
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 17 February 2023
Sec. Hematology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1083242
 Areez Shafqat†
Areez Shafqat† Ahmed Noor Eddin†
Ahmed Noor Eddin† Ghaith Adi
Ghaith Adi Mohammed Al-Rimawi
Mohammed Al-Rimawi Saleha Abdul Rab*
Saleha Abdul Rab* Mylia Abu-Shaar
Mylia Abu-Shaar Kareem Adi
Kareem Adi Khaled Alkattan
Khaled Alkattan Ahmed Yaqinuddin
Ahmed Yaqinuddin
Neutrophils are the first cells to be recruited to sites of acute inflammation and contribute to host defense through phagocytosis, degranulation and neutrophil extracellular traps (NETs). Neutrophils are rarely found in the brain because of the highly selective blood-brain barrier (BBB). However, several diseases disrupt the BBB and cause neuroinflammation. In this regard, neutrophils and NETs have been visualized in the brain after various insults, including traumatic (traumatic brain injury and spinal cord injury), infectious (bacterial meningitis), vascular (ischemic stroke), autoimmune (systemic lupus erythematosus), neurodegenerative (multiple sclerosis and Alzheimer’s disease), and neoplastic (glioma) causes. Significantly, preventing neutrophil trafficking into the central nervous system or NET production in these diseases alleviates brain pathology and improves neurocognitive outcomes. This review summarizes the major studies on the contribution of NETs to central nervous system (CNS) disorders.
Loss of blood-brain barrier (BBB) integrity and neuroinflammation are central to the pathogenesis of central nervous system (CNS) pathologies. Neutrophils are the most abundant leukocyte population, which adhere to activated endothelium, transmigrate to tissues, and contribute to inflammatory processes. Neutrophil extracellular traps (NETs) produced by activated neutrophils are composed of a DNA scaffold on which various proteins are deposited (1). The release of NETs was traditionally related to neutrophil death (termed NETosis), but neutrophils also undergo vital and mitochondrial NET production by extruding their contents through blebbing from the cell membrane (2), remaining viable and retaining effector functions (Figure 1).
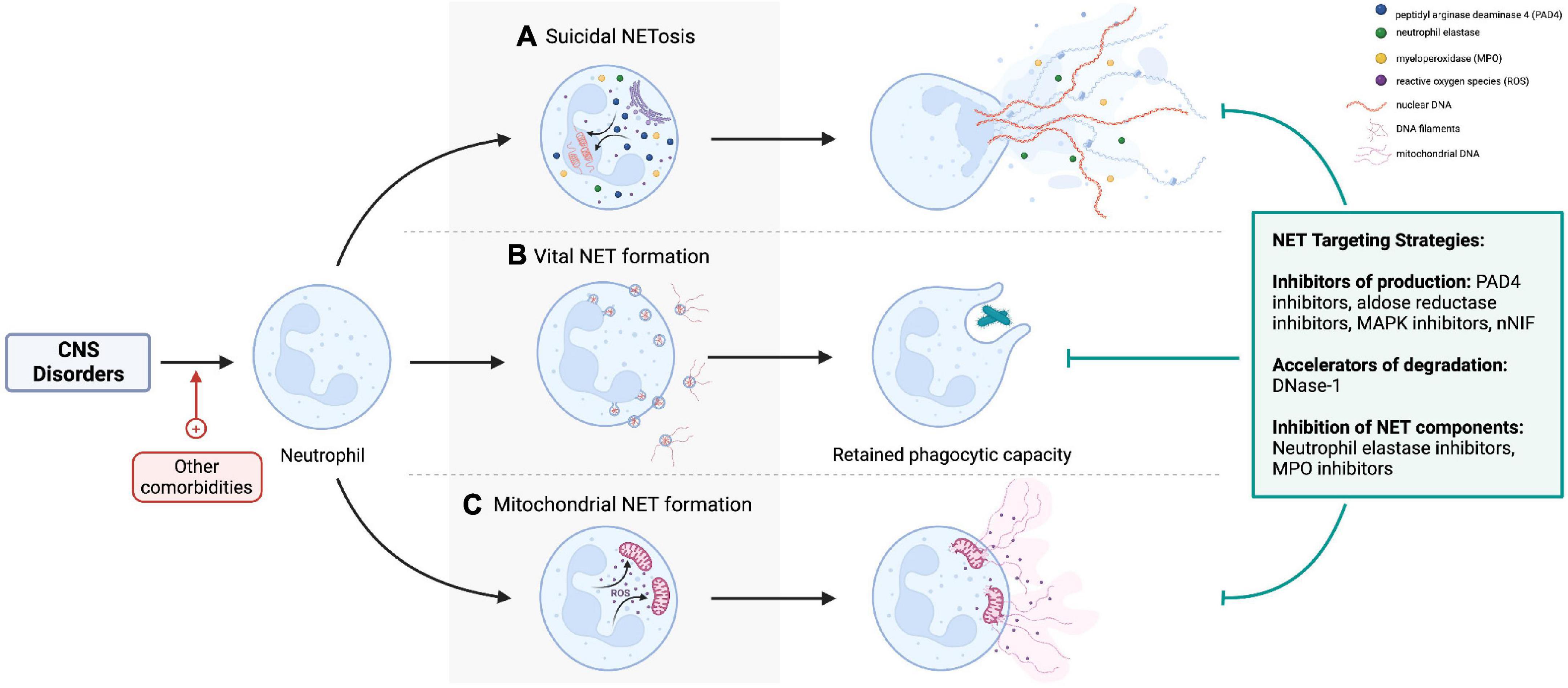
Figure 1. (A) Lytic NETosis describes neutrophil cell death with release of intracellular contents as neutrophil extracellular traps (NETs). By contrast, neutrophils retain viability and phagocytic effector functions after (B) vital and (C) mitochondrial NET formation. Nicotinamide adenine dinucleotide phosphate oxidase activation, reactive oxygen species, neutrophil elastase (NE), myeloperoxidase (MPO), and peptidyl-arginine deaminase-4 (PAD-4) are important cellular mediators of NETs production. NETs can be targeted pharmacologically by preventing their formation (e.g., PAD4 inhibitors, NE inhibitors), accelerating degradation by DNase, or inhibiting specific NET components [e.g., NE inhibitors, matrix metalloproteinase-9 (MMP-9) inhibitors]. Created with BioRender.com.
The production of different types of NETs is context-dependent, varying with the nature of the NET-inducing stimuli and disease process (3). For example, lytic NETosis is induced by infections (4), pro-inflammatory cytokines (5), damage-associated molecular patterns (DAMPs) (6, 7), activated platelets (8, 9), complement (10, 11), autoantibodies (12, 13), and immune complexes; vital NETs are mainly seen in infections (2, 14, 15); and mitochondrial NETs during infections and autoimmune diseases (16, 17). The molecular mechanisms governing NET production in each of these instances are beyond the scope of this review and are discussed elsewhere (18–20). Regulators of this heterogeneity of NETs and whether different types of NETs exert variable, context-specific effects (beneficial versus harmful) are outstanding questions in the field of NETs research (21).
Despite their apparent benefit in infectious disease (4), NETs cause significant collateral tissue damage (22). For example, cell-free DNA (16), histones (23–25), matrix metalloproteinase-9 (MMP-9) (26), and LL-37 (27), have all been implicated in various tissue-damaging effects, including direct cytotoxicity (28), thrombosis (29), and chronic inflammation (24). Several excellent reviews on NETs in defense and disease have been published (30–36). Recent studies have shown neutrophil recruitment in the CNS and NETs to contribute to various CNS pathologies. Table 1 summarizes seminal studies implicating NETs as pathogenic in brain CNS diseases. This review provides a concise summary of recent data on the role of NETs in various brain disorders.

Table 1. Evidence of neutrophil extracellular traps (NETs) in central nervous system (CNS) diseases.
Brain microvascular endothelial cells (BMECs) are connected by various adherens and tight junctions, forming the highly selective BBB. Neutrophils thus cannot readily cross the BBB and are rarely found in a healthy brain. However, BBB permeability can increase secondary to trauma, inflammation, ischemia, and degenerative changes. Pro-inflammatory cytokines released by activated astrocytes and microglia cells upregulate adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) on BMECs, facilitating neutrophil adhesion (37). Subsequently, neutrophil-endothelial cell interactions without transmigration increase BBB permeability (Figure 2; 38).
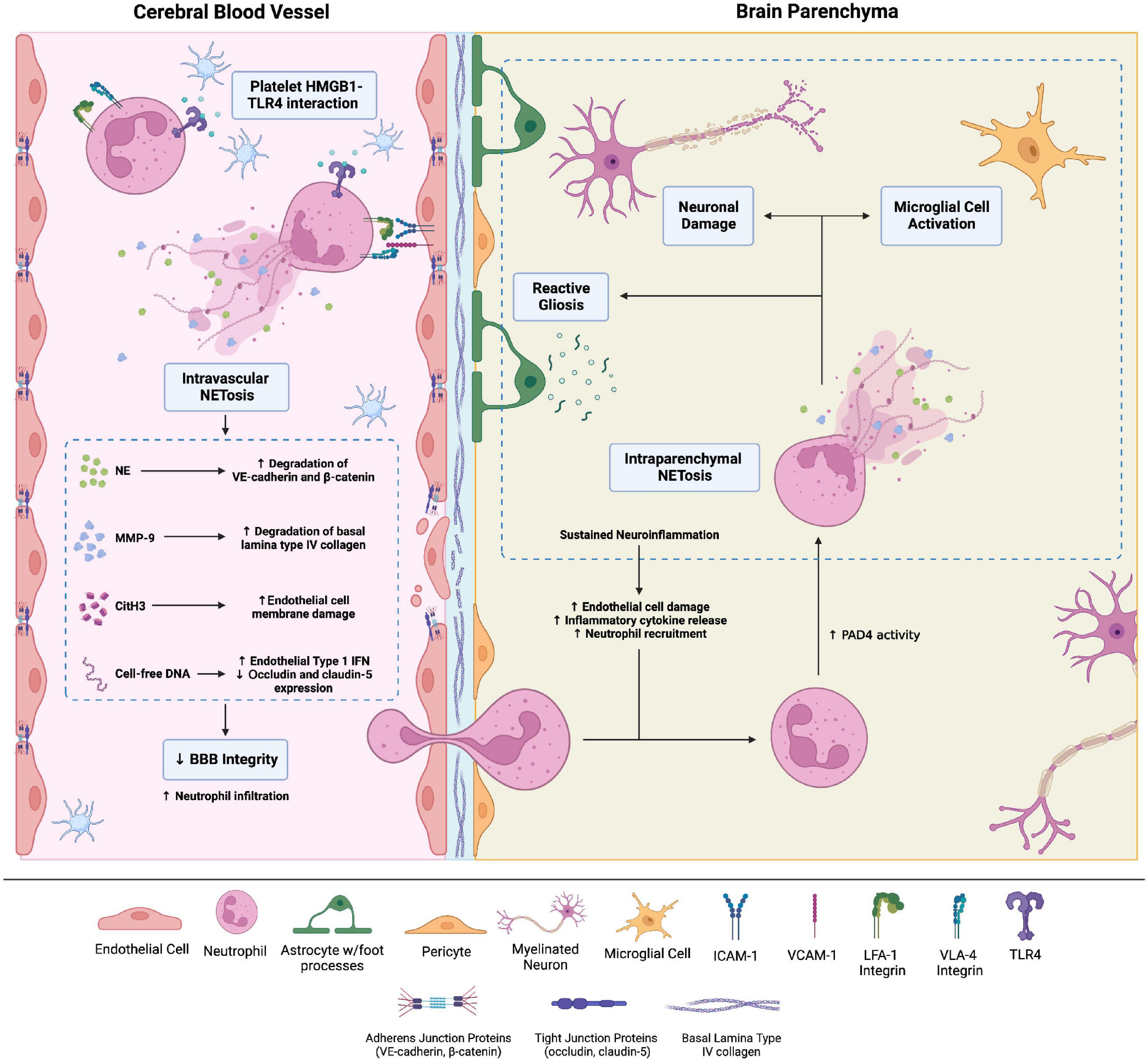
Figure 2. Brain injuries of various etiologies cause neutrophil adhesion to the brain microvascular endothelial cells (BMECs) via integrins, namely LFA-1 and VLA-4 integrins, which bind endothelial surface intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1, respectively. Adhesion to the BMECs activates neutrophils. Neutrophil adhesion to platelet derived high-mobility group box-1 (HMGB1) through toll-like receptor-4 (TLR4) causes neutrophil activation and intravascular neutrophil extracellular trap (NET) production. NETs comprise neutrophil elastase, citrullinated histone H3, matrix metalloproteinases (MMPs), and cell-free DNA. These NET components increase blood-brain barrier (BBB) permeability through a variety of mechanisms. For instance, MMP-9 degrades type IV collagen of the basal lamina of cerebral blood vessels, compromising blood-brain barrier (BBB) integrity. Intraparenchymal neutrophils also undergo NETosis. The ensuing neuronal damage and microglial cell activation amplify neuroinflammation and cause neuronal loss. Created with Biorender.com.
Multiple mechanisms explain neutrophil adhesion-dependent BBB disruption. Firstly, neutrophil adhesion promotes blood flow stasis, leading to vascular obstruction; neutrophil depletion enhances CNS perfusion and decreases brain damage after stroke (39). Neutrophil adhesion to BMECs via the β2 integrins LFA-1 and MAC-1 also activates neutrophils, increasing oxidative stress and NETosis (40). Activated neutrophils release neutrophil elastase (NE)—possibly within NETs—which disrupts adherens junction proteins VE-cadherin and β-catenin, increasing BBB permeability (41). Agaphelin—an NE inhibitor—reduces BBB permeability in stroke, decreasing infarct volume, improving neurologic function, and reducing mortality in mice (42). NET-associated MMP-9 in the cerebral microvessels degrades type IV collagen of the basal lamina to disrupt BBB integrity (43). Histones also increase BBB permeability, particularly in the hippocampus, by disrupting adherens and tight junctions (44).
Extracellular DNA, microbial DNA, and damaged intracellular self-DNA are major inducers of inflammation through cGMP-AMP synthase (cGAS)/STING-dependent type I IFN and pro-inflammatory cytokine production (45–48). DNA contained within NETs also activates cGAS and enhances type I IFN production and pro-inflammatory cytokine production (16, 49). Therefore, in the CNS, NETs may promote type I IFN and pro-inflammatory cytokine responses in microglia, which are known to express cGAS and contribute to CNS pathology (50–53). NET-induced microglial activation via cGAS has been noted in murine models of ischemic stroke (54) and tissue plasminogen activator (tPA)-induced intracerebral hemorrhage (55). A recent preprint study demonstrated NET-induced cGAS activation in microglia in TBI, associated with neuroinflammation and neurological deficits (56). Depleting neutrophils or inhibiting peptidyl-arginine demaniase-4 (PAD-4) significantly blunt the IFN response and improve neurologic deficits (54). Together, these findings indicate that NETs mediate BBB disruption, either directly via histones and various proteases or indirectly by augmenting type I IFN responses.
Analysis of cerebrospinal fluid (CSF) from humans and rodents with pneumococcal meningitis reveals the presence of neutrophils and externalized neutrophil proteins such as myeloperoxidase (MPO), histones, NE, and proteinase 3 (PR3) (57), which may imply NET production. Combining penicillin with DNase-1 to treat pneumococcal meningitis accelerates NET degradation, enhances bacterial killing, lowers IL-1β levels, and decreases patient mortality compared to a penicillin regimen alone (58, 59).
Despite these findings, the beneficial effect of neutrophils in CNS infection must not be underestimated. Neutrophil depletion in mouse models of pneumococcal meningitis results in an elevated CSF bacterial load, higher IL-1β, lower TNF-α, and poor survival (60). Therefore, neutrophil and NET dynamics in CNS infections need to be studied further for therapeutic strategies to attenuate pathogenic functions of NETs without interfering with CNS protective mechanisms.
Sepsis-induced encephalopathy has an abysmal prognosis. Lipopolysaccharide (LPS) causes neutrophil transmigration into brain parenchyma via CXCL1-CXCR2 interactions, whereas SB225002, a selective CXCR2 antagonist, mitigated neutrophil migration into the brain (61). However, this study did not directly explore NETs, neither were associations between SB225002 administration and improvement in neurologic parameters studied. Although NET production was not directly studied, CXCL1 stimulates ROS-dependent NET formation in COVID-19 (62), deep vein thrombosis (63), and cancer (64), and inhibiting the CXCL1-CXCR2 axis in experimental human and murine sepsis models by reparixin attenuates NET formation, multi-organ injury, and mortality (65). Sepsis-induced NET production has indeed been studied extensively in other organ systems, such as the lungs (65, 66). Mechanistically, extracellular histones exert significant pathologic effects in sepsis (23), and circulating cit-H3 significantly correlates with the severity of septic shock (67).
Traumatic brain injury (TBI) carries a high mortality risk, and complications such as cognitive impairment, memory deficits, post-traumatic stress disorder, post-traumatic encephalopathy, and neuroinflammation in survivors are frequent (68, 69). Neutrophils adhere to cerebral vessels, infiltrate hypoxic brain tissue, and produce NETs in murine models of TBIs (70, 71), associated with worse TBI outcomes, including cerebral edema, cognitive deficits, and paroxysmal sympathetic hyperactivity (72, 73). Activation of toll-like receptor-4 (TLR4) on neutrophils activates NET formation after TBI, which correlates with higher intracranial pressure (ICP), suggesting that neutrophils cause cerebral edema by NET production (70). TLR4-knockout, the NET formation inhibitor Cl-amidine, and DNase-1 reduce NET formation in the brain post-TBI and better neurologic and behavioral outcomes (70). Together, these findings indicate that NET-targeted therapies may be beneficial in alleviating hypoxia and cerebral edema after TBI.
Neutrophils infiltrate the CNS following cerebral ischemia (74) and correlate positively with neuronal loss, infarct size, and cognitive dysfunction (75). Circulating neutrophils from ischemic stroke patients exhibit higher NET formation than healthy control neutrophils (76). Moreover, CNS-infiltrating neutrophils in ischemic stroke patients form NETs (77, 78). Higher serum cell-free DNA levels in acute ischemic stroke patients correlate with worse clinical outcomes using the modified Rankin scale, and lower serum DNase levels are found in patients who developed stroke-associated infections (79). Platelet-derived high-mobility group box-1 (HMGB1) is a major inducer of NET production in ischemic stroke, and HMGB1-depleted attenuates NET formation after stroke and betters neurologic outcomes (78, 80). Along similar lines, treating mice with neonatal NET-inhibitory factor (nNIF) ameliorates NET production and decreased infarct size (78). NETs may also protect thrombi from degradation by tPA (81), and adding DNase-1 to tPA regimens significantly accelerates ex vivo lysis of stroke thrombi (82). Furthermore, NETs can contribute to tPA-induced intracerebral hemorrhage and BBB disruption (discussed above) (55). Since BBB disruption underlies the narrow therapeutic window of tPA administration (83), whether inhibiting NETs delays BBB disruption and increases the therapeutic window of tPA administration warrants further investigation.
Neuropsychiatric manifestations of SLE (NPSLE) are commonly attributed to neuroinflammation; leukocyte infiltration precedes NPSLE-related cognitive deficits in mice (84). Neutrophils isolated from SLE patients display impaired phagocytosis, increased platelet aggregation, reactive oxygen species (ROS) production, and NET formation (85, 86). SLE skews the composition of the neutrophil compartment toward low-density granulocytes (LDGs), which readily produce NETs (87, 88). SLE patient plasma induces NET production by neutrophils isolated from healthy controls, indicating that autoantibodies and immune complexes in SLE plasma induce NET formation (16, 89). NETs released by LDGs upon stimulation by anti-ribonucleoprotein antibodies and immune complexes cause endothelial dysfunction—inducing apoptosis and impairing vasorelaxation—through components like MMP-2 (90). LDG NETs also contain various immunostimulatory proteins and autoantigens, like LL-37, IL-33, IL-17, and dsDNA, which can worsen inflammation and introduce neoepitopes, fostering an amplification of autoimmunity (91–93).
The degradation of NETs may be impaired in SLE, particularly during disease flares. Auto-antibodies against NET components—e.g., anti-dsDNA and anti-histone antibodies—are thought to protect NETs against degradation by circulating DNase (94). Furthermore, NET-activated C1q can directly inhibit DNase to prevent NET degradation (95). Genetic variations in DNase activity can also increase the risk of SLE. For example, individuals with DNASE1L3 gene mutations develop childhood SLE (96). These observations suggest that DNase-1 as a therapeutic strategy may not improve the clinical severity of SLE, as one study demonstrated in lupus nephritis patients (97).
Serum NET levels have not yet been studied as prognostic markers in NPSLE, nor has the effect of inhibiting neutrophil CNS trafficking or lowering serum and CSF NET markers on cognitive function been tested experimentally. The fundamental importance of the type I IFN response in SLE is well-established. Type I IFNs have indeed been linked to NPSLE (93, 98), but mechanistic links between type I IFNs and NPSLE manifestations are lacking. Neutrophils are perhaps major downstream mediators of the type I IFN response in SLE. They exhibit the highest expression of interferon-stimulated genes (ISGs) out of the myeloid cells (99). Type I IFNs induce mitochondrial NET formation by LDGs, and mitochondrial DNA within LDG NETs can activate cGAS-STING signaling in plasmacytoid dendritic cells to drive type I IFN production (16, 100). Therefore, whether NETs—cGAS/STING axis increases BBB permeability and neuroinflammation in NPSLE is an important question for future research.
Multiple sclerosis (MS) is a demyelinating disease of the CNS resulting in neuronal impairment. Elevated neutrophil-to-lymphocyte ratios, NETs, CXCL1, and CXCL5 are detected in blood samples of relapsing-remitting MS (RRMS) patients, but not other forms of MS (101–103). Neutrophils isolated from MS patients exhibit a primed phenotype characterized by reduced apoptosis and higher degranulation, ROS production, and NET formation (104). Males with MS show higher NETs levels than females (102), hinting at gender-specific differences in MS pathogenesis. The role of neutrophils and the contribution of various neutrophil components to MS pathogenesis have recently been reviewed (105). However, the mechanisms behind NET production in RRMS are not yet elucidated. Furthermore, NET components are yet to be physically associated with active MS lesions. Therefore, studies on NETs in MS are still in their infancy, and we caution against drawing any causal relationships.
The adaptive immune response plays a crucial role in the development of MS, particularly IL-17-secreting Th17 cells (106–108). Activation of the transcription factor retinoic acid-related orphan receptor γt (RORγt) is key to the differentiation of Th cells into Th17 cells and subsequent IL-17 production. Activators of RORγt have thus been the subject of intense research, as they constitute therapeutic targets to mitigate the Th17 response and ameliorate MS severity. Wilson et al. (109) recently demonstrated that NETs-derived histones promote Th17 cell differentiation and IL-17 production by engaging TLR2 on the surface of undifferentiated Th cells and activating RORγt. The histone inhibitor mCBS abrogated histone-induced Th17 differentiation (109). These mechanistic links need to be investigated in experimental models of MS; whether histone inhibition mitigates the Th17 response and betters MS outcomes is important to determine. Other components of the pathological CNS environment in MS, such as members of the chondroitin sulfate proteoglycan (CSPG) family, also promote Th17 polarization, which are toxic to oligodendrocyte precursor cells and impair remyelination (110). These data open new avenues to explore the potential interactions of innate immune cells such as neutrophils with extracellular matrix components and their collective impact in shaping the T-cell differentiation phenotype.
Alzheimer’s disease is a debilitating form of dementia and a significant cause of patient and caregiver morbidity and health expenditures. Although amyloid-β (Aβ) deposition is a pathologic hallmark of AD, amyloid-targeting therapies have been largely ineffective in treating or delaying AD progression in clinical trials (111). Therefore, new biomarkers and treatment strategies against AD are needed.
Neuroinflammation has been demonstrated to be a hallmark feature of AD and contributes to disease progression (112–115). Recent advances in understanding neutrophil biology have reignited interest in their role in AD. Initial studies demonstrated that circulating neutrophils in AD upregulate CD11b and ROS production, both markers of neutrophil activation (116). Furthermore, neutrophils are seen in the brains of AD patients and correlate with the burden of neurofibrillary tangles and Aβ (117). Neutrophils isolated from AD patients exhibit a gene expression signature indicative of mitochondrial dysfunction, energy hypometabolism, leukocyte adhesion, and cytokine signaling, suggesting that neutrophils contribute to neuroinflammation in AD (117, 118). Lastly, the role of neutrophil granule proteins—which can be found in NETs—in AD has recently been reviewed by Stock et al. (119).
Studies using 3xFAD and 3xTg AD mouse models have provided further insights into neutrophils and NETs in AD (119). Neutrophils adhere to BMECs in AD, transmigrate into the brain parenchyma and co-localize with Aβ plaques in 5xFAD and 3xTg-AD mice (120, 121). Neutrophil accumulation in AD mice brains precedes cognitive dysfunction, indicating that neutrophil recruitment contributes to AD symptomatology. Mechanistically, Aβ induces LFA-1 expression on neutrophils, responsible for their adhesion to BMECs and subsequent extravasation (121). Subsequently, Aβ amyloid fibrils from various sources induce nicotinamide adenine dinucleotide phosphate-oxidase-dependent lytic NETosis (122, 123). NE within NETs can then cleave amyloid fibrils and form cytotoxic oligomers (122). In short, these findings indicate that Aβ mediates neutrophil recruitment and transmigration through upregulation of LFA-1 on neutrophils and subsequently stimulates NETosis, the components of which can fragment Aβ to oligomers that exert neurotoxic or neuroinflammatory effects. LFA-1 blockade reduces CNS neutrophil recruitment, microgliosis, and Aβ load in the brain, and improves long-term memory (121). Intriguingly, a case report administered DNase-1 to an AD patient and noticed considerable cognitive improvement (124).
Amyloid-β may also induce intravascular and intraparenchymal NETosis through indirect mechanisms by fostering a neuroinflammatory microenvironment (125). For example, Aβ activates NLRP3 inflammasome signaling in microglial cells and the release of IL-1β, which has been demonstrated to induce NETosis in cancer (126) and gout (127, 128). However, while aberrant innate immune responses in AD were well documented, their direct relation to AD neurodegeneration was only recently shown (129). In AD, Aβ co-localizes with microglia and is endocytosed, subsequently damaging mitochondrial DNA and causing double-stranded DNA breaks (130, 131). Damaged DNA activates cGAS and promotes downstream type I IFN and pro-inflammatory cytokine responses (129). These proinflammatory, M1 microglia in turn foster neurotoxic astrocyte phenotypes, which contribute to neurodegeneration (129). Furthermore, BMECs in AD upregulate type I IFN receptors and interferon-stimulated genes, associated with downregulation of VE-cadherin and tight junction proteins occludin and claudin-5, increasing BBB leakiness (132), indicating that microglia-derived IFNs disrupt the BBB. Like microglia, neutrophils also co-localize with Aβ plaques, associated with NETosis (121, 125). Furthermore, DNA within NETs is recognized by cGAS in microglia (discussed above). On this basis, we propose that a positive feedback loop may exist, where Aβ and microglia-initiated neuroinflammation mediate neurotoxicity through astrocytes but also recruit and activate neutrophils to release NETs, that, in turn, worsen microglial activation. Testing such associations could implicate NET inhibition or degradation as a viable strategy to attenuate AD pathology and symptoms.
Glioma tumor cells produce granulocyte colony-stimulating factor that drives granulopoiesis in the bone marrow (133, 134). Consequently, neutrophilia and an elevated neutrophil-to-lymphocyte ratio are commonly seen in glioma patients (135), are more pronounced in high-grade (IV) tumors, and confer a poor prognosis (136). In agreement with these findings, depleting neutrophils by using an anti-Ly6G monoclonal antibody prolongs survival in mice with gliomas (137). Glioma cells also produce IL-8 that recruits neutrophils, termed tumor-associated neutrophils (TANs) (138). IL-8 induces NETosis in TANs through PI3K-signaling and ROS production. Indeed, TANs and NETs have been visualized within glioma lesions (138). NET components such as HMGB1 bind RAGE receptors on the surface of glioma tumor cells and stimulate proliferation, invasion, and IL-8 production in vitro (138). These findings suggest that a self-reinforcing NETs/IL-8 axis may collectively amplify tumor inflammation in glioma. In glioblastoma multiforme (GBM), TANs stimulate GBM tumor cell proliferation and epithelial-to-mesenchymal transition through S100A4 (139). NETs also contribute to a hypercoagulable state in high-grade glioma by inducing endothelial cell dysfunction, alleviated by DNase1 + protein C treatment in vitro (140). Given these findings, exploring specific pro-tumorigenic NET components in glioma may reveal novel disease markers and therapeutic targets in glioma. However, it is important to mention that neutrophils also exert numerous anti-tumor functions by killing tumor cells and T enhancing anti-tumor -cell responses (141). Therefore, the dilemma of inhibiting NETs and inadvertently attenuating beneficial—in this case, tumor suppressive—neutrophil functions must be carefully considered. Only rigorous mechanistic analyses can dissect beneficial versus detrimental effects of NETs in the context of cancer. We refer readers to in-depth reviews on neutrophils, NETs and cancer for more information (142–147).
Considerable data exist on TANs and NETs as mediators of chemotherapy, immunotherapy, and radiotherapy resistance (148–150). In glioma, NET-derived S100A4 mediates resistance to anti-VEGF therapy, whereas inhibition of S100A4 enhances response to treatment (139). Silencing S100A4 gene expression in endothelial cells by S100A4 small-interfering RNA induces an anti-angiogenic gene signature, and administering S100A4 siRNA into human prostate cancer xenografts significantly decreased tumor vascularity and inhibited tumor growth (151). These studies have set the stage for S100A4-targeted anti-cancer therapies. Since S100A4 may be derived from NETs, future research must ascertain whether inhibiting NET production normalizes the tumor vasculature and constitutes a potential anti-angiogenic cancer treatment strategy.
Single-cell technologies have revealed that the neutrophil population comprises functionally distinct subsets. Understanding how the neutrophil population is skewed in CNS diseases is essential to identifying context-dependent neutrophil transcriptomic alterations and novel disease-specific therapeutic strategies. Therefore, the composition and function of NETs may also vary in different CNS diseases; lytic NETs, vital NETs, and mitochondrial NETs are released in response to distinct stimuli and have varying compositions. However, studies investigating NETs in CNS disease thus far do not account for NET heterogeneity; the specific type of NETs seen are rarely reported. Therefore, if NETs are to be considered a future therapeutic target in CNS disease, they need to be studied in greater resolution. Future studies into the topic may reveal novel disease markers and cause-specific therapeutic targets to improve the care of patients suffering from CNS diseases. Interestingly, while the activation of canonical and non-canonical inflammasome pathways was previously linked to NETosis (152), a recent study showed that inflammasome-related NET formation does not require cell death (153). Therefore, the field of NETs research is wide open; new research will likely reveal new NET-inducing stimuli, important signaling pathways worth targeting to mitigate NET formation, new NET components, and yet unknown beneficial and harmful effects.
AS and AY: conceptualization. AS, ANE, GA, MA-R, SAR, and KAd: writing—original draft preparation. AS, KAd, and AY: writing—review and editing. KAd and AY: supervision. All authors have read and agreed to the published version of the manuscript.
Figures were created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Urban C, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
2. Yipp B, Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
3. Jorch S, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
4. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss D, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
5. Keshari R, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One. (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
6. Sofoluwe A, Bacchetta M, Badaoui M, Kwak B, Chanson M. ATP amplifies NADPH-dependent and-independent neutrophil extracellular trap formation. Sci Rep. (2019) 9:16556. doi: 10.1038/s41598-019-53058-9
7. Huang H, Tohme S, Al-Khafaji A, Tai S, Loughran P, Chen L. Damage-associated molecular pattern–activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. (2015) 62:600–14. doi: 10.1002/hep.27841
8. Clark S, Ma A, Tavener S, McDonald B, Goodarzi Z, Kelly M, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
9. Jiao Y, Li W, Wang W, Tong X, Xia R, Fan J, et al. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit Care. (2020) 24:380. doi: 10.1186/s13054-020-03082-3
10. Palmer L, Damgaard C, Holmstrup P, Nielsen C. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodont Res. (2016) 51:70–6. doi: 10.1111/jre.12284
11. de Bont C, Boelens W, Pruijn G. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. (2019) 16:19–27. doi: 10.1038/s41423-018-0024-0
12. Huang Y, Wang H, Wang C, Chen M, Zhao M. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthr Rheumatol. (2015) 67:2780–90. doi: 10.1002/art.39239
13. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross W, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
14. Slaba I, Wang J, Kolaczkowska E, McDonald B, Lee W, Kubes P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology. (2015) 62:1593–605. doi: 10.1002/hep.28003
15. Yipp B, Petri B, Salina D, Jenne C, Scott B, Zbytnuik L, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
16. Lood C, Blanco L, Purmalek M, Carmona-Rivera C, De Ravin S, Smith C, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
17. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon H. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
18. Sollberger G, Tilley D, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. (2018) 44:542–53. doi: 10.1016/j.devcel.2018.01.019
19. Rosazza T, Warner J, Sollberger G. NET formation – mechanisms and how they relate to other cell death pathways. FEBS J. (2021) 288:3334–50. doi: 10.1111/febs.15589
20. Tan C, Aziz M, Wang P. The vitals of NETs. J Leukocyte Biol. (2021) 110:797–808. doi: 10.1002/JLB.3RU0620-375R
21. Boeltz S, Amini P, Anders H, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. (2019) 26:395–408. doi: 10.1038/s41418-018-0261-x
22. Brinkmann V. Neutrophil Extracellular Traps in the Second Decade. J Innate Immun. (2018) 10:414–21. doi: 10.1159/000489829
23. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo C, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
24. Silvestre-Roig C, Braster Q, Wichapong K, Lee E, Teulon J, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. (2019) 569:236–40. doi: 10.1038/s41586-019-1167-6
25. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. (2017) 8:e2812. doi: 10.1038/cddis.2017.52
26. Albrengues J, Shields M, Ng D, Park C, Ambrico A, Poindexter M, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
27. Herster F, Bittner Z, Archer N, Dickhöfer S, Eisel D, Eigenbrod T, et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat Commun. (2020) 11:105. doi: 10.1038/s41467-019-13756-4
28. Saffarzadeh M, Juenemann C, Queisser M, Lochnit G, Barreto G, Galuska S, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
29. Massberg S, Grahl L, von Bruehl M, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
30. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
31. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
32. Wigerblad G, Kaplan M. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. (2022) doi: 10.1038/s41577-022-00787-0 [Epub ahead of print].
33. Zhu S, Yu Y, Ren Y, Xu L, Wang H, Ling X, et al. The emerging roles of neutrophil extracellular traps in wound healing. Cell Death Dis. (2021) 12:984. doi: 10.1038/s41419-021-04294-3
34. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. (2021) 18:666–82. doi: 10.1038/s41569-021-00552-1
35. Apel F, Zychlinsky A, Kenny E. The role of neutrophil extracellular traps in rheumatic diseases. Nat Rev Rheumatol. (2018) 14:467–75. doi: 10.1038/s41584-018-0039-z
36. Mutua V, Gershwin LJA. Review of Neutrophil Extracellular Traps (NETs) in disease: potential Anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
37. Greenwood J, Etienne-Manneville S, Adamson P, Couraud P. Lymphocyte migration into the central nervous system: implication of ICAM-1 signalling at the blood–brain barrier. Vasc Pharmacol. (2002) 38:315–22. doi: 10.1016/S1537-1891(02)00199-4
38. Sienel R, Kataoka H, Kim S, Seker F, Plesnila N. Adhesion of leukocytes to cerebral venules precedes neuronal cell death and is sufficient to trigger tissue damage after cerebral ischemia. Front Neurol. (2021) 12:807658. doi: 10.3389/fneur.2021.807658
39. Cruz Hernández J, Bracko O, Kersbergen C, Muse V, Haft-Javaherian M, Berg M, et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci. (2019) 22:413–20. doi: 10.1038/s41593-018-0329-4
40. Santos-Lima B, Pietronigro E, Terrabuio E, Zenaro E, Constantin G. The role of neutrophils in the dysfunction of central nervous system barriers. Front Aging Neurosci. (2022) 14:965169. doi: 10.3389/fnagi.2022.965169
41. Johnson-Léger C, Aurrand-Lions M, Imhof B. The parting of the endothelium: miracle, or simply a junctional affair? J Cell Sci. (2000) 113:921–33. doi: 10.1242/jcs.113.6.921
42. Leinweber J, Mizurini D, Francischetti I, Fleischer M, Hermann D, Kleinschnitz C, et al. Elastase inhibitor agaphelin protects from acute ischemic stroke in mice by reducing thrombosis, blood–brain barrier damage, and inflammation. Brain Behav Immun. (2021) 93:288–98. doi: 10.1016/j.bbi.2020.12.027
43. Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo E, Montaner J. MMP-9–Positive neutrophil infiltration is associated to blood–brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. (2008) 39:1121–6. doi: 10.1161/STROKEAHA.107.500868
44. Villalba N, Baby S, Cha B, Yuan S. Site-specific opening of the blood-brain barrier by extracellular histones. J Neuroinflamm. (2020) 17:281. doi: 10.1186/s12974-020-01950-x
45. Zhou C, Wang B, Wu Q, Lin P, Qin S, Pu Q, et al. Identification of cGAS as an innate immune sensor of extracellular bacterium Pseudomonas aeruginosa. iScience. (2021) 24:101928. doi: 10.1016/j.isci.2020.101928
46. de Mingo Pulido Á, Hänggi K, Celias D, Gardner A, Li J, Batista-Bittencourt B, et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity. (2021) 54:1154–67.e7. doi: 10.1016/j.immuni.2021.04.019
47. Chen Q, Sun L, Chen Z. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol. (2016) 17:1142–9. doi: 10.1038/ni.3558
48. Li T, Chen Z. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
49. Apel F, Andreeva L, Knackstedt L, Streeck R, Frese C, Goosmann C, et al. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci Signal. (2021) 14:eaax7942. doi: 10.1126/scisignal.aax7942
50. Cox D, Field R, Williams D, Baran M, Bowie A, Cunningham C, et al. DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia. (2015) 63:812–25. doi: 10.1002/glia.22786
51. Jiang G, Yang X, Zhou H, Long J, Liu B, Zhang L, et al. cGAS knockdown promotes microglial M2 polarization to alleviate neuroinflammation by inhibiting cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Res Bull. (2021) 171:183–95. doi: 10.1016/j.brainresbull.2021.03.010
52. Jeffries A, Marriott I. Human microglia and astrocytes express cGAS-STING viral sensing components. Neurosci Lett. (2017) 658:53–6. doi: 10.1016/j.neulet.2017.08.039
53. Ding R, Li H, Liu Y, Ou W, Zhang X, Chai H, et al. Activating cGAS-STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J Neuroinflammation. (2022) 19:137. doi: 10.1186/s12974-022-02511-0
54. Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun. (2020) 11:2488. doi: 10.1038/s41467-020-16191-y
55. Wang R, Zhu Y, Liu Z, Chang L, Bai X, Kang L, et al. Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood. (2021) 138:91–103. doi: 10.1182/blood.2020008913
56. Liu L, Cao Y, Min X, Jia H, Mi L, Zhang Y, et al. Neutrophil extracellular traps exacerbate microglia/macrophages-mediated neuroinflammation via cGAS in mice with traumatic brain injury. Res Square. (2022). doi: 10.21203/rs.3.rs-2252334/v1
57. de Buhr N, Reuner F, Neumann A, Stump-Guthier C, Tenenbaum T, Schroten H, et al. Neutrophil extracellular trap formation in the Streptococcus suis-infected cerebrospinal fluid compartment. Cell Microbiol. (2017) 19:e12649. doi: 10.1111/cmi.12649
58. Mohanty T, Fisher J, Bakochi A, Neumann A, Cardoso J, Karlsson C, et al. Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat Commun. (2019) 10:1667. doi: 10.1038/s41467-019-09040-0
59. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. (2006) 16:401–7. doi: 10.1016/j.cub.2006.01.056
60. Too L, Mitchell A, McGregor I, Hunt N. Antibody-induced neutrophil depletion prior to the onset of pneumococcal meningitis influences long-term neurological complications in mice. Brain Behav Immun. (2016) 56:68–83. doi: 10.1016/j.bbi.2016.01.021
61. Wu F, Chen X, Zhai L, Wang H, Sun M, Song C, et al. CXCR2 antagonist attenuates neutrophil transmigration into brain in a murine model of LPS induced neuroinflammation. Biochem Biophys Res Commun. (2020) 529:839–45. doi: 10.1016/j.bbrc.2020.05.124
62. Kaiser R, Leunig A, Pekayvaz K, Popp O, Joppich M, Polewka V, et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. JCI Insight. (2021) 6:e150862. doi: 10.1172/jci.insight.150862
63. Yago T, Liu Z, Ahamed J, McEver R. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. (2018) 132:1426–37. doi: 10.1182/blood-2018-05-850859
64. Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. (2020) 52:856.–871. doi: 10.1016/j.immuni.2020.03.001
65. Alsabani M, Abrams S, Cheng Z, Morton B, Lane S, Alosaimi S, et al. Reduction of NETosis by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br J Anaesth. (2022) 128:283–93. doi: 10.1016/j.bja.2021.10.039
66. Qu M, Chen Z, Qiu Z, Nan K, Wang Y, Shi Y, et al. Neutrophil extracellular traps-triggered impaired autophagic flux via METTL3 underlies sepsis-associated acute lung injury. Cell Death Discov. (2022) 8:375. doi: 10.1038/s41420-022-01166-3
67. Tian Y, Russo R, Li Y, Karmakar M, Liu B, Puskarich M, et al. Serum citrullinated histone H3 concentrations differentiate patients with septic verses non-septic shock and correlate with disease severity. Infection. (2021) 49:83–93. doi: 10.1007/s15010-020-01528-y
68. Simon D, McGeachy M, Bayır H, Clark R, Loane D, Kochanek P. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. (2017) 13:171–91. doi: 10.1038/nrneurol.2017.13
69. Blennow K, Brody D, Kochanek P, Levin H, McKee A, Ribbers G, et al. Traumatic brain injuries. Nat Rev Dis Primers. (2016) 2:16084. doi: 10.1038/nrdp.2016.84
70. Vaibhav K, Braun M, Alverson K, Khodadadi H, Kutiyanawalla A, Ward A, et al. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci Adv. (2020) 6:eaax8847. doi: 10.1126/sciadv.aax8847
71. Carlos T, Clark R, Franicola-Higgins D, Schiding J, Kochanek P. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukocyte Biol. (1997) 61:279–85. doi: 10.1002/jlb.61.3.279
72. Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation. (2012) 9:17. doi: 10.1186/1742-2094-9-17
73. Zhu K, Zhu Y, Hou X, Chen W, Qu X, Zhang Y, et al. NETs Lead to Sympathetic Hyperactivity After Traumatic Brain Injury Through the LL37-Hippo/MST1 Pathway. Front Neurosci. (2021) 15:621477. doi: 10.3389/fnins.2021.621477
74. Chu H, Kim H, Lee S, Moore J, Chan C, Vinh A, et al. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab. (2014) 34:450–9. doi: 10.1038/jcbfm.2013.217
75. Allen C, Thornton P, Denes A, McColl B, Pierozynski A, Monestier M, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. (2012) 189:381–92. doi: 10.4049/jimmunol.1200409
76. Datsi A, Piotrowski L, Markou M, Köster T, Kohtz I, Lang K, et al. Stroke-derived neutrophils demonstrate higher formation potential and impaired resolution of CD66b + driven neutrophil extracellular traps. BMC Neurol. (2022) 22:186. doi: 10.1186/s12883-022-02707-0
77. Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. (2015) 129:239–57. doi: 10.1007/s00401-014-1381-0
78. Denorme F, Portier I, Rustad J, Cody M, de Araujo C, Hoki C, et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Investig. (2022) 132:e154225. doi: 10.1172/JCI154225
79. Grosse G, Blume N, Abu-Fares O, Götz F, Ernst J, Leotescu A, et al. Endogenous deoxyribonuclease activity and cell-free deoxyribonucleic acid in acute ischemic stroke: a cohort study. Stroke. (2022) 53:1235–44. doi: 10.1161/STROKEAHA.121.036299
80. Kim S, Lee H, Lee H, Kim I, Lee J. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun. (2019) 7:94. doi: 10.1186/s40478-019-0747-x
81. Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, et al. Thrombus neutrophil extracellular traps content impair TPA-induced thrombolysis in acute ischemic stroke. Stroke. (2018) 49:754–7. doi: 10.1161/STROKEAHA.117.019896
82. Laridan E, Denorme F, Desender L, François O, Andersson T, Deckmyn H, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. (2017) 82:223–32. doi: 10.1002/ana.24993
83. Ma Y, Li L, Kong L, Zhu Z, Zhang W, Song J, et al. Pinocembrin protects blood-brain barrier function and expands the therapeutic time window for tissue-type plasminogen activator treatment in a rat thromboembolic stroke model. Biomed Res Int. (2018) 2018:8943210. doi: 10.1155/2018/8943210
84. Duarte-Delgado N, Vásquez G, Ortiz-Reyes B. Blood-brain barrier disruption and neuroinflammation as pathophysiological mechanisms of the diffuse manifestations of neuropsychiatric systemic lupus erythematosus. Autoimmun Rev. (2019) 18:426–32. doi: 10.1016/j.autrev.2018.12.004
85. Kaplan M. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
86. Knight J, Kaplan M. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr Opin Rheumatol. (2012) 24:441–50. doi: 10.1097/BOR.0b013e3283546703
87. Bennett L, Palucka A, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. (2003) 197:711–23. doi: 10.1084/jem.20021553
88. Carmona-Rivera C, Kaplan M. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
89. Yu Y, Su K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J Clin Cell Immunol. (2013) 4:139. doi: 10.4172/2155-9899.1000139
90. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan M. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
91. Villanueva E, Yalavarthi S, Berthier C, Hodgin J, Khandpur R, Lin A, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
92. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
93. Georgakis S, Gkirtzimanaki K, Papadaki G, Gakiopoulou H, Drakos E, Eloranta M, et al. NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFN-α production in patients with SLE. JCI Insight. (2021) 6:e147671. doi: 10.1172/jci.insight.147671
94. Hakkim A, Fürnrohr B, Amann K, Laube B, Abed U, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U.S.A. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
95. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
96. Al-Mayouf S, Sunker A, Abdwani R, Abrawi S, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
97. Davis J Jr, Manzi S, Yarboro C, Rairie J, McInnes I, Averthelyi D, et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus. (1999) 8:68–76. doi: 10.1191/096120399678847380
98. Zeng J, Meng X, Zhou P, Yin Z, Xie Q, Zou H, et al. Interferon-α exacerbates neuropsychiatric phenotypes in lupus-prone mice. Arthr Res Therapy. (2019) 21:205. doi: 10.1186/s13075-019-1985-9
99. Mistry P, Nakabo S, O’Neil L, Goel R, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U.S.A. (2019) 116:25222–8. doi: 10.1073/pnas.1908576116
100. Garcia-Romo G, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
101. Bisgaard A, Pihl-Jensen G, Frederiksen J. The neutrophil-to-lymphocyte ratio as disease actvity marker in multiple sclerosis and optic neuritis. Mult Scler Relat Disord. (2017) 18:213–7. doi: 10.1016/j.msard.2017.10.009
102. Tillack K, Naegele M, Haueis C, Schippling S, Wandinger K, Martin R, et al. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J Neuroimmunol. (2013) 261:108–19. doi: 10.1016/j.jneuroim.2013.05.004
103. Rumble J, Huber A, Krishnamoorthy G, Srinivasan A, Giles D, Zhang X, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. (2015) 212:23–35. doi: 10.1084/jem.20141015
104. Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. (2012) 242:60–71. doi: 10.1016/j.jneuroim.2011.11.009
105. De Bondt M, Hellings N, Opdenakker G, Struyf S. Neutrophils: underestimated players in the pathogenesis of Multiple Sclerosis (MS). Int J Mol Sci. (2020) 21:4558. doi: 10.3390/ijms21124558
106. Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. (1999) 5:101–4. doi: 10.1177/135245859900500206
107. Tzartos J, Friese M, Craner M, Palace J, Newcombe J, Esiri M, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. (2008) 172:146–55. doi: 10.2353/ajpath.2008.070690
108. Hedegaard C, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. (2008) 125:161–9. doi: 10.1111/j.1365-2567.2008.02837.x
109. Wilson A, Randall K, Pettitt J, Ellyard J, Blumenthal A, Enders A, et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun. (2022) 13:528. doi: 10.1038/s41467-022-28172-4
110. Ghorbani S, Jelinek E, Jain R, Buehner B, Li C, Lozinski B, et al. Versican promotes T helper 17 cytotoxic inflammation and impedes oligodendrocyte precursor cell remyelination. Nat Commun. (2022) 13:2445. doi: 10.1038/s41467-022-30032-0
111. Jackson M, Hewitt E. Why are Functional Amyloids Non-Toxic in Humans? Biomolecules. (2017) 7:71. doi: 10.3390/biom7040071
112. Griciuc A, Patel S, Federico A, Choi S, Innes B, Oram M, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. (2019) 103:820–35.e7. doi: 10.1016/j.neuron.2019.06.010
113. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
114. Zhang B, Gaiteri C, Bodea L, Wang Z, McElwee J, Podtelezhnikov A, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. (2013) 153:707–20. doi: 10.1016/j.cell.2013.03.030
115. Rajendran L, Paolicelli R. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J Neurosci. (2018) 38:2911–9. doi: 10.1523/JNEUROSCI.1136-17.2017
116. Scali C, Prosperi C, Bracco L, Piccini C, Baronti R, Ginestroni A, et al. Neutrophils CD11b and fibroblasts PGE(2) are elevated in Alzheimer’s disease. Neurobiol Aging. (2002) 23:523–30. doi: 10.1016/S0197-4580(01)00346-3
117. Song L, Yang Y, Guo Q, Zhao X. Cellular transcriptional alterations of peripheral blood in Alzheimer’s disease. BMC Med. (2022) 20:266. doi: 10.1186/s12916-022-02472-4
118. Amorim J, Coppotelli G, Rolo A, Palmeira C, Ross J, Sinclair D. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. (2022) 18:243–58. doi: 10.1038/s41574-021-00626-7
119. Stock A, Kasus-Jacobi A, Pereira H. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J Neuroinflammation. (2018) 15:240. doi: 10.1186/s12974-018-1284-4
120. Baik S, Cha M, Hyun Y, Cho H, Hamza B, Kim D, et al. Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol Aging. (2014) 35:1286–92. doi: 10.1016/j.neurobiolaging.2014.01.003
121. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
122. Azevedo E, Guimarães-Costa A, Torezani G, Braga C, Palhano F, Kelly J, et al. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J Biol Chem. (2012) 287:37206–18. doi: 10.1074/jbc.M112.369942
123. Munir H, Jones J, Janowitz T, Hoffmann M, Euler M, Martins C, et al. Stromal-driven and Amyloid β-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat Commun. (2021) 12:683. doi: 10.1038/s41467-021-20982-2
124. Tetz V, Tetz G. Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: a case report. J Med Case Rep. (2016) 10:131. doi: 10.1186/s13256-016-0931-6
125. Pietronigro E, Della Bianca V, Zenaro E, Constantin G. NETosis in Alzheimer’s Disease. Front Immunol. (2017) 8:211. doi: 10.3389/fimmu.2017.00211
126. Gomes T, Várady C, Lourenço A, Mizurini D, Rondon A, Leal A, et al. IL-1β Blockade attenuates thrombosis in a neutrophil extracellular trap-dependent breast cancer model. Front Immunol. (2019) 10:2088. doi: 10.3389/fimmu.2019.02088
127. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One. (2011) 6:e29318. doi: 10.1371/journal.pone.0029318
128. Sil P, Wicklum H, Surell C, Rada B. Macrophage-derived IL-1β enhances monosodium urate crystal-triggered NET formation. Inflamm Res. (2017) 66:227–37. doi: 10.1007/s00011-016-1008-0
129. Xie X, Ma G, Li X, Zhao J, Zhao Z, Zeng J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nat Aging. (2023). doi: 10.1038/s43587-022-00337-2
130. Moya G, Rivera P, Dittenhafer-Reed K. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int J Mol Sci. (2021) 22:7030. doi: 10.3390/ijms22137030
131. Mao P, Reddy P. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim Biophys Acta. (2011) 1812:1359–70. doi: 10.1016/j.bbadis.2011.08.005
132. Jana A, Wang X, Leasure J, Magana L, Wang L, Kim Y, et al. Increased Type I interferon signaling and brain endothelial barrier dysfunction in an experimental model of Alzheimer’s disease. Sci Rep. (2022) 12:16488. doi: 10.1038/s41598-022-20889-y
133. Nitta T, Sato K, Allegretta M, Brocke S, Lim M, Mitchell D, et al. Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Res. (1992) 571:19–25. doi: 10.1016/0006-8993(92)90505-4
134. Albulescu R, Codrici E, Popescu I, Mihai S, Necula L, Petrescu D, et al. Cytokine patterns in brain tumour progression. Mediators Inflamm. (2013) 2013:979748. doi: 10.1155/2013/979748
135. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy L, Maiti S, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. (2016) 1:e85841. doi: 10.1172/jci.insight.85841
136. Massara M, Persico P, Bonavita O, Mollica Poeta V, Locati M, Simonelli M, et al. Neutrophils in Gliomas. Front Immunol. (2017) 8:1349. doi: 10.3389/fimmu.2017.01349
137. Fujita M, Scheurer M, Decker S, McDonald H, Kohanbash G, Kastenhuber E, et al. Role of type 1 IFNs in antiglioma immunosurveillance–using mouse studies to guide examination of novel prognostic markers in humans. Clin Cancer Res. (2010) 16:3409–19. doi: 10.1158/1078-0432.CCR-10-0644
138. Zha C, Meng X, Li L, Mi S, Qian D, Li Z, et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. (2020) 17:154–68. doi: 10.20892/j.issn.2095-3941.2019.0353
139. Liang J, Piao Y, Holmes L, Fuller G, Henry V, Tiao N, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. (2014) 20:187–98. doi: 10.1158/1078-0432.CCR-13-1279
140. Zhang S, Guo M, Liu Q, Liu J, Cui Y. Neutrophil extracellular traps induce a hypercoagulable state in glioma. Immun Inflamm Dis. (2021) 9:1383–93. doi: 10.1002/iid3.488
141. Pylaeva E, Korschunow G, Spyra I, Bordbari S, Siakaeva E, Ozel I, et al. During early stages of cancer, neutrophils initiate anti-tumor immune responses in tumor-draining lymph nodes. Cell Rep. (2022) 40:111171. doi: 10.1016/j.celrep.2022.111171
142. Hedrick C, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. (2022) 22:173–87. doi: 10.1038/s41577-021-00571-6
143. McFarlane A, Fercoq F, Coffelt S, Carlin L. Neutrophil dynamics in the tumor microenvironment. J Clin Investig. (2021) 131:e143759. doi: 10.1172/JCI143759
144. Sounbuli K, Mironova N, Alekseeva L. Diverse neutrophil functions in cancer and promising neutrophil-based cancer therapies. Int J Mol Sci. (2022) 23:15827. doi: 10.3390/ijms232415827
145. Mantovani A, Marchesi F, Jaillon S, Garlanda C, Allavena P. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol. (2021) 18:566–78. doi: 10.1038/s41423-020-00613-4
146. Veglia F, Sanseviero E, Gabrilovich D. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
147. Zhao J, Jin J. Neutrophil extracellular traps: New players in cancer research. Front Immunol. (2022) 13:937565. doi: 10.3389/fimmu.2022.937565
148. Khan S, Mittal S, McGee K, Alfaro-Munoz K, Majd N, Balasubramaniyan V, et al. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int J Mol Sci. (2020) 21:1954. doi: 10.3390/ijms21061954
149. Mir H, Singh S. Neutrophils: a roadblock for immunotherapy. Nat Rev Cancer. (2022) 22:378–9. doi: 10.1038/s41568-022-00464-3
150. Zhou S, Zhou Z, Hu Z, Huang X, Wang Z, Chen E, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. (2016) 150:1646–58.e17. doi: 10.1053/j.gastro.2016.02.040
151. Ochiya T, Takenaga K, Endo H. Silencing of S100A4, a metastasis-associated protein, in endothelial cells inhibits tumor angiogenesis and growth. Angiogenesis. (2014) 17:17–26. doi: 10.1007/s10456-013-9372-7
152. Sollberger G, Choidas A, Burn G, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
153. Stojkov D, Claus M, Kozlowski E, Oberson K, Schären O, Benarafa C, et al. NET formation is independent of gasdermin D and pyroptotic cell death. Sci Signal. (2023) 16:eabm0517. doi: 10.1126/scisignal.abm0517
154. Appelgren D, Enocsson H, Skogman B, Nordberg M, Perander L, Nyman D, et al. Neutrophil Extracellular Traps (NETs) in the cerebrospinal fluid samples from children and adults with central nervous system infections. Cells. (2019) 9:43. doi: 10.3390/cells9010043
Keywords: neutrophil extracellular traps, blood-brain barrier, neuroinflammation, stroke, neurodegeneration, traumatic brain injury, Alzheimer’s disease, neutrophils (PMNs)
Citation: Shafqat A, Noor Eddin A, Adi G, Al-Rimawi M, Abdul Rab S, Abu-Shaar M, Adi K, Alkattan K and Yaqinuddin A (2023) Neutrophil extracellular traps in central nervous system pathologies: A mini review. Front. Med. 10:1083242. doi: 10.3389/fmed.2023.1083242
Received: 28 October 2022; Accepted: 06 February 2023;
Published: 17 February 2023.
Edited by:
Ahmet Emre Eskazan, Istanbul University-Cerrahpaşa, TürkiyeReviewed by:
Abbas Azadmehr, Babol University of Medical Sciences, IranCopyright © 2023 Shafqat, Noor Eddin, Adi, Al-Rimawi, Abdul Rab, Abu-Shaar, Adi, Alkattan and Yaqinuddin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saleha Abdul Rab,  c2FiZHVscmFiQGFsZmFpc2FsLmVkdQ==
c2FiZHVscmFiQGFsZmFpc2FsLmVkdQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.