 10.1016/j.parepi.2022.e00244
131. Patil V, Hernandez-Franco JF, Yadagiri G, Bugybayeva D, Dolatyabi S, Feliciano-Ruiz N, et al. A split influenza vaccine formulated with a combination adjuvant composed of alpha-d-glucan nanoparticles and a STING agonist elicits cross-protective immunity in pigs. J Nanobiotechnol. (2022) 20:1–20. doi: 10.1186/s12951-022-01677-2
132. Aruleba RT, Carter KC, Brombacher F, Hurdayal R. Can we harness immune responses to improve drug treatment in leishmaniasis? Microorganisms. (2020) 8:1–20. doi: 10.3390/microorganisms8071069
133. Murray HW, Lu CM, Brooks EB, Fichtl RE, DeVecchio JL, Heinzel FP, et al. Modulation of T-cell costimulation as immunotherapy or immunochemotherapy in experimental visceral leishmaniasis. Infect Immun. (2003) 71:6453–62. doi: 10.1128/IAI.71.11.6453-6462.2003
134. Gupta AK, Das S, Kamran M, Ejazi SA, Ali N. The pathogenicity and virulence of Leishmania-interplay of virulence factors with host defenses. Virulence. (2022) 13:903–35. doi: 10.1080/21505594.2022.2074130
135. Murray HW. Interleukin 10 receptor blockade—pentavalent antimony treatment in experimental visceral leishmaniasis. Acta Trop. (2005) 93:295–301. doi: 10.1016/j.actatropica.2004.11.008
136. Samant M, Sahu U, Pandey SC, Khare P. Role of cytokines in experimental and human visceral Leishmaniasis. Front Cell Infect Microbiol. (2021) 11:624009. doi: 10.3389/fcimb.2021.624009
137. Osero BOO, Aruleba RT, Brombacher F, Hurdayal R. Unravelling the unsolved paradoxes of cytokine families in host resistance and susceptibility to leishmania infection. Cytokine: X. (2020) 2:100043. doi: 10.1016/j.cytox.2020.100043
138. Singh A, Yadagiri G, Negi M, Kushwaha AK, Singh OP, Sundar S, et al. Carboxymethyl chitosan modified lipid nanoformulations as a highly efficacious and biocompatible oral anti-leishmanial drug carrier system. Int J Biol Macromol. (2022) 204:373–85. doi: 10.1016/j.ijbiomac.2022.02.006
139. Yasmin H, Adhikary A, Al-Ahdal MN, Roy S, Kishore U. Host–pathogen interaction in leishmaniasis: immune response and vaccination strategies. Immuno. (2022) 2:218–54. doi: 10.3390/immuno2010015
140. Parvez S, Yadagiri G, Arora K, Javaid A, Kushwaha AK, Singh OP, et al. Coalition of biological agent (Melatonin) with chemotherapeutic agent (Amphotericin b) for combating visceral leishmaniasis via oral administration of modified solid lipid nanoparticles. ACS Biomater Sci Eng. (2021). doi: 10.1021/acsbiomaterials.1c00859
141. Dinc R. Leishmania vaccines: the current situation with its promising aspect for the future. Korean J Parasitol. (2022) 60:379–91. doi: 10.3347/kjp.2022.60.6.379
142. Mente KA, Mcgowan SE, Wilson ME, Young BM, Davidson BL. Leishmaniasis in murine visceral β the importance of TGF. (2021).
Keywords: visceral leishmaniasis, chemotherapy, drug resistance, immunotherapy, immunochemotherapy
Citation: Yadagiri G, Singh A, Arora K and Mudavath SL (2023) Immunotherapy and immunochemotherapy in combating visceral leishmaniasis. Front. Med. 10:1096458. doi: 10.3389/fmed.2023.1096458
Received: 12 November 2022; Accepted: 14 April 2023;
Published: 17 May 2023.
Edited by:
Fangli Lu, Sun Yat-sen University, ChinaReviewed by:
Dalia Ashour, Tanta University, EgyptFrédéric Frézard, Federal University of Minas Gerais, Brazil
Copyright © 2023 Yadagiri, Singh, Arora and Mudavath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shyam Lal Mudavath, c2h5YW1sYWxAaW5zdC5hYy5pbg==; c2hhdnMwNTAyQGdtYWlsLmNvbQ==