 Fabio Casciano1,2*†
Fabio Casciano1,2*† Enrico Zauli3†
Enrico Zauli3† Erika Rimondi1Marco Mura4
Erika Rimondi1Marco Mura4 Maurizio Previati1
Maurizio Previati1 Massimo Busin3
Massimo Busin3 Giorgio Zauli4
Giorgio Zauli4- 1Department of Translational Medicine and LTTA Centre, University of Ferrara, Ferrara, Italy
- 2Interdepartmental Research Center for the Study of Multiple Sclerosis and Inflammatory and Degenerative Diseases of the Nervous System, University of Ferrara, Ferrara, Italy
- 3Department of Translational Medicine, University of Ferrara, Ferrara, Italy
- 4Research Department, King Khaled Eye Specialist Hospital, Riyadh, Saudi Arabia
The retina, the part of the eye, translates the light signal into an electric current that can be sent to the brain as visual information. To achieve this, the retina requires fine-tuned vascularization for its energy supply. Diabetic retinopathy (DR) causes alterations in the eye vascularization that reduce the oxygen supply with consequent retinal neurodegeneration. During DR, the mammalian target of rapamycin (mTOR) pathway seems to coordinate retinal neurodegeneration with multiple anabolic and catabolic processes, such as autophagy, oxidative stress, cell death, and the release of pro-inflammatory cytokines, which are closely related to chronic hyperglycemia. This review outlines the normal anatomy of the retina and how hyperglycemia can be involved in the neurodegeneration underlying this disease through over activation or inhibition of the mTOR pathway.
Introduction
The retina is an anatomical site where microvascular architecture and neuronal organization are strictly related for proper visual function. Among ocular diseases, diabetic retinopathy (DR) is an increasingly prevalent degenerative disease and is the leading cause of blindness in developed countries. DR is one of the disorders related to diabetes mellitus (DM). DM is a chronic metabolic disease with multiple homeostatic alterations leading to the disruption of redox regulation, the activation of immune responses, and systemic inflammation (1). During DM, sustained hyperglycemia is a well-recognized cause of retinal microvascular/neuronal rewiring. As a consequence of hyperglycemia-induced energy imbalance, cells quickly alter their biochemical activity by enhancing the expression of advanced glycation end products (AGEs) and reactive oxygen species (ROS) (2). The kinase, mammalian target of rapamycin (mTOR), resides at the interface between hyperglycemia and biochemical modifications. mTOR is a sensor of nutrient availability and growth factors, and has been implicated in multiple diseases like cancer, diabetes, and aging (3). mTOR and its related pathways, namely, mTOR complexes (mTORCs), control tissue homeostasis to manage cell growth, proliferation, autophagy, and apoptotic events by virtue of its role. Indeed, changes in retinal morphology are driven by mTOR pathways, mainly by ROS production and dysregulated autophagic processes (4, 5).
This review addresses how hyperglycemia alters mTOR pathways during DR and provides useful tools for understanding normal retinal anatomy and the role of mTOR in tissue generation and in the pathophysiology of DR.
Functional anatomy of the retina and blood–Retina barrier
The retina is the innermost of the three layers that constitute the wall of the eyeball. The retina internally covers the choroid and externally coats the vitreous body. Based on histological sections, the retina contains 10 different conventionally recognized morphological layers, composed of a complex array of neurons, glial cells, and blood vessels.
The first and outermost layer consists of the retinal pigment epithelium (RPE), which separates the retina from the innermost layer of the choroid, Bruch's membrane. More internally, three different types of neural cells are connected in series. Photoreceptors, rods, and cones convert the absorbed light into a neural signal, accounting for roughly 110–130 million cells in the entire retina. Photoreceptors, which account for roughly 30 million cells, are radially connected to bipolar cells, and transfer the neural signal to the innermost layer of ganglion cells whose axon forms the optic nerve, and are thought to number between 0.6 and 1.2 million. Physiological convergence of the signal from the more abundant photoreceptors to fewer ganglion cells is modulated by two different types of cells embedded in the retinal wall, horizontal cells that mediate the interaction between photoreceptors and bipolar cells, and amacrine cells interposed between bipolar cells and ganglion cells (6).
In addition to these five different types of neurons, the retina contains a large number of glial cells, namely, Müller cells, astrocytes, and microglia (7). Müller cells are the most representative and abundant among retinal glial cells. They constitute a larger part of the volume of the retina and fill the remaining space among neurons. Müller cells run radially from the inner to the outer limiting membrane, traversing all layers up to the outer nuclear layer and isolating neurons from each other except at their synaptic contacts. In correspondence with the internal limiting membrane, Müller cells contribute to the formation of this membrane with a footplate formed by their basal expansions. Astrocytes are much less abundant than Müller cells and are instead confined to ganglion cell and nerve fiber layers, while microglial cells are distributed throughout the whole retinal thickness but are particularly found near the vessels. It has also been demonstrated that glial cells are involved in retinal inflammation. Indeed, Müller cells have different receptors for cytokines and release cytokines to regulate inflammation (8). In physiological conditions, microglial cells maintain the homeostasis of the retina, undertake phagocytosis, clear debris, and control inflammation. Prolonged stress conditions, such as hyperglycemia associated with DR, can increase the number of microglial cells and release stress proteins and cytokines (9).
Among human tissues, the retina shows the highest oxygen consumption per unit weight to sustain elevated aerobic metabolism (10). In physiological conditions, the elevated blood flow is guaranteed by a dual arterial supply that allows independent vascularization of the outer and inner parts of the retina. Indeed, retinal layers are sandwiched between the outer and inner blood–retina barriers (oBRB and iBRB), which exhibit a very different anatomical structure (Figure 1). The retina from the external limiting membrane to the RPE is avascular and nourished by diffusion from the choroidal capillaries (11). Here, the oBRB is composed of the choroidal capillaries, the RPE, and Bruch's membrane, which are located between the basement membranes of the choroidal capillaries and RPE (6). The choroidal capillaries are fenestrated to provide a sustained intake of nutrients and adequate removal of waste products. In contrast, Bruch's membrane is formed by collagen and elastic sheets, and regulates the diffusion of nutrients based on their molecular weight. In addition, Bruch's membrane limits cell migration and controls intraocular pressure to randomize physical forces, thereby stabilizing the RPE layer. As a result, the RPE provides a wide exchange surface by means of an elevated number of microvilli, which extend between the outer parts of rods and cones, regulate nutrient supply, and recycle intracellular metabolites derived from the phagocytosed outer parts of photoreceptors. In addition, the RPE stabilizes the oBRB by providing vascular endothelial growth factor (VEGF) and other trophic factors to maintain the choroidal capillaries and their fenestrations. In a healthy individual, the most significant function of the choroidal circulation and the oBRB is to supply oxygen to photoreceptors, which are thought to consume more than 75% of retinal oxygen (12, 13). Notably, the pathological hallmark of DR is reduced oxygen exchange and consumption, followed by a low arteriovenous difference and abnormal venous oxygen saturation (14).
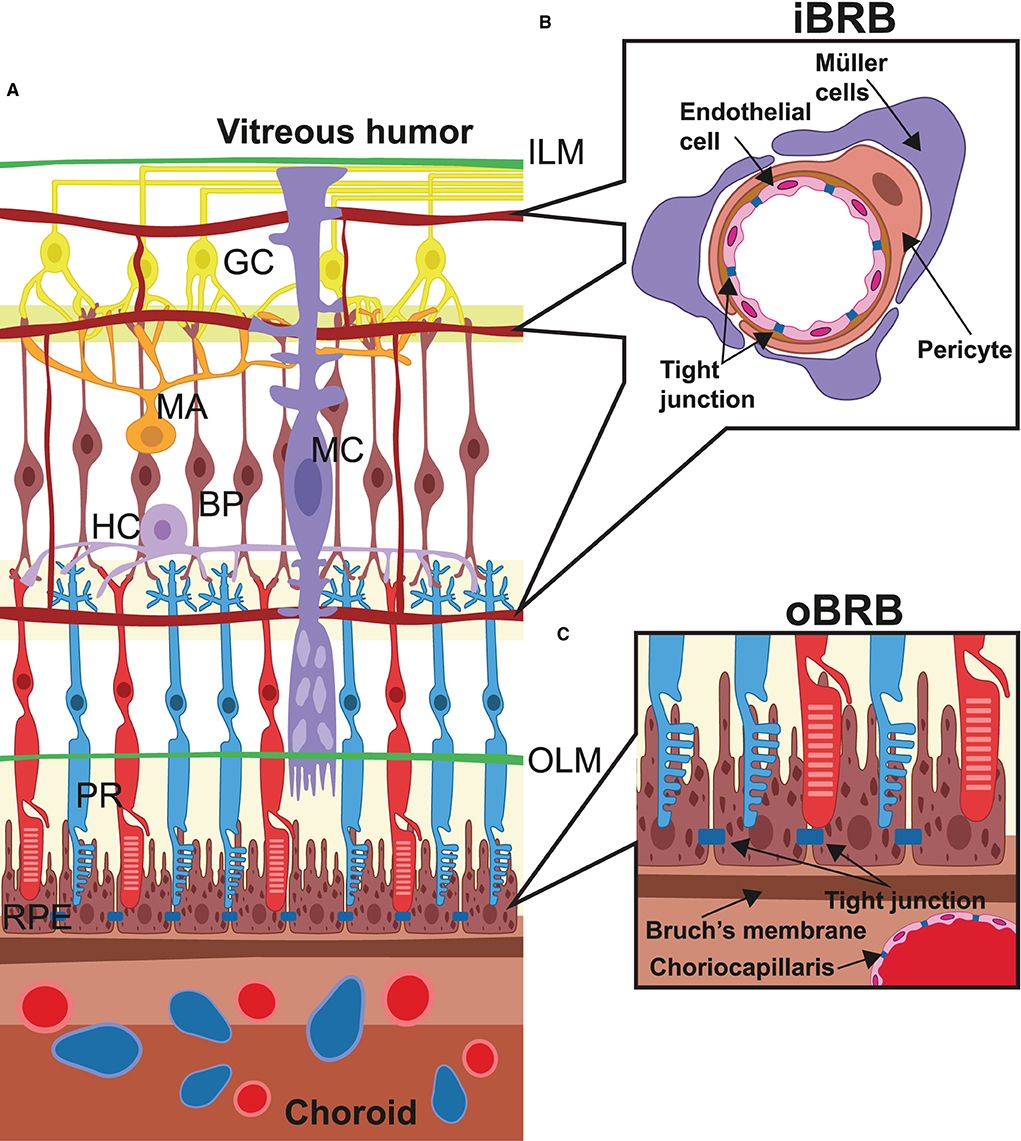
Figure 1. Retinal microanatomy and blood–retina barriers. (A) This image depicts the retinal structure situated in between the internal limiting membrane (ILM) and the choroidal layer (choroid), which externally lines the retinal pigment epithelium (RPE) cells. Over the RPE layer, neurons cross the whole retinal thickness and are radially connected (PR, photoreceptors; BP, bipolar cells; GC, ganglion cells). Other neural cells are amacrine cells (MA), horizontal cells (HC), and Müller cells (MC), which, in particular, terminate at the level of the outer limiting membrane (OLM). Vessels derived from the central retina artery form three different capillary beds were placed alongside the layers of nerve/ganglion layer, internal plexiform, and outer plexiform and interconnected by arterioles and capillaries. In (B), the structure of the internal blood–retina barrier is given in detail, which consists of several histological elements: the adherent and tight junctions among the capillaries, the pericyte layers around endothelial cells, and extensions from the Müller cells, all of which take part in the control of metabolites and liquid exchange and endothelial proliferation. In (C), the structure of the outer blood–retina barrier (oBRB) is represented and made from Bruch's membrane and the RPE layer, which play a pivotal role in controlling several parameters including internal pressure and ocular shape, oxygen exchange, and recycling of materials from the photoreceptor layer.
Blood, supplied from the central vessels of the retina which pass through the optic nerve, emerge at the papilla, and reach the internal surface of the retina. Unlike the outermost retinal layer, the innermost retinal layer is vascularized by three different plexuses organized alongside the layers of nerve/ganglion, internal plexiform, and outer plexiform. Similarly, the internal blood–retina barrier, which resembles the blood–brain barrier more, is consistently different from the oBRB (15). The capillaries do not show fenestrations but are continuous, and vicinal endothelial cells are joined together by adherent and tight junctions (Figure 1). In addition, endothelial cells are surrounded by pericytes even more covered by Müller cells, astrocytes, and microglial cells, which participate in the formation of the iBRB and collectively regulate endothelial cell proliferation and blood flow, providing additional trophic factors, antioxidants, and cytokines (16).
Diabetic retinopathy
Diabetes mellitus is a leading pathology in the industrialized country, resulting in several serious life-threatening complications and death. It has been estimated that 1–5 Americans could be affected in 2050 (17). DM affects multiple organs like the retina, kidney, peripheral nerves, and blood vessels due to prolonged exposure to hyperglycemia caused by chronic and/or relative insulin insufficiency (18, 19). Vascular complications of diabetes are grouped into “macrovascular diseases,” associated with an increase in myocardial infarction and stroke, and “microvascular diseases” such as diabetic nephropathy, retinopathy, and neuropathy (20, 21). Of these, DR is the most prevalent illness among elderly people with diabetes living in developed countries. The prevalence of DR has reached 50% of patients with type 2 diabetes and 75% of patients with type 1 diabetes and remains a leading cause of moderate-to-severe vision loss and blindness worldwide (22). Other microvascular complications, such as diabetic nephropathy, have been shown to be major risk factors for macrovascular complications such as heart attacks and strokes (23–25).
In DR, prolonged hyperglycemia changes the structure of the retina and induces alterations to both neuronal and vascular cells, resulting in vision loss and blindness (26–28). In DR, new vessels grow in the normally avascular outer retina and in the subretinal space. Moreover, older patients with diabetes show impaired macular blood flow regulation that exacerbates diabetic retinal damage (29–31). In addition to vascular remodeling, inflammation, typically associated with type 2 diabetes, also seems to play a role in DR. This is suggested by the finding that only half of the patients were successfully treated with a specific anti-VEGF treatment (32). Accordingly, it is possible to detect an increase in several inflammatory markers in the blood serum, aqueous or vitreous humor, and ocular tissue of patients with DR. In particular, intercellular adhesion molecule 1 (ICAM-1), interleukin-1β (IL-1β), IL-6, IL-8, tumor necrosis factor α (TNF-α), and monocyte chemoattractant protein-1 (MCP-1) have been found to be increased in patients with DR (33, 34). Both neurons and glial cells are involved in the release of these inflammatory mediators, which can recruit leucocytes at the ocular level, further promoting the shift toward a pro-inflammatory environment. In addition, lymphokines and chemokines can also directly target endothelial cells, stimulating cell death and vasculature rearrangements.
The development of new diagnostic techniques has led to powerful improvements in the visualization of retinal structures and vasculature and, consequently, in the diagnosis of DR. Classically, the diagnosis of DR has been based on color fundus photographs and fluorescein angiography, injecting dye into a vein in the arm during a dilated eye examination. Nowadays, optical coherence tomography (OCT) imaging offers a rapid and non-invasive test by imaging cross-sectional pictures of the macula layers to detect retinal alteration that heralds the onset of DR (35).
Clinical examination of the retinal microvasculature defines the two major types of DR. Non-proliferative DR (NPDR) is characterized by the presence of microaneurysms, dot and blot hemorrhages, exudates, cotton wool spots, and intraretinal microvascular abnormalities, while proliferative DR (PDR) involves more extensive ischemia, neovascularization, and tractional retinal detachment, which is a high-risk factor for severe vision loss (36).
Effect of diabetes and hyperglycemia on microvasculature and neurovascular units
The causative events underlying the pathogenesis of this disease are not completely understood. One of the most accredited hypotheses is that diabetes and hyperglycemia directly increase ROS production and alter the structure of the iBRB and oBRB events underlying vascular rearrangements (37, 38).
Briefly, the exposure of retinal cells to hyperglycemia triggers several related events, including massive glycation of cellular proteins, the formation of advanced glycation end products (AGEs), the formation of ROS, the release of pro-inflammatory cytokines followed by cell death—particularly in cells that are more exposed to hyperglycemia, such as pericytes (39). The loss of pericytes in the iBRB represents an early hallmark and is a breakthrough for DR because the disruption of the iBRB corresponds to deregulation in endothelial cell proliferation, leading to an outgrowth of dilated capillaries and microaneurysms, followed by vascular leakage and edema. Alternatively, it is frequently possible to find non-perfused or obliterated vessels with subsequent impaired flow and ischemia, followed by hypoxia-driven altered capillary regrowth (40).
Glycolysis is a metabolic pathway that produces adenosine triphosphate (ATP) under conditions that normally prevent ATP production by mitochondria. In healthy people with normal glycemia, glycolysis is fine-tuned to keep the intermediates stable. In diabetic patients with DR, hyperglycemia boosts glycolysis, leading to the accumulation of intermediates such as sorbitol via aldose reductase, as well as diacylglycerol (DAG) and AGEs (41). Increased DAG production during hyperglycemia activates protein kinase C-β (PKCβ), an isoform belonging to the PKC family active in vascular tissue (42), which consequently induces endothelial permeability, VEGF secretion, and inflammation (43). AGEs then bind to the ACE receptors, triggering VEGF expression and sustaining the pro-inflammatory event (44). In this view, VEGF has a central role during the inflammatory response in DR (45), as seen in other pathologies such as cardiovascular diseases like heart failure (46).
Being vascular remodeling one of the most dramatic events in DR, it is not surprising that in recent years, treatment of this pathology has been targeted at the retinal vessels: corticosteroids, laser photocoagulation, and anti-VEGF. Corticosteroids control inflammation and modulate genes encoding multiple inflammatory and anti-inflammatory proteins, leading to an amelioration of the BRB, even though it has also been demonstrated that corticosteroids can induce ocular hypertension and glaucoma (47). Because of the pivotal role of vascular outgrowth, laser photocoagulation has been a well-established treatment option for DR for more than half a century. Retinal oxygen demand is a regulator of angiogenesis, and this metabolic requirement can be reduced by laser photocoagulation, indicating that it could be an effective treatment for PDR (48). Potential side effects associated with this technique are moderate vision loss, diminished visual field, reduced color vision, and contrast sensitivity (49). In recent years, intravitreal injections of anti-VEGF drugs, such as ranibizumab, aflibercept, brolucizumab, aflibercept, or off-label drugs like bevacizumab, have become a common treatment for macular and retinal pathologies (50–52). However, these drugs have a short half-life and require regular intravitreal injections to maintain their efficacy, amplifying the risk of developing endophthalmitis caused by intravitreal injections of anti-VEGF agents (53, 54).
In addition to vascular remodeling, it is claimed that hyperglycemia can act directly and independently on neural cells. Using OTC analysis in addition to visual function tests [i.e., contrast sensitivity, perimetry testing, multifocal electroretinogram (mfERG), and dark adaptation], retinal thinning and visual dysfunction can be identified before the onset of DR, as has been demonstrated in diabetic patients without DR or with very early DR (55–57). This suggests a role for retinal neurodegeneration in the pathophysiology of DR. Retinal neurodegenerative events are common among species, as confirmed by animal models of diabetes. In mice and rats with autoimmune diabetes induced by the β-cell toxin streptozotocin, OTC analysis reveals thinning of the ganglion cell layer or inner plexiform layer, the inner/outer nuclear layer, as well as the entire retina (58, 59). Along with this neurodegeneration, vascular manifestation can be independent or at least concurrent. Indeed, in the ob/ob mouse model of type 2 diabetes, there was overall glial activation with leukostasis and a shift in microglia/macrophage phenotype before microvascular degeneration (60).
The breakdown of the inner BRB leads to loss of this complex neural environment and contributes to increased retinal vascular permeability and vision loss (61). Moreover, retinal microglia lose their motile cellular processes, became unresponsive to injuries, became denser, and had a smaller dendritic arbor (62). Retinal damage during DR further implicates retinal Müller glial cells and microglia as initiators of retinal inflammation. Purinergic signaling may explain this activation because of its well-established role in the immune-mediated inflammatory response in cardiovascular-related diseases (63). Purinergic signaling relies on the expression of receptors, i.e., purinergic P1 and P2 receptors, which recognize ATP, ADP, UTP, UDP, and nucleoside adenosine (ADO) molecules. Along with their function in the cell, these purine and pyrimidine molecules act as intercellular messengers. Indeed, after triggering of these receptors, subsequent cell signal transduction modulates tissue metabolism and normal physiology, but also the onset of pathological states of retinal diseases (64). Consistently, in DR, Müller cells amplify inflammation by releasing ATP in a CD40-dependent way, resulting in the activation of P2X7 purinergic receptors on retinal microglia, with subsequent expression of inflammatory cytokines, leading to neuroinflammation, vascular damage, and leakage (65).
mTOR: An overview
The mechanistic (mammalian) target of rapamycin is a serine/threonine protein kinase involved in different diseases such as cancer, diabetes, and cardiac hypertrophy (66). The name derives from the identification of mTOR as the target of rapamycin, a macrolide antibiotic extracted from Streptomyces hygroscopicus in the 1970's. mTOR is a 289-kDa protein with multiple domains: HEATS repeats, the FAT domain, the FKBP12-rapamycin binding domain [FKBP–rapamycin-binding (FRB); the core domain that belongs to the phosphatidylinositol 3-kinase-related kinase family of protein kinase], and the focal adhesion targeting C-terminal (FATC) domain. The N-terminus HEATS is a docking site for the regulatory-associated protein (Raptor) and the rapamycin-insensitive companion of TOR (Rictor). The FAT domain binds to the regulatory protein Deptor; the FRB domain is the domain responsible for mTOR inhibition via the FKB-12-rapamycin complex; the C-terminus FATC domain is for substrate recognition and catalytic activity. mTOR interacts with several proteins to form two distinct signaling complexes, namely, mTORC1 and mTORC2, which are implicated in many cellular functions like cellular growth, metabolism, and autophagy in response to environmental cues (67). mTORC1 regulates metabolic pathways involving macromolecular synthesis, cell growth, and autophagy, while mTORC2 controls cell proliferation, survival, cytoskeletal remodeling, neovascularization, and autophagy (68, 69). mTORC1 is characterized by the association of mTOR with Raptor, together with other companion proteins, and by its sensitivity to rapamycin, while mTORC2 is insensitive to rapamycin (70), and Raptor has been replaced by Rictor, which is necessary for the mTORC2 substrate interaction (Figure 2).
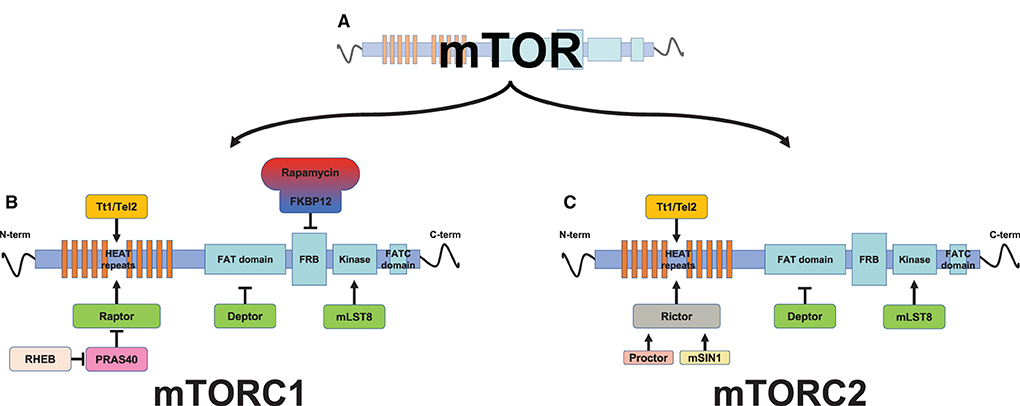
Figure 2. Schematic representation of mammalian target of rapamycin (mTOR) protein complexes. The mTOR protein (A) is associated with other proteins to form two distinct multiprotein complexes, mTOR complex 1 (mTORC1) (B) and mTORC2 (C). mTOR is a multidomain protein kinase. From the N-terminus, the first domain of the mTOR protein is formed by a HEAT repeat (tandem-repeat protein domains) domain, which functions as protein-protein interaction surfaces for the substrate's recruitment proteins, Raptor (regulatory-associated protein of TOR, which define mTOR complex 1, mTORC1) and Rictor (rapamycin-insensitive companion of TOR, which define mTOR complex 2, mTORC2). Proctor (protein observed with Rictor) and mSin1 (mammalian stress activated protein kinase interacting protein 1) act as a Rictor activator. HEAT repeats further interaction with the Tt1/Tel2 complex (Tt1, Tel two interacting protein 1; Tel2, telomere maintenance 2, also described as HCLK2), thus stabilizing the mTOR protein. The Tt1/Tel2 complex (Tt1, Tel two interacting protein 1; Tel2, telomere maintenance 2, also described as HCLK2) interacts with the HEAT repeats, thus stabilizing mTOR. The FAT (FRAP, ATM, TRRAP) domain is the binding site for the mTOR inhibitor Deptor (the DEP domain containing a mTOR-interacting protein). The FRB (FKBP–rapamycin-binding) domain, which precedes the kinase domain, interacts with the inhibitory rapamycin via the immunophilin FKBP-12 (FK506-binding protein 12 kD). The mTOR kinase domain resides between the FRB and focal adhesion targeting C-terminal (FATC) domains, and shares a characteristic of both PI3K and canonical protein kinase families. Its kinase activity is enhanced by mLST8 (mammalian lethal with SEC13 protein 8). The latter domain of the mTOR protein is the FATC (FAT C-terminus) domain, located at the C-terminus of the protein.
Mammalian target of rapamycin complex 1 responds to growth factors, amino acids, and ATP levels (67). Growth factors, such as insulin, activate the phosphatidyl inositol 3-kinase (PI3K)/Akt cell signaling pathway. Akt phosphorylation by PDK1 carries out two events: the inactivation of the GTPase tuberous sclerosis protein (TCS)1/2 complex, which allows the Ras homolog enriched in the brain (Rheb) to accumulate in a GTP-bound form capable of binding to mTOR (71), and the disassociation of the inhibitor PRAS40 from Raptor, both of which events allow the activation of mTORC1 (72). Moreover, mTORC1 acts as a sensor of nutrients (amino acids) and energy (ATP) that enables protein synthesis, only when these components are available to support the metabolic requirement. Indeed, amino acids trigger the translocation of mTORC1 to the lysosomal surface where Rheb is located, activating mTORC1 via the Regulator complex (73). AMPK is a metabolic enzyme that acts as an indirect mTORC1 regulator, sensing the cellular AMP/ATP ratio. In the absence of an adequate amount of intracellular ATP, AMPK promotes the formation of the TSC1/2 complex, thereby increasing the inactive Rheb GDP-bound form and consequently inducing the inhibition of mTORC1 (74).
Mammalian target of rapamycin complex 2 responds to growth factors via phosphatidylinositol (3–5)-trisphosphate (PIP3), which is generated by PI3K in the cell membrane. The inhibition of mTORC2 is relieved upon the binding of PIP3 to the Pleckstrin homology domain of mSin1. By targeting Ser/Thr protein kinases (i.e., the AGC family of protein kinases and Akt), mTORC2 regulates cell migration through cytoskeletal remodeling and cell proliferation as well as apoptosis (67, 75, 76).
By virtue of their role as a sensor for the availability of nutrients and growth factors, mTORC1 and mTORC2 play physiological roles during embryonic development and tissue regeneration. Studies on animal models have shown that hematopoietic stem cells (HSCs) undergo long-term exhaustion as a result of mTOR ablation, enhancing their transition from G0 to G1 (77, 78). This exhaustion is higher in the high HSC ROS population where p38 inhibitor or rapamycin was able to restore HSC function (79).
Moreover, mTOR signaling plays a critical role in neuronal development, particularly in adult neurogenesis and neuronal atrophy (80). In the central nervous system, the mTOR pathway prevents apoptotic cell death, and this function is strictly linked with trophic factor activity (81). mTOR signaling is also involved in neurogenesis of the eye, and is highly activated in embryonic stages. Indeed, its temporal regulation is essential for the normal development of the retina and the optic nerve (82). Upon the deletion of TSC1, the activation of the mTOR pathway triggers the regeneration of adult retinal ganglion cells after optic nerve injury (83). This activity seems to be dependent on mTORC1 (84). Interestingly, in addition to its main role of regulating the production of red blood cells, erythropoietin (EPO) also has a role in neuroplasticity and neurogenesis after functional hypoxia (85), and this role seems to be linked to mTOR activity. Indeed, after oxygen-glucose deprivation, a condition that could cause some ischemia at the retinal level, EPO intimately regulates the mTOR pathway, preventing cellular injury and apoptotic events via the inhibition of PRAS40 (86). The relationship between EPO and mTOR could dependent on the Wnt pathway (87), as it has been documented that WISP1 activates mTORC1 through the phosphorylation of PRAS40 and TSC2 during microglial oxidative stress (88, 89). Moreover, the treatment of EPO also decreases mTOR expression and orchestrates the autophagy-related signaling pathways, suppressing cell injury in a rotenone-induced neurotoxicity model (90). In some cases, under hypoxic and superoxide stress, EPO promotes the survival of retinal progenitor cells by reducing autophagy (91) and becoming a promising neuroprotective agent for optic nerve protection and repair (92).
mTOR and retinopathy
The retina is a high-demand site for oxygen and nutrients. During DR, in response to metabolic insults such as hypoxia, the retina undergoes morphological changes characterized by neovascularization with epiretinal vascular proliferation and subsequent vascular leakage and tractional retinal detachment. Early retinal pathophysiological modifications occur within the first few weeks of diabetes (93), and sustained hyperglycemia alters the distribution of oxygen around the retinal arterioles, inducing retinal vasculature rewiring, as is shown in a diabetes-induced rat model (94). Hypoxia modulates the angiogenic factor HIF-1α, an oxygen-sensitive transcription factor, to support retinal neovascularization. VEGF-α is a hypoxia-inducible gene target of HIF-1α. These proteins are downstream targets of mTORC1, as shown by rapamycin-directed suppression of hypoxia-inducible factors and vascular endothelial growth factors, followed by a reduction in vascularized tumor volume (95). In hyperglycemic rats for 8 weeks, an intraperitoneal injection of rapamycin reduces diabetes-induced VEGF overexpression that controls vascular permeability and angiogenesis (96). These observations are in line with the studies by Liu et al., in which the degree of retinopathy was mTORC1 dependent according to the expression of VEGF and PEDF proteins induced via the p-S6 protein in a DR rat model (97). Furthermore, VEGF may subsequently activate mTOR. Indeed, after the binding of VEGF to its receptor VEGFR-2, the PI3K/Akt pathway is activated and subsequently activates mTOR (98). This highlights that targeting the PI3K/Akt/mTOR signaling pathway could be a strategy to improve DR, as seen in human acute lymphoblastic leukemia (99).
Immunolocalization studies on human, rat, and mouse retinae have shown that the inner retina expresses mTORC pathways, with mTORC1 mainly localized to retinal ganglion cells and mTORC2 primarily relying on glial cells (100). As previously stated, the retina is a high-demand site for energy, and mTOR not only drives the perception of multiple upstream stimuli but also the regulation of cell metabolism and growth as downstream targets of PI3K. Indeed, the concurrent loss of mTORC1 and mTORC2 leads to inner and outer retinal morphology changes with a concurrent reduction in cone function, thus explaining the photoreceptor function loss observed during diabetes (101). In the retina, glial cells exert trophic support and influence programmed cell death, potentiating the neurodegeneration observed in retinal diseases (102). Experiments in an in vitro immortalized human Müller glial cell line and in an in vivo mouse-induced diabetic model show that the blockade of PPP1CA/YAP/GS/Gln/mTORC1 inhibits Müller cell proliferation and activation, suggesting a potential way to mitigate the development of DR (103).
The crosstalk between mTOR and ROS in DR
Despite a wide body of literature focusing on ROS-induced microvasculature alterations, many studies report that retinal neurons can be directly targeted by diabetes, become an independent source of ROS, and undergo cell death, independently of and even before microvasculature alterations (104, 105).
It is well-known that hyperglycemia, as a consequence of nutrient overload, can promote oxidative stress through various metabolic pathways (5). Excessive amounts of ROS alter lipids, proteins, or deoxyribonucleic acid (DNA). NF-κB, acting as a redox sensor, plays a critical role in the regulation of the inflammatory response and programmed cell death (apoptosis) (5). In the retina, increased ROS production causes the activation of microglia with the expression of inflammatory cytokines, including IL-6, TNF-α, IL-1β, and IL-8, and adhesion molecules like ICAM-I and vascular cell adhesion molecule 1 (VCAM-1). Overall, these factors contribute to leukostasis and vascular leakage (106).
Yoshida et al. have shown that in a mouse model, hypoxia activates NF-κB in various retinal cell types (107). Furthermore, a recent in vitro experiment showed that NF-κB is activated in human retinal cells, including endothelial cells, pericytes, and astrocytes, under high glucose conditions (16). The overall effect of ROS can be ameliorated by oral administration of the natural phenol resveratrol, which reduces the level of inflammatory TNF-α and IL-6, in addition to activating NF-κB, in diabetic rat models (108, 109). This is in line with the effects observed by Bucolo et al. where the administration of antioxidant natural compounds, such as curcumin, carnosine, and α-lipoic acid, reduced the TNF-α and VEGF levels in the retinae of diabetic rats (110, 111). The mechanism of this inhibition could be explained by the downregulation of phosphorylation of NF-κB and the MAPK family in a mTOR-dependent manner, as shown in in vitro experiments conducted in lipopolysaccharide- (LPS-) stimulated microglial cells (112). Furthermore, it is well-known that ARPE-19, a human RPE cell line, under hyperglycemia presents metabolic changes including oxidative stress mediated by ROS (113). Yahg et al. have shown that the combined treatment of antidiabetic drugs, semaglutide, and rosiglitazone reduces high glucose-induced inflammatory injury by inhibiting ROS/PI3K/Akt/mTOR signaling pathway-related proteins TNF-α, IL-6, and IL-1β in ARPE-19, and enhances overall antioxidant capacity in a DR rat model by downregulating and upregulating, respectively, the levels of serum lipid peroxidation and superoxide dismutase (SOD) (114).
It is well-known that RPE cells play an essential role in maintaining the viability and functionality of photoreceptors, and that their loss of function results in alterations that are potentially causative of various retinal diseases (115). During pathogenic conditions, fully differentiated epithelial cells, via a process known as epithelial–mesenchymal transition (EMT), could reverse their phenotype to mesenchymal cells with invasive and migratory behavior toward the neuroretina, which in turn differentiate into fibroblasts/myofibroblasts. Next, the latter cell types could secrete excessive amounts of extracellular matrix components such as collagen (types I, III, IV, V, and VI) and fibronectin, resulting in fibrosis (116). Of note, during DR, EMT seems to be linked with the mTOR pathway and ROS, mainly driven by TGF-β. TGF-β has been found to be upregulated in the postmortem eyes of patients with ocular diseases and EMT, revealing a relevant role during the generation of DR (115, 117). Indeed, Kim et al. suggested a mechanism for mTOR activation and ROS generation with TGF-β, which contributes to EMT and fibrosis in retinal pigment epithelial cells (118).
Furthermore, recent findings in support of the involvement of the mTOR pathway showed that an mTOR inhibitor can modulate the expression of VEGF in the diabetic rat retina and VEGF-induced ROS enhancement in the Müller cell line (TR-MUL5) (96). These results are in line with the studies by Kim et al. where resveratrol limits the increase of VEGF, reducing early vascular lesions in diabetes-induced mouse retinae (119). L-glutamate is a major excitatory neurotransmitter in the nervous system, but excess extracellular glutamate may lead to neuronal and non-neuronal death and/or damage (120). In this context, ROS reduces glutamate clearance in the retina via the inhibition of glutamate intake in Müller cells, ultimately inducing retinal neurodegeneration (121). These effects are in line with the aforementioned relationship between ROS and mTOR, in the light of glutamate transporter 1 expression being a downstream target of mTOR (122).
From this point of view, it is clear that the observed relationship between oxidative stress and mTOR pathway in DR could be used to unveil potential new therapeutic opportunities to treat this illness.
The crosstalk between mTOR and MicroRNAs in DR
MicroRNAs (miRNAs) are small, non-coding ribonucleic acids (RNAs) that regulate gene expression by pairing with complementary DNA sites and/or interfering with mRNA translation and stability (123). Although several studies have highlighted the crosstalk between the mTOR pathway and miRNA gene targeting (124, 125), few articles have investigated the role of miRNAs during DR. Among them, Li et al. have shown that the presence of ROS modulates the expression of miRNA-34a, increasing oxidative stress-related markers and cell apoptosis in ARPE-19 treated with high glucose (126). These findings are in line with a study by Liao et al. that miR-34a upregulates the phosphorylation of mTOR, which further reduces autophagy and enhances apoptosis in prostate cancer cells (127). However, further studies are needed to address the direct target(s) of miR-34 in regulating the mTOR pathway during DR.
Furthermore, in the treatment of ARPE-19 under high glucose conditions (50 mM), the overexpression of miR-130a exerts an antioxidant role by increasing the scavenger SOD1 levels in a TNF-α-dependent manner, as confirmed by the upregulation of TNF-α or knockdown of SOD1 (128). Although there is no clear evidence of the crosstalk between miR-130a and the mTOR pathway during DR, it is noteworthy that miR-130a is a negative regulator of TSC1, capable of upregulating the mTOR pathway in high-grade serous ovarian carcinoma (129). Then, the consequent aforementioned TNF-α upregulation and SOD1 phosphorylation could be the result of mTORC1 activation (114, 130). These findings could open up attractive new research areas for researchers involved in the study of DR. Currently, other miRNAs seem to be involved in the mTOR pathway during DR. During hypoxia, miR-7 is a critical mediator of the cellular response, reversing hypoxia-induced inhibition of mTOR signaling (131). Furthermore, it has been found that miR-7 can modulate cell proliferation by downregulating the expression of Hoxb3, mTOR, p-PI3K, and p-AKT in retinal epithelial cells (132).
Recent findings showed that the retinae of mice with DR notably decreased the expression of miRNA-29, and this event was associated with the inhibition of AMPK phosphorylation, with AMPK being the target protein of miR-29, and increased the expression of p-mTOR, thereby leading to excessive apoptosis observed during DR (133).
Lastly, the role of miRNA and mTOR pathways in DR needs further study, and bioinformatic analysis could be a useful tool to highlight their contribution to the pathophysiology of DR. Indeed, according to bioinformatic analysis, miRNA-204 with its three target genes Rictor, Dlg1, and SYNJ2BP, is associated with retinal diseases, suggesting that it has a relevant role in regulating Wnt signaling, the blood–retinal barrier, and angiogenesis (134).
The crosstalk between mTOR and autophagy in DR
Autophagy is a catabolic process in which damaged cellular components are sequestered within a vacuole and degraded by fusing with lysosomes. Autophagy also allows cells to obtain free fatty and amino acids to sustain protein synthesis, and occurs as a selective process against specific organelles for disposal and tissue remodeling (135). Therefore, autophagy maintains adequate cellular homeostasis and energy levels. Autophagy can be distinguished into three main types: macroautophagy, microautophagy, and chaperone-mediated autophagy. Furthermore, autophagy discriminates targets in a specific and non-specific manner. Selective autophagy requires one or more receptors that tag targets for engulfment in the autophagosome, while non-selective autophagy is a bulk process that randomly picks up any kind of cytoplasmic proteins and ships it into the lysosome. In macro- and microautophagy, cytosolic components are engulfed in vacuoles and lysosomes through selective and non-selective mechanisms, while in chaperone-mediated autophagy, lysosome-associated membrane protein type 2A (LAMP-2A) first binds the substrate protein to the lysosomal membrane (136, 137).
Notably, the modulation of autophagy processes has been shown to represent an effective approach to the treatment of several human pathologies including neurodegenerative diseases (138, 139).
On the other hand, the modulation of autophagy is strictly dependent on the specific illness. Autophagy can play contrasting roles in different neurodegenerative diseases, playing an ameliorative role in some illnesses and contributing to the course of the disease in others.
In some neurodegenerative diseases, autophagy can act as a scavenger of misfolded and abnormally aggregated proteins, and in this context, autophagy stimulation can have a positive therapeutic role. Indeed, mTOR inhibition and autophagy activation have been shown to play a critical role in Alzheimer's disease. In Alzheimer's disease, mTOR activation promotes the production and accumulation of amyloid-β in the brain, and this event is linked with a direct inhibition of the autophagy-lysosomal system (140). Moreover, autophagy reduces the production of amyloid-β and ameliorates memory function in some animal models of Alzheimer's disease (141).
Similar findings are shown in neurogenerative Parkinson's disease where autophagic processes are dysfunctional with related accumulation of α-synuclein and other polyubiquitinated proteins (142).
Ischemia causes disorders related to nutritional needs and metabolic demands, and autophagy restores energy production via a catabolic process that allows neuronal cells to survive the nutrient depletion (143). In a neonatal model of hypoxia/ischemia, the inhibition of mTOR can activate autophagy (144). Moreover, in a model of spinal cord injury, rapamycin drives neuronal cell protection, promoting autophagy by inhibiting mTOR signaling (145).
Recently, Patergnani et al. reported alterations in glucose metabolism, impairment in mitochondrial functions, and excess of autophagy and mitophagy related to alterations in the mTOR/ULK1 pathway, in in vitro, ex vivo, and in vivo models of multiple sclerosis. The inhibition of autophagy with FDA-approved drugs strongly ameliorated axonal remyelination in all models and in vivo behavioral tests (146).
Similarly, in a mouse model of spinal cord injury, treatment with bisperoxovanadium was shown to activate the Akt/mTOR pathway, reduce autophagy, and rescue motor neurons from death (147).
Neurodegenerative disorders are significantly increasing worldwide. DR is now widely recognized as a neurodegenerative disorder (148), and its pathophysiology is closely related to the regulation of autophagy. Non-neuronal cells like RPE cells may also play a critical role in DR. RPE is part of the oBRB and regulates the transport of nutrients, water, and solutes from the choroid to the retina. In addition, RPE sustains photoreceptors and ensures the recycling of cones and rods that need to be replaced upon light absorption. Therefore, the autophagy of these cells appears to be relevant during DR. Consistently, Zhang et al. showed that high glucose conditions mediate the damage to ARPE-19 and increase its autophagy as well as apoptotic markers (p-p53, Bcl-2, and p62), and that these damages can be reversed by the autophagy inhibitor 3-methyladenine (3-MA), indicating a dysregulation of the autophagic process (149). This last observation was further confirmed by the same group, pointing out the beneficial effect of procyanidin, a member of the flavonoids, which inhibits autophagy (150). Interestingly, these beneficial effects were reversed when the autophagy agonist rapamycin was added to procyanidin treatment.
In DR, hypoxia and nutrient starvation increase circulating adipokines (i.e., leptin and adiponectin) to overcome metabolic deficiency. Recent findings indicate that adipokines may contribute to neovascularization during DR (151), a phenomenon linked with the mTOR pathway and the autophagy process. Li et al. found that in the rhesus choroid-retinal endothelial (RF-6A) cell model, during high glucose treatment, adiponectin promotes the expression of p-PI3K, p-AKT, and p-mTOR, increasing cell viability and lowering the autophagic process, therefore inhibiting high glucose-induced angiogenesis (152). Moreover, in the retina of DR mouse models, hypoxia and high glucose increase the expression of AGGF1 (an angiogenic protein with a function similar to VEGF-A) and promote autophagy with related angiogenesis. These phenomena were further confirmed in vitro using RF/6A cells, where the inhibition of the PI3K/AKT/mTOR pathway and the activation of autophagy-induced AGGF1-driven cell proliferation and tube-like structure formation (153).
As previously mentioned, hyperglycemia, acting as a major mechanism of DR pathology, causes neurodegeneration earlier than the detectable microvascular damage in which the mTOR/autophagy pathway is prominent. Recent studies on streptozotocin-induced diabetic models suggest that prolonged hyperglycemia downregulates mTOR-related proteins and GLUT1, with an increase of apoptotic markers as well as autophagic proteins in the ganglion cell layer. Blockage of autophagy by phlorizin (an insulin-independent glycemic control) and MHY1485 (an mTOR activator) normally rescues neuronal cells, suggesting that the mTOR pathway plays a relevant role associated with the damage to retinal ganglion cell (4).
Collectively, these observations highlight the dual nature of autophagy. In some circumstances, autophagy behaves as a protective mechanism, regulating inflammation, reducing starvation stress, and destroying noxious proteins. In others, it can worsen mitochondrial activity and energy replenishment, definitely targeting cells for death. This makes the dysregulation of autophagy a very interesting target in attempts to prevent the worsening of several illnesses, including DR.
Cellular responses like autophagy and senescence are closely related because many stresses including DNA damage, oxidative stress, and oncogenic stress can activate them. Both cellular responses prevent further proliferation of damaged cells, triggering cytotoxic or cytoprotective effects (154). Indeed, according to the level of autophagy, cells are driven to cell death or cellular senescence. It has been well-described that the exposure of ARPE-19 cells to a high concentration of glucose alters metabolism and increases overall ROS production and lipid accumulation, contributing to senescence (155, 156). Thus, Chae et al. reported interesting results: in a doxorubicin-induced mouse model of RPE senescence, they found that selective targeting of senescent RPE cells by Nutlin-3a ameliorates age-related macular degeneration (157). This finding may appear tricky, but it is noteworthy that although p53 is an autophagy agonist via the inhibition of mTORC1, Nutlin-3a causes quiescence and senescence program suppression (158, 159). Overall, in RPE, given that mTOR and p53 are key mediators of autophagy and senescence responses, this may represent an attractive target to eliminate senescent cells in DR.
Along with non-retinal cells that are affected by DR, Müller cells and retinal microvascular endothelial cells have been implicated in altering autophagic processes. Müller cells respond to vascular injury, trauma, and metabolic stresses by releasing trophic factors (i.e., VEGF) and phagocytosing degenerated cells to maintain retinal homeostasis (160). In vitro experiments have shown that upon high glucose stress, Müller cells increase autophagic markers with the accumulation of p62/SQTSM1. Despite this process, glial cells undergo programmed cell death and release massive amounts of VEGF. On the other hand, rapamycin restores the autophagic machinery and protects cells from apoptosis, thus highlighting the role of autophagic dysfunction in these cells during DR (161). Along with these alterations, Müller cells undergo the dysregulation of mitophagy and become more susceptible to redox stress (162).
Therefore, the induction of autophagy seems to play a relevant role in maintaining cell survival in the nervous system, and mTOR is a conductor for autophagic activity in the cells, making it a candidate for the crosstalk between the mTOR pathway and autophagy as an attractive option to manage DR.
Conclusion
Diabetic retinopathy is a complex disease without a completely clarified etiology. Diabetes at the intracellular level prompts oxidative stress and redox equilibrium imbalance through different cellular and mitochondrial pathways. The successive cellular alterations and death lead to profound changes in the histology of the retina, with malfunction and loss of photoreceptors and other neural cells. In parallel, weakening the BRB leads to microvascular changes that reduce the availability of oxygen to photoreceptors. This impacts the survival of neurons and, in the meantime, induces a marked trophic factor-dependent redrawing of the retinal microvascular structure, as is typical of DR.
Mammalian target of rapamycin coordinates multiple anabolic and catabolic processes involved in promoting cell growth and acts as a sensor for growth factors and nutrients. The finding reported in this review highlights that in neurodegenerative diseases like retinopathy, the mTOR pathway can be over activated or inhibited. Furthermore, corticosteroids, laser photocoagulation, and anti-VEGF therapy are becoming the standard of care for the treatment of DR, but can have adverse problems or encounter non-responding subjects, preventing their use in some patients.
Concurrently, this review highlights that in DR, the mTOR pathway seems to be involved in a plethora of effects linked to oxidative stress, autophagy dysregulation, and cell death, as seen in various experimental models (Figure 3). In this opinion, although knowledge gaps deserve further elucidation, mTOR targeting in particular could be an attractive target for researchers to postulate novel therapies to treat DR.
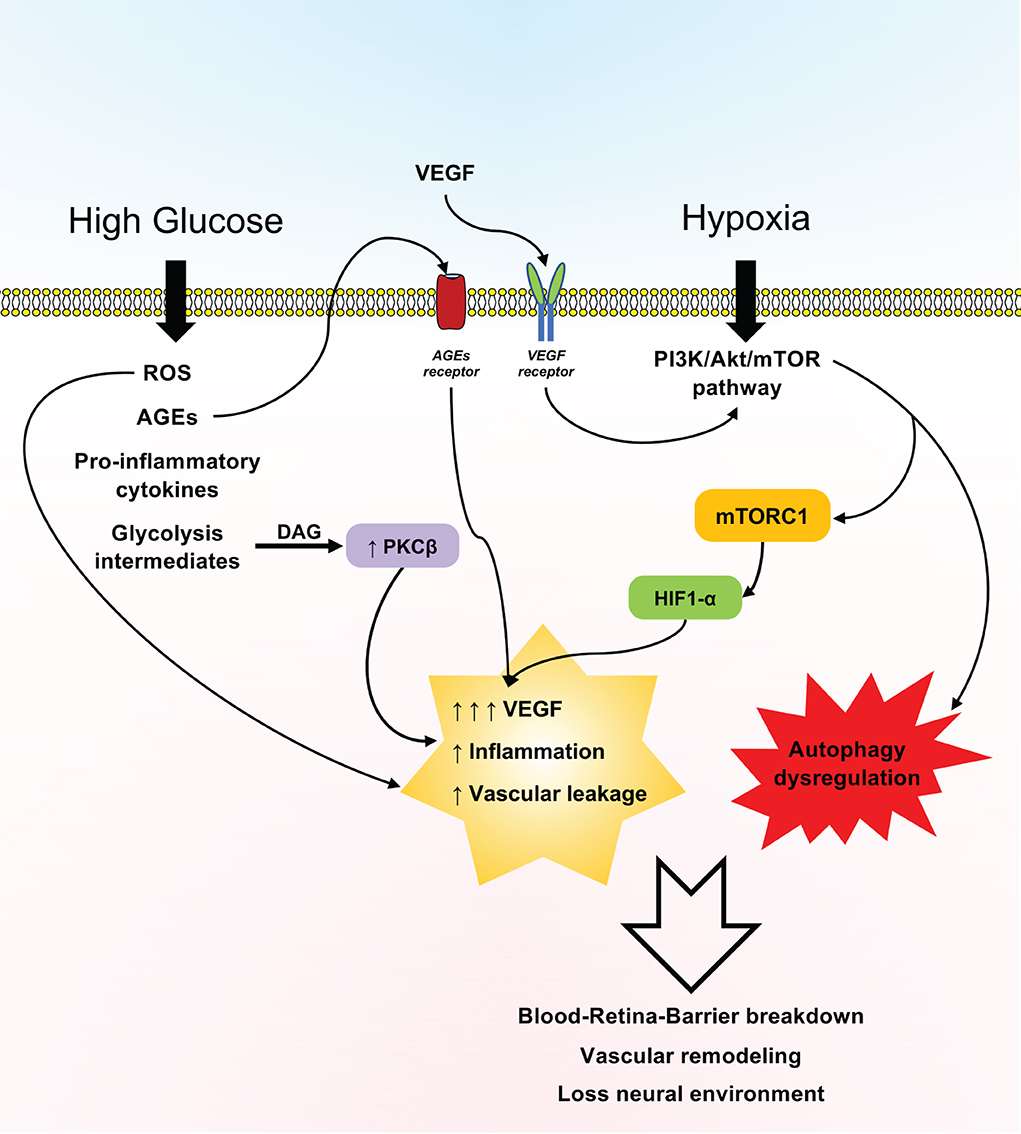
Figure 3. Schematic representation of the effects of the mTOR pathway in diabetic retinopathy (DR). Hyperglycemia and hypoxia prompt a plethora of effects associated with the expression of the PI3K/Akt/mTOR pathway, reactive oxygen species (ROS), advanced glycation end products (AGEs), inflammatory cytokines, and glycolysis intermediates, which in turn sustain vascular leakage and overall inflammation of the retina, with the loss of the blood–retina barrier and the neural microenvironment being mainly driven by increased vascular endothelial growth factor (VEGF) production and dysregulated autophagy.
Author contributions
GZ and FC: conceptualization. FC, EZ, and MP: writing—original draft preparation. FC, ER, and MP: writing—review and editing. FC: figures preparation. FC, ER, MP, MM, MB, and GZ: revision process and final editing. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was supported by local funds from the University of Ferrara grant numbers: 2020-FAR.L-CF_003 and 2021-FAR.L-CF_002.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Daryabor G, Atashzar MR, Kabelitz D, Meri S, Kalantar K. The effects of type 2 diabetes mellitus on organ metabolism and the immune system. Front Immunol. (2020) 11:1582. doi: 10.3389/fimmu.2020.01582
2. ValdezGuerrero AS, Quintana-Perez JC, Arellano-Mendoza MG, Castaneda-Ibarra FJ, Tamay-Cach F, Aleman-Gonzalez-Duhart D. Diabetic retinopathy: important biochemical alterations and the main treatment strategies. Can J Diabetes. (2021) 45:504–11. doi: 10.1016/j.jcjd.2020.10.009
3. Fernandes SA, Demetriades C. The multifaceted role of nutrient sensing and Mtorc1 signaling in physiology and aging. Front Aging. (2021) 2:707372. doi: 10.3389/fragi.2021.707372
4. Madrakhimov SB, Yang JY, Kim JH, Han JW, Park TK. Mtor-dependent dysregulation of autophagy contributes to the retinal ganglion cell loss in streptozotocin-induced diabetic retinopathy. Cell Commun Signal. (2021) 19:29. doi: 10.1186/s12964-020-00698-4
5. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
6. Purnyn H. The mammalian retina: structure and blood supply. Neurophysiology. (2013) 45:266–76. doi: 10.1007/s11062-013-9365-6
7. Reichenbach A, Bringmann A. Glia of the human retina. Glia. (2020) 68:768–96. doi: 10.1002/glia.23727
8. Chen Y, Xia Q, Zeng Y, Zhang Y, Zhang M. Regulations of retinal inflammation: focusing on Muller glia. Front Cell Dev Biol. (2022) 10:898652. doi: 10.3389/fcell.2022.898652
9. Kinuthia UM, Wolf A, Langmann T. Microglia and inflammatory responses in diabetic retinopathy. Front Immunol. (2020) 11:564077. doi: 10.3389/fimmu.2020.564077
10. O'Leary F, Campbell M. The blood-retina barrier in health and disease. FEBS J. (2021) 2021:1–14. doi: 10.1111/febs.16330
11. Pournaras CJ, Rungger-Brandle E, Riva CE, Hardarson SH, Stefansson E. Regulation of retinal blood flow in health and disease. Prog Retin Eye Res. (2008) 27:284–330. doi: 10.1016/j.preteyeres.2008.02.002
12. Country MW. Retinal metabolism: a comparative look at energetics in the retina. Brain Res. (2017) 1672:50–7. doi: 10.1016/j.brainres.2017.07.025
13. Feenstra DJ, Drawnel FM, Jayagopal A. Imaging of Hypoxia in Retinal Vascular Disease. London: IntechOpen (2018). doi: 10.5772/intechopen.72252
14. Stefansson E, Olafsdottir OB, Eliasdottir TS, Vehmeijer W, Einarsdottir AB, Bek T, et al. Retinal oximetry: metabolic imaging for diseases of the retina and brain. Prog Retin Eye Res. (2019) 70:1–22. doi: 10.1016/j.preteyeres.2019.04.001
15. Diaz-Coranguez M, Ramos C, Antonetti DA. The inner blood-retinal barrier: cellular basis and development. Vision Res. (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
16. Fresta CG, Fidilio A, Caruso G, Caraci F, Giblin FJ, Leggio GM, et al. A new human blood-retinal barrier model based on endothelial cells, pericytes, and astrocytes. Int J Mol Sci. (2020) 21:51636. doi: 10.3390/ijms21051636
17. Halaweish I, Alam HB. Changing demographics of the American population. Surg Clin North Am. (2015) 95:1–10. doi: 10.1016/j.suc.2014.09.002
18. Li J, Cao Y, Liu W, Wang Q, Qian Y, Lu P. Correlations among diabetic microvascular complications: a systematic review and meta-analysis. Sci Rep. (2019) 9:3137. doi: 10.1038/s41598-019-40049-z
19. Zhang AMY, Wellberg EA, Kopp JL, Johnson JD. Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab J. (2021) 45:285–311. doi: 10.4093/dmj.2020.0250
20. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
21. Wei J, Tian J, Tang C, Fang X, Miao R, Wu H, et al. The influence of different types of diabetes on vascular complications. J Diabetes Res. (2022) 2022:3448618. doi: 10.1155/2022/3448618
22. Cohen SR, Gardner TW. Diabetic retinopathy and diabetic macular edema. Dev Ophthalmol. (2016) 55:137–46. doi: 10.1159/000438970
23. Kosiborod M, Gomes MB, Nicolucci A, Pocock S, Rathmann W, Shestakova MV, et al. Vascular complications in patients with type 2 diabetes: prevalence and associated factors in 38 countries (the discover study program). Cardiovasc Diabetol. (2018) 17:150. doi: 10.1186/s12933-018-0787-8
24. Jawa A, Kcomt J, Fonseca VA. Diabetic nephropathy and retinopathy. Med Clin North Am. (2004) 88:1001–36. doi: 10.1016/j.mcna.2004.04.012
25. Huang D, Refaat M, Mohammedi K, Jayyousi A, Al Suwaidi J, Abi Khalil C. Macrovascular complications in patients with diabetes and prediabetes. Biomed Res Int. (2017) 2017:7839101. doi: 10.1155/2017/7839101
26. Al-Maskari F, El-Sadig M. Prevalence of diabetic retinopathy in the United Arab Emirates: a cross-sectional survey. BMC Ophthalmol. (2007) 7:11. doi: 10.1186/1471-2415-7-11
27. Hendrick AM, Gibson MV, Kulshreshtha A. Diabetic retinopathy. Prim Care. (2015) 42:451–64. doi: 10.1016/j.pop.2015.05.005
28. Homme RP, Singh M, Majumder A, George AK, Nair K, Sandhu HS, et al. Remodeling of retinal architecture in diabetic retinopathy: disruption of ocular physiology and visual functions by inflammatory gene products and pyroptosis. Front Physiol. (2018) 9:1268. doi: 10.3389/fphys.2018.01268
29. Wiacek MP, Modrzejewska M, Zaborski D. Age-related changes in retrobulbar circulation: a literature review. Int Ophthalmol. (2020) 40:493–501. doi: 10.1007/s10792-019-01176-8
30. Hanaguri J, Yokota H, Watanabe M, Yamagami S, Kushiyama A, Kuo L, et al. Retinal blood flow dysregulation precedes neural retinal dysfunction in type 2 diabetic mice. Sci Rep. (2021) 11:18401. doi: 10.1038/s41598-021-97651-3
31. Shoshani Y, Harris A, Shoja MM, Arieli Y, Ehrlich R, Primus S, et al. Impaired ocular blood flow regulation in patients with open-angle glaucoma and diabetes. Clin Exp Ophthalmol. (2012) 40:697–705. doi: 10.1111/j.1442-9071.2012.02778.x
32. Singer MA, Kermany DS, Waters J, Jansen ME, Tyler L. Diabetic macular edema: it is more than just Vegf. F1000Res. (2016) 5:1. doi: 10.12688/f1000research.8265.1
33. Funatsu H, Noma H, Mimura T, Eguchi S, Hori S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology. (2009) 116:73–9. doi: 10.1016/j.ophtha.2008.09.037
34. Boss JD, Singh PK, Pandya HK, Tosi J, Kim C, Tewari A, et al. Assessment of neurotrophins and inflammatory mediators in vitreous of patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. (2017) 58:5594–603. doi: 10.1167/iovs.17-21973
35. Figurska M, Robaszkiewicz J, Wierzbowska J. Optical coherence tomography in imaging of macular diseases. Klin Oczna. (2010) 112:138–46.
36. Singh R, Ramasamy K, Abraham C, Gupta V, Gupta A. Diabetic retinopathy: an update. Indian J Ophthalmol. (2008) 56:178–88. doi: 10.4103/0301-4738.40355
37. Arredondo Zamarripa D, Díaz-Lezama N, Meléndez García R, Chávez Balderas J, Adán N, Ledesma-Colunga MG, et al. Vasoinhibins regulate the inner and outer blood-retinal barrier and limit retinal oxidative stress. Front Cell Neurosci. (2014) 8:333. doi: 10.3389/fncel.2014.00333
38. Mei X, Zhang T, Ouyang H, Lu B, Wang Z, Ji L. Scutellarin alleviates blood-retina-barrier oxidative stress injury initiated by activated microglia cells during the development of diabetic retinopathy. Biochem Pharmacol. (2019) 159:82–95. doi: 10.1016/j.bcp.2018.11.011
39. Ghanian Z, Mehrvar S, Jamali N, Sheibani N, Ranji M. Time-lapse microscopy of oxidative stress demonstrates metabolic sensitivity of retinal pericytes under high glucose condition. J Biophotonics. (2018) 11:e201700289. doi: 10.1002/jbio.201700289
40. Bandello F, Lattanzio R, Zucchiatti I, Del Turco C. Pathophysiology and treatment of diabetic retinopathy. Acta Diabetol. (2013) 50:1–20. doi: 10.1007/s00592-012-0449-3
41. Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: pathophysiology, screening, and novel therapies. Diabetes Care. (2003) 26:2653–64. doi: 10.2337/diacare.26.9.2653
42. Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. (2007) 55:498–510. doi: 10.1016/j.phrs.2007.04.016
43. Aiello LP. The potential role of Pkc beta in diabetic retinopathy and macular edema. Surv Ophthalmol. (2002) 47(Suppl.2):S263–9. doi: 10.1016/S0039-6257(02)00391-0
44. Xu J, Chen LJ, Yu J, Wang HJ, Zhang F, Liu Q, et al. Involvement of advanced glycation end products in the pathogenesis of diabetic retinopathy. Cell Physiol Biochem. (2018) 48:705–17. doi: 10.1159/000491897
45. Semeraro F, Cancarini A, dell'Omo R, Rezzola S, Romano MR, Costagliola C. Diabetic retinopathy: vascular and inflammatory disease. J Diabetes Res. (2015) 2015:582060. doi: 10.1155/2015/582060
46. Voltan R, Zauli G, Rizzo P, Fucili A, Pannella M, Marci R, et al. In vitro endothelial cell proliferation assay reveals distinct levels of proangiogenic cytokines characterizing sera of healthy subjects and of patients with heart failure. Mediators Inflamm. (2014) 2014:257081. doi: 10.1155/2014/257081
47. Fini ME, Schwartz SG, Gao X, Jeong S, Patel N, Itakura T, et al. Steroid-induced ocular hypertension/glaucoma: focus on pharmacogenomics and implications for precision medicine. Prog Retin Eye Res. (2017) 56:58–83. doi: 10.1016/j.preteyeres.2016.09.003
48. Japanese Society of Ophthalmic Diabetology SotSoDRT, Sato Y, Kojimahara N, Kitano S, Kato S, Ando N, et al. Multicenter randomized clinical trial of retinal photocoagulation for preproliferative diabetic retinopathy. Jpn J Ophthalmol. (2012) 56:52–9. doi: 10.1007/s10384-011-0095-2
49. Fong DS, Girach A, Boney A. Visual side effects of successful scatter laser photocoagulation surgery for proliferative diabetic retinopathy: a literature review. Retina. (2007) 27:816–24. doi: 10.1097/IAE.0b013e318042d32c
50. Wallsh JO, Gallemore RP. Anti-Vegf-resistant retinal diseases: a review of the latest treatment options. Cells. (2021) 10:51049. doi: 10.3390/cells10051049
51. Plyukhova AA, Budzinskaya MV, Starostin KM, Rejdak R, Bucolo C, Reibaldi M, et al. Comparative safety of bevacizumab, ranibizumab, and aflibercept for treatment of neovascular age-related macular degeneration (AMD): a systematic review and network meta-analysis of direct comparative studies. J Clin Med. (2020) 9:51522. doi: 10.3390/jcm9051522
52. Adamis AP, Brittain CJ, Dandekar A, Hopkins JJ. Building on the success of anti-vascular endothelial growth factor therapy: a vision for the next decade. Eye. (2020) 34:1966–72. doi: 10.1038/s41433-020-0895-z
53. Sachdeva MM, Moshiri A, Leder HA, Scott AW. Endophthalmitis following intravitreal injection of anti-Vegf agents: long-term outcomes and the identification of unusual micro-organisms. J Ophthalmic Inflamm Infect. (2016) 6:2. doi: 10.1186/s12348-015-0069-5
54. Stewart MW, Rosenfeld PJ, Penha FM, Wang F, Yehoshua Z, Bueno-Lopez E, et al. Pharmacokinetic rationale for dosing every 2 weeks versus 4 weeks with intravitreal ranibizumab, bevacizumab, and aflibercept (vascular endothelial growth factor trap-eye). Retina. (2012) 32:434–57. doi: 10.1097/IAE.0b013e31822c290f
55. McAnany JJ, Park JC, Liu K, Liu M, Chen YF, Chau FY, et al. Contrast sensitivity is associated with outer-retina thickness in early-stage diabetic retinopathy. Acta Ophthalmol. (2020) 98:e224–e31. doi: 10.1111/aos.14241
56. van de Kreeke JA, Darma S, Chan Pin Yin J, Tan HS, Abramoff MD, Twisk JWR, et al. The spatial relation of diabetic retinal neurodegeneration with diabetic retinopathy. PLoS ONE. (2020) 15:e0231552. doi: 10.1371/journal.pone.0231552
57. van Dijk HW, Verbraak FD, Stehouwer M, Kok PH, Garvin MK, Sonka M, et al. Association of visual function and ganglion cell layer thickness in patients with diabetes mellitus type 1 and no or minimal diabetic retinopathy. Vision Res. (2011) 51:224–8. doi: 10.1016/j.visres.2010.08.024
58. Zeng XX, Ng YK, Ling EA. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. (2000) 17:463–71. doi: 10.1017/S0952523800173122
59. Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. (2004) 45:3330–6. doi: 10.1167/iovs.04-0247
60. Lee VK, Hosking BM, Holeniewska J, Kubala EC, Lundh von Leithner P, Gardner PJ, et al. Btbr Ob/Ob mouse model of type 2 diabetes exhibits early loss of retinal function and retinal inflammation followed by late vascular changes. Diabetologia. (2018) 61:2422–32. doi: 10.1007/s00125-018-4696-x
61. Ivanova E, Alam NM, Prusky GT, Sagdullaev BT. Blood-retina barrier failure and vision loss in neuron-specific degeneration. JCI Insight. (2019) 5:126747. doi: 10.1172/jci.insight.126747
62. Ma W, Wong WT. Aging changes in retinal microglia and their relevance to age-related retinal disease. Adv Exp Med Biol. (2016) 854:73–8. doi: 10.1007/978-3-319-17121-0_11
63. Ferrari D, la Sala A, Milani D, Celeghini C, Casciano F. Purinergic signaling in controlling macrophage and T cell functions during atherosclerosis development. Front Immunol. (2020) 11:617804. doi: 10.3389/fimmu.2020.617804
64. Platania CBM, Drago F, Bucolo C. The P2x7 receptor as a new pharmacological target for retinal diseases. Biochem Pharmacol. (2022) 198:114942. doi: 10.1016/j.bcp.2022.114942
65. Portillo JC, Lopez Corcino Y, Miao Y, Tang J, Sheibani N, Kern TS, et al. Cd40 in retinal Muller cells induces P2x7-dependent cytokine expression in macrophages/microglia in diabetic mice and development of early experimental diabetic retinopathy. Diabetes. (2017) 66:483–93. doi: 10.2337/db16-0051
66. Zhou H, Huang S. The complexes of mammalian target of rapamycin. Curr Protein Pept Sci. (2010) 11:409–24. doi: 10.2174/138920310791824093
67. Saxton RA, Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell. (2017) 169:361–71. doi: 10.1016/j.cell.2017.03.035
68. El Hiani Y, Egom EE, Dong XP. Mtor signalling: jack-of-all-trades (1). Biochem Cell Biol. (2019) 97:58–67. doi: 10.1139/bcb-2018-0004
69. Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal Mtorc2/Phlpp1/Akt regulate chaperone-mediated autophagy. Mol Cell. (2015) 59:270–84. doi: 10.1016/j.molcel.2015.05.030
70. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of Mtor, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. (2004) 14:1296–302. doi: 10.1016/j.cub.2004.06.054
71. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, tuberin and hamartin, control Mtor signaling by acting as a Gtpase-activating protein complex toward Rheb. Curr Biol. (2003) 13:1259–68. doi: 10.1016/S0960-9822(03)00506-2
72. Wang L, Harris TE, Roth RA, Lawrence JC Jr. Pras40 regulates Mtorc1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. (2007) 282:20036–44. doi: 10.1074/jbc.M702376200
73. Yao Y, Jones E, Inoki K. Lysosomal regulation of Mtorc1 by amino acids in mammalian cells. Biomolecules. (2017) 7:30051. doi: 10.3390/biom7030051
74. Lin SC, Hardie DG. Ampk: sensing glucose as well as cellular energy status. Cell Metab. (2018) 27:299–313. doi: 10.1016/j.cmet.2017.10.009
75. Ebner M, Sinkovics B, Szczygiel M, Ribeiro DW, Yudushkin I. Localization of Mtorc2 activity inside cells. J Cell Biol. (2017) 216:343–53. doi: 10.1083/jcb.201610060
76. Baffi TR, Lorden G, Wozniak JM, Feichtner A, Yeung W, Kornev AP, et al. Mtorc2 controls the activity of Pkc and Akt by phosphorylating a conserved tor interaction Motif. Sci Signal. (2021) 14:abe4509. doi: 10.1126/scisignal.abe4509
77. Guo F, Zhang S, Grogg M, Cancelas JA, Varney ME, Starczynowski DT, et al. Mouse gene targeting reveals an essential role of Mtor in hematopoietic stem cell engraftment and hematopoiesis. Haematologica. (2013) 98:1353–8. doi: 10.3324/haematol.2012.080424
78. Fan C, Zhao C, Zhang F, Kesarwani M, Tu Z, Cai X, et al. Adaptive responses to Mtor gene targeting in hematopoietic stem cells reveal a proliferative mechanism evasive to Mtor inhibition. Proc Natl Acad Sci USA. (2021) 118:e2020102118. doi: 10.1073/pnas.2020102118
79. Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. (2007) 110:3056–63. doi: 10.1182/blood-2007-05-087759
80. Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of Mtor. Neuroscience. (2017) 341:112–53. doi: 10.1016/j.neuroscience.2016.11.017
81. Maiese K. Targeting the core of neurodegeneration: Foxo, Mtor, and Sirt1. Neural Regen Res. (2021) 16:448–55. doi: 10.4103/1673-5374.291382
82. Avet-Rochex A, Carvajal N, Christoforou CP, Yeung K, Maierbrugger KT, Hobbs C, et al. Unkempt is negatively regulated by Mtor and uncouples neuronal differentiation from growth control. PLoS Genet. (2014) 10:e1004624. doi: 10.1371/journal.pgen.1004624
83. Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, et al. Promoting axon regeneration in the adult Cns by modulation of the Pten/Mtor pathway. Science. (2008) 322:963–6. doi: 10.1126/science.1161566
84. Pita-Thomas W, Mahar M, Joshi A, Gan D, Cavalli V. Hdac5 promotes optic nerve regeneration by activating the Mtor pathway. Exp Neurol. (2019) 317:271–83. doi: 10.1016/j.expneurol.2019.03.011
85. Wakhloo D, Scharkowski F, Curto Y, Javed Butt U, Bansal V, Steixner-Kumar AA, et al. Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat Commun. (2020) 11:1313. doi: 10.1038/s41467-020-15041-1
86. Chong ZZ, Shang YC, Wang S, Maiese K. Pras40 is an integral regulatory component of erythropoietin Mtor signaling and cytoprotection. PLoS ONE. (2012) 7:e45456. doi: 10.1371/journal.pone.0045456
87. Shang YC, Chong ZZ, Wang S, Maiese K. Erythropoietin and Wnt1 govern pathways of Mtor, Apaf-1, and Xiap in inflammatory microglia. Curr Neurovasc Res. (2011) 8:270–85. doi: 10.2174/156720211798120990
88. Shang YC, Chong ZZ, Wang S, Maiese K. Tuberous Sclerosis Protein 2 (Tsc2) modulates Ccn4 cytoprotection during apoptotic amyloid toxicity in microglia. Curr Neurovasc Res. (2013) 10:29–38. doi: 10.2174/156720213804806007
89. Shang YC, Chong ZZ, Wang S, Maiese K. Wnt1 inducible signaling pathway protein 1 (Wisp1) targets Pras40 to govern beta-amyloid apoptotic injury of microglia. Curr Neurovasc Res. (2012) 9:239–49. doi: 10.2174/156720212803530618
90. Jang W, Kim HJ, Li H, Jo KD, Lee MK, Yang HO. The neuroprotective effect of erythropoietin on rotenone-induced neurotoxicity in Sh-Sy5y cells through the induction of autophagy. Mol Neurobiol. (2016) 53:3812–21. doi: 10.1007/s12035-015-9316-x
91. Sanghera KP, Mathalone N, Baigi R, Panov E, Wang D, Zhao X, et al. The Pi3k/Akt/Mtor pathway mediates retinal progenitor cell survival under hypoxic and superoxide stress. Mol Cell Neurosci. (2011) 47:145–53. doi: 10.1016/j.mcn.2011.03.010
92. Lai YF, Lin TY, Ho PK, Chen YH, Huang YC, Lu DW. Erythropoietin in optic neuropathies: current future strategies for optic nerve protection and repair. Int J Mol Sci. (2022) 23:7143. doi: 10.3390/ijms23137143
93. Curtis TM, Gardiner TA, Stitt AW. Microvascular lesions of diabetic retinopathy: clues towards understanding pathogenesis? Eye. (2009) 23:1496–508. doi: 10.1038/eye.2009.108
94. Wanek J, Teng PY, Blair NP, Shahidi M. Inner retinal oxygen delivery and metabolism in streptozotocin diabetic rats. Invest Ophthalmol Vis Sci. (2014) 55:1588–93. doi: 10.1167/iovs.13-13537
95. Dodd KM, Yang J, Shen MH, Sampson JR, Tee AR. Mtorc1 drives Hif-1alpha and Vegf-a signalling via multiple mechanisms involving 4e-Bp1, S6k1 and Stat3. Oncogene. (2015) 34:2239–50. doi: 10.1038/onc.2014.164
96. Kida T, Oku H, Osuka S, Horie T, Ikeda T. Hyperglycemia-induced Vegf and Ros production in retinal cells is inhibited by the Mtor inhibitor, rapamycin. Sci Rep. (2021) 11:1885. doi: 10.1038/s41598-021-81482-3
97. Liu Y, Zheng Y, Zhou Y, Liu Y, Xie M, Meng W, et al. The expression and significance of Mtorc1 in diabetic retinopathy. BMC Ophthalmol. (2020) 20:297. doi: 10.1186/s12886-020-01553-3
98. Karar J, Maity A. Pi3k/Akt/Mtor pathway in angiogenesis. Front Mol Neurosci. (2011) 4:51. doi: 10.3389/fnmol.2011.00051
99. Simioni C, Ultimo S, Martelli AM, Zauli G, Milani D, McCubrey JA, et al. Synergistic effects of selective inhibitors targeting the Pi3k/Akt/Mtor pathway or Nup214-Abl1 fusion protein in human acute lymphoblastic leukemia. Oncotarget. (2016) 7:79842–53. doi: 10.18632/oncotarget.13035
100. Losiewicz MK, Elghazi L, Fingar DC, Rajala RVS, Lentz SI, Fort PE, et al. Mtorc1 and Mtorc2 expression in inner retinal neurons and glial cells. Exp Eye Res. (2020) 197:108131. doi: 10.1016/j.exer.2020.108131
101. Ma S, Venkatesh A, Langellotto F, Le YZ, Hall MN, Ruegg MA, et al. Loss of Mtor signaling affects cone function, cone structure and expression of cone specific proteins without affecting cone survival. Exp Eye Res. (2015) 135:1–13. doi: 10.1016/j.exer.2015.04.006
102. Silverman SM, Wong WT. Microglia in the retina: roles in development, maturity, and disease. Annu Rev Vis Sci. (2018) 4:45–77. doi: 10.1146/annurev-vision-091517-034425
103. Guo Y, Cang X, Zhu L, Zhu M, Li A, Wang Z, et al. Ppp1ca/Yap/Gs/Gln/Mtorc1 pathway activates retinal Muller cells during diabetic retinopathy. Exp Eye Res. (2021) 210:108703. doi: 10.1016/j.exer.2021.108703
104. Valverde AM, Miranda S, Garcia-Ramirez M, Gonzalez-Rodriguez A, Hernandez C, Simo R. Proapoptotic and survival signaling in the neuroretina at early stages of diabetic retinopathy. Mol Vis. (2013) 19:47–53.
105. Szabo K, Enzsoly A, Dekany B, Szabo A, Hajdu RI, Radovits T, et al. Histological evaluation of diabetic neurodegeneration in the retina of zucker diabetic fatty (ZDF) rats. Sci Rep. (2017) 7:8891. doi: 10.1038/s41598-017-09068-6
106. Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr Pharm Des. (2013) 19:5695–703. doi: 10.2174/1381612811319320005
107. Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H. Suppression of retinal neovascularization by the Nf-Kappab inhibitor pyrrolidine dithiocarbamate in mice. Invest Ophthalmol Vis Sci. (1999) 40:1624–9.
108. Szkudelska K, Okulicz M, Hertig I, Szkudelski T. Resveratrol ameliorates inflammatory and oxidative stress in type 2 diabetic Goto-Kakizaki rats. Biomed Pharmacother. (2020) 125:110026. doi: 10.1016/j.biopha.2020.110026
109. Soufi FG, Vardyani M, Sheervalilou R, Mohammadi M, Somi MH. Long-term treatment with resveratrol attenuates oxidative stress pro-inflammatory mediators and apoptosis in streptozotocin-nicotinamide-induced diabetic rats. Gen Physiol Biophys. (2012) 31:431–8. doi: 10.4149/gpb_2012_039
110. Bucolo C, Drago F, Maisto R, Romano GL, D'Agata V, Maugeri G, et al. Curcumin prevents high glucose damage in retinal pigment epithelial cells through Erk1/2-mediated activation of the Nrf2/Ho-1 pathway. J Cell Physiol. (2019) 234:17295–304. doi: 10.1002/jcp.28347
111. Bucolo C, Marrazzo G, Platania CB, Drago F, Leggio GM, Salomone S. Fortified extract of red berry, ginkgo biloba, and white willow bark in experimental early diabetic retinopathy. J Diabetes Res. (2013) 2013:432695. doi: 10.1155/2013/432695
112. Zhong LM, Zong Y, Sun L, Guo JZ, Zhang W, He Y, et al. Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS ONE. (2012) 7:e32195. doi: 10.1371/journal.pone.0032195
113. Haranahalli Shivarudrappa A, Gopal SS, Ponesakki G. An in vitro protocol to study the effect of hyperglycemia on intracellular redox signaling in human retinal pigment epithelial (arpe-19) cells. Mol Biol Rep. (2019) 46:1263–74. doi: 10.1007/s11033-019-04597-x
114. Yang X, Wu S, Feng Z, Yi G, Zheng Y, Xia Z. Combination therapy with semaglutide and rosiglitazone as a synergistic treatment for diabetic retinopathy in rodent animals. Life Sci. (2021) 269:119013. doi: 10.1016/j.lfs.2020.119013
115. Zhou M, Geathers JS, Grillo SL, Weber SR, Wang W, Zhao Y, et al. Role of epithelial-mesenchymal transition in retinal pigment epithelium dysfunction. Front Cell Dev Biol. (2020) 8:501. doi: 10.3389/fcell.2020.00501
116. Kobayashi Y, Tokuda K, Yamashiro C, Higashijima F, Yoshimoto T, Ota M, et al. Inhibition of epithelial-mesenchymal transition in retinal pigment epithelial cells by a retinoic acid receptor-alpha agonist. Sci Rep. (2021) 11:11842. doi: 10.1038/s41598-021-90618-4
117. Bonfiglio V, Platania CBM, Lazzara F, Conti F, Pizzo C, Reibaldi M, et al. Tgf-beta serum levels in diabetic retinopathy patients and the role of Anti-Vegf therapy. Int J Mol Sci. (2020) 21:9558. doi: 10.3390/ijms21249558
118. Kim SJ, Kim YS, Kim JH, Jang HY, Ly DD, Das R, et al. Activation of Erk1/2-Mtorc1-Nox4 mediates Tgf-Beta1-induced epithelial-mesenchymal transition and fibrosis in retinal pigment epithelial cells. Biochem Biophys Res Commun. (2020) 529:747–52. doi: 10.1016/j.bbrc.2020.06.034
119. Kim YH, Kim YS, Roh GS, Choi WS, Cho GJ. Resveratrol blocks diabetes-induced early vascular lesions and vascular endothelial growth factor induction in mouse retinas. Acta Ophthalmol. (2012) 90:e31–7. doi: 10.1111/j.1755-3768.2011.02243.x
120. Al-Nasser MN, Mellor IR, Carter WG. Is L-glutamate toxic to neurons and thereby contributes to neuronal loss and neurodegeneration? a systematic review. Brain Sci. (2022) 12:50577. doi: 10.3390/brainsci12050577
121. Ma M, Zhao S, Zhang J, Sun T, Fan Y, Zheng Z. High glucose-induced Trpc6 channel activation decreases glutamate uptake in rat retinal Muller cells. Front Pharmacol. (2019) 10:1668. doi: 10.3389/fphar.2019.01668
122. Ji YF, Zhou L, Xie YJ, Xu SM, Zhu J, Teng P, et al. Upregulation of glutamate transporter Glt-1 by Mtor-Akt-Nf-Small Ka, cyrillicb cascade in astrocytic oxygen-glucose deprivation. Glia. (2013) 61:1959–75. doi: 10.1002/glia.22566
123. Singh G, Storey KB. Microrna cues from nature: a roadmap to decipher and combat challenges in human health and disease? Cells. (2021) 10:123374. doi: 10.3390/cells10123374
124. Zhang Y, Huang B, Wang HY, Chang A, Zheng XFS. Emerging role of micrornas in Mtor signaling. Cell Mol Life Sci. (2017) 74:2613–25. doi: 10.1007/s00018-017-2485-1
125. Tabak S, Schreiber-Avissar S, Beit-Yannai E. Crosstalk between microrna and oxidative stress in primary open-angle glaucoma. Int J Mol Sci. (2021) 22:52421. doi: 10.3390/ijms22052421
126. Li W, Xiao H. Dihydromyricetin alleviates high glucose-induced oxidative stress and apoptosis in human retinal pigment epithelial cells by downregulating Mir-34a expression. Diabetes Metab Syndr Obes. (2021) 14:387–97. doi: 10.2147/DMSO.S290633
127. Liao H, Xiao Y, Hu Y, Xiao Y, Yin Z, Liu L, et al. Methylation-induced silencing of Mir-34a enhances chemoresistance by directly upregulating Atg4b-induced autophagy through Ampk/Mtor pathway in prostate cancer. Oncol Rep. (2016) 35:64–72. doi: 10.3892/or.2015.4331
128. Xi X, Yang Y, Ma J, Chen Q, Zeng Y, Li J, et al. Mir-130a alleviated high-glucose induced retinal pigment epithelium (RPE) death by modulating Tnf-Alpha/Sod1/Ros cascade mediated pyroptosis. Biomed Pharmacother. (2020) 125:109924. doi: 10.1016/j.biopha.2020.109924
129. Wang Y, Zhang X, Tang W, Lin Z, Xu L, Dong R, et al. Mir-130a upregulates Mtor pathway by targeting Tsc1 and is transactivated by Nf-Kappab in high-grade serous ovarian carcinoma. Cell Death Differ. (2017) 24:2089–100. doi: 10.1038/cdd.2017.129
130. Tsang CK, Chen M, Cheng X, Qi Y, Chen Y, Das I, et al. Sod1 phosphorylation by Mtorc1 couples nutrient sensing and redox regulation. Mol Cell. (2018) 70:502–15 e8. doi: 10.1016/j.molcel.2018.03.029
131. Seong M, Lee J, Kang H. Hypoxia-induced regulation of Mtor signaling by Mir-7 targeting Redd1. J Cell Biochem. (2019) 120:4523–32. doi: 10.1002/jcb.27740
132. Yang Z, Hu H, Zou Y, Luo W, Xie L, You Z. Mir-7 reduces high glucose induced-damage via Hoxb3 and Pi3k/Akt/Mtor signaling pathways in retinal pigment epithelial cells. Curr Mol Med. (2020) 20:372–8. doi: 10.2174/1566524019666191023151137
133. Zhao BW, Dai HY, Hao LN, Liu YW. Mir-29 regulates retinopathy in diabetic mice via the Ampk signaling pathway. Eur Rev Med Pharmacol Sci. (2019) 23:3569–74. doi: 10.26355/eurrev_201905_17778
134. Bereimipour A, Najafi H, Mirsane ES, Moradi S, Satarian L. Roles of Mir-204 in retinal development and maintenance. Exp Cell Res. (2021) 406:112737. doi: 10.1016/j.yexcr.2021.112737
135. Perrotta C, Cattaneo MG, Molteni R, De Palma C. Autophagy in the regulation of tissue differentiation and homeostasis. Front Cell Dev Biol. (2020) 8:602901. doi: 10.3389/fcell.2020.602901
136. Gubas A, Dikic I. A guide to the regulation of selective autophagy receptors. FEBS J. (2022) 289:75–89. doi: 10.1111/febs.15824
137. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
138. Rodolfo C, Campello S, Cecconi F. Mitophagy in neurodegenerative diseases. Neurochem Int. (2018) 117:156–66. doi: 10.1016/j.neuint.2017.08.004
139. Giorgi C, Bouhamida E, Danese A, Previati M, Pinton P, Patergnani S. Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases. Biomedicines. (2021) 9:20149. doi: 10.3390/biomedicines9020149
140. Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, et al. Autophagy and Alzheimer's disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. (2018) 10:4. doi: 10.3389/fnagi.2018.00004
141. Zhu Z, Yan J, Jiang W, Yao XG, Chen J, Chen L, et al. Arctigenin effectively ameliorates memory impairment in Alzheimer's disease model mice targeting both beta-amyloid production and clearance. J Neurosci. (2013) 33:13138–49. doi: 10.1523/JNEUROSCI.4790-12.2013
142. Navarro-Romero A, Montpeyo M, Martinez-Vicente M. The emerging role of the lysosome in Parkinson's disease. Cells. (2020) 9:112399. doi: 10.3390/cells9112399
143. Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. (2020) 15:1388–96. doi: 10.4103/1673-5374.274331
144. Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy. (2009) 5:221–3. doi: 10.4161/auto.5.2.7363
145. Sekiguchi A, Kanno H, Ozawa H, Yamaya S, Itoi E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J Neurotrauma. (2012) 29:946–56. doi: 10.1089/neu.2011.1919
146. Patergnani S, Bonora M, Ingusci S, Previati M, Marchi S, Zucchini S, et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc Natl Acad Sci USA. (2021) 118:e2020078118. doi: 10.1073/pnas.2020078118
147. Walker CL, Walker MJ, Liu NK, Risberg EC, Gao X, Chen J, et al. Systemic bisperoxovanadium activates Akt/Mtor, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS ONE. (2012) 7:e30012. doi: 10.1371/journal.pone.0030012
148. Lynch SK, Abramoff MD. Diabetic retinopathy is a neurodegenerative disorder. Vision Res. (2017) 139:101–7. doi: 10.1016/j.visres.2017.03.003
149. Zhang Q, Li HS, Li R, Du JH, Jiao C. Autophagy dysregulation mediates the damage of high glucose to retinal pigment epithelium cells. Int J Ophthalmol. (2021) 14:805–11. doi: 10.18240/ijo.2021.06.04
150. Li R, Li H, Zhang Q. Procyanidin protects human retinal pigment epithelial cells from high glucose by inhibiting autophagy. Environ Toxicol. (2022) 37:201–11. doi: 10.1002/tox.23389
151. Fu Z, Gong Y, Lofqvist C, Hellstrom A, Smith LE. Review: adiponectin in retinopathy. Biochim Biophys Acta. (2016) 1862:1392–400. doi: 10.1016/j.bbadis.2016.05.002
152. Li R, Du J, Yao Y, Yao G, Wang X. Adiponectin inhibits high glucose-induced angiogenesis via inhibiting autophagy in Rf/6a cells. J Cell Physiol. (2019) 234:20566–76. doi: 10.1002/jcp.28659
153. Li R, Yao G, Zhou L, Zhang M, Yan J, Wang X, et al. Autophagy is required for the promoting effect of angiogenic factor with G patch domain and forkhead-associated domain 1 (Aggf1) in retinal angiogenesis. Microvasc Res. (2021) 138:104230. doi: 10.1016/j.mvr.2021.104230
154. Rajendran P, Alzahrani AM, Hanieh HN, Kumar SA, Ben Ammar R, Rengarajan T, et al. Autophagy and senescence: a new insight in selected human diseases. J Cell Physiol. (2019) 234:21485–92. doi: 10.1002/jcp.28895
155. Zhang W, Song J, Zhang Y, Ma Y, Yang J, He G, et al. Intermittent high glucose-induced oxidative stress modulates retinal pigmented epithelial cell autophagy and promotes cell survival via increased Hmgb1. BMC Ophthalmol. (2018) 18:192. doi: 10.1186/s12886-018-0864-5
156. Chen Q, Tang L, Xin G, Li S, Ma L, Xu Y, et al. Oxidative stress mediated by lipid metabolism contributes to high glucose-induced senescence in retinal pigment epithelium. Free Radic Biol Med. (2019) 130:48–58. doi: 10.1016/j.freeradbiomed.2018.10.419
157. Chae JB, Jang H, Son C, Park CW, Choi H, Jin S, et al. Targeting senescent retinal pigment epithelial cells facilitates retinal regeneration in mouse models of age-related macular degeneration. Geroscience. (2021) 43:2809–33. doi: 10.1007/s11357-021-00457-4
158. Hasty P, Sharp ZD, Curiel TJ, Campisi J. Mtorc1 and P53: clash of the gods? Cell Cycle. (2013) 12:20–5. doi: 10.4161/cc.22912
159. Kucheryavenko O, Nelson G, von Zglinicki T, Korolchuk VI, Carroll B. The Mtorc1-autophagy pathway is a target for senescent cell elimination. Biogerontology. (2019) 20:331–5. doi: 10.1007/s10522-019-09802-9
160. Rubsam A, Parikh S, Fort PE. Role of inflammation in diabetic retinopathy. Int J Mol Sci. (2018) 19:942. doi: 10.3390/ijms19040942
161. Lopes de Faria JM, Duarte DA, Montemurro C, Papadimitriou A, Consonni SR, Lopes de Faria JB. Defective autophagy in diabetic retinopathy. Invest Ophthalmol Vis Sci. (2016) 57:4356–66. doi: 10.1167/iovs.16-19197
Keywords: mammalian target of rapamycin (mTOR), diabetic retinopathy, hyperglycemia, reactive oxygen species (ROS), oxidative stress, inflammation, autophagy, retinae
Citation: Casciano F, Zauli E, Rimondi E, Mura M, Previati M, Busin M and Zauli G (2022) The role of the mTOR pathway in diabetic retinopathy. Front. Med. 9:973856. doi: 10.3389/fmed.2022.973856
Received: 20 June 2022; Accepted: 05 September 2022;
Published: 01 November 2022.
Edited by:
Mohamed Abu-Farha, Dasman Diabetes Institute, KuwaitReviewed by:
Ranjan Das, Oregon Health and Science University, United StatesClaudio Bucolo, University of Catania, Italy
Sandra Marmiroli, University of Modena and Reggio Emilia, Italy
Copyright © 2022 Casciano, Zauli, Rimondi, Mura, Previati, Busin and Zauli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Casciano, ZmFiaW8uY2FzY2lhbm9AdW5pZmUuaXQ=
†These authors have contributed equally to this work