 Ryu Watanabe
Ryu Watanabe Manami Kiji
Manami Kiji Motomu Hashimoto
Motomu Hashimoto
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 15 August 2022
Sec. Rheumatology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.983939
This article is part of the Research TopicExploring the Link between Autoinflammatory Diseases and VasculitisView all 4 articles
Vasculitis is an inflammatory disorder of the blood vessels that causes damage to a wide variety of organs through tissue ischemia. Vasculitis is classified according to the size (large, medium, or small) of the blood vessels. In 2020, VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome, a novel autoinflammatory syndrome, was described. Somatic mutations in methionine-41 of UBA1, the major E1 enzyme that initiates ubiquitylation, are attributed to this disorder. This new disease entity connects seemingly unrelated conditions: inflammatory syndromes (relapsing chondritis, Sweet's syndrome, or neutrophilic dermatosis) and hematologic disorders (myelodysplastic syndrome or multiple myeloma). Notably, such patients sometimes develop vasculitis, such as giant cell arteritis and polyarteritis nodosa, and fulfill the corresponding classification criteria for vasculitis. Thus, vasculitis can be an initial manifestation of VEXAS syndrome. In this research topic exploring the link between autoinflammatory diseases and vasculitis, we first provide an overview of the disease mechanisms and clinical phenotypes of VEXAS syndrome. Then, a literature review using the PubMed database was performed to delineate the clinical characteristics of vasculitis associated with VEXAS syndrome. Finally, the therapeutic options and unmet needs of VEXAS syndrome are discussed.
In 1997, gain-of-function mutations in MEFV were reported to cause familial Mediterranean fever (FMF) (1). Based on this discovery, the concept of autoinflammation was proposed to delineate a group of disorders characterized by recurrent episodes of fever and systemic inflammation (2). Since then, advances in genomic techniques have identified multiple monogenic disorders and their corresponding signaling pathways (3–5). Accordingly, the disease concept of autoinflammation has shifted from monogenic disorders to complex multifactorial conditions (6).
Autoimmunity and autoinflammation were considered distinct disease entities, but it has become clear that they form a spectrum of diseases, with monogenic autoinflammatory diseases and autoimmune diseases characterized by multiple autoantibodies representing the two ends (7, 8). Systemic lupus erythematosus is a representative autoimmune disease that depends on acquired immunity, whereas FMF and tumor necrosis factor (TNF) receptor-associated periodic syndromes are the best described autoinflammatory diseases in which innate immunity considerably contributes to pathogenesis (9, 10). Most rheumatic and musculoskeletal diseases show a mixed pattern of autoinflammation and autoimmunity.
In 2020, a novel autoinflammatory syndrome [vacuoles, E1 enzyme, X-linked, autoinflammatory, and somatic (VEXAS) syndrome] was reported (11). Hereafter, we refer to this paper as “the initial report”. This new disease is attributable to somatic mutations in methionine-41 of UBA1, the major E1 enzyme that initiates ubiquitylation. Surprisingly, this syndrome connects seemingly unrelated conditions, such as inflammatory syndromes (relapsing chondritis, Sweet's disease, or neutrophilic dermatosis) and hematologic disorders (myelodysplastic syndrome or multiple myeloma). Each organ manifestation often fulfills the corresponding diagnostic or classification criteria. Thus, this syndrome is considered to exhibit a mixed pattern of autoinflammation and autoimmunity.
In addition, patients with VEXAS syndrome sometimes develop vasculitis such as giant cell arteritis (GCA) and polyarteritis nodosa (PAN) (11). However, as case reports and series of VEXAS syndrome have been accumulated, various types of coexisting vasculitis have been reported, including leukocytoclastic vasculitis (LCV), immunoglobulin A (IgA) vasculitis, and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) (12–14). Moreover, this disease was considered to be found only in males, since UBA1 lies on the X chromosome. However, several female cases of inherited or acquired monosomy of the X chromosome have been reported (15–18). Thus, the clinical picture of VEXAS syndrome is heterogeneous and continuously expanding.
In this review, we provide an overview of the disease mechanisms and clinical phenotypes of VEXAS syndrome. We focused on the clinical characteristics of vasculitis associated with VEXAS syndrome through a literature review using the PubMed database. Finally, we discuss the therapeutic options and unmet needs of this syndrome.
Ubiquitylation is a multi-step post-translational modification that triggers proteasomal degradation (19). In this process, ubiquitin covalently binds to the substrate sequentially. Ubiquitylation is essential for multiple cellular processes such as cell cycle progression, DNA damage response, and immune signaling pathways (20, 21). Thus, dysregulation of the ubiquitin-proteasome system results in many disease states, such as infantile neurodegeneration, susceptibility to infection, lymphoproliferative disorders, and malignancy (21–23). Several autoinflammatory diseases have also been linked to alterations in the ubiquitylation system (24).
Ubiquitylation is initiated by the attachment of a single ubiquitin molecule to a target protein through a three-step enzymatic cascade that includes ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes (25). The concerted action of E1, E2, and E3 enzymes, as well as deubiquitylases, generates specific ubiquitylation patterns, which trigger the recognition and degradation of substrates by the proteasome. Ubiquitin-like modifier-activating enzyme 1 (UBA1), the major E1 enzyme, has two isoforms: UBA1a and UBA1b (26). UBA1a is the long isoform starting from codon 1 (Met1) of UBA1 and is localized in the nucleus, whereas UBA1b is the short isoform starting from codon 41 (Met41) and is localized in the cytoplasm without a nuclear localization signal.
Individuals with VEXAS syndrome acquire missense mutations at or around the start codon for UBA1b (Met41), which abrogates the expression of UBA1b. More than half of myeloid and erythroid precursor cells harbor such loss-of-function mutations (11). Therefore, somatic mutations found in VEXAS syndrome lead to a reduction in cytoplasmic UBA1 function, and the resultant decreased ubiquitylation activates the unfolded protein response and type I interferon production (21). Indeed, transcriptome analysis of the peripheral blood from patients with VEXAS syndrome showed highly activated inflammatory signatures in multiple pathways, including TNF, interleukin-6 (IL-6), interferon-γ, and IL-8 (11). Neutrophils may also participate in exacerbating the inflammatory response by enhancing neutrophil extracellular trap (NET) formation (27).
In the initial report, 25 patients were diagnosed with VEXAS syndrome based on the confirmation of somatic mutations in codon 41 of UBA1 (p.Met41Val, p.Met41Thr, or p.Met41Leu) (11). The median age at disease onset was 64 years, and all patients were male. Auricular and/or nasal chondritis was one of the most common organ manifestations (Figure 1), with 15 (60%) patients meeting the classification criteria for relapsing polychondritis (RP). According to other cohort studies, the incidence of chondritis was 36–50% (28–30).
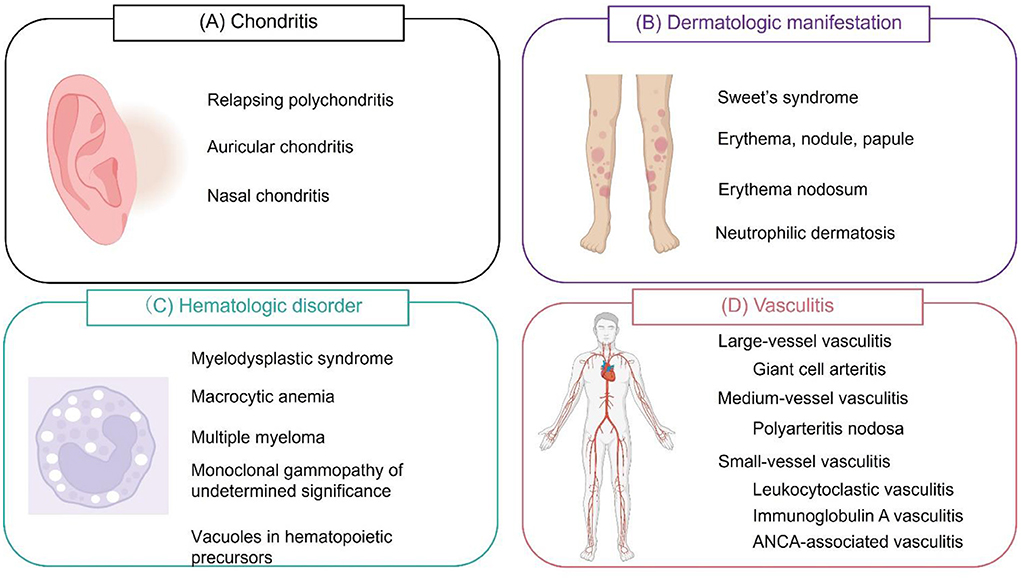
Figure 1. Symptoms of VEXAS syndrome. (A) Relapsing polychondritis (auricular and/or nasal chondritis) is one of the most common manifestations of VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. (B) Dermatological manifestations include Sweet's syndrome, erythema, nodules, papules, erythema nodosum, and panniculitis. These lesions are histologically characterized by neutrophilic dermatosis. (C) Hematological manifestations include myelodysplastic syndrome, macrocytic anemia, multiple myeloma, and monoclonal gammopathy of undetermined significance. Vacuole formation is characteristic of hematopoietic and myeloid precursor cells. (D) VEXAS syndrome can cause inflammation of blood vessels of any size. Cranial or extracranial giant cell arteritis, polyarteritis nodosa, leukocytoclastic vasculitis, immunoglobulin A vasculitis, and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis can be observed. Leukocytoclastic vasculitis is the most common type of vasculitis.
The frequency of UBA1 mutations in patients with RP has varied greatly in previous reports, ranging from 7.6% (7/92 patients) to 72.7% (8/11 patients) (17, 31). The reason for this difference remains unclear but could be due to racial differences. RP patients with UBA1 mutations (VEXAS-RP) had significantly higher mortality rates than those without, necessitating the early identification of VEXAS-RP (31). A decision tree algorithm based on male sex, mean corpuscular volume >100 fl, and platelet count <200 ×103/μl may help differentiate VEXAS-RP from RP (31).
Skin manifestations are also common in VEXAS syndrome, including Sweet's syndrome, erythema, nodules, papules, erythema nodosum, and panniculitis (14, 29, 32–36). The histological hallmark of skin lesions is neutrophilic dermatosis, which is often accompanied by LCV. The most commonly affected sites are the neck and trunk, but they also appear in the extremities (37). According to a literature review, approximately 90% (126/141) of published cases of VEXAS syndrome had cutaneous signs (14).
Zakine et al. performed molecular analyses of skin tissue samples and demonstrated that dermal infiltrates are derived from pathological myeloid clones with UBA1 mutations (34). However, Lacombe et al. identified UBA1 mutations only in neutrophilic dermatosis, but not in non-neutrophilic dermatosis, suggesting a distinction between “clonal” (neutrophilic dermatosis) and “paraclonal” (LCV and panniculitis) cutaneous involvements (37).
Hematologic disorders are also prevalent in VEXAS syndrome, including myelodysplastic syndrome (MDS), macrocytic anemia, multiple myeloma, and monoclonal gammopathy of undetermined significance (38). The name “V”EXAS syndrome stems from “vacuoles” in myeloid and erythroid cells, but not in T lymphocytes, B lymphocytes, or fibroblasts (11). Of note, myeloid and erythroid vacuolization is not specific to VEXAS syndrome, but has been described in other disorders, including copper deficiency (39), acute myeloid leukemia (40), Menkes disease (41), and transcobalamin II deficiency (42).
Thrombotic manifestations occur in approximately 40% of patients with VEXAS syndrome (43). The reported incidence of venous thromboembolism (36.4%) is much higher than that of arterial thrombosis (1.6%) (44). Crosstalk between aberrantly activated immune cells, platelets, and endothelial cells may result in the dysregulation of hemostasis and endothelial dysfunction.
Lung involvement, such as pulmonary infiltration, organizing pneumonia, and pleural effusion, can be found in approximately 50–70% of patients with VEXAS syndrome (11, 29, 30). Other symptoms include recurrent fever, arthritis, lymphadenopathy, and ocular manifestations. A wide array of clinical manifestations necessitate generalists, primary care providers, and rheumatologists to familiarize themselves with this syndrome (45).
In the initial report, 1 of 25 patients (4%) was diagnosed with GCA based on temporal artery biopsy. A 77-year-old male with a UBA1 mutation (p.Met41Val) presented with fever, pulmonary infiltrates, deep vein thrombosis, and MDS. Despite treatment with glucocorticoids, the patient died at 78 years old (11). As of June 25, 2022, 102 papers regarding VEXAS syndrome have been published in the PubMed database when the search term “VEXAS” was applied, and all papers were carefully reviewed. Table 1 summarizes the vasculitides associated with VEXAS syndrome. To the best of our knowledge, another case was diagnosed as extra-cranial GCA (46). This patient was a male in his sixties presenting with fever, pulmonary infiltrates, ear and nose chondritis, macrocytic anemia, and thrombocytopenia with a UBA1 mutation (p.Met41Thr). 18F-fluorodeoxyglucose positron emission tomography showed increased uptake in the aortic wall of the ascending aorta and aortic arch. The patient showed resistance to glucocorticoids and multiple biological disease-modifying antirheumatic drugs (bDMARDs) such as tocilizumab, anakinra, and infliximab, and died 8 years later (Table 1). Since only two cases of VEXAS-GCA have been reported, sufficient examination to discriminate GCA and VEXAS-GCA could not be conducted. The differential diagnoses of large vessel vasculitis are diverse (53), and these cases illustrate that VEXAS syndrome could be a potential mimicker of large vessel vasculitis (54).
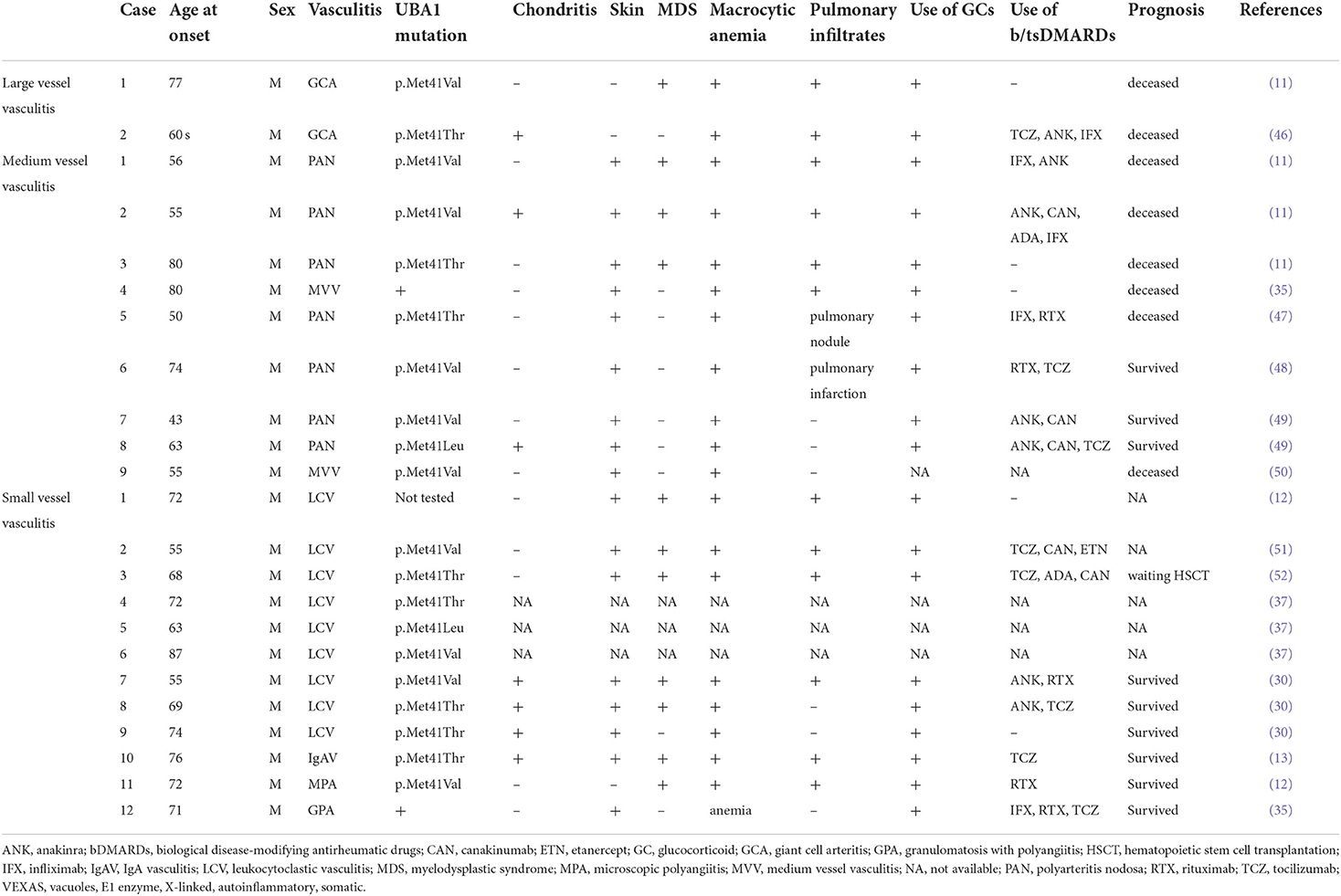
Table 1. Vasculitis associated with VEXAS syndrome identified by our literature review.
The incidence of GCA in reported cases with VEXAS syndrome seems rare. The question is how often UBA1 mutations are found in patients with GCA. To address this, Poulter et al. sequenced UBA1 in 612 male samples obtained from the UK GCA Consortium. No samples showed UBA1 mutations, whereas the mutation was identified in seven out of 1,055 samples from the cytopenic cohorts (55). The authors concluded that VEXAS syndrome is rarely misdiagnosed as GCA in the UK. However, as the incidence of GCA varies greatly by race, perhaps due to HLA differences (56, 57), race-specific UBA1 mutations in patients with GCA need to be assessed.
In the initial report, 3 (12%) patients with VEXAS syndrome fulfilled the classification criteria for PAN (58), which is a medium-sized vessel vasculitis (59). Our literature review found nine cases of medium vessel vasculitis (Table 1) (11, 35, 47–50). The average age at disease onset was 61.8 years (ranging from 43 to 80 years) and all were male. Seven of the nine patients fulfilled the classification criteria for PAN. Macrocytic anemia and skin lesions were observed in all cases, while chondritis was found in only two of the nine cases. Despite the use of high-dose glucocorticoids and multiple bDMARDs, such as infliximab, anakinra, and rituximab, six of the nine patients died during the treatment course. Notably, two of the three survivors successfully underwent allogeneic hematopoietic stem cell transplantation (49).
The most common vasculitis is small-vessel vasculitis, particularly LCV. The reason stems from the fact that skin manifestations, which are observed in 80–90% of patients, often reveal the histopathology of LCV (14, 37, 60). LCV is histologically characterized by angiocentric segmental inflammation, fibrinoid necrosis, and neutrophilic infiltration around the blood vessel walls (14, 61). In the initial report, LCV was histologically confirmed in seven of 25 patients (28%) with VEXAS syndrome and in seven of 22 patients (31.8%) with dermatologic manifestations. Although the demographics of patients who developed LCV was unclear in the initial report, an additional nine cases of LCV were identified in our literature review (Table 1) (12, 30, 37, 51, 52). The average age at disease onset was 68.3 years (ranging from 55 to 87 years) and all were male. Clinical features of these patients seem not different from patients without LCV.
Furthermore, it has become clear that VEXAS syndrome can be associated with small vessel vasculitides other than LCV, such as IgA vasculitis (13) and AAV (microscopic polyangiitis and granulomatosis with polyangiitis) (12, 35). It remains unclear whether the complications of these diseases are coincidental or causative; however, aberrant neutrophil activation with excessive NETs formation is a common feature of both VEXAS syndrome and AAV (27, 62). In AAV, ANCA activates neutrophils to produce reactive oxygen species and extrude chromosomal DNA in the form of NETs. The neutrophil activation highly depends on the priming by TNF, lipopolysaccharide, or complement factor 5a (C5a) (62). The efficacy and safety of C5 receptor blockade by avacopan has been demonstrated in patients with AAV (63), but has not yet been tested in VEXAS syndrome.
Considering the inflammatory aspect, immunosuppressive therapies are a sensible treatment option for this syndrome. Glucocorticoids, cyclophosphamide, conventional synthetic DMARDs (methotrexate, mycophenolate mofetil, azathioprine), and bDMARDs [including anti-IL-1 (anakinra, canakinumab), anti-TNF (infliximab and adalimumab), anti-IL-6 (tocilizumab), anti-CD20 (rituximab), anti-IL-17 (secukinumab), anti-IL-12/IL-23 (ustekinumab) therapies, and abatacept] have been administered to patients with VEXAS syndrome (11, 47, 64–66). In addition, janus kinase (JAK) inhibitors, such as ruxolitinib, tofacitinib, and baricitinib, have been introduced to some patients to block intracellular cytokine signalings (67–70). Given that multiple cytokines are involved in the disease mechanism of VEXAS syndrome, use of JAK inhibitors seems better therapeutic strategy rather than single cytokine blockade. However, these therapies work great for some patients and not for others, yielding varying results. Biomarkers that distinguish patients who are responsive to these therapies and those who are not should be established.
Since abnormal DNA methylation patterns drive the disease mechanisms of MDS, standard therapies for MDS include hypomethylating agents such as azacytidine (71). Considering the high prevalence of MDS in VEXAS syndrome, azacytidine could be a good candidate for this syndrome (67). Indeed, there have been a case report of successful treatment with azacytidine (72). However, in the French registry, 11 patients with VEXAS syndrome and MDS were treated with azacytidine, and the clinical response was achieved only in five patients (73), suggesting that this agent does not necessarily guarantee sufficient therapeutic effect and long-term prognosis.
If above-mentioned therapies fail, allogeneic hematopoietic stem cell transplantation (ASCT) may be the last treatment option for VEXAS syndrome. Although the incidence of treatment-related mortality remains high, given that irreversible somatic mutations have already been introduced in myeloid precursor cells in VEXAS syndrome, rejuvenation of the immune system by ASCT may be an ideal treatment. In fact, a series of cases showing the successful treatment courses with ASCT has been accumulated (49, 74).
VEXAS syndrome was discovered using an innovative genotype-driven approach. Currently, the diagnosis of this syndrome depends solely on the presence of UBA1 mutations confirmed by Sanger sequencing, the gold standard for genetic sequencing (75). However, Sanger sequencing method is time-consuming and has several limitations such as the missed recognition of variations (76). Therefore, it remains unclear whether this is sufficient. Use of next-generation sequencing (NGS), including whole-genome sequencing and whole-exome sequencing, may favorably serve in the faster and more accurate diagnosis.
In addition, classification criteria for the syndrome should be established to conduct clinical studies. In addition to genetic sequencing, a point-based system that weighs each clinical manifestation may be required.
As described, VEXAS syndrome shows resistance to multiple therapeutic agents, resulting in high mortality rates. Early identification of this syndrome by NGS and classification criteria may alter the treatment course. Furthermore, if the precise mechanism by which somatic mutations in UBA1 occur in middle-aged or elderly males is elucidated, a more disease-specific therapeutic approach for this syndrome, and even prevention of this mutation, could be possible.
In conclusion, VEXAS syndrome has led to the recognition that somatic mutations may be a more frequent cause of human disease than previously recognized (77). Further studies are required to provide appropriate diagnosis and treatment.
MK and RW conducted a literature review and generated Figure and Table. RW drafted the manuscript. MH finalized the manuscript. All authors contributed to the article and approved the submitted version.
This work was in part supported by JSPS KAKENHI Grant Numbers 20K17418 and 22K08569, a grant-in-aid of the Cardiovascular Research Fund, Tokyo, Japan, and a Grant for Promoting Research and Survey in Rheumatic Diseases by Japan Rheumatism Foundation to RW.
We would like to thank https://BioRender.com for the schematic Figure and Editage for the language review (https://www.editage.jp/).
Author RW receives speaker's fee from Eli Lilly. Author MH receives research grants from AbbVie, Asahi-Kasei, Brystol-Meyers, Eisai, Eli Lilly, and Novartis Pharma.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bernot A, Clepet C, Dasilva C, Devaud C, Petit JL, Caloustian C, et al. A candidate gene for familial Mediterranean fever. Nat Genet. (1997) 17:25–31. doi: 10.1038/ng0997-25
2. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
3. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. (2010) 140:784–90. doi: 10.1016/j.cell.2010.03.002
4. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. (2017) 18:832–42. doi: 10.1038/ni.3777
5. Stone DL, Beck DB, Manthiram K, Park YH, Chae JJ, Remmers E, et al. The systemic autoinflammatory diseases: coming of age with the human genome. J Allergy Clin Immunol. (2020) 146:997–1001. doi: 10.1016/j.jaci.2020.09.014
6. Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol. (2020) 146:925–37. doi: 10.1016/j.jaci.2020.08.017
7. Savic S, Caseley EA, McDermott MF. Moving towards a systems-based classification of innate immune-mediated diseases. Nat Rev Rheumatol. (2020) 16:222–37. doi: 10.1038/s41584-020-0377-5
8. Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benko S, Szucs G, et al. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. (2021) 17:585–95. doi: 10.1038/s41584-021-00652-9
9. Koga T, Kawakami A. Diagnosis and treatment of autoinflammatory diseases in adults: a clinical approach from rheumatologists. Immunol Med. (2018) 41:177–80. doi: 10.1080/25785826.2018.1524105
10. Migita K, Asano T, Sato S, Koga T, Fujita Y, Kawakami A, et al. Familial Mediterranean fever: overview of pathogenesis, clinical features and management. Immunol Med. (2018) 41:55–61. doi: 10.1080/13497413.2018.1481579
11. Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
12. Muratore F, Marvisi C, Castrignanò P, Nicoli D, Farnetti E, Bonanno O, et al. VEXAS syndrome: a case series from a single-center cohort of Italian patients with vasculitis. Arthritis Rheumatol. (2022) 74:665–70. doi: 10.1002/art.41992
13. Pàmies A, Ferràs P, Bellaubí-Pallarés N, Giménez T, Raventós A, Colobran R, et al. VEXAS syndrome: relapsing polychondritis and myelodysplastic syndrome with associated immunoglobulin A vasculitis. Rheumatology. (2022) 61:e69–71. doi: 10.1093/rheumatology/keab782
14. Sterling D, Duncan ME, Philippidou M, Salisbury JR, Kulasekararaj AG, Basu TN, et al. VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) for the dermatologist. J Am Acad Dermatol. (2022) S0190-9622(22)00181–5. doi: 10.1016/j.jaad.2022.01.042
15. Arlet JB, Terrier B, Kosmider O. Mutant UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2021) 384:2163. doi: 10.1056/NEJMc2102124
16. Barba T, Jamilloux Y, Durel CA, Bourbon E, Mestrallet F, Sujobert P, et al. VEXAS syndrome in a woman. Rheumatology. (2021) 60:e402–3. doi: 10.1093/rheumatology/keab392
17. Tsuchida N, Kunishita Y, Uchiyama Y, Kirino Y, Enaka M, Yamaguchi Y, et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis. (2021) 80:1057–61. doi: 10.1136/annrheumdis-2021-220089
18. Stubbins RJ, McGinnis E, Johal B, Chen LY, Wilson L, Cardona DO, et al. VEXAS syndrome in a female patient with constitutional 45, X (Turner syndrome). Haematologica. (2022) 107:1011–3. doi: 10.3324/haematol.2021.280238
19. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
20. Craney A, Rape M. Dynamic regulation of ubiquitin-dependent cell cycle control. Curr Opin Cell Biol. (2013) 25:704–10. doi: 10.1016/j.ceb.2013.07.004
21. Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol. (2022) 18:435–47. doi: 10.1038/s41584-022-00778-4
22. Oh E, Akopian D, Rape M. Principles of ubiquitin-dependent signaling. Annu Rev Cell Dev Biol. (2018) 34:137–62. doi: 10.1146/annurev-cellbio-100617-062802
23. Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. (2018) 19:59–70. doi: 10.1038/nrm.2017.83
24. Aksentijevich I, Zhou Q. NF-kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. doi: 10.3389/fimmu.2017.00399
25. Morreale FE, Walden H. Types of ubiquitin ligases. Cell. (2016) 165:248–248. doi: 10.1016/j.cell.2016.03.003
26. Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. (2009) 10:319–31. doi: 10.1038/nrm2673
27. Lacombe V, Prevost M, Bouvier A, Thépot S, Chabrun F, Kosmider O, et al. Vacuoles in neutrophil precursors in VEXAS syndrome: diagnostic performances and threshold. Br J Haematol. (2021) 195:286–9. doi: 10.1111/bjh.17679
28. Koster MJ, Kourelis T, Reichard KK, Kermani TA, Beck DB, Cardona DO, et al. Clinical heterogeneity of the VEXAS syndrome: a case series. Mayo Clin Proc. (2021) 96:2653–9. doi: 10.1016/j.mayocp.2021.06.006
29. Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. (2022) 186:564–74. doi: 10.1111/bjd.20805
30. van der Made CI, Potjewijd J, Hoogstins A, Willems HP, Kwakernaak AJ, de Sevaux RG, et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J Allergy Clin Immunol. (2022) 149:432–9. doi: 10.1016/j.jaci.2021.05.014
31. Ferrada MA, Sikora KA, Luo Y, Wells KV, Patel B, Groarke EM, et al. Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheumatol. (2021) 73:1886–95. doi: 10.1002/art.41743
32. Alhomida F, Beck DB, George TI, Shaffer A, Lebiedz-Odrobina D, Kovacsovics T, et al. Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome-clinical presentation of a newly described somatic, autoinflammatory syndrome. JAAD Case Rep. (2021) 14:111–3. doi: 10.1016/j.jdcr.2021.06.010
33. Dehghan N, Marcon KM, Sedlic T, Beck DB, Dutz JP, Chen LYC, et al. Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome: fevers, myalgia, arthralgia, auricular chondritis, and erythema nodosum. Lancet. (2021) 398:621. doi: 10.1016/S0140-6736(21)01430-6
34. Zakine E, Schell B, Battistella M, Vignon-Pennamen MD, Chasset F, Mahévas T, et al. UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS syndrome. JAMA Dermatol. (2021) 157:1349–54. doi: 10.1001/jamadermatol.2021.3344
35. Afsahi V, Christensen RE, Alam M. VEXAS syndrome in dermatology. Arch Dermatol Res. (2022). doi: 10.1007/s00403-022-02340-4. [Epub ahead of print].
36. Khosravi-Hafshejani T, O'Connor M, To F, Sreenivasan G, Shojania K, Au S, et al. The spectrum of skin disease in VEXAS syndrome: a report of a novel clinico-histopathologic presentation. J Eur Acad Dermatol Venereol. (2022) 36:e435–7. doi: 10.1111/jdv.17924
37. Lacombe V, Beucher A, Urbanski G, Le Corre Y, Cottin L, Croué A, et al. Distinction between clonal and paraclonal cutaneous involvements in VEXAS syndrome. Exp Hematol Oncol. (2022) 11:6. doi: 10.1186/s40164-022-00262-5
38. Grayson PC, Patel BA, Young NS. VEXAS syndrome. Blood. (2021) 137:3591–4. doi: 10.1182/blood.2021011455
39. Lazarchick J. Update on anemia and neutropenia in copper deficiency. Curr Opin Hematol. (2012) 19:58–60. doi: 10.1097/MOH.0b013e32834da9d2
40. Gajendra S, Gupta R, Sharma A, Gupta R, Gogia A. Acute myeloid leukaemia with Pseudo-Chediak-Higashi granules and intracytoplasmic vacuoles. Eur J Haematol. (2011). doi: 10.1111/j.1600-0609.2011.01668.x
41. Sayin S, Unal S, Cetin M, Gumruk F. Vacuolization in myeloid and erythroid precursors in a child with menkes disease. Turk J Haematol. (2019) 36:203–4. doi: 10.4274/tjh.galenos.2018.2018.0104
42. Unal S, Karahan F, Arikoglu T, Akar A, Kuyucu S. Different presentations of patients with transcobalamin II deficiency: a single-center experience from Turkey. Turk J Haematol. (2019) 36:37–42. doi: 10.4274/tjh.galenos.2018.2018.0230
43. Groarke EM, Dulau-Florea AE, Kanthi Y. Thrombotic manifestations of VEXAS syndrome. Semin Hematol. (2021) 58:230–8. doi: 10.1053/j.seminhematol.2021.10.006
44. Oo TM, Koay JTJ, Lee SF, Lee SMS, Lim XR, Fan BE, et al. Thrombosis in VEXAS syndrome. J Thromb Thrombolysis. (2022) 53:965–70. doi: 10.1007/s11239-021-02608-y
45. Koster MJ, Warrington KJ. VEXAS within the spectrum of rheumatologic disease. Semin Hematol. (2021) 58:218–25. doi: 10.1053/j.seminhematol.2021.10.002
46. Midtvedt Ø, Stray-Pedersen A, Andersson H, Gunnarsson R, Tveten K, Ali MM, et al. A man in his sixties with chondritis and bone marrow failure. Tidsskr Nor Laegeforen. (2022) 142. doi: 10.4045/tidsskr.21.0370
47. Kao RL, Jacobsen AA, Billington CJ Jr, Yohe SL, Beckman AK, Vercellotti GM, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. (2022) 93:102636. doi: 10.1016/j.bcmd.2021.102636
48. Itagane M, Teruya H, Kato T, Tsuchida N, Maeda A, Uchiyama Y, et al. VEXAS syndrome presenting as treatment-refractory polyarteritis nodosa. Arthritis Rheumatol. (2022) 371:1653. doi: 10.1002/art.42257. [Epub ahead of print].
49. Diarra A, Duployez N, Fournier E, Preudhomme C, Coiteux V, Magro L, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. (2022) 6:998–1003. doi: 10.1182/bloodadvances.2021004749
50. Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. (2021) 5:3203–15. doi: 10.1182/bloodadvances.2021004976
51. Takahashi N, Takeichi T, Nishida T, Sato J, Takahashi Y, Yamamura M, et al. Extensive multiple organ involvement in VEXAS syndrome. Arthritis Rheumatol. (2021) 73:1896–7. doi: 10.1002/art.41775
52. Lötscher F, Seitz L, Simeunovic H, Sarbu AC, Porret NA, Feldmeyer L, et al. Case report: genetic double strike: VEXAS and TET2-positive myelodysplastic syndrome in a patient with long-standing refractory autoinflammatory disease. Front Immunol. (2021) 12:800149. doi: 10.3389/fimmu.2021.800149
53. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of giant cell arteritis and takayasu arteritis-similarities and differences. Curr Rheumatol Rep. (2020) 22:68. doi: 10.1007/s11926-020-00948-x
54. Ramon A, Greigert H, Ornetti P, Bonnotte B, Samson M. Mimickers of large vessel giant cell arteritis. J Clin Med. (2022) 11. doi: 10.3390/jcm11030495. [Epub ahead of print].
55. Poulter J, Consortium UV, Morgan A, Cargo C, Savic S. A high-throughput amplicon screen for somatic UBA1 variants in cytopenic and giant cell arteritis cohorts. J Clin Immunol. (2022). doi: 10.1101/2021.12.08.21266919
56. Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med. (2014) 371:1653. doi: 10.1056/NEJMc1409206
57. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Cellular signaling pathways in medium and large vessel vasculitis. Front Immunol. (2020) 11:587089. doi: 10.3389/fimmu.2020.587089
58. Lightfoot RW Jr, Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. (1990) 33:1088–93. doi: 10.1002/art.1780330805
59. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65;1–11. doi: 10.1002/art.37715
60. Stubbins RJ, Cherniawsky H, Chen LYC, Nevill TJ. Innovations in genomics for undiagnosed diseases: vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome. CMAJ. (2022) 194:E524–e527. doi: 10.1503/cmaj.211770
61. Iglesias-Gamarra A, Restrepo JF, Matteson EL. Small-vessel vasculitis. Curr Rheumatol Rep. (2007) 9:304–11. doi: 10.1007/s11926-007-0049-3
62. Michailidou D, Mustelin T, Lood C. Role of Neutrophils in systemic vasculitides. Front Immunol. (2020) 11:619705. doi: 10.3389/fimmu.2020.619705
63. Jayne DRW, Merkel PA, Schall TJ, Bekker P, Group AS. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. (2021) 384:599–609. doi: 10.1056/NEJMoa2023386
64. Kirino Y, Takase-Minegishi K, Tsuchida N, Hirahara L, Kunishita Y, Yoshimi R, et al. Tocilizumab in VEXAS relapsing polychondritis: a single-center pilot study in Japan. Ann Rheum Dis. (2021) 80:1501–2. doi: 10.1136/annrheumdis-2021-220876
65. Magnol M, Couvaras L, Degboé Y, Delabesse E, Bulai-Livideanu C, Ruyssen-Witrand A, et al. VEXAS syndrome in a patient with previous spondyloarthritis with a favourable response to intravenous immunoglobulin and anti-IL17 therapy. Rheumatology. (2021) 60:e314–5. doi: 10.1093/rheumatology/keab211
66. Pathmanathan K, Taylor E, Balendra J, Lim A, Carroll G. VEXAS syndrome: favourable clinical and partial haematological responses to subcutaneous abatacept therapy with 30-month follow up. Rheumatology. (2022) 61:e174–7. doi: 10.1093/rheumatology/keac054
67. Heiblig M, Patel BA, Groarke EM, Bourbon E, Sujobert P. Toward a pathophysiology inspired treatment of VEXAS syndrome. Semin Hematol. (2021) 58:239–46. doi: 10.1053/j.seminhematol.2021.09.001
68. Fu Y, Wu W, Chen Z, Gu L, Wang X, Ye S, et al. Trisomy 8 Associated Clonal Cytopenia Featured With Acquired Auto-Inflammation and Its Response to JAK Inhibitors. Front Med. (2022) 9:895965. doi: 10.3389/fmed.2022.895965
69. Heiblig M, Ferrada MA, Koster MJ, Barba T, Gerfaud-Valentin M, Mékinian A, et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: a retrospective multi-center study. Blood. (2022). doi: 10.1182/blood.2022016642
70. Watanabe R, Hashimoto M. Perspectives of JAK Inhibitors for large vessel vasculitis. Front Immunol. (2022) 13:881705. doi: 10.3389/fimmu.2022.881705
71. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. (2002) 20:2429–40. doi: 10.1200/JCO.2002.04.117
72. Cordts I, Hecker JS, Gauck D, Park J, Härtl J, Günthner R, et al. Successful treatment with azacitidine in VEXAS syndrome with prominent myofasciitis. Rheumatology. (2022) 61:e117–9. doi: 10.1093/rheumatology/keab866
73. Comont T, Heiblig M, Rivière E, Terriou L, Rossignol J, Bouscary D, et al. Azacitidine for patients with vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol. (2022) 196:969–74. doi: 10.1111/bjh.17893
74. Loschi M, Roux C, Sudaka I, Ferrero-Vacher C, Marceau-Renaut A, Duployez N, et al. Allogeneic stem cell transplantation as a curative therapeutic approach for VEXAS syndrome: a case report. Bone Marrow Transplant. (2022) 57:315–8. doi: 10.1038/s41409-021-01544-y
75. Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, et al. A brief history of human disease genetics. Nature. (2020) 577:179–89. doi: 10.1038/s41586-019-1879-7
76. Mahmud S, Biswas S, Afrose S, Mita MA, Hasan MR, Shimu MSS, et al. Use of next-generation sequencing for identifying mitochondrial disorders. Curr Issues Mol Biol. (2022) 44:1127–48. doi: 10.3390/cimb44030074
Keywords: autoinflammatory disease, giant cell arteritis, leukocytoclastic vasculitis, vasculitis, VEXAS syndrome
Citation: Watanabe R, Kiji M and Hashimoto M (2022) Vasculitis associated with VEXAS syndrome: A literature review. Front. Med. 9:983939. doi: 10.3389/fmed.2022.983939
Received: 01 July 2022; Accepted: 27 July 2022;
Published: 15 August 2022.
Edited by:
Tomohiro Koga, Nagasaki University, JapanReviewed by:
Yuichiro Fujieda, Hokkaido University, JapanCopyright © 2022 Watanabe, Kiji and Hashimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryu Watanabe, ZG9jdG9yd2F0YW5hYmVyeXVAeWFob28uY28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.