
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 22 August 2022
Sec. Intensive Care Medicine and Anesthesiology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.971882
 Thomas Datzmann1,2*†
Thomas Datzmann1,2*† David Alexander Christian Messerer1,2,3†Franziska Münz1,2
David Alexander Christian Messerer1,2,3†Franziska Münz1,2 Andrea Hoffmann1Michael Gröger1René Mathieu4Simon Mayer4Holger Gässler5
Andrea Hoffmann1Michael Gröger1René Mathieu4Simon Mayer4Holger Gässler5 Fabian Zink1
Fabian Zink1 Oscar McCook1
Oscar McCook1 Tamara Merz1,2Angelika Scheuerle6
Tamara Merz1,2Angelika Scheuerle6 Eva-Maria Wolfschmitt1Timo Thebrath1Stefan Zuech1
Eva-Maria Wolfschmitt1Timo Thebrath1Stefan Zuech1 Enrico Calzia1Pierre Asfar7
Enrico Calzia1Pierre Asfar7 Peter Radermacher1Thomas Kapapa8
Peter Radermacher1Thomas Kapapa8Controversial evidence is available regarding suitable targets for the arterial O2 tension (PaO2) after traumatic brain injury and/or hemorrhagic shock (HS). We previously demonstrated that hyperoxia during resuscitation from hemorrhagic shock attenuated cardiac injury and renal dysfunction in swine with coronary artery disease. Therefore, this study investigated the impact of targeted hyperoxemia in a long-term, resuscitated model of combined acute subdural hematoma (ASDH)-induced brain injury and HS. The prospective randomized, controlled, resuscitated animal investigation consisted of 15 adult pigs. Combined ASDH plus HS was induced by injection of 0.1 ml/kg autologous blood into the subdural space followed by controlled passive removal of blood. Two hours later, resuscitation was initiated comprising re-transfusion of shed blood, fluids, continuous i.v. noradrenaline, and either hyperoxemia (target PaO2 200 – 250 mmHg) or normoxemia (target PaO2 80 – 120 mmHg) during the first 24 h of the total of 54 h of intensive care. Systemic hemodynamics, intracranial and cerebral perfusion pressures, parameters of brain microdialysis and blood biomarkers of brain injury did not significantly differ between the two groups. According to the experimental protocol, PaO2 was significantly higher in the hyperoxemia group at the end of the intervention period, i.e., at 24 h of resuscitation, which coincided with a higher brain tissue PO2. The latter persisted until the end of observation period. While neurological function as assessed using the veterinary Modified Glasgow Coma Score progressively deteriorated in the control group, it remained unaffected in the hyperoxemia animals, however, without significant intergroup difference. Survival times did not significantly differ in the hyperoxemia and control groups either. Despite being associated with higher brain tissue PO2 levels, which were sustained beyond the intervention period, targeted hyperoxemia exerted neither significantly beneficial nor deleterious effects after combined ASDH and HS in swine with pre-existing coronary artery disease. The unavailability of a power calculation and, thus, the limited number of animals included, are the limitations of the study.
Post-traumatic mortality depends on the presence of hemorrhagic shock (HS) and/or traumatic brain injury (TBI). Hemorrhagic shock accounts for 30% of trauma-related mortality (1), TBI is the most prevalent cause of death after polytrauma (2).
During HS, supplemental O2 is used because increasing the amount of physically dissolved O2 during blood loss-related impairment of O2 transport may faster repay a tissue O2 debt (3, 4). Despite its vasoconstrictor properties (3, 4), pure O2 ventilation improved tissue PO2 during HS (5). After near-lethal hemorrhagic shock, pure O2 ventilation also attenuated organ dysfunction (6). However, hyperox(em)ia enhances reactive oxygen species (ROS) formation, especially during I/R and/or hypoxia/re-oxygenation, e.g., resuscitation from trauma-and-hemorrhage (3, 4). After TBI, any hyperoxemia-related vasoconstriction may exert beneficial effects due to reduced intracranial pressure without compromised tissue oxygenation.
Current recommendations suggest that PaO2 > 300 mmHg (40 kPa) should be avoided (3, 4), but a putative “optimal level” after TBI and/or HS remains open: in trauma patients, retrospective analyzes showed that PaO2 > 150 and > 200 mmHg, respectively, within the first 24 h coincided with improved survival (7, 8). Moreover, PaO2≈150–250 mmHg during the first 24 h of intensive care unit (ICU) stay were associated with the lowest mortality and the best neurological outcome after severe TBI (9).
Therefore, this study was to investigate the impact of targeted hyperoxemia in a randomized, controlled, resuscitated, large animal model of combined acute subdural hematoma (ASDH) -induced TBI and HS over 56 h post-injury. The main outcome parameters were whether this approach (i) ameliorates brain tissue oxygenation, (ii) improves neurologic function, and (iii) is safe with respect to parameters of oxidative stress.
After ethical approval by the Animal Care Committee of the Universität Ulm and the Federal Authorities for Animal Research (Regierungspräsidium Tübingen, #1316) experiments were conducted in adherence to the European Union Directive 2010/63/EU on the protection of animals used for scientific purposes. 15 adult Familial hypercholesterolemia-Bretoncelles-Meishan (FBM (10) pigs (median[interquartile range] body weight 63[56; 71]kg; age 38[36; 41] months) of either sex (8 castrated males, 7 females) were used. This pig strain is a cross-bread of Rapacz farm pigs homozygous for the R84C low density lipoprotein receptor mutation with the smaller Meishan and Bretoncelles strains, and at the age of at least 9 – 12 months, it presents with atherosclerosis and coronary artery disease due to a cholesterol-enriched diet administered for at least 3 – 4 months (11). The animals in our study had received the atherogenic diet for at least 9 months, and we had previously confirmed typical coronary artery lesions in animals of similar age that had undergone this treatment (12, 13). FBM swine were studied because ASDH as a major contributor to TBI (14) assumes particular importance in the elderly and co-morbid patient (15). Moreover, in previous experiments investigating therapeutic interventions during hemorrhage-and-resuscitation we had shown that either hyperoxemia or infusion of Na2S2O3 had exerted organ-protective effects in swine with coronary artery disease (6, 16), while no benefit was obtained in cardiovascular healthy animals undergoing the same treatment protocol (17, 18). Due to one drop-out because of uncontrollable bleeding during the surgical instrumentation data refer to 14 animals.
Anesthesia and (neuro)surgical instrumentation have been described in detail previously (10). Briefly, during the 12 h preceding the experiments, animals received a nutritional solution (Fresubin®, Fresenius Kabi) and had free access to water. This approach is based on our previous experiments (6, 10, 16, 18): administration of a nutritional solution instead of a regular diet allows for feeding the animals and thus avoiding stress to hunger until the induction of anesthesia while at the same time the potential danger of regurgitation of solid food particles and their aspiration into the trachea is prevented during induction of anesthesia and prior to endotracheal intubation. Prior to induction of anesthesia, premedication consisted of intramuscular 5 mg/kg azaperone and 1–2 mg/kg midazolam. Anesthesia was induced with intravenous i.v. propofol (1–2 mg/kg) and ketamine (1 mg/kg). Pigs were intubated endotracheally and mechanically ventilated (ventilator settings: tidal volume 8 ml/kg, respiratory rate 8–12 breaths/minute adapted to an arterial PCO2 [PaCO2] = 35–40 mmHg, inspiratory/expiratory ratio [I/E] = 1:1.5, fraction of inspiratory O2 [FIO2]) = 0.3, positive end-expiratory pressure [PEEP] = 10 cmH2O to prevent formation of atelectasis). Anesthesia was maintained with continuous i.v. propofol (10 mg/kg/h) and remifentanil (5 μg/kg initial bolus, followed by 15–20 μg kg–1 h–1). A balanced electrolyte solution (10 ml kg–1 h–1, Jonosteril 1/1®, Fresenius Kabi) was infused as maintenance fluid. After surgical exposure, a 9-F catheter sheath was inserted into the right femoral vein for placement of a 4-lumen venous catheter (Arrow International). Both femoral arteries were exposed for placement of a 4-F PiCCO catheter (PULSION Medical Systems) for continuous cardiac output, pulse pressure and stroke volume variation measurement, and a 10-F catheter for blood sampling and passive blood removal to induce HS. A catheter was placed in the urinary bladder via midline mini-laparotomy. Thereafter, the animal was turned into the prone position, the skull was exposed, and a craniotomy was drilled over the left and right parietal cortices (10). After exposing the dura, a small incision was made, and a catheter was inserted approximately 5 mm into the subdural space. On the contralateral side, a similar burr hole was placed. Afterward, microdialysis catheters and multimodal brain monitoring probes (Neurovent-PTO§, Raumedic) were inserted about 10–15 mm into the brain parenchyma in both hemispheres. The multimodal probes were placed under visual control for intracranial pressure (ICP), brain tissue O2 partial pressure (PbtO2), and temperature measurements. The probes were calibrated before insertion according to the manufacturer’s specifications. After placement, all catheters were allowed to equilibrate for about 1 h, and recording was started when PbtO2 values were stable. At the end of the neurosurgical procedure, the burr holes were closed using bone wax, which also served for catheter, microdialysis and probe fixation. Bilateral neurosurgery was performed to avoid sham experiments, i.e., to comply with the 3R principle the hemisphere without ASDH served as the control for the hemisphere with ASDH. During surgery, hydroxyethyl starch 6%–130/0.42 (Vitafusal§, Serumwerk, Bernburg) was used to maintain pulse pressure variation, with a maximum dose of 30 ml/kg according to the manufacturer’s specifications.
Figure 1 shows the experimental protocol. In order to mimic the clinical situation where resuscitation measures are initiated only after trauma-and-hemorrhage, maintenance fluid infusion rate was reduced to 100 ml/h, ventilator settings were tidal volume 8 ml/kg, PEEP = 0 cmH2O, I/E ratio = 1:2, FIO2 = 0.21. Thereafter, 0.1 ml/kg body weight of autologous blood was injected over 15 min via the subdural catheter using a syringe pump. This subdural blood volume was injected because ≈10% of the intracranial volume represents the threshold for supra-tentorial volume tolerance (19). Immediately thereafter, HS was initiated by passive removal of blood over 30 min via the 10F-arterial catheter targeting 30% of the calculated blood volume (6, 16). The removed blood was stored at 4–8°C in citrate-anticoagulated bags (6, 16). Blood removal was slowed if necessary to maintain cerebral perfusion pressure (CPP), i.e., the difference between mean arterial pressure (MAP) and ICP, ≥ 50 mmHg. After 105 min of HS, i.e., 2 h of combined ASDH and HS, resuscitation was initiated comprising re-transfusion of shed blood within 30 min, fluid resuscitation (20 ml kg–1 h–1; reduced to 10 ml kg–1h–1 if venous pressure > 16 mmHg), and continuous i.v. noradrenaline titrated to maintain MAP at pre-shock levels and CPP ≥ 75 mmHg over a maximum of 54 h (20). Temperature management aimed to achieve brain normothermia, i.e., external cooling with ice-cold water-filled bags was applied if brain temperature reached 39°C. Animals were randomly assigned to control (normoxemia, PaO2 ≈ 80 – 120 mmHg throughout the whole observation period) vs. targeted hyperoxemia (PaO2 ≈ 200 – 250 mmHg during the first 24 h of treatment, thereafter similar to the control group).
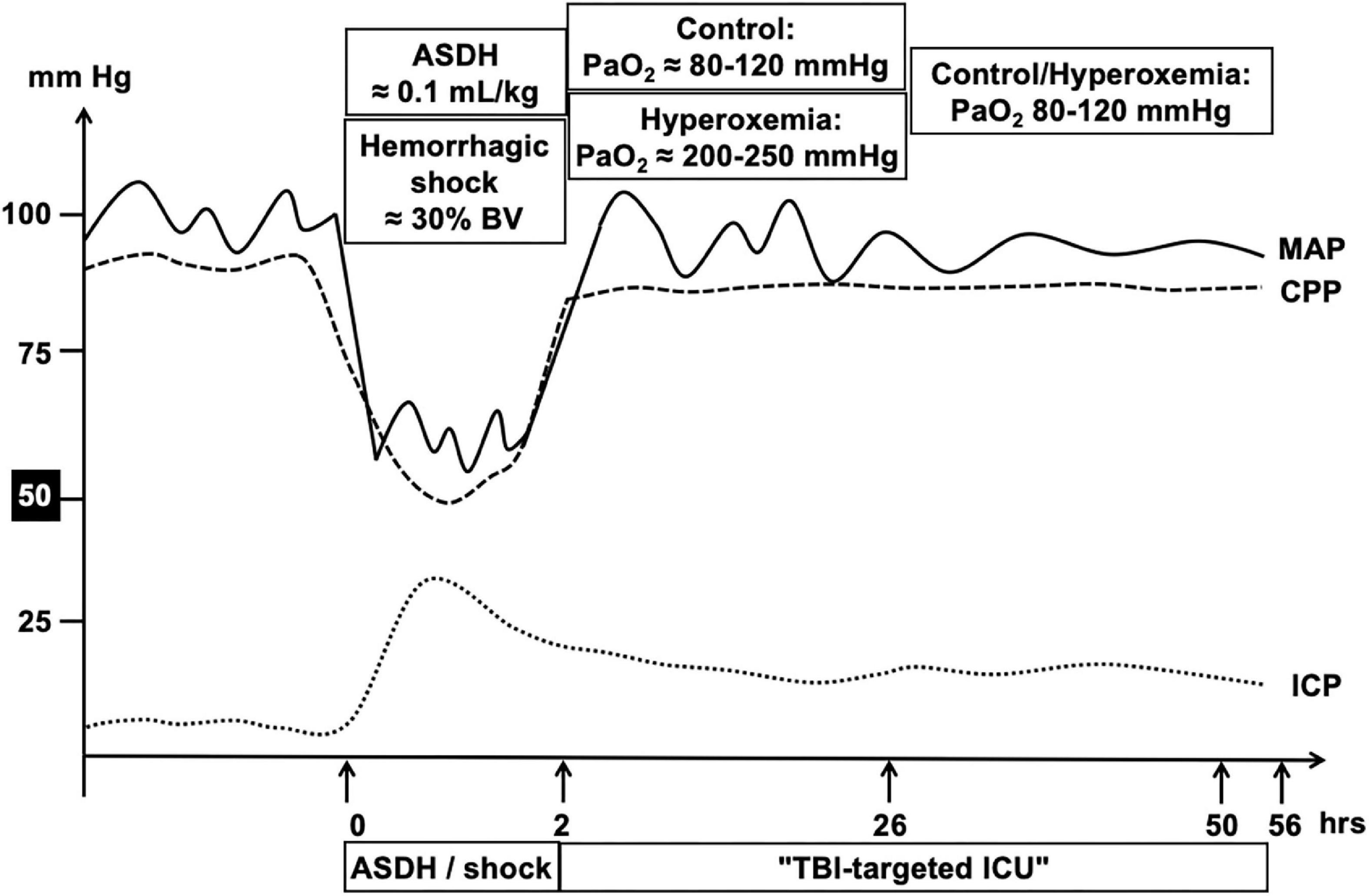
Figure 1. Schematic summary of the experimental design. After instrumentation and a resting period, ASDH was induced by injection of 0.1 ml/kg autologous blood into the subdural space. Immediately after induction of ASDH, hemorrhagic shock was initiated by passive removal of blood targeting 30% of the calculated blood volume. Blood removal was slowed down if necessary to maintain cerebral perfusion pressure (CPP), i.e., the difference between the simultaneously measured mean arterial (MAP) and intracranial (ICP) pressures, ≥ 50 mmHg. At 2 h of combined ASDH and hemorrhagic shock, resuscitation was initiated comprising re-transfusion of shed blood, fluid resuscitation, and continuous i.v. noradrenaline titrated to maintain MAP at pre-shock levels and CPP ≥ 75 mmHg over a maximum of 54 h. Animals were randomly assigned to control (normoxemia, PaO2 ≈ 80 – 120 mmHg throughout the whole observation period) vs. targeted hyperoxemia (PaO2 ≈ 200 – 250 mmHg during the first 24 h of treatment, thereafter similar to the control group).
Measurements of systemic hemodynamics and gas exchange, arterial blood sampling for PaO2, PaCO2, acid-base-status, electrolyte (N+, K+) levels and metabolic parameters (lactate, glucose, urea) were performed together with ICP, CPP, PbtO2, and brain temperature at baseline, i.e., before initiation of ASDH and HS, at the end of the 2-h of combined ASDH and HS, and at 24 and 48 h of treatment. At baseline as well as at 24 and 48 h of treatment, anesthesia depth was reduced by transitorily stopping the continuous i.v. propofol and remifentanil until adequate spontaneous breathing had resumed for 30 min, and neurological function was assessed using a swine-adapted, veterinary Modified Glasgow Coma Scale (MGCS) as described in detail previously (10). Thereafter, continuous i.v. anesthesia was resumed. Upon initiation of resuscitation, baseline ventilator settings were reinstalled, except for FIO2: during the first 24 h of treatment, after drawing ballots from a box, animals were randomly assigned to hyperoxemia (PaO2 = 200–250 mmHg) vs. control (normoxemia, PaO2 = 80–120 mmHg). This PaO2 target window was chosen, because it had coincided with the lowest mortality and the best neurological outcome after severe TBI (16). Thereafter, both groups were treated with the same ventilator settings until the end of the experiment. After 54 h of intensive care, i.e., after treatment had been continued for another 6 h after the last neurological assessment, the pigs were euthanized with KCl after further anesthesia deepening. Animals were euthanized before the end of the programmed 54-h period if (i) CPP < 60 mmHg despite maximum vasopressor dose, limited to a heart rate of ≤ 160/min to prevent tachycardia-induced myocardial injury, and/or the presence of (ii) refractory acute respiratory failure with PaO2 < 60 mmHg at FIO2 = 1.0, and/or (iii) anuric acute kidney failure with K+ > 6 mmol/L. Target PaO2 values during the hyperoxemia period never required FIO2 levels > 0.4, values during the control periods were always achieved using 0.23 < FIO2 < 0.3, and kaliemia always remained below 4 mmol/L (see Supplementary Table 1). Hence, all experiments terminated pre-schedule were due to sudden ICP increases and a refractory fall of CPP < 60 mmHg. To extrapolate the “missing values” for MGCS at the respective time points, in these animals the minimum MGCS score possible (= 3) was used.
Intracerebral tissue metabolites were determined using microdialysis (CMA 600 Microdialysis Analyzer®, CMA/Microdialysis AB) (10). Microdialysis catheters (70 microdialysis bolt catheter®, M Dialysis AB) were perfused (perfusion fluid, CMA/Microdialysis AB) by a microdialysis pump (CMA/102 microdialysis pump®, CMA/Microdialysis AB), and after calibration according to the manufacturer, microdialysate samples were collected in microvials (M Dialysis AB) over 3 h (i.e., from 1 h before until 2 h after each measurement time point) and immediately analyzed for glutamate, lactate, pyruvate, and glucose.
Serum concentrations of cytokines (interleukin-6 and 10) and the “brain injury markers” S100β (21), microtubule-associated protein 2 (MAP-2) (22), glial fibrillary acidic protein (GFAP) (22), and neuron-specific enolase (NSE) (21) were determined using commercially available porcine-specific kits as described previously (10, 16).
Immediately after sampling arterial whole blood samples were analyzed for superoxide anion (O2⋅–) concentrations by electron spin resonance (ESR) as described previously (23). Briefly, whole blood freshly drawn was mixed with the O2⋅–-specific spin probe 1-Hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine and the metal chelators deferoxamine-methanesulfonate and diethyldithiocarbamic-acid in Krebs-Hepes-Buffer (all chemicals from Noxygen). After transferral to a glass capillary tube and incubation, the sample was analyzed using an ESR spectrometer (EMXnano®, Bruker) equipped with a temperature controller (BIO-III®, Noxygen). Averaging three scans, O2⋅– concentrations were derived from comparison of the sample spectrum amplitude with that of standard dilution series of the stable radical 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine after subtraction of blank solution values.
At the end of the experiment, immediate post mortem brain tissue specimens from regions adjacent to the brain area next to the ASDH ipsilateral and corresponding contralateral area were analyzed for mitochondrial respiratory activity using “high-resolution respirometry” (Oxygraph-2K®, Oroboros Instruments) as described previously (10, 23). After supplementation of substrates for complexes I, II and ADP, the oxidative phosphorylation (coupled state, OXPHOS) was assessed. Maximal respiratory capacity of the electron transfer system (ETS) in the uncoupled state was measured after addition of 4-(trifluoromethoxy)phenylhydrazone. Data reported is normalized for tissue wet weight.
We had predefined MGCS as the main outcome parameter. The MGCS had already been validated by others in dogs (Lit Platt) (24). Moreover, we have previously used the MGCS in swine undergoing ASDH alone without hemorrhagic shock and subsequent ICU care for 54 h (10). In that study, the absence of hemorrhagic shock allowed for maintenance of CPP and PbtO2 throughout the whole observational period, which prevented impairment of tissue energy metabolism and, subsequently, major neurological dysfunction. Consequently, a power analysis on the possible effect of hyperoxemia after ASDH plus hemorrhage was not feasible based on the available data, and, hence, we did not calculate a number of animals to include to observe a difference in GCS. Therefore, the Animal Care Committee of the Universität Ulm and the Federal Authorities for Animal Research (Regierungspräsidium Tübingen) deemed our study as “exploratory” allowing for n = 7 per group only. Consequently, we did not calculate a number of animals to include to observe a difference in GCS. Survival was analyzed using a Kaplan-Meyer-graph followed by Log-rank (Mantel-Cox) Test. Experimental data were considered to be non-parametric. Intergroup comparison was conducted with a Mann-Whitney U-test. Kruskal-Wallis test with post hoc Dunn’s multiple comparison was used for multiple within-group testing. Data in the figures is presented as individual data points as well as median and interquartile range. Table data is reported as median and interquartile range. Statistical analysis was conducted with GraphPad Prism5® (GraphPad Software Inc., San Diego, CA, United States).
Median survival times after initiation of resuscitation were 54 (54; 54) and 36 (26; 54) hours in the hyperoxemia and control groups and did not differ significantly between groups (p = 0.11) (Figure 2). No matter the group assignment, all experiments terminated pre-schedule were due to sudden ICP increases and consecutive fall of CPP < 60 mmHg.
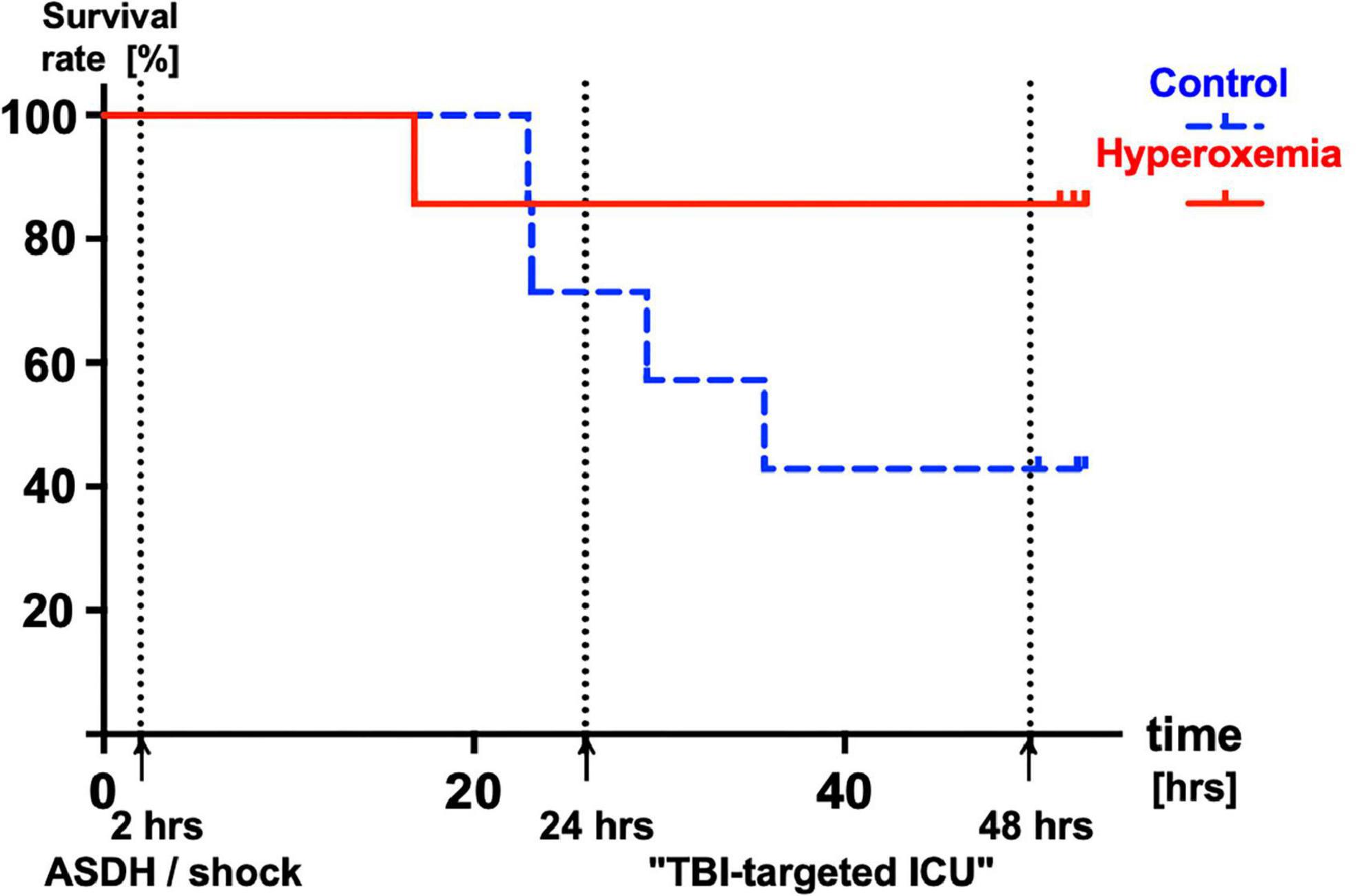
Figure 2. Kaplan-Meyer survival analysis of control (PaO2 ≈ 80 – 120 mmHg, n = 7, broken blue line) and animals treated with targeted hyperoxemia (PaO2 ≈ 200 – 250 mmHg, n = 7, solid red line) during the first 24 h of resuscitation. Median (interquartile range) survival times 54 (54; 54) and 36 (26; 54) hours in the hyperoxemia and control groups, respectively (p = 0.11).
Supplementary Table 1 summarizes the parameters of systemic hemodynamics, gas exchange, acid-base status, and metabolism. None of these parameters showed any significant intergroup difference throughout the experiment, except for significantly lower glycemia and hemoglobin content values in the hyperoxemia animals at the end of the 2 h of combined ASDH plus HS. Severity of shock was also comparable, the amount of blood withdrawn and the noradrenaline infusion rates needed to achieve hemodynamic targets significantly differed. Neither during the first 24 h (i.e., during the targeted hyperoxemia period) nor during the second half of the observation period diuresis showed any significant intergroup difference.
Table 1 and Figure 3 show the parameters of brain perfusion, oxygenation and microdialysis. After combined ASDH and HS, ICP progressively rose and CPP fell until the end of the experiment. Figure 3 shows that target hyperoxemia was associated with significantly higher PbtO2 levels at the end of the 24-h intervention period both in the blood-injected and in the contralateral hemisphere, and this effect persisted until the end of the experiment. While microdialysis measurements did not show any significant effect for lactate or pyruvate, glucose concentrations progressively dropped, however, without intergroup difference. Glutamate concentrations also progressively declined in the hyperoxemia-group, while they remained unchanged in the control group (Table 1). Ultimately, in the hyperoxemia-group, this coincided with well-maintained neurological function, as suggested by the unaffected MGCS. In contrast, MGCS progressively fell in the control animals until the end of the experiment (Table 2).
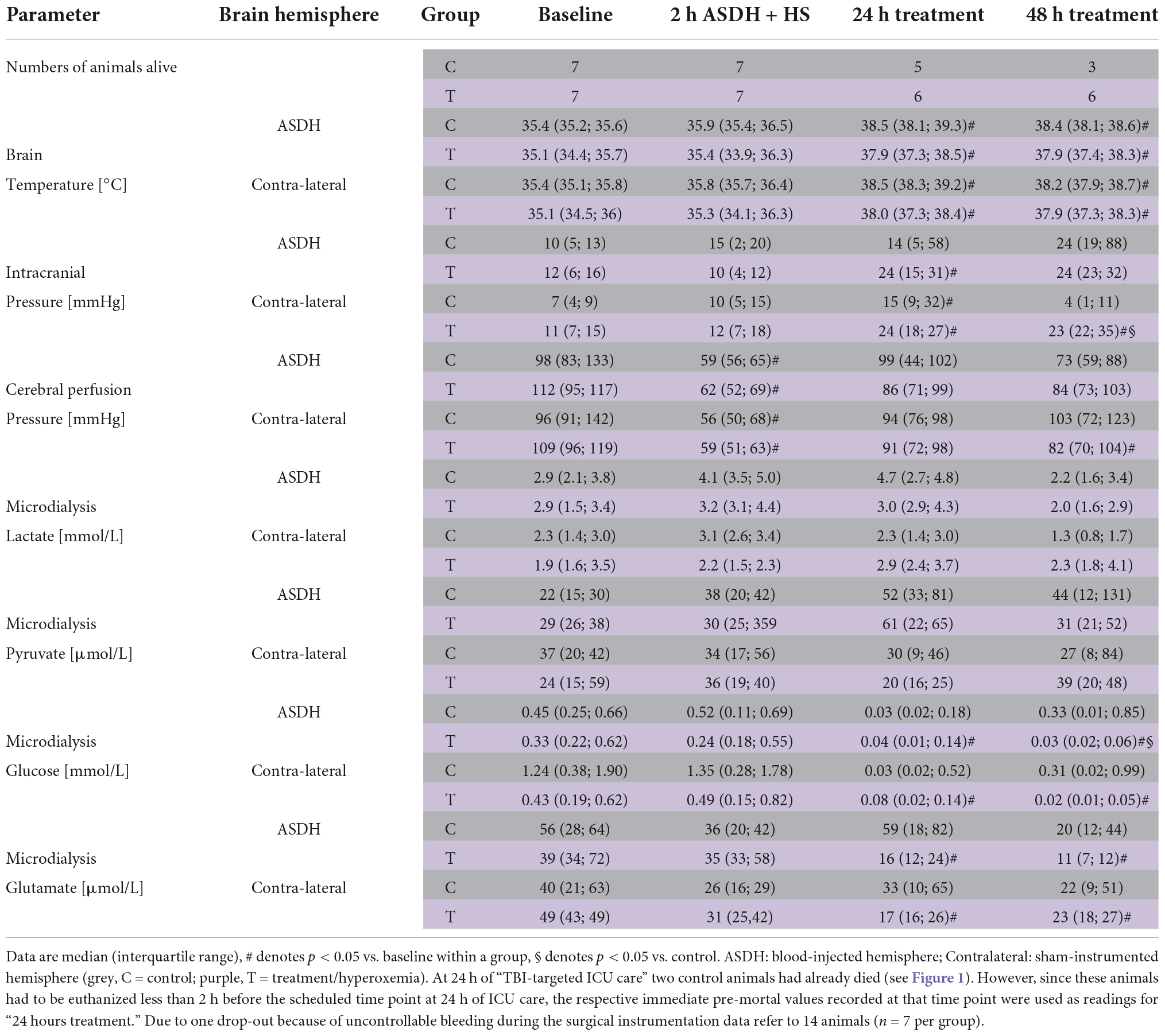
Table 1. Brain perfusion, oxygenation, microdialysis-derived metabolism and mitochondrial respiration.
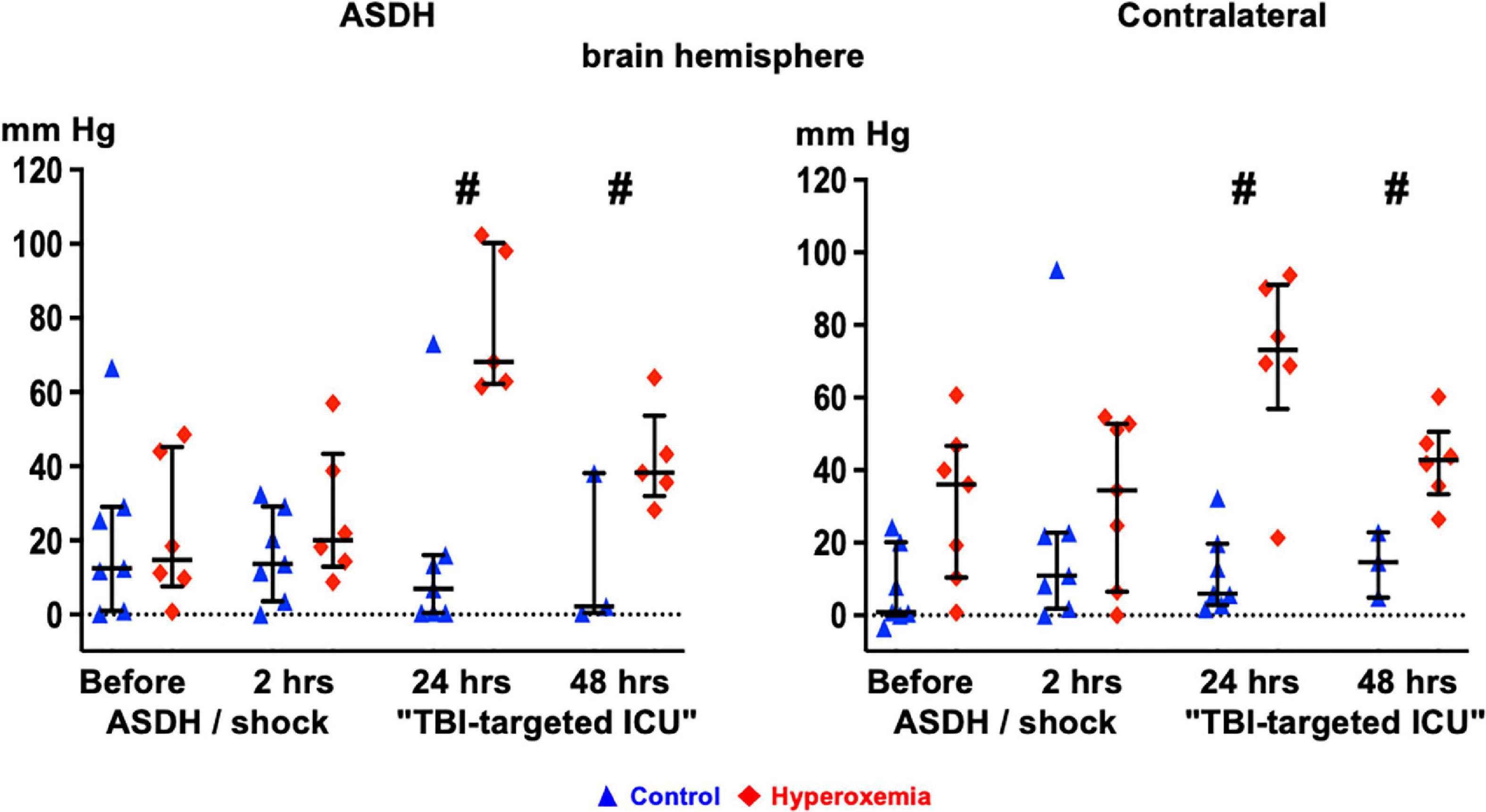
Figure 3. Time course of brain tissue O2 partial pressure (PbtO2) in the blood-injected (“ASDH”, left panel) and the contralateral (right panel) brain hemisphere in the control animals (blue triangles) and the targeted hyperoxemia group (red diamonds). Presented are individual data points, median and interquartile range; # denotes p < 0.05 vs. control. Note: At 24 h of “TBI-targeted ICU care” two control animals had already died. However, since these animals had to be euthanized at less than 2 h before the scheduled time point at 24 h of ICU care, the immediate pre-mortal PbtO2 values recorded at that time point were used as readings for “24 hours of TBI-targeted ICU care”.
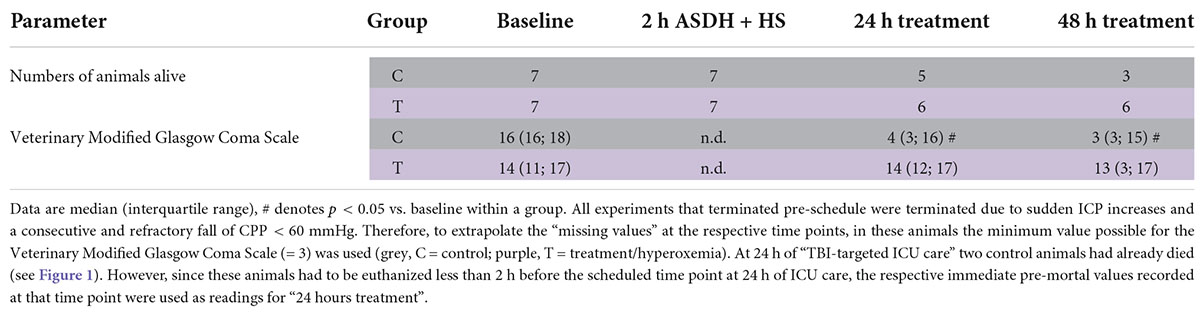
Table 2. Assessment of the veterinary Modified Glasgow Coma Scale.
Table 3 summarizes the plasma biomarkers of inflammation, oxidative stress and brain injury. While IL-6 concentrations decreased over time without intergroup difference, IL-10 levels showed the opposite pattern, however, without intergroup difference. Hyperoxemia did not aggravate oxidative stress as evaluated by the superoxide anion and isoprostane concentrations (Table 3), which was confirmed by the unaffected mitochondrial respiratory activity (both oxidative phosphorylation and maximum electron transport capacity of the respiratory chain in the uncoupled state) in post mortem brain specimen (Supplementary Table 2). Plasma concentrations of all brain injury biomarkers (GFAP, MAP-2, NSE, and protein S100β) progressively decreased in the hyperoxemia group, whereas MAP-2 only showed this pattern in the control animals (Table 3).
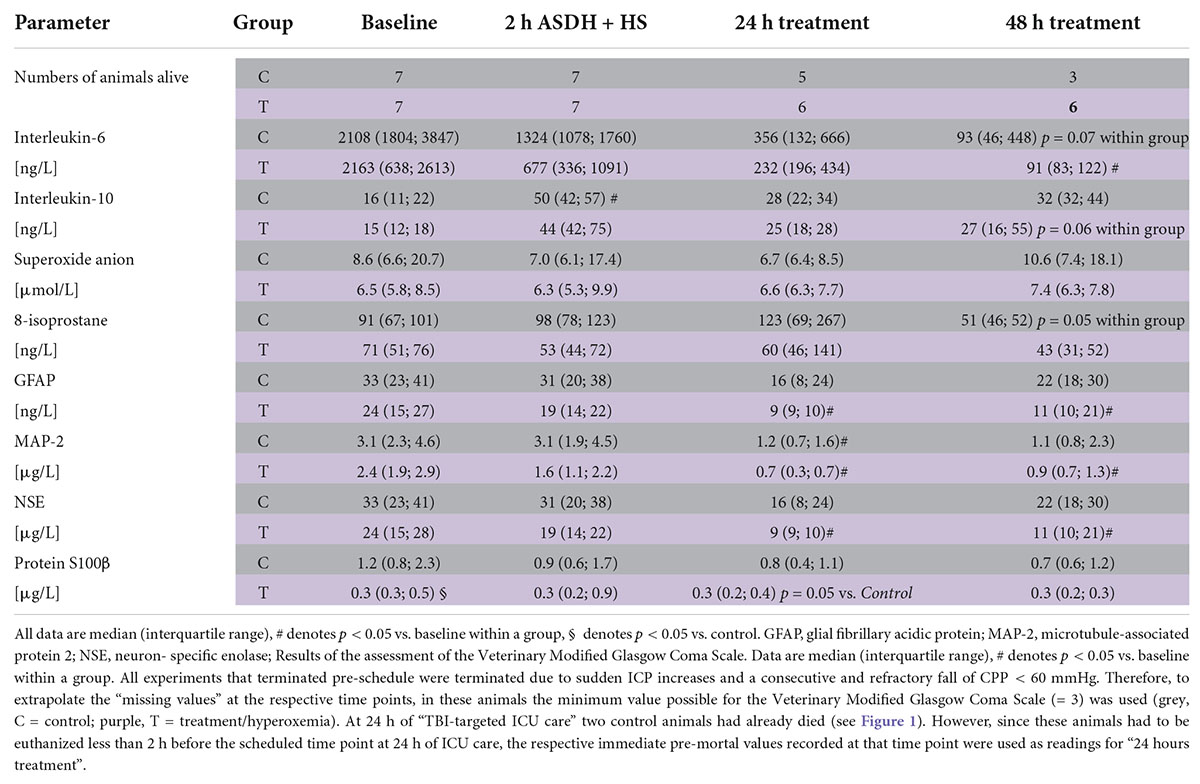
Table 3. Biomarkers of inflammation, oxidative stress, and brain injury.
The main results of this study were that in a long-term, resuscitated, animal model of combined ASDH and HS over 56 h post-injury, targeted hyperoxemia (i) ameliorated brain tissue oxygenation, which persisted beyond the intervention period; (ii) attenuated neurologic dysfunction; and (iii) was safe with respect to parameters of oxidative stress. This porcine model mimics a frequent scenario of severe combined injury, namely hemorrhagic shock and ASDH, the latter being a major contributor to TBI (14). The model makes use of “FBM” swine characterized by atherosclerosis and coronary artery disease, and, thus, mirrors the elderly and co-morbid patient (15).
While it seems unequivocal that excessive hyperoxemia with PaO2 > 300 mmHg should be avoided (3, 4), it remains open whether there is a “sweet spot” for PaO2 after TBI and/or HS (3, 4). This discussion is best characterized that for possible neuroprotective properties of hyperox(em)ia, there is “… probably a narrow effective dose, and benefit may be limited to at-risk tissue…” (25), and the “ … need to identify optimal approaches to improve O2 delivery without exacerbating… oxidative stress or injury…” (26). Our findings of possible beneficial effects of a target window of PaO2 = 200–250 mmHg during the first 24 h of resuscitation agree with retrospective data after severe multiple trauma showing that PaO2 > 150 and > 200 mmHg, respectively, within the first 24 h of ICU stay coincided with improved survival (7, 8). The interval chosen in our study was also associated with the lowest mortality and the best neurological outcome after severe TBI during the first 24 h of ICU (9). It is noteworthy that albeit not significantly different, the two survival curves cleaved at approx. 24 h of resuscitation (p = 0.11). Again, this agrees with retrospective data in TBI patients concluding that moderate hyperoxemia with “…a PaO2 above 12 kPa and higher may improve oxidative cerebral energy metabolism and pressure autoregulation, particularly in cases of limited energy substrate supply in the early phase of TBI…” (27).
It could be argued that hyperox(em)ia-induced systemic vasoconstriction should have caused reduced norepinephrine infusion rates (3, 4). Moreover, due to its preferential vasoconstrictor effect in the cerebral circulation, target hyperox(em)ia might have been expected to lower ICP values (3, 4). It should be noted, however, that in the HYPER2S multicenter trial in patients with norepinephrine-dependent septic shock, even 24 h of FIO2 = 1.0 resulting in mean PaO2 levels of 227–272 mmHg during the treatment period had not influenced the norepinephrine infusion rates (28). In mechanically ventilated patients with severe TBI and requiring FIO2 levels < 0.4 to maintain normoxemia, normobaric hyperoxia (FIO2 = 1.0) resulting in PbtO2 levels of 86 ± 12 mm Hg, i.e., even slightly higher than in our study, had not affected ICP (29). Hence, our findings well agree with existing clinical data.
PbtO2 monitoring has been referred to as a promising tool after TBI, in particular when combined with ICP measurements and/or microdialysis data (30). In contrast to our study, Hawryluk et al. reported that in swine undergoing controlled cortical impact, hyperoxia increased PbtO2 values distal to, but if so, had only little effect near the site of injury. It should be noted, however, that these authors’ study lasted for 15.2 h only, animals received no hemodynamic support to maintain perfusion pressure, and FIO2 = 100% was used, with PaO2 as high as ≈ 560 mmHg (31). Albeit PbtO2 correlates with PaO2 (32, 33), equivocal results are available whether a hyperoxemia-induced PbtO2 increase improves cerebral energy metabolism (34–37). Strikingly, the effect of targeted hyperoxemia on PbtO2 values was sustained until the end of our experiment, i.e., beyond the intervention period. It could be argued that increased microcirculatory O2 content under conditions of comparable macrocirculatory O2 availability might reflect impaired cellular O2 utilization rather than improved energy metabolism. However, albeit there was no significant intergroup difference, microdialysis data favored the hyperoxemia-treated animals: glutamate concentrations, a marker of excitotoxic secondary brain injury in patients (38) and swine (39, 40), progressively declined in the hyperoxemia-group, while they remained unchanged in the control group. At first glance, this finding is in contrast to retrospective patient data demonstrating a direct relation between hyperoxia and microdialysis-derived glutamate concentrations, i.e., suggesting hyperoxia-induced aggravation of excitotoxicity (41). However, in that study, increased glutamate concentrations under conditions of “compromised” PbtO2 values (< 20 mmHg, i.e., comparable to those in our control animals) required an FIO2 > 60%. Since in that study the “…majority of patients had no pulmonary dysfunction…,” PaO2 levels had most likely been markedly higher than in our swine with maximum FIO2 = 40%.
Targeted hyperoxemia affected neither blood O2⋅– (a marker of ROS formation) nor isoprostane (a marker of lipid peroxidation) concentrations. Immediate post mortem brain tissue mitochondrial respiration was not affected either. Both in experimental animals and in patients, equivocal data is available on the impact of hyperox(em)ia on oxidative stress after TBI: aggravated (42), unchanged (43, 44) or attenuated (45) markers of radical damage were reported. In mice with TBI and HS, Blasiole et al. found that despite enhanced oxidative stress and neuro-inflammation, hyperoxic resuscitation improved hippocampal neuronal survival (46). The authors concluded that benefits of enhanced O2 supply may outweigh detrimental effects of enhanced radical production. It is noteworthy that most of the above-mentioned studies investigated short-term (up to 4 h) pure O2 ventilation with PaO2≈300–450 mmHg rather than moderate hyperoxemia within a well-defined target window. According to the clinical data available (9) we had avoided extreme hyperoxemia: even hyperoxemia comparable to our study for up to 14 days to did not affect serum markers of oxidative stress, inflammation or neurological injury (44).
The major limitation of this study is that we used MGCS as the main outcome parameter, but that a power analysis had not been possible prior to the study due to the lacking available data on the time course of this parameter in a model of combined ASDH plus hemorrhage. Therefore, we had been incapable to calculate a number of animals to include to observe a difference in MGCS, and, consequently, the number of animals studies was limited, rendering the study merely exploratory. Moreover, the experimental setting clearly is established for either ASDH or HS alone (10, 17), but no comparable data are available in the current literature for the combined trauma. Finally, the approach is limited due to its duration of 56 h of injury and resuscitation for reasons of ethical acceptability and feasibility.
In summary, in a resuscitated model of combined ASDH and HS over 56 h post-injury in swine with pre-existing atherosclerosis target hyperoxemia was associated with higher PbtO2 levels, which was sustained beyond the intervention period. Target hyperoxemia exerted neither significantly beneficial nor deleterious effects. A benefit for neurological outcome may have been missed due to the tendency toward lower survival in the normoxemia-treated animals.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Ethical approval by the Animal Care Committee of the Universität Ulm and the Federal Authorities for Animal Research (Regierungspräsidium Tübingen, #1316) experiments were conducted in adherence to the European Union Directive 2010/63/EU on the protection of animals used for scientific purposes.
PR and TK: conceptualization. PR, TK, and AH: methodology. MG, FZ, OM, TM, E-MW, EC, TD, DM, and PR: formal analysis. TD, DM, HG, AH, MG, RM, SM, TT, SZ, EC, PR, and TK: investigation. PR: resources and supervision. TD and PR: writing – original draft. TD, DM, FM, AH, MG, RM, SM, HG, FZ, OM, TM, AS, E-MW, TT, SZ, EC, PA, PR, and TK: writing – review and editing. TD, DM, TT, and SZ: visualization. All authors have read and approved the final version of the article.
This study was funded by grants to PR (Collaborative Research Center 1149 project number 251293561) by the Deutsche Forschungsgemeinschaft. DM received funding by means of a “Gerok Rotation” (rotation as clinician scientist) by the Collaborative Research Center 1149 (project number 251293561), Deutsche Forschungsgemeinschaft. The funders had no role in the design of this study, data collection, or interpretation, or the decision to submit the results. Ethical approval was granted by the Local Animal Care Committee of Ulm University and the Federal Authorities for Animal Research (#1316).
We are indebted to Tanja Schulz, Sandra Kress, and Andrea Seifritz, for skillful technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.971882/full#supplementary-material
2. Niemeyer M, Jochems D, Houwert RM, van Es MA, Leenen L, van Wessem K. Mortality in Polytrauma Patients with Moderate to Severe Tbi on Par with Isolated Tbi Patients: Tbi as Last Frontier in Polytrauma Patients. Injury. (2022) 53:1443–8. doi: 10.1016/j.injury.2022.01.009
3. Demiselle J, Calzia E, Hartmann C, Messerer DAC, Asfar P, Radermacher P, et al. Target Arterial Po(2) according to the underlying pathology: a mini-review of the available data in mechanically ventilated patients. Ann Intensive Care. (2021) 11:88. doi: 10.1186/s13613-021-00872-y
4. Singer M, Young PJ, Laffey JG, Asfar P, Taccone FS, Skrifvars MB, et al. Dangers of Hyperoxia. Crit Care. (2021) 25:440. doi: 10.1186/s13054-021-03815-y
5. Dyson A, Simon F, Seifritz A, Zimmerling O, Matallo J, Calzia E, et al. Bladder tissue oxygen tension monitoring in pigs subjected to a range of cardiorespiratory and pharmacological challenges. Intensive Care Med. (2012) 38:1868–76. doi: 10.1007/s00134-012-2712-z
6. Hartmann C, Loconte M, Antonucci E, Holzhauser M, Hölle T, Katzsch D, et al. Effects of hyperoxia during resuscitation from hemorrhagic shock in swine with preexisting coronary artery disease. Crit Care Med. (2017) 45:e1270–9. doi: 10.1097/CCM.0000000000002767
7. Baekgaard JS, Abback PS, Boubaya M, Moyer JD, Garrigue D, Raux M, et al. Early hyperoxemia is associated with lower adjusted mortality after severe trauma: results from a french registry. Crit Care. (2020) 24:604. doi: 10.1186/s13054-020-03274-x
8. Duclos G, Rivory A, Rességuier N, Hammad E, Vigne C, Meresse Z, et al. Effect of early hyperoxemia on the outcome in servere blunt chest trauma: a propensity score-based analysis of a single-center retrospective cohort. J Crit Care. (2021) 63:179–86. doi: 10.1016/j.jcrc.2020.09.008
9. Alali AS, Temkin N, Vavilala MS, Lele AV, Barber J, Dikmen S, et al. Matching early arterial oxygenation to long-term outcome in severe traumatic brain injury: target values. J Neurosurg. (2019) 132:537–44. doi: 10.3171/2018.10.Jns18964
10. Datzmann T, Kapapa T, Scheuerle A, McCook O, Merz T, Unmuth S, et al. In-Depth characterization of a long-term, resuscitated model of acute subdural hematoma-induced brain injury. J Neurosurg. (2021) 134:223–34. doi: 10.3171/2019.9.Jns191789
11. Thim T, Hagensen MK, Drouet L, Bal Dit Sollier C, Bonneau M, Granada JF, et al. Familial hypercholesterolaemic downsized pig with human-like coronary atherosclerosis: a model for preclinical studies. EuroIntervention. (2010) 6:261–8.
12. Nußbaum BL, McCook O, Hartmann C, Matallo J, Wepler M, Antonucci E, et al. Left Ventricular function during porcine-resuscitated septic shock with pre-existing atherosclerosis. Intensive Care Med Exp. (2016) 4:14. doi: 10.1186/s40635-016-0089-y
13. Merz T, Stenzel T, Nussbaum B, Wepler M, Szabo C, Wang R, et al. Cardiovascular disease and resuscitated septic shock lead to the downregulation of the H2s-producing enzyme cystathionine-gamma-lyase in the porcine coronary artery. Intensive Care Med Exp. (2017) 5:17. doi: 10.1186/s40635-017-0131-8
14. Lee JJ, Segar DJ, Morrison JF, Mangham WM, Lee S, Asaad WF. Subdural hematoma as a major determinant of short-term outcomes in traumatic brain injury. J Neurosurg. (2018) 128:236–49. doi: 10.3171/2016.5.Jns16255
15. Newcombe VFJ, Chow A. The features of the typical traumatic brain injury patient in the icu are changing: what will this mean for the intensivist? Curr Opin Crit Care. (2021) 27:80–6. doi: 10.1097/mcc.0000000000000814
16. Datzmann T, Hoffmann A, McCook O, Merz T, Wachter U, Preuss J, et al. Effects of sodium thiosulfate (Na(2)S(2)O(3)) during resuscitation from hemorrhagic shock in swine with preexisting atherosclerosis. Pharmacol Res. (2020) 151:104536. doi: 10.1016/j.phrs.2019.104536
17. Datzmann T, Wepler M, Wachter U, Vogt JA, McCook O, Merz T, et al. Cardiac effects of hyperoxia during resuscitation from hemorrhagic shock in swine. Shock. (2019) 52:e52–9. doi: 10.1097/shk.0000000000001283
18. Messerer DAC, Gaessler H, Hoffmann A, Gröger M, Benz K, Huhn A, et al. The H(2)S Donor Sodium Thiosulfate (Na(2)S(2)O(3)) does not improve inflammation and organ damage after hemorrhagic shock in cardiovascular healthy swine. Front Immunol. (2022) 13:901005. doi: 10.3389/fimmu.2022.901005
19. Timaru-Kast R, Meissner A, Heimann A, Hoelper B, Kempski O, Alessandri B. Acute subdural hematoma in pigs: role of volume on multiparametric neuromonitoring and histology. J Neurotrauma. (2008) 25:1107–19. doi: 10.1089/neu.2008.0517
20. Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery. (2017) 80:6–15. doi: 10.1227/neu.0000000000001432
21. Vogt N, Herden C, Roeb E, Roderfeld M, Eschbach D, Steinfeldt T, et al. Cerebral alterations following experimental multiple trauma and hemorrhagic shock. Shock. (2018) 49:164–73. doi: 10.1097/shk.0000000000000943
22. Teranishi K, Scultetus A, Haque A, Stern S, Philbin N, Rice J, et al. Traumatic brain injury and severe uncontrolled haemorrhage with short delay pre-hospital resuscitation in a swine model. Injury. (2012) 43:585–93. doi: 10.1016/j.injury.2010.09.042
23. Zink F, Vogt J, Wachter U, Hartert J, Horchler M, Zhang X, et al. Effects of acute subdural hematoma-induced brain injury on energy metabolism in peripheral blood mononuclear cells. Shock. (2021) 55:407–17. doi: 10.1097/shk.0000000000001642
24. Platt SR, Radaelli ST, McDonnell JJ. The prognostic value of the modified glasgow coma scale in head trauma in dogs. J Vet Intern Med. (2001) 15:581–4.
25. Stocchetti N, Taccone FS, Citerio G, Pepe PE, Le Roux PD, Oddo M, et al. Neuroprotection in acute brain injury: an up-to-date review. Crit Care. (2015) 19:186. doi: 10.1186/s13054-015-0887-8
26. Kochanek PM, Bayır H. Titrating the dose of oxygen after severe traumatic brain injury in the era of precision medicine. J Neurotrauma. (2017) 34:3067–9. doi: 10.1089/neu.2017.5159
27. Wettervik TS, Engquist H, Howells T, Lenell S, Rostami E, Hillered L, et al. Arterial oxygenation in traumatic brain injury-relation to cerebral energy metabolism, autoregulation, and clinical outcome. J Intensive Care Med. (2021) 36:1075–83. doi: 10.1177/0885066620944097
28. Asfar P, Schortgen F, Boisrame-Helms J, Charpentier J, Guerot E, Megarbane B, et al. Hyperoxia and hypertonic saline in patients with septic shock (hypers2s): a two-by-two factorial, multicentre, randomised, clinical trial. Lancet Respir Med. (2017) 5:180–90. doi: 10.1016/s2213-2600(17)30046-2
29. Rockswold SB, Rockswold GL, Zaun DA, Zhang X, Cerra CE, Bergman TA, et al. A prospective, randomized clinical trial to compare the effect of hyperbaric to normobaric hyperoxia on cerebral metabolism, intracranial pressure, and oxygen toxicity in severe traumatic brain injury. J Neurosurg. (2010) 112:1080–94. doi: 10.3171/2009.7.Jns09363
30. Leach MR, Shutter LA. How much oxygen for the injured brain - can invasive parenchymal catheters help? Curr Opin Crit Care. (2021) 27:95–102. doi: 10.1097/mcc.0000000000000810
31. Hawryluk GW, Phan N, Ferguson AR, Morabito D, Derugin N, Stewart CL, et al. Brain tissue oxygen tension and its response to physiological manipulations: influence of distance from injury site in a swine model of traumatic brain injury. J Neurosurg. (2016) 125:1217–28. doi: 10.3171/2015.7.Jns15809
32. Rangel-Castilla L, Lara LR, Gopinath S, Swank PR, Valadka A, Robertson C. Cerebral hemodynamic effects of acute hyperoxia and hyperventilation after severe traumatic brain injury. J Neurotrauma. (2010) 27:1853–63. doi: 10.1089/neu.2010.1339
33. Ghosh A, Highton D, Kolyva C, Tachtsidis I, Elwell CE, Smith M. Hyperoxia results in increased aerobic metabolism following acute brain injury. J Cereb Blood Flow Metab. (2017) 37:2910–20. doi: 10.1177/0271678x16679171
34. Reinert M, Barth A, Rothen HU, Schaller B, Takala J, Seiler RW. Effects of cerebral perfusion pressure and increased fraction of inspired oxygen on brain tissue oxygen, lactate and glucose in patients with severe head injury. Acta Neurochir (Wien). (2003) 145:341–9. doi: 10.1007/s00701-003-0027-0
35. Magnoni S, Ghisoni L, Locatelli M, Caimi M, Colombo A, Valeriani V, et al. Lack of improvement in cerebral metabolism after hyperoxia in severe head injury: a microdialysis study. J Neurosurg. (2003) 98:952–8. doi: 10.3171/jns.2003.98.5.0952
36. Nortje J, Coles JP, Timofeev I, Fryer TD, Aigbirhio FI, Smielewski P, et al. Effect of hyperoxia on regional oxygenation and metabolism after severe traumatic brain injury: preliminary findings. Crit Care Med. (2008) 36:273–81. doi: 10.1097/01.Ccm.0000292014.60835.15
37. Diringer MN, Aiyagari V, Zazulia AR, Videen TO, Powers WJ. Effect of hyperoxia on cerebral metabolic rate for oxygen measured using positron emission tomography in patients with acute severe head injury. J Neurosurg. (2007) 106:526–9. doi: 10.3171/jns.2007.106.4.526
38. Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma. (2016) 33:1775–83. doi: 10.1089/neu.2015.4226
39. Hwabejire JO, Jin G, Imam AM, Duggan M, Sillesen M, Deperalta D, et al. Pharmacologic modulation of cerebral metabolic derangement and excitotoxicity in a porcine model of traumatic brain injury and hemorrhagic shock. Surgery. (2013) 154:234–43. doi: 10.1016/j.surg.2013.04.008
40. Hwabejire JO, Imam AM, Jin G, Liu B, Li Y, Sillesen M, et al. Differential effects of fresh frozen plasma and normal saline on secondary brain damage in a large animal model of polytrauma, hemorrhage and traumatic brain injury. J Trauma Acute Care Surg. (2013) 75:968–74. doi: 10.1097/TA.0b013e31829a021a
41. Quintard H, Patet C, Suys T, Marques-Vidal P, Oddo M. Normobaric hyperoxia is associated with increased cerebral excitotoxicity after severe traumatic brain injury. Neurocrit Care. (2015) 22:243–50. doi: 10.1007/s12028-014-0062-0
42. Ahn ES, Robertson CL, Vereczki V, Hoffman GE, Fiskum G. Normoxic ventilatory resuscitation following controlled cortical impact reduces peroxynitrite-mediated protein nitration in the hippocampus. J Neurosurg. (2008) 108:124–31. doi: 10.3171/jns/2008/108/01/0124
43. Puccio AM, Hoffman LA, Bayir H, Zullo TG, Fischer M, Darby J, et al. Effect of short periods of normobaric hyperoxia on local brain tissue oxygenation and cerebrospinal fluid oxidative stress markers in severe traumatic brain injury. J Neurotrauma. (2009) 26:1241–9. doi: 10.1089/neu.2008.0624
44. Lång M, Skrifvars MB, Siironen J, Tanskanen P, Ala-Peijari M, Koivisto T, et al. A Pilot study of hyperoxemia on neurological injury, inflammation and oxidative stress. Acta Anaesthesiol Scand. (2018) 62:801–10. doi: 10.1111/aas.13093
45. Vidal-Jorge M, Sánchez-Guerrero A, Mur-Bonet G, Castro L, Rãdoi A, Riveiro M, et al. Does normobaric hyperoxia cause oxidative stress in the injured brain? A Microdialysis Study Using 8-Iso-Prostaglandin F2α as a Biomarker. J Neurotrauma. (2017) 34:2731–42. doi: 10.1089/neu.2017.4992
Keywords: traumatic brain injury, multimodal brain monitoring, GFAP, MAP-2, protein S100β, neuron-specific enolase, mitochondrial respiration, oxidative stress hyperoxemia after acute subdural hematoma and hemorrhagic shock
Citation: Datzmann T, Messerer DAC, Münz F, Hoffmann A, Gröger M, Mathieu R, Mayer S, Gässler H, Zink F, McCook O, Merz T, Scheuerle A, Wolfschmitt E-M, Thebrath T, Zuech S, Calzia E, Asfar P, Radermacher P and Kapapa T (2022) The effect of targeted hyperoxemia in a randomized controlled trial employing a long-term resuscitated, model of combined acute subdural hematoma and hemorrhagic shock in swine with coronary artery disease: An exploratory, hypothesis-generating study. Front. Med. 9:971882. doi: 10.3389/fmed.2022.971882
Received: 22 June 2022; Accepted: 04 August 2022;
Published: 22 August 2022.
Edited by:
Raghavan Pillai Raju, Augusta University, United StatesReviewed by:
Tobias Jan Krämer, Johannes Gutenberg University Mainz, GermanyCopyright © 2022 Datzmann, Messerer, Münz, Hoffmann, Gröger, Mathieu, Mayer, Gässler, Zink, McCook, Merz, Scheuerle, Wolfschmitt, Thebrath, Zuech, Calzia, Asfar, Radermacher and Kapapa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Datzmann, VGhvbWFzLmRhdHptYW5uQHVuaS11bG0uZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.