 Geoffrey P. Dobson
Geoffrey P. Dobson Jodie L. Morris
Jodie L. Morris Hayley L. Letson
Hayley L. Letson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 30 August 2022
Sec. Intensive Care Medicine and Anesthesiology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.968453
This article is part of the Research TopicTranslational Immunology in Trauma - to Provide New Insights for Improving OutcomesView all 11 articles
When a traumatic injury exceeds the body’s internal tolerances, the innate immune and inflammatory systems are rapidly activated, and if not contained early, increase morbidity and mortality. Early deaths after hospital admission are mostly from central nervous system (CNS) trauma, hemorrhage and circulatory collapse (30%), and later deaths from hyperinflammation, immunosuppression, infection, sepsis, acute respiratory distress, and multiple organ failure (20%). The molecular drivers of secondary injury include damage associated molecular patterns (DAMPs), pathogen associated molecular patterns (PAMPs) and other immune-modifying agents that activate the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic stress response. Despite a number of drugs targeting specific anti-inflammatory and immune pathways showing promise in animal models, the majority have failed to translate. Reasons for failure include difficulty to replicate the heterogeneity of humans, poorly designed trials, inappropriate use of specific pathogen-free (SPF) animals, ignoring sex-specific differences, and the flawed practice of single-nodal targeting. Systems interconnectedness is a major overlooked factor. We argue that if the CNS is protected early after major trauma and control of cardiovascular function is maintained, the endothelial-glycocalyx will be protected, sufficient oxygen will be delivered, mitochondrial energetics will be maintained, inflammation will be resolved and immune dysfunction will be minimized. The current challenge is to develop new systems-based drugs that target the CNS coupling of whole-body function.
Globally, over one billion people sustain traumatic injuries, and over six million die annually (1). Mortality is twofold higher in low- and middle-income countries compared to high-income countries, and up to 5-times higher in resource-limited rural and remote regions (1, 2). In patients who survive the first few hours of hospital admission, complications can occur at different times (Figure 1) (3). The first window is 3–6 to 24 h where CNS dysfunction (∼50% of early deaths) and circulatory collapse (usually manifesting as shock) lead to early deaths (40% of early deaths) (Figure 1) (4–6). The second window occurs over the next few weeks and typically involves infectious complications with a prolonged indolent form of multiple organ failure, immunosuppression and sepsis, referred to as Persistent Inflammation, Immunosuppression and Catabolism Syndrome (PIICS) (∼20% of deaths) (Figure 1) (4, 7, 8). Sepsis develops in ∼10% of these patients and multiple organ dysfunction syndrome (MODS) in around 70% (3, 9, 10). Despite decades of research, little progress has been made in the development of effective drugs to treat the polytrauma patient (2, 11, 12). The lack of progress in drug development may reflect the way we think about the problem (13, 14). In this review, our aim is to discuss the inflammatory and immune mechanisms that are believed to be responsible for early and late secondary injuries and death following traumatic injury, and possible ways to reduce morbidity and mortality from a systems-based perspective. Before doing so, we will briefly discuss the physiological importance of the system.

Figure 1. After hospital admission, complications occur at different times and depend upon type and severity of injury. These have been characterized as early and late Deaths. Modified after Rauf et al. (3) and Brohi et al. (4). Patients who died at the point-of-injury or in the first few hours upon arrival to hospital have not been included. CNS, central nervous system; TBI, traumatic brain injury.
After a traumatic injury, defined as one or more sudden injuries requiring immediate medical attention, the body activates a series of defense mechanisms to restore homeostatic balance. The concept of homeostatic balance was introduced into medicine in 1916 by Cannon (15). Cannon’s genius was to combine the ideas of Pfluger’s “natural adjustments” (1877), Bernard’s concept of “milieu intérieur” (1878), and Richet’s “living beings were stable but modifiable” (1900) into a unified scheme (16). Cannon proposed that every living organism was in a dynamic state of constancy, with its constituent parts and processes being actively maintained in balance despite external fluctuations (15). The system is not an equilibrium system as it requires a continual flow of matter, energy and exchange with the environment (16, 17). In the mid-1930s, Cannon’s concept was refined to include negative and positive feedback circuits (18), and the system’s steady-state was now viewed as the net sum of negative and positive feedback mechanisms that operate within a range of tolerances, which differ from person to person, and from species to species. The system has evolved such that small injury perturbations are self-limiting and quickly resolved. However, when the trauma overwhelms the system, it triggers a CNS stress response that typically involves excessive sympathetic and neuroendocrine outflows from the brain’s central control, hyperinflammation, immune dysregulation, coagulopathy, endothelial activation and metabolic dysfunction (13, 19, 20). If homeostatic balance is not restored early, secondary injury processes will amplify and may become life-threatening (14).
When I first put forward the biological theory of inflammation 8 years ago, I expressed the idea that this reaction is affected by the intermediation of a physiological continuity between “the cells of the connective tissue, those of the endothelial wall and the leucocytes, which form a complete chain and play the principal part in the inflammation of vertebrates.” The connective tissue cells which are first attacked, would, I thought, transmit the action to the vascular wall, the cells of which would contract to facilitate the passage of the white corpuscles.
Metchnikoff (21) p. 191.
Any trauma to the body inflicts a barrier breach in three-dimensional space and one in time. Damage signals from cellular, vascular and nerve injury are sent around the body, and to the CNS via nerve afferents and resident damage control mechanisms to begin the process of tissue repair and remodeling (14, 22). Recovery begins by rapidly closing the breach, activating immuno-inflammatory processes, removing damaged cells and killing any invading microbes (Figure 2). Immune defense occurs in two parts: First, there is a local frontline defense from patrolling resident immune cells in tissues, and second, from deployment of additional leukocyte subsets from the circulation. Early defense includes activation of tissue resident macrophages, dendritic cells (DCs), neutrophils (PMNs), mast cells, a subset of memory B cells, natural killer (NK) cells, complement (22–24) and recently characterized resident T cells, referred to as innate lymphoid cells (ILCs), which are believed to interact with other resident cells, and trigger the early adaptive immune response and recruitment of cells from the circulation to repair and restore tissue function (25–29) (Figure 2).
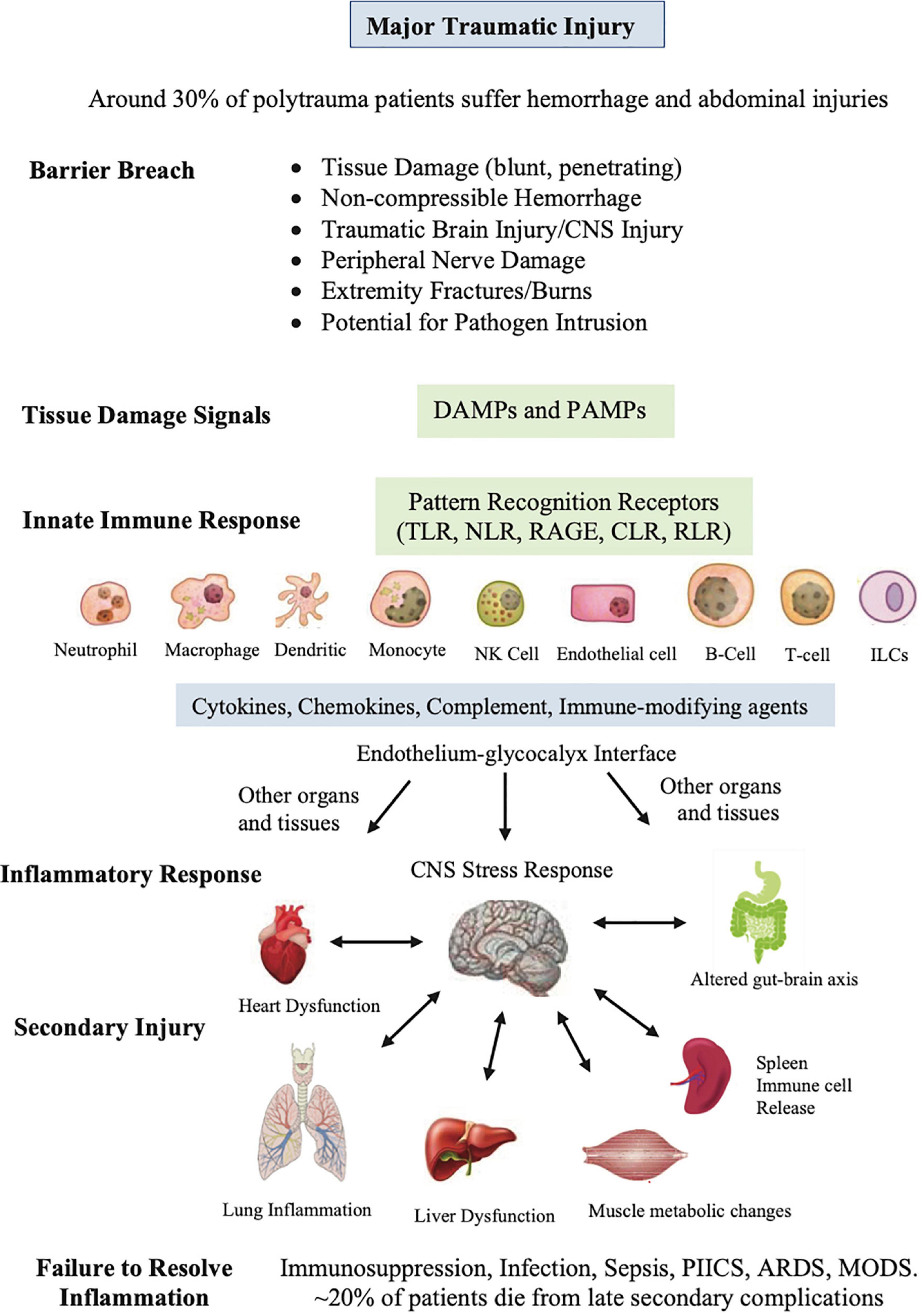
Figure 2. Sequence of events that occur after major traumatic injury. This diverse group of innate cells resident in the tissues detect and respond to changes in the local environment, including damage associated molecular patterns (DAMPs), pathogen associated molecular patterns (PAMPs), neural signals, and other immune-modifying triggers. The pattern recognition receptors on these cells include Toll-like receptors (TLRs), C-type lectin receptors (CLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic-acid-inducible gene-I (RIG-I)-like receptors (RLRs), and receptor for advanced glycation end products (RAGE). PAMPs can be derived from viruses, opportunistic bacteria, fungi, and protozoa and helminths. The innate cells orchestrate an immune response to response to the barrier breach by releasing different inflammatory factors. If dysregulated, the response can lead to secondary injury to the CNS and major organs of the body. The spleen has been included as it is reservoir of platelets, peripheral macrophages, undifferentiated monocytes and other immune cells. ILCs, innate lymphoid cells; NK, natural killer cells, PIICS; Persistent Inflammation, Immunosuppression and Catabolism Syndrome, ARDS; acute respiratory distress syndrome, MODS; multiple organ dysfunction.
This diverse group of resident innate cells have evolved different pattern-recognition receptors that detect and respond to changes in the local environment, including damage associated molecular patterns (DAMPS), pathogen associated molecular patterns (PAMPs) and other immune-modifying triggers (Figure 2) (30–32). DAMPs are released from damaged, stressed or dying cells, including extracellular and cell membrane, cytosolic, cytoskeleton, nuclear mitochondrial, endothelial and blood components (30, 33), while PAMPs are signature proteins, lipoproteins, nucleic acids and saccharides located on the cell surface or released from invading pathogens. Together, they activate the body’s early immune and inflammatory systems to dial in the right response to repair and restore function (Figure 2). Early post-traumatic DAMP markers include high mobility group box protein 1 (HMGB1), mitochondrial DNA (mtDNA), S100, cell fragments, and many other molecules from injured or dying cells and proteoglycans and glycoproteins from endothelial-glycocalyx shedding (34). Importantly, DAMPs and PAMPs are not mutually exclusive and may share co-receptors and accessory molecules, and form partnerships to coordinate the right response (35).
A 2011 landmark study of Xiao and collaborators shed light on the early activation patterns of the immune system following severe blunt trauma and burn injuries. The group reported there was ∼80% activation of the leukocyte transcriptome in the circulation, which they termed a genomic storm (36). This storm developed within 4–12 h and lasted days to weeks. Importantly, in Xiao’s study, what separated patients who developed secondary complications was not the magnitude of the storm, rather the time to resolve it (36). Prolonged resolution times led to worse outcomes. Moreover, both pro-inflammatory and anti-inflammatory pathways were activated early, which challenges the older two-hit and other sequential pro-inflammatory and compensatory anti-inflammatory models of trauma (37). On a cautionary note, although transcriptomic analysis establishes early temporal patterns of change, it provides little or no knowledge into the molecular mechanisms. Future studies should include proteomic and pathway-level analysis to establish the different roles of the early innate (and adaptive systems) to amplify inflammation after severe trauma.
Inflammation is universal, beneficial and restorative. However, after major trauma, it can be lethal. As mentioned earlier, the massive release of DAMPs can overwhelm the system and trigger a hyperinflammatory state that, if not resolved in a timely manner, can lead to immune dysfunction, immunosuppression, infection, sepsis and MODS (4, 13, 19, 38–41). The disruption can lead to pathological interactions between monocyte, macrophage, NK and DCs, T cell dysfunction, and the development of persistent lymphopenia (8–10, 34, 38, 40, 42–45). Persistent lymphopenia carries a high mortality. Brohi’s group recently reported a 45% mortality rate in trauma patients when the lymphocyte count was ≤ 0.5 × 109/L at 48 h after hospital admission (38). In addition, the type of trauma determines a patient’s susceptibility to persistent lymphopenia and infection, with traumatic brain injury (TBI) patients having disproportionally worse outcomes compared to those with burns, polytrauma or major surgery (43). A recent study of Campbell et al. reported that 37% of TBI patients were lymphopenic on hospital admission, and its persistence was associated with increased risk of mortality and pneumonia (46). Wang further reported that up to 83% of severe TBI patients contracted a respiratory infection within 3 days following injury (43, 47).
The mechanisms responsible for persistent lymphopenia and immunosuppression are not well understood (38, 48). The difficulty is that immunosuppression is a highly heterogeneous response involving differential T cell loss, T-cell exhaustion, T-helper 1 (Th1) depression, receptor shedding, and expansion of myeloid-derived suppressor cells (MDSCs) that have suppressive activity (44, 49). Separating the relative contributions of different immune cell subsets to post-traumatic immunosuppression has been a challenge. In a ground-breaking study, Mansen and colleagues examined early changes in circulating lymphocytes and showed that trauma patients who developed MODS within 24 h had nearly 2-fold higher CD56dim NK cells, 80% lower gamma delta (γδ)-low T cells and 4-fold higher IFN-γ upon hospital admission, compared to patients who did not (38). CD56dim NK cells are potent mediators of natural and antibody-dependent cytotoxicity and only weakly secrete cytokines (50). Moreover, the group showed that the patients who developed MODS also developed lymphopenia within 24 h of injury, which if persisted to 48 h led to high mortality (38). The association between lymphopenia, MODS and decreased frequencies and functional responses of innate T cells in trauma patients suggests that early immuno-inflammatory events may “predetermine” late secondary complications. The rise in NK cells and early fall in γδ-low T cells seen in patients who developed MODS may be clinically significant and predict risk for late complications, however, further studies are required (38, 48).
Another early driver of immune complications is HMGB1, which is a major DAMP that induces inflammation via TNF-α, IL-6, and IL-1β that in turn stimulate pattern recognition receptors TLR4 and RAGE on immune cells (Figure 2) (44, 51). In a rat polytrauma model (femoral osteotomy, blunt chest contusion and burn injury), Muire and colleagues showed that HMGB1 was an early contributor to the onset of lymphopenia and the loss of CD4+, CD8+, and γδ-T cells (34). Interestingly, the decrease in T cells was partly attenuated when HMGB1 was neutralized immediately post-trauma, however, the γδ-T cell population was not affected (51). The authors proposed that diminished levels of surface expression of RAGE and TLR4 on T cells, via ectodomain shedding, may be responsible for suppression in vivo (51). HMGB1 has also been shown to activate MDSCs after trauma and cancer (44), and is a late mediator of sepsis (44, 51), which further highlights the complexity of the system.
Apoptosis is believed to play a central role in persistent lymphopenia (52–58). Three main mechanisms for inducing lymphocyte apoptosis include: (1) cell-autonomous T-cell death (ACAD), (2) stress-related activation-induced cell death (AICD), and (3) newly discovered inflammasome-dependent monocyte activation (52–58) (Table 1). Persistently elevated plasma interleukin (IL)-10 levels have further been correlated with monocyte deactivation, reduced T cell activation and secondary infectious complications (8, 39, 40, 42, 59). Platelets also modulate T cell subsets via PAR4 that may link the innate and adaptive systems via pro-inflammatory cytokines (58). The interconnectedness of the T cell subsets and potential drivers of immunosuppression requires further research. Interestingly, post-injury immunosuppression shares many similarities with non-traumatic, sepsis-induced immunosuppression (41, 57).
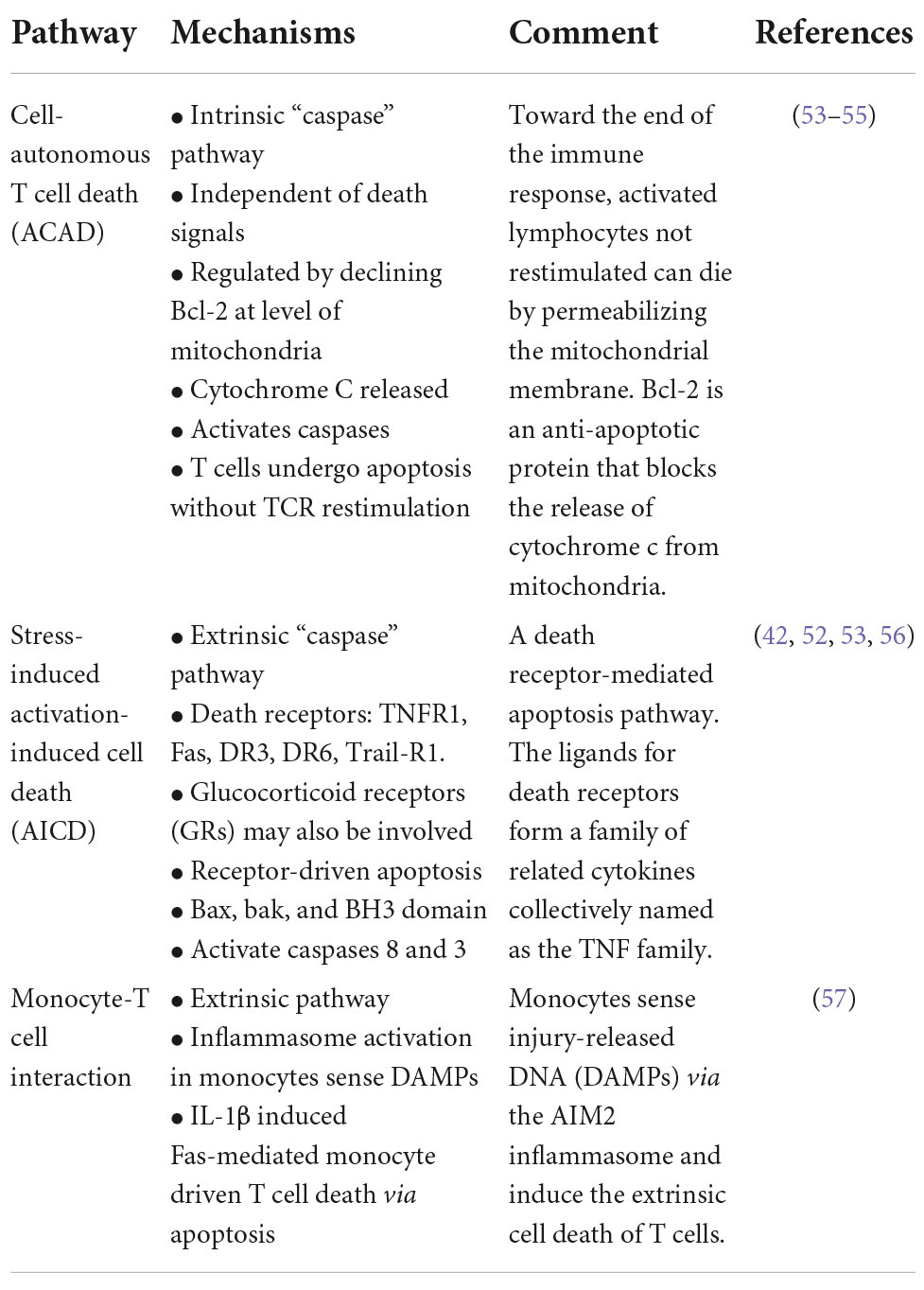
Table 1. Possible mechanisms for T-cell apoptosis and immunosuppression after major trauma.
The defense of the organism against deleterious agencies, which is at first confined to the phagocytic mechanisms and the somatic system of nerves, by and by spreads to and is undertaken by the psychical nervous apparatus … One function of these psychical cells has been to develop a complete science for the defense of the organism against hostile influences.
Metchnikoff (21) p. 195.
Metchnikoff had it right over 130 years ago. Activation of the “psychical cells” of the CNS following severe trauma results are important, and involve the release of norepinephrine, epinephrine and hormones (ACTH and glucocorticoids) from the adrenal medulla into the circulation and from the postganglionic nerve endings innervating the heart, and other organs of the body (14, 60–65). Traditionally, this is known as the whole-body stress response which dates back to Cannon (20, 66). The link between CNS injury, the immune system and immunosuppression is less well known. Yang and colleagues recently showed in a rat model of TBI that activation of sympathetic nervous system upregulated the expression of programmed cell death-1 (PD-1) on CD4+ and CD8+ T cells, and subsequently contributed to immunosuppression (43). The group speculated that immunosuppression may be partly mediated by stress hormones targeting β-adrenergic receptors (β-AR) on T cells (and indirectly B cells), because propranolol, a β-AR blocker, restored dysfunction in vitro, although they acknowledge it was more complex in the intact animal (43).
CNS modulation of the immune system occurs via the central hypothalamic-pituitary-adrenal (HPA) axis and the brainstem’s nucleus tractus solitarius (NTS) (67–70). After major trauma, the CNS balance switches to a sympathetic dominance and suppression of the parasympathetic system that normally counters inflammation via activation of the parasympathetic vagal cholinergic neurons and splanchnic/splenic nerves, known as the inflammatory reflex (71–73). The shift in CNS balance also impacts other organs, such as the heart and vasculature, and the gut microbiome via the gut-brain axis, which can alter blood flow to the gut wall and cause ischemia and increased permeability, where bacteria and/or their active metabolic products (lipopolysaccharides, cytokines, neuropeptides, and protein messengers) can enter the blood stream or lymph vessels and increase PAMPs and a patient’s susceptibility to infection (74, 75). Together, all these factors may contribute to predetermining the extent and resolvability of the immuno-inflammatory response after major trauma.
Another unappreciated fact in the polytrauma patient is that many undergo a second trauma from the corrective surgery itself (20). At all times, from the prehospital setting to after major surgery, the brain remains “wide awake” to changes in circulating DAMPs and PAMPs, inflammatory cytokines and immune cells circulating in the body (20, 76). Even the anesthetized brain remains “awake” because the blood brain barrier (BBB) is disrupted from the trauma and changes in cerebral blood flow and shear stress, which is part of the injury phenotype (77), and this is further amplified in the patient with a TBI (20, 74, 78, 79). Following any major trauma, the brain loses its “immune privilege” as it is no longer “separated” from the rest of the body (77). This is a research area in its infancy. We recently showed in a rat model of a laparotomy, designed to simulate a penetrating wound, that profound changes in gene expression occurred in brain, heart and other organs (80). Abdominal trauma was associated with 10–20-fold increases in plasma corticosterone, pro-inflammatory cytokines, endothelial injury markers, neutrophils (6 h), lactate (3 days), and coagulopathy (80). Lymphocytes decreased by ∼70% at 6 h and 3 days, and IL-10 dramatically increased from undetectable baseline levels to 483 pg/ml after 6 h and again at 3 days (1,149 pg/ml). Cortical excitability was high over 3 days with 30-fold increases in M1 muscarinic receptor expression and α-1A-adrenergic expression, and similar in heart with 8-fold increases in β-1-adrenergic receptor expression, and up to 6-fold increases in M2 and M1 muscarinic receptors after 6 h despite no changes in hemodymanics (80). These “silent” changes are remarkable given that there was only one incision, with no further injury to brain or heart. Unfortunately, we did not examine changes in the different T-cell subsets to further understand changes in immune activation.
Except on few occasions, the patient appears to die from the body’s response to infection rather than from it.
William Osler (81)
Osler’s point cannot be overemphasized. It is not the infection that kills you it’s the body’s response to the trauma. The shift in homeostatic balance toward extreme limits and death led us to develop the Systems Hypothesis of Trauma (SHOT) (82) (Figure 3), which has undergone a number of iterations to include hemorrhagic trauma and the trauma of surgery (12, 13, 16, 20). SHOT has three pillars of protection.
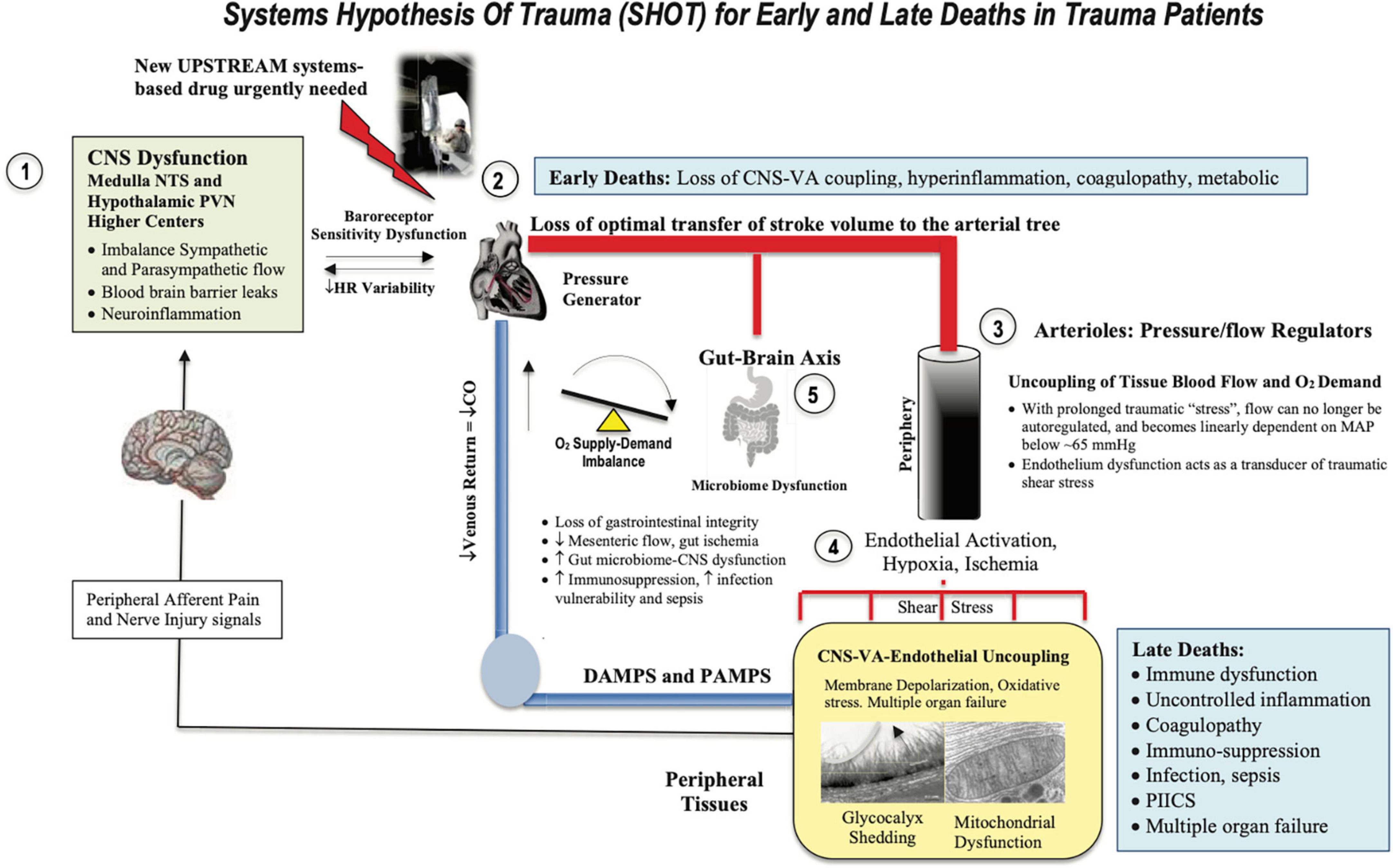
Figure 3. Systems Hypothesis Of Trauma (SHOT) showing key inter-connected sites of uncoupling during early and late deaths: (1) brain, (2) heart, (3) vasculature, (4) endothelial-glycocalyx-mitochondrial unit, and (5) gut barrier breaches. Our hypothesis is that if central and local control of cardiac output and ventriculo-arterial (VA) coupling are improved, endothelial and microvascular function will be improved and tissue O2 delivery will be maintained. An uncoupling is reflected by an increase in stress hormones, sympathetic discharge, loss of baroreceptor sensitivity, reduced heart rate variability (183, 184), unresolved inflammation, immune dysfunction, coagulopathy and mitochondrial dysfunction. New drugs are required to prevent inflammatory hyperdrive and support a VA-coupled, high flow, vasodilatory system with endothelial-glycocalyx protection and tissue oxygenation (185). HR, heart rate; ATP, adenosine triphosphate; CNS, central nervous system; NTS, nucleus tractus solitarius; PVN, paraventricular nucleus of the hypothalamus; MAP, mean arterial pressure. PIICS, Persistent Inflammation, Immunosuppression and Catabolism Syndrome, see text for description of early and late deaths.
1. CNS-cardiovascular coupling (Central Controller)
2. Endothelial glycocalyx material exchange (Systemic Integrator)
3. Mitochondrial integrity (Systemic Regulator)
If the CNS can be protected early after trauma and the “hyperdrive” response can be suppressed, we argue that the immuno-inflammatory storms and lymphopenia will be reduced (20). According to SHOT, shifting autonomic balance toward parasympathetic outflows in the first minutes to hours after trauma would assist to maintain ventriculo-arterial (VA) coupling close to unity. VA coupling is a metric rarely discussed or measured in major trauma patients. It is the ratio of arterial elastance (Ea) to left-ventricular (LV) elastance (Ees) and can be measured from routine echocardiography (83–88). When the ratio is close to unity, the efficiency of the system is considered optimal. If the ratio is excessively high or low, the heart as a pump and vascular load become uncoupled with adverse downstream clinical outcomes (86, 89, 90). The clinical advantage of VA coupling over ejection fraction (EF) or cardiac output (CO) is that it provides arterial load properties in addition to LV function (86, 87). If the proximal arterial vessels become stiff, as a result of the CNS stress response, it increases load on the pump (91), whereas if the heart becomes stiff it cannot relax optimally to fill and eject blood into the conduit vessels (87). If both occur, they lead to VA uncoupling, tissue hypoperfusion, mitochondrial damage (92, 93) and subsequent immuno-inflammatory dysfunction (Figure 3). In the case of a high VA coupling ratio, vasodilator therapies can lower Ea and reduce the Ea/Ees ratio toward 1.0, and in the case of a low ratio, inotropes can increases Ees to improve VA coupling (92).
We predict further that maintaining VA coupling would improve immune function by reducing gut-brain axis dysfunction and preventing the gut wall from becoming ischemic and leaky which exacerbates immuno-inflammatory conditions, coagulopathy, immunosuppression, infection and sepsis (75). Howard and colleagues reported in trauma patients rapid changes in the microbiome during resuscitation and stabilization (94), although further studies are required to understand the role of the gut in exacerbating systemic inflammation and infectious complications after major trauma.
The second pillar of SHOT is to maintain the health of the endothelium (95). The endothelium is located at the nexus of the blood and tissues and controls the transfer of O2, metabolic fuels, hormones, immune cells and factors, inflammatory regulators and fluids (96–102). Trauma-induced damage to this organ is termed the endotheliopathy of trauma (EoT), which is characterized by endothelial activation, vasoactivity, loss of barrier function, leukocyte adhesion, coagulopathy, inflammation and organ dysfunction (103–110). In addition, the endothelium, like the BBB, is highly sensitive to changes in blood flow and shear stress, which can alter vascular tone, tissue perfusion, exchange and permeability (111).
The endothelial surface area (SA) has been estimated to be 3,000–7,000 m2 (98, 112). However, this estimate ignores the SA of the glycocalyx mesh that is synthesized and secreted by the endothelium and anchored to its cellular lining. As mentioned, the function of the endothelial glycocalyx is dynamic and diverse and it also acts as a vascular filter overlying the endothelial cell-cell junctions as it contains a large volume of non-circulating plasma (1–1.7 L) (113–115). We have estimated for the first time the SA of glycocalyx and found it was at least 10-fold higher than the endothelium (SAglycocalyx = 46,120 m2) (see Figure 4). This is equivalent to a SA of over ∼200 tennis courts or 8 USA football fields, and given its central role has major implications to immune function and secondary injury progression post-trauma.
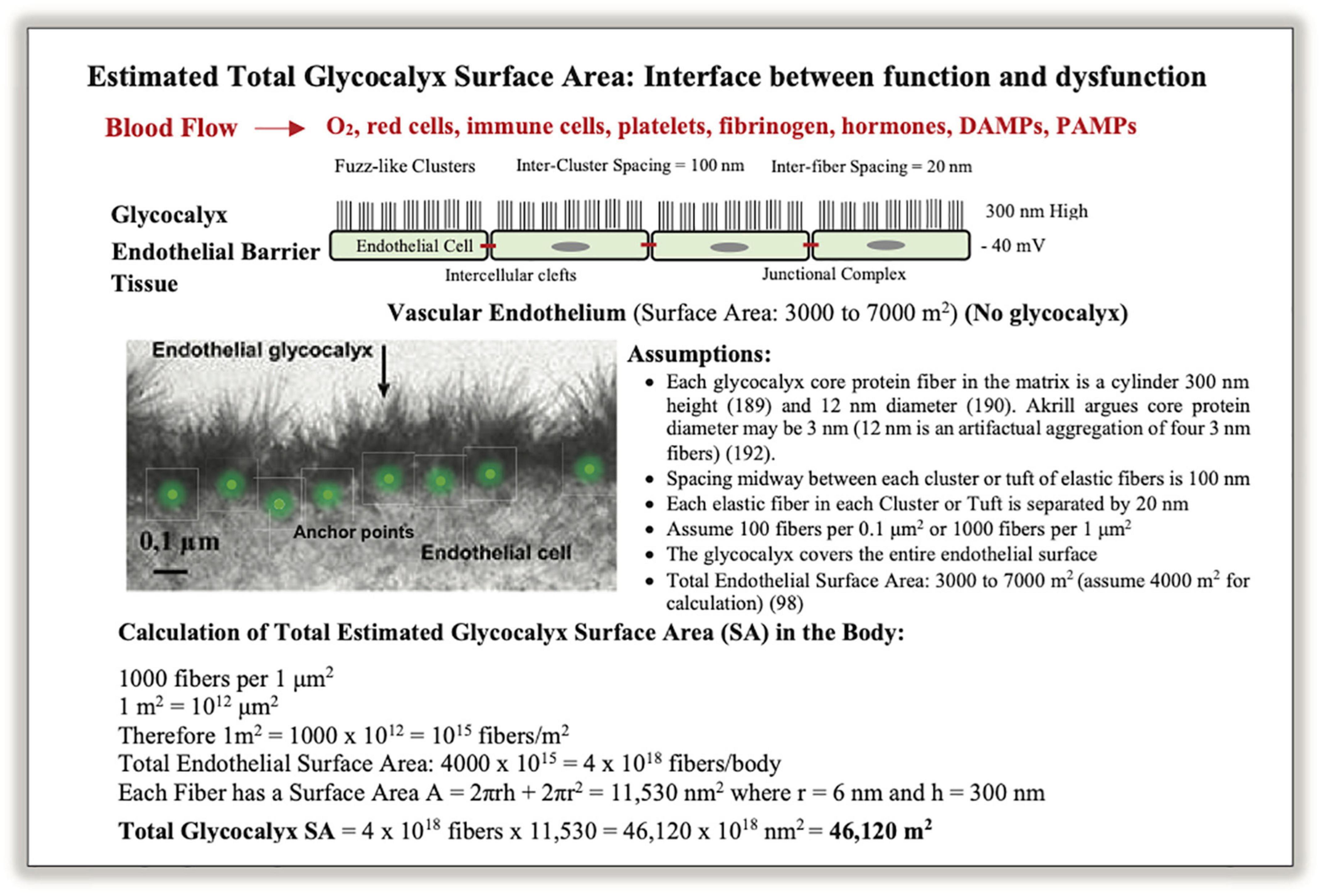
Figure 4. A schematic of the vascular endothelium and calculation of total glycocalyx surface area (SA) in humans. Photo insert was modified from Chappell et al. (186). Glycocalyx fibers appear in clusters composed of proteoglycans, glycosaminoglycans, and glycoproteins, which are anchored into endothelial cells by core-proteins (113, 115, 187, 188). Together they form a dynamic structure that participates in shear stress regulation, barrier protection, vascular permeability, inflammation, coagulopathy, fibrinolysis, mechanotransduction, immune function and cytokine signaling (97, 101, 104, 116). The glycocalyx is difficult to characterize because of its fragility and instability, and its structural dimensions critically depend on the method of ultrastructural visualization (97, 116, 189–193). The SA calculation should be viewed as approximate. A glycocalyx SA of 44,120 m2 for material exchange equates to ∼200 tennis courts or over 8 US football fields (see text).
When damaged by inflammatory mechanisms, the endothelium can rapidly shed its glycocalyx “fuzz” via sheddases, and release nanoscale bioactives and DAMPs, such as thrombomodulin, syndecan-1, heparan sulfate, hyaluronic acid, and other proteoglycans and glycoproteins, into the circulation (96, 104, 107, 116–119). This degradation is believed to perpetuate immuno-inflammation and coagulopathy (13, 120–125), immunosuppression (102, 126, 127) and mitochondrial dysfunction (115, 128, 129). SHOT predicts if VA coupling is close to unity and tissue perfusion and O2 exchange can be maintained, damage to the endothelium-glycocalyx will be minimized and these secondary injury processes reduced (Figure 3). Remarkably, if adequate tissue perfusion can be restored, the damaged glycocalyx has the capacity to repair itself (130, 131). Timing of repair appears to depend upon the duration and extent of hypoperfusion and ischemia, and the type and severity of trauma (130, 131).
Maintaining the functional integrity of mitochondria post-trauma is essential for a good outcome. Mitochondria are sensor organelles of ancient bacterial origins involved in ATP production, substrate regulation, immune cell signaling, calcium homeostasis, endoplasmic reticulum communication, and cell death regulation (32, 132–135). After severe trauma, prolonged hypoperfusion leads to mitochondrial damage. However, before damage occurs there is a switch from aerobic mitochondrial oxidative and to anaerobic glycolytic metabolism, which can only be sustained for short periods of time (136). Damage occurs from depletion of local glycogen stores, depolarization of the sarcolemma membrane, increased lactate, reduced pH, increases in cell Ca2+ loading, a fall in ATP phosphorylation and redox potentials, increased reactive oxygen species, reduced inner mitochondrial membrane proton pumping, opening of the permeability transition pore, collapse of mitochondrial membrane potential and finally the release of cytochrome C, and other DAMPs (32, 134, 135, 137–142). DAMPs from damaged mitochondria exacerbate CNS injury, cardiovascular dysfunction and secondary injury (143–145). According to SHOT, improving CNS protection and CNS-cardiovascular-endothelium coupling will improve tissue perfusion and protect mitochondrial integrity (146) (Figure 3).
In 2017, Johansson and colleagues introduced a model of SHock-INduced Endotheliopathy (SHINE) to better understand the underlying pathophysiological mechanisms for critically ill patients (147). Like SHOT, they propose that shock-induced sympatho-adrenal hyperactivation is a critical driver of endothelial cell and glycocalyx damage, hypoperfusion, and subsequent hemostatic aberrations and multiorgan dysfunction (110). More recently, Henriksen reported that patients with identical trauma severity developed significantly different degrees of endothelial dysfunction, as measured by syndecan-1, and proposed a minimum of four shock-induced endotheliopathy phenotypes (148) with the differences most likely driven by a genetic component (148). Moreover, they introduced a new research tool in trauma by using metabolic systems biology, which should be encouraged. A major difference between SHOT and SHINE is the functional linkage between CNS and VA coupling, which is testable. SHINE does not include this key linkage, which describes the coupling of cardiac and arterial vascular reactivity to optimally propel blood to deliver sufficient oxygen from the lungs to tissue mitochondria and prevent and/or reduce ongoing immuno-inflammatory dysfunction (discussed above).
How do we switch the injury phenotype of a polytrauma patient to a survival one? Why are there no effective drugs to treat immune dysregulation in the early hours to days following major trauma or in the critically ill patient? We argue the main reasons for lack of progress in drug development include:
1. Failure to replicate the heterogeneity of humans.
2. Poorly designed trials lacking diversity.
3. Inappropriate use of pathogen-free animals.
4. Ignoring sex-specific differences.
5. The flawed practice of single-nodal targeting.
The heterogeneity of the human condition is a major variable when conducting animal experiments to solve a medical problem (149). Preclinical models typically use animals from relatively homogeneous breeding colonies whereas humans are genetically, epigenetically, biologically and physiologically heterogeneous (149, 150). Large animals, such as pigs and sheep, do have some advantages with similar physiologies and/or anatomies as humans, however, they are more costly than using rodents (149, 150). A second confounding variable are poorly designed human trials that are either not sufficiently powered or recruit patients who do not adequately represent the wider population for which the drug therapy is intended (151–153).
Similar problems apply to preclinical models that use specific pathogen-free (SPF) animals. SPF animals were introduced in the early 1960s to minimize disease or infection as an unwanted variable in experimental design (151, 154). However, SPF animals have different gut microbiota that can profoundly influence basic physiology, stress behaviors and the immuno-inflammatory response to trauma (151–153, 155). Beura et al. showed that SPF adult mice, for example, have “immature” immune systems that were more prone to infection than conventionally bred mice (156). SPF animals may be useful for studying specific questions in biochemical mechanisms, but they do not mimic the patient following trauma (152). The current consensus is that conventionally bred animals are the animals of choice if translation of a new drug therapy is the end-game (151). In addition, the mouse model may be problematic for trauma studies because unlike rats, guinea-pigs, pigs, sheep, dogs, and humans, mice can enter a dormant state, called torpor, when subjected to traumatic stress (157, 158). Torpor itself can profoundly change the animal’s immune system by reducing the numbers of circulating leukocytes, lowering complement levels, and changing the animal’s response to infection (159).
The other important variable in preclinical and clinical studies is sex. An increasing number of animal and human studies show sex-specific differences in pathophysiological responses to polytrauma, hemorrhagic shock, TBI and burns (160–162). Chaudry and colleagues have been emphasizing the importance of sex in biochemical research for over two decades. They showed that administration of female sex hormone 17β-estradiol in males and ovariectomized females after trauma-hemorrhage prevented the suppression of immune response (163, 164). On the basis of accumulated data, greater inclusion of females in preclinical modeling and translation has been earmarked by the National Institutes of Health (NIH), European Commission, US Department of Defense and FDA (151–153).
Lastly, the practice of single-nodal targeting is another factor for why there are no effective systems-acting drugs for the polytrauma patient. Past drug development efforts have focused more on alleviating symptoms rather than addressing an underlying problem. The current practice of treat-as-you-go using sequential, single-target therapies leads to what US surgeon William C. Shoemaker considered: “an uncoordinated and sometimes contradictory therapeutic outcome” (165). Targeting individual pro-inflammatory cytokines, or any single step along a signaling pathway, ignores the critical importance of the system. Single-nodal thinking rarely solves a medical problem unless the site is believed to be a central hub or upstream intersection point. The IL-1 receptor has been proposed to be such a target, and while anakinra (IL-1 antagonist) has an excellent safety record, further trials are required to demonstrate its clinical efficacy after trauma or infection (166, 167). Reductionism in scientific discovery is important in breaking a system into its constituent parts, however, it does not do away with the system (151–153). This flawed way of thinking, we believe, is a major contributor for the high failure rate of translating promising new drugs in clinical trials (168). Choosing the right model and experimental design, a systems approach is much more likely to increase animal-to-human translational success to improve trauma care.
If you control hemorrhage and infection, the patient will do the recovery, since every cell in his body is working hard in that direction already. But you must understand what those cells are doing so that you can help them.
Walter B. Cannon [Moore, (169) p. 816].
We have been developing a small-volume intravenous (IV) ALM fluid therapy to treat polytrauma for civilian and military use (12, 16). In different animal models, ALM confers a survival advantage after hemorrhagic shock (12, 16, 170, 171), traumatic injury (170–174), sepsis (175, 176) and endotoxin insult (177). The ALM survival phenotype is not replicated with individual actives adenosine, lidocaine or magnesium (12, 16). ALM confers its benefit by shifting CNS function from sympathetic to parasympathetic dominance (178), blunting inflammation (172), correcting coagulopathy (179), maintaining VA coupling, improving tissue blood flow, lowering energy demand and protecting mitochondria (178). Studies carried out by US Army Institute of Surgical Research also showed that ALM therapy restored 97% of endothelial glycocalyx after severe hemorrhagic shock (180). Currently, we don’t know how and when the “switch” from an injury phenotype to a survival phenotype occurs, however, we suspect it is early because the same 5 h therapy confers dual protection against trauma and infection (12, 14, 16). It is possible ALM may act in the first minutes to hours after administration to assist the body to develop a “normal” immune response with timely resolution of the immuno-inflammatory genomic storms. While the preclinical ALM data appear promising, translation to humans remains challenging given the failure rate of translating new drugs into humans exceeds 95% (181), and of those that do obtain FDA approval, around 30% show postmarket safety concerns (182). Understanding the underlying mechanisms of action of ALM is vital for safe translation.
Trauma is a leading cause of death and disability worldwide. Currently there are no effective drug therapies to reduce hyperinflammation and immune dysfunction, immunosuppression, infection and MODS following major trauma. The present treat-as-you-go approaches fail to appreciate that immuno-inflammatory complications are a systems failure, and not a single nodal failure. New therapies are required to target the CNS control of cardiovascular function, endothelial-glycocalyx shedding, tissue O2 supply and its mitochondrial circuitry in both homeostatic and pathophysiological processes to prevent those complications.
GD: concept. GD, JM, and HL: data collection, data analyses, interpretation, and manuscript preparation and editing. All authors contributed equally to the design, implementation, literature analysis and writing of the manuscript.
This work was supported by the US Department of Defense (Award No. W81XWH-21-MBRP-IDA; Log No. MB210101).
We thank the College of Medicine and Dentistry, James Cook University, and the US Department of Defense for their continued support.
GD is the sole inventor of the ALM concept for cardiac surgery, trauma and sepsis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the US Department of the Navy, Department of the Army, Department of Defense, nor the U.S. Government.
1. Bedard AF, Mata LV, Dymond C, Moreira F, Dixon J, Schauer SG, et al. A scoping review of worldwide studies evaluating the effects of prehospital time on trauma outcomes. Int J Emerg Med. (2020) 13:64. doi: 10.1186/s12245-020-00324-7
2. Dobson GP, Morris JL, Letson HL. Why are bleeding trauma patients still dying? Towards a systems hypothesis of trauma. Front Physiol. (2022) 13:990903. doi: 10.3389/fphys.2022.990903
3. Rauf R, Von Matthey F, Croenlein M, Zyskowski M, Van Griensven M, Biberthaler P, et al. Changes in the temporal distribution of in-hospital mortality in severely injured patients-an analysis of the TraumaRegister DGU. PLoS One. (2019) 14:e0212095. doi: 10.1371/journal.pone.0212095
4. Brohi K, Gruen RL, Holcomb JB. Why are bleeding trauma patients still dying? Intensive Care Med. (2019) 45:709–11. doi: 10.1007/s00134-019-05560-x
5. Lee C, Rasmussen TE, Pape HC, Gary JL, Stannard JP, Haller JM. The polytrauma patient: Current concepts and evolving care. OTA Int. (2021). 4:e108–14.
6. Lansink KW, Gunning AC, Leenen LP. Cause of death and time of death distribution of trauma patients in a Level I trauma centre in the Netherlands. Eur J Trauma Emerg Surg. (2013) 39:375–83. doi: 10.1007/s00068-013-0278-2
7. Rosenthal MD, Moore FA. Persistent Inflammation, Immunosuppression, and catabolism: evolution of multiple organ dysfunction. Surg Infect (Larchmt). (2016) 17:167–72. doi: 10.1089/sur.2015.184
8. Efron PA, Mohr AM, Bihorac A, Horiguchi H, Hollen MK, Segal MS, et al. Persistent inflammation, immunosuppression, and catabolism and the development of chronic critical illness after surgery. Surgery. (2018) 164:178–84. doi: 10.1016/j.surg.2018.04.011
9. Osuka A, Ogura H, Ueyama M, Shimazu T, Lederer JA. Immune response to traumatic injury: harmony and discordance of immune system homeostasis. Acute Med Surg. (2014) 1:63–9. doi: 10.1002/ams2.17
10. Mas-Celis F, Olea-Lopez J, Parroquin-Maldonado JA. Sepsis in trauma: a deadly complication. Arch Med Res. (2021) 52:808–16. doi: 10.1016/j.arcmed.2021.10.007
11. Kotwal RS, Staudt AM, Trevino JD, Valdez-Delgado KK, Le TD, Gurney JM, et al. A review of casualties transported to role 2 medical treatment facilities in Afghanistan. Mil Med. (2018) 183(Suppl. 1):134–45. doi: 10.1097/01.ccm.0000528322.39346.3a
12. Dobson GP, Letson HL. Far forward gaps in hemorrhagic shock and prolonged field care: an update of ALM fluid therapy for field use. J Spec Oper Med. (2020) 20:78–84. doi: 10.55460/06VT-9IH4
13. Dobson GP, Morris JL, Davenport LM, Letson HL. Traumatic-induced coagulopathy as a systems failure: a new window into hemostasis. Semin Thromb Hemost. (2020) 46:199–214. doi: 10.1055/s-0039-1701018
14. Dobson GP, Biros E, Letson HL, Morris JL. Living in a hostile world: inflammation, new drug development and coronavirus. Front Immunol. (2021) 11:610131. doi: 10.3389/fimmu.2020.610131
15. Cannon WB. Organisation for physiological homeostasis. Physiol Rev. (1929) 9:399–431. doi: 10.1152/physrev.1929.9.3.399
16. Dobson GP, Letson HL. Adenosine, lidocaine and Mg2+ (ALM): from cardiac surgery to combat casualty care: teaching old drugs new tricks. J Trauma Acute Care Surg. (2016) 80:135–45. doi: 10.1097/TA.0000000000000881
17. Dobson GP. Organ arrest, protection and preservation: natural hibernation to cardiac surgery: a review. Comp Biochem Physiol Part B. (2004) 139:469–85. doi: 10.1016/j.cbpc.2004.06.002
18. Egiazaryan GG, Sudakov KV. Theory of functional systems in the scientific school of P.K. Anokhin. J Hist Neurosci. (2007) 16:194–205. doi: 10.1080/09647040600602805
19. Carlson DL, Horton JW. Cardiac molecular signaling after burn trauma. J Burn Care Res. (2006) 27:669–75. doi: 10.1097/01.BCR.0000237955.28090.41
20. Dobson GP. Trauma of major surgery: a global problem that is not going away. Int J Surg. (2020) 81:47–54. doi: 10.1016/j.ijsu.2020.07.017
21. Metchnikoff E. Lectures on the comparative pathology of inflammation, delivered at the Pasteur institute in 1891. In: E Metchnikoff editor. Lectures on the Comparative Pathology of Inflammation. (Translated by F. A. Starling and E. H. Starling.). New York, NY: Dover Publications (1968).
22. Chou C, Li MO. Tissue-resident lymphocytes across innate and adaptive lineages. Front Immunol. (2018) 9:2104. doi: 10.3389/fimmu.2018.02104
23. Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell (2016) 164:1198–211. doi: 10.1016/j.cell.2016.02.048
24. Roehr B. Tissue resident memory cells emerging as key player in health and disease. Proc Natl Acad Sci USA. (2017) 114:12092–3. doi: 10.1073/pnas.1715754114
25. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
26. Klose CSN, Artis D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res. (2020) 30:475–91. doi: 10.1038/s41422-020-0323-8
27. Galli SJ, Gaudenzio N. Human mast cells as antigen-presenting cells: when is this role important in vivo? J Allergy Clin Immunol. (2018) 141:92–3. doi: 10.1016/j.jaci.2017.05.029
28. Dorrington MG, Fraser IDC. NF-kappaB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol. (2019) 10:705. doi: 10.3389/fimmu.2019.00705
29. Allie SR, Randall TD. Resident memory B cells. Viral Immunol. (2020). [Epub ahead of print]. doi: 10.1089/vim.2019.0141
30. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. (2007) 81:1–5. doi: 10.1189/jlb.0306164
31. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
32. Hauser CJ, Otterbein LE. Danger signals from mitochondrial DAMPS in trauma and post-injury sepsis. Eur J Trauma Emerg Surg. (2018) 44:317–24. doi: 10.1007/s00068-018-0963-2
33. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
34. Muire PJ, Schwacha MG, Wenke JC. Systemic T cell exhaustion dynamics is linked to early high mobility group box protein 1 (HMGB1) driven hyper-inflammation in a polytrauma rat model. Cells. (2021) 10:1646. doi: 10.3390/cells10071646
35. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. (2010) 2010:1–21. doi: 10.1155/2010/672395
36. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. (2011) 208:2581–90. doi: 10.1084/jem.20111354
37. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, Mckinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. (2012) 72:1491–501. doi: 10.1097/TA.0b013e318256e000
38. Manson J, Cole E, De’ath HD, Vulliamy P, Meier U, Pennington D, et al. Early changes within the lymphocyte population are associated with the development of multiple organ dysfunction syndrome in trauma patients. Crit Care. (2016) 20:176. doi: 10.1186/s13054-016-1341-2
39. Thompson KB, Krispinsky LT, Stark RJ. Late immune consequences of combat trauma: a review of trauma-related immune dysfunction and potential therapies. Mil Med Res. (2019) 6:11. doi: 10.1186/s40779-019-0202-0
40. Sturm R, Xanthopoulos L, Heftrig D, Oppermann E, Vrdoljak T, Dunay IR, et al. Regulatory T cells modulate CD4 proliferation after severe trauma via IL-10. J Clin Med. (2020) 9:1052. doi: 10.3390/jcm9041052
41. Vazquez AC, Arriaga-Pizano L, Ferat-Osorio E. Cellular markers of immunosuppression in sepsis. Arch Med Res. (2021) 52:828–35. doi: 10.1016/j.arcmed.2021.10.001
42. Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Miyazaki M. Immunosuppression following surgical and traumatic injury. Surg Today. (2010) 40:793–808. doi: 10.1007/s00595-010-4323-z
43. Yang Y, Ye Y, Chen C, Kong C, Su X, Zhang X, et al. Acute traumatic brain injury induces CD4+ and CD8+ T cell functional impairment by upregulating the expression of PD-1 via the activated sympathetic nervous system. Neuroimmunomodulation. (2019) 26:43–57. doi: 10.1159/000495465
44. Ruan X, Darwiche SS, Cai C, Scott MJ, Pape HC, Billiar TR. Anti-HMGB1 monoclonal antibody ameliorates immunosuppression after peripheral tissue trauma: attenuated T-lymphocyte response and increased splenic CD11b (+) Gr-1 (+) myeloid-derived suppressor cells require HMGB1. Mediators Inflamm. (2015) 2015:458626. doi: 10.1155/2015/458626
45. Islam MN, Bradley BA, Ceredig R. Sterile post-traumatic immunosuppression. Clin Transl Immunol. (2016) 5:e77. doi: 10.1038/cti.2016.13
46. Campbell B, Budreau D, Williams-Perez S, Chakravarty S, Galet C, Mcgonagill P. Admission lymphopenia predicts infectious complications and mortality in traumatic brain injury victims. Shock. (2022) 57:189–98. doi: 10.1097/SHK.0000000000001872
47. Wang J-W, Li J-P, Song Y-L, Zhao Q-H. Humoral and cellular immunity changed after traumatic brain injury in human patients. Ann Clin Lab Sci. (2017) 47:10–6.
48. Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. (2010) 10:467–78. doi: 10.1038/nri2781
49. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
50. Muller-Heck RM, Bosken B, Michiels I, Dudda M, Jager M, Flohe SB. Major surgical trauma impairs the function of natural killer cells but does not affect monocyte cytokine synthesis. Life (Basel). (2021) 12:13. doi: 10.3390/life12010013
51. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. (2020) 11:484. doi: 10.3389/fimmu.2020.00484
52. Kumar R, Herbert PE, Warrens AN. An introduction to death receptors in apoptosis. Int J Surg. (2005) 3:268–77. doi: 10.1016/j.ijsu.2005.05.002
53. Arnold R, Brenner D, Becker M, Frey CR, Krammer PH. How T lymphocytes switch between life and death. Eur J Immunol. (2006) 36:1654–8. doi: 10.1002/eji.200636197
54. Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. (2007) 7:532–42. doi: 10.1038/nri2115
55. Brenner D, Krammer PH, Arnold R. Concepts of activated T cell death. Crit Rev Oncol Hematol. (2008) 66:52–64. doi: 10.1016/j.critrevonc.2008.01.002
56. Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. (2010) 2010:214074. doi: 10.1155/2010/214074
57. Roth S, Cao J, Singh V, Tiedt S, Hundeshagen G, Li T, et al. Post-injury immunosuppression and secondary infections are caused by an AIM2 inflammasome-driven signaling cascade. Immunity. (2021) 54:648–659.e8. doi: 10.1016/j.immuni.2021.02.004
58. Bock M, Bergmann CB, Jung S, Biberthaler P, Heimann L, Hanschen M. Platelets differentially modulate CD4(+) Treg activation via GPIIa/IIIb-, fibrinogen-, and PAR4-dependent pathways. Immunol Res. (2022) 70:185–96. doi: 10.1007/s12026-021-09258-5
59. Abe R, Hirasawa H, Oda S, Sadahiro T, Nakamura M, Watanabe E, et al. Up-regulation of interleukin-10 mRNA expression in peripheral leukocytes predicts poor outcome and diminished human leukocyte antigen-DR expression on monocytes in septic patients. J Surg Res. (2008) 147:1–8. doi: 10.1016/j.jss.2007.07.009
60. Desborough JP. The stress response to trauma and surgery. Br J Anaesth. (2000) 85:109–17. doi: 10.1093/bja/85.1.109
61. Ballard-Croft C, Maass DL, Sikes P, White JM, Horton J. Activation of stress-responsive pathways by the sympathetic nervous system in burn trauma. Shock. (2002) 18:38–45. doi: 10.1097/00024382-200207000-00008
62. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. (2006) 6:318–28. doi: 10.1038/nri1810
63. Hotamisligil GS, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. (2016) 8:a006072. doi: 10.1101/cshperspect.a006072
64. Powell K, Shah K, Hao C, Wu YC, John A, Narayan RK, et al. Neuromodulation as a new avenue for resuscitation in hemorrhagic shock. Bioelectron Med. (2019) 5:17. doi: 10.1186/s42234-019-0033-z
65. Gans IM, Coffman JA. Glucocorticoid-mediated developmental programming of vertebrate stress responsivity. Front Physiol. (2021) 12:812195. doi: 10.3389/fphys.2021.812195
66. Prete A, Yan Q, Al-Tarrah K, Akturk HK, Prokop LJ, Alahdab F, et al. The cortisol stress response induced by surgery: a systematic review and meta-analysis. Clin Endocrinol (Oxf). (2018) 89:554–67. doi: 10.1111/cen.13820
67. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialog Clin Neurosci. (2006) 8:383–95. doi: 10.31887/DCNS.2006.8.4/ssmith
68. Charkoudian N, Wallin BG. Sympathetic neural activity to the cardiovascular system: integrator of systemic physiology and interindividual characteristics. Compr Physiol. (2014) 4:825–50. doi: 10.1002/cphy.c130038
69. Burford NG, Webster NA, Cruz-Topete D. Hypothalamic-pituitary-adrenal axis modulation of glucocorticoids in the cardiovascular system. Int J Mol Sci. (2017) 18:2150. doi: 10.3390/ijms18102150
70. Barman SM. 2019 Ludwig lecture: rhythms in sympathetic nerve activity are a key to understanding neural control of the cardiovascular system. Am J Physiol Regul Integr Comp Physiol. (2020) 318:R191–205. doi: 10.1152/ajpregu.00298.2019
71. Pavlov VA, Tracey KJ. Neural regulators of innate immune responses and inflammation. Cell Mol Life Sci. (2004) 61:2322–31. doi: 10.1007/s00018-004-4102-3
72. Huston JM. The vagus nerve and the inflammatory reflex: wandering on a new treatment paradigm for systemic inflammation and sepsis. Surg Infect (Larchmt). (2012) 13:187–93. doi: 10.1089/sur.2012.126
74. Sudo N. Microbiome, HPA axis and production of endocrine hormones in the gut. In: M Lyte, J Cryan editors. Microbiology Endocrinology: The Microbiota-Gut Brain Axis in Health and Disease. New York, NY: Springer (2014). doi: 10.1007/978-1-4939-0897-4_8
75. Mayer EA, Tillisch K, Gupta A. Gut/brain axis and the microbiota. J Clin Invest. (2015) 125:926–38. doi: 10.1172/JCI76304
76. Crile G. Nitrous oxide anaesthesia and a note on anoci-association, a new principle in operative surgery. Surg Gynecol Obstet. (1911) 13:170–3.
77. Montagne A, Toga AW, Zlokovic BV. Blood-brain barrier permeability and gadolinium: benefits and potential pitfalls in research. JAMA Neurol. (2016) 73:13–4. doi: 10.1001/jamaneurol.2015.2960
78. Turnbull AV, Rivier C. Regulation of the HPA axis by cytokines. Brain Behav Immun. (1995) 9:253–75. doi: 10.1006/brbi.1995.1026
79. Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. (2020) 17:69. doi: 10.1186/s12987-020-00230-3
80. Dobson GP, Morris JL, Biros E, Davenport LM, Letson HL. Major surgery leads to a proinflammatory phenotype: differential gene expression following a laparotomy. Ann Med Surg (Lond). (2021) 71:102970. doi: 10.1016/j.amsu.2021.102970
81. Osler W. The Evolution of Modern Medicine. New Haven, CT: Yale, University Press (1921). p. 243.
82. Dobson GP. Addressing the global burden of trauma in major surgery. Front Surg. (2015) 2:43. doi: 10.3389/fsurg.2015.00043
83. Suga H, Goto Y, Kawaguchi O, Hata K, Takasago T, Saeki TW, et al. Ventricular perspective on efficiency. In: D Burkhoff, J Schaefer, K Schaffner, DT Yue editors. Myocardial Optimization and Efficiency, Evolutionary Aspects and Philosophy of Science Considerations. New York, NY: Springer-Verlag (1993). p. 43–65.
84. London GM. Role of arterial wall properties in the pathogenesis of systolic hypertension. Am J Hypertens. (2005) 18(1 Pt 2):19S–22S. doi: 10.1016/j.amjhyper.2004.10.001
85. Kass DA. Ventricular arterial stiffening: integrating the pathophysiology. Hypertension. (2005) 46:185–93. doi: 10.1161/01.HYP.0000168053.34306.d4
86. Guarracino F, Baldassarri R, Pinsky MR. Ventriculo-arterial decoupling in acutely altered hemodynamic states. Crit Care. (2013) 17:213–20. doi: 10.1186/cc12522
87. Cholley B, Le Gall A. Ventriculo-arterial coupling: the comeback? J Thorac Dis. (2016) 8:2287–9. doi: 10.21037/jtd.2016.08.34
88. Dobson GP, Arsyad A, Letson HL. The adenosine hypothesis revisited: a possible role for arterial compliance and its implications to coronary perfusion. Front Physiol. (2017) 8:824. doi: 10.3389/fphys.2017.00824
89. Onorati F, Santini F, Dandale R, Ucci G, Pechlivanidis K, Menon T, et al. “Polarizing” microplegia improves cardiac cycle efficiency after CABG for unstable angina. Int J Cardiol. (2013) 167:2739–46. doi: 10.1016/j.ijcard.2012.06.099
90. Granfeldt A, Letson HL, Hyldebrandt JA, Wang ER, Salcedo PA, Nielson TK, et al. Small-volume 7.5% NaCl adenosine, lidocaine and Mg2+ has multiple benefits during hypotensive and blood resuscitation in the pig following severe blood loss: rat to pig translation. Crit Care Med. (2014) 42:e329–44. doi: 10.1097/CCM.0000000000000225
91. Ye Z, Coutinho T, Pellikka PA, Villarraga HR, Borlaug BA, Kullo IJ. Associations of alterations in pulsatile arterial load with left ventricular longitudinal strain. Am J Hypertens. (2015) 28:1325–31. doi: 10.1093/ajh/hpv039
92. Antonini-Canterin F, Poli S, Vriz O, Pavan D, Bello VD, Nicolosi GL. The ventricular-arterial coupling: from basic pathophysiology to clinical application in the echocardiography laboratory. J Cardiovasc Echogr. (2013) 23:91–5. doi: 10.4103/2211-4122.127408
93. Ky B, French B, May Khan A, Plappert T, Wang A, Chirinos JA, et al. Ventricular-arterial coupling, remodeling, and prognosis in chronic heart failure. J Am Coll Cardiol. (2013) 62:1165–72. doi: 10.1016/j.jacc.2013.03.085
94. Howard BM, Kornblith LZ, Christie SA, Conroy AS, Nelson MF, Campion EM, et al. Characterizing the gut microbiome in trauma: significant changes in microbial diversity occur early after severe injury. Trauma Surg Acute Care Open. (2017) 2:e000108. doi: 10.1136/tsaco-2017-000108
95. Wiel E, Vallet B, ten Cate H. The endothelium in intensive care. Crit Care Clin. (2005) 21:403–16. doi: 10.1016/j.ccc.2005.03.001
96. Bennett HS. Morphological aspects of extracellular polysaccharides. J Histochem Cytochem. (1963) 11:14–23. doi: 10.1177/11.1.14
97. Luft JH. The structure and properties of the cell surface coat. Int Rev Cytol. (1976) 45:291–382. doi: 10.1016/S0074-7696(08)60081-9
98. Aird WC. Spatial and temporal dynamics of the endothelium. J Thromb Haemost. (2005) 3:1392–406. doi: 10.1111/j.1538-7836.2005.01328.x
99. Johansson PI, Henriksen HH, Stensballe J, Gybel-Brask M, Cardenas JC, Baer LA, et al. Traumatic endotheliopathy: a prospective observational study of 424 severely injured patients. Ann Surg. (2017) 265:597–603. doi: 10.1097/SLA.0000000000001751
100. Gonzalez Rodriguez E, Ostrowski SR, Cardenas JC, Baer LA, Tomasek JS, Henriksen HH, et al. Syndecan-1: a quantitative marker for the endotheliopathy of trauma. J Am Coll Surg. (2017) 225:419–27. doi: 10.1016/j.jamcollsurg.2017.05.012
101. Halbgebauer R, Braun CK, Denk S, Mayer B, Cinelli P, Radermacher P, et al. Hemorrhagic shock drives glycocalyx, barrier and organ dysfunction early after polytrauma. J Crit Care. (2018) 44:229–37. doi: 10.1016/j.jcrc.2017.11.025
102. Richards JE, Samet RE, Grissom TE. Scratching the surface: endothelial damage in traumatic hemorrhagic shock. Adv Anesth. (2021) 39:35–51. doi: 10.1016/j.aan.2021.07.003
103. Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc Pharm. (2003) 39:173–85. doi: 10.1016/S1537-1891(03)00007-7
104. Reitsma S, Slaaf DW, Vink H, Van Zandvoort MA, Oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. (2007) 454:345–59. doi: 10.1007/s00424-007-0212-8
105. Chappell D, Westphal M, Jacob M. The impact of the glycocalyx on microcirculatory oxygen distribution in critical illness. Curr Opin Anaesthesiol. (2009) 22:155–62. doi: 10.1097/ACO.0b013e328328d1b6
106. Biddle C. Like a slippery fish, a little slime is a good thing: the glycocalyx revealed. AANA J. (2013) 81:473–80.
107. Aditianingsih D, George YWH. Guiding principles of fluid and volume therapy. Best Pract Res Clin Anaesthesiol. (2014) 28:249–60. doi: 10.1016/j.bpa.2014.07.002
108. Gall LS, Vulliamy P, Gillespie S, Jones TF, Pierre RSJ, Breukers SE, et al. The S100A10 pathway mediates an occult hyperfibrinolytic subtype in trauma patients. Ann Surg. (2019) 269:1184–91. doi: 10.1097/SLA.0000000000002733
109. Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers. (2021) 7:30. doi: 10.1038/s41572-021-00264-3
110. Krocker JD, Lee KH, Henriksen HH, Wang Y-WW, Schoof EM, Karvelsson ST, et al. Exploratory investigation of the plasma proteome associated with the endotheliopathy of trauma. Int J Mol Sci. (2022) 23:6213. doi: 10.3390/ijms23116213
111. Fu BM, Tarbell JM. Mechano-sensing and transduction by endothelial surface glycocalyx: composition, structure, and function. Wiley Interdiscip Rev Syst Biol Med. (2013) 5:381–90. doi: 10.1002/wsbm.1211
112. van Hinsbergh VW. Endothelium–role in regulation of coagulation and inflammation. Semin Immunopathol. (2012) 34:93–106. doi: 10.1007/s00281-011-0285-5
113. Schott U, Solomon C, Fries D, Bentzer P. The endothelial glycocalyx and its disruption, protection and regeneration: a narrative review. Scand J Trauma Resusc Emerg Med. (2016) 24:48. doi: 10.1186/s13049-016-0239-y
114. Hahn RG. Water content of the endothelial glycocalyx layer estimated by volume kinetic analysis. Intensive Care Med Exp. (2020) 8:29. doi: 10.1186/s40635-020-00317-z
115. Moore KH, Murphy HA, George EM. The glycocalyx: a central regulator of vascular function. Am J Physiol Regul Integr Comp Physiol. (2021) 320:R508–18. doi: 10.1152/ajpregu.00340.2020
116. Luft JH. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed Proc. (1966) 25:1773–83.
117. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. (2004) 84:869–901. doi: 10.1152/physrev.00035.2003
118. Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth. (2012) 108:384–94. doi: 10.1093/bja/aer515
119. Chappell D, Jacob M. Role of the glycocalyx in fluid management: small things matter. Best Pract Res Clin Anaesthesiol. (2014) 28:227–34. doi: 10.1016/j.bpa.2014.06.003
120. Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. (2007) 245:812–8. doi: 10.1097/01.sla.0000256862.79374.31
121. Holcomb JB, Jenkins D, Rhee P, Johannigman J, Mahoney PF, Mehta S, et al. Damage control resuscitation: directly addressing the early coagulopathy of trauma. J Trauma. (2007) 62:307–10. doi: 10.1097/TA.0b013e3180324124
122. Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, et al. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. (2014) 77:811–7. doi: 10.1097/TA.0000000000000341
123. Dobson GP, Letson HL, Sharma R, Sheppard F, Cap AP. Mechanisms of early traumatic-induced coagulopathy (TIC): the clot thickens or not? J Trauma Acute Care Surg. (2015) 79:301–9. doi: 10.1097/TA.0000000000000729
124. D’elia RV, Harrison K, Oyston PC, Lukaszewski RA, Clark GC. Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol. (2013) 20:319–27. doi: 10.1128/CVI.00636-12
125. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA. (2015) 72:355–62. doi: 10.1001/jamaneurol.2014.3558
126. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin. (2017) 33:245–58. doi: 10.1016/j.ccc.2016.12.001
127. Qasim Z, Butler FK, Holcomb JB, Kotora JG, Eastridge BJ, Brohi K, et al. Selective prehospital advanced resuscitative care – developing a strategy to prevent prehospital deaths from noncompressible torso hemorrhage. Shock. (2022) 57:7–14. doi: 10.1097/SHK.0000000000001816
128. Moore EE, Moore FA, Harken AH, Johnson JL, Ciesla D, Banerjee A. The two-event construct of postinjury multiple organ failure. Shock. (2005) 24(Suppl. 1):71–4. doi: 10.1097/01.shk.0000191336.01036.fe
129. Dewar DC, Butcher NE, King KL, Balogh ZJ. Post injury multiple organ failure. Trauma. (2011) 13:81–91. doi: 10.1177/1460408610386657
130. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One. (2014) 9:e86249. doi: 10.1371/journal.pone.0086249
131. Hahn RG, Patel V, Dull RO. Human glycocalyx shedding: systematic review and critical appraisal. Acta Anaesthesiol Scand. (2021) 65:590–606. doi: 10.1111/aas.13797
132. Dobson GP. On being the right size: heart design, mitochondrial efficiency, and lifespan potential. Clin Exp Pharm Physiol. (2003) 30:590–7. doi: 10.1046/j.1440-1681.2003.03876.x
133. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. (2013) 112:1171–88. doi: 10.1161/CIRCRESAHA.111.300233
134. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders – a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
135. Berry BJ, Trewin AJ, Amitrano AM, Kim M, Wojtovich AP. Use the protonmotive force: mitochondrial uncoupling and reactive oxygen species. J Mol Biol. (2018) 430:3873–91. doi: 10.1016/j.jmb.2018.03.025
136. Canyon SJ, Dobson GP. The effect of adenosine and lidocaine infusion on myocardial high energy phosphates and pH during regional ischemia in the rat model in vivo. Can J Physiol Pharmacol. (2006) 84:903–12. doi: 10.1139/y06-035
137. Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, et al. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. (2005) 38:367–74. doi: 10.1016/j.yjmcc.2004.12.001
138. Baines CP. The mitochondrial permeability transition pore as a target of cardioprotective signaling. Am J Physiol Heart Circ Physiol. (2007) 293:H903–4. doi: 10.1152/ajpheart.00575.2007
139. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. (2011) 11:389–402. doi: 10.1038/nri2975
140. Dehne N, Brüne B. Sensors, transmitters, and targets in mitochondrial oxygen shortage-a hypoxia-inducible factor relay story. Antioxid Redox Signal. (2014) 20:339–52. doi: 10.1089/ars.2012.4776
141. Kunkel GH, Chaturvedi P, Tyagi SC. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail Rev. (2016) 21:499–517. doi: 10.1007/s10741-016-9561-8
142. Cherry AD, Piantados CA. Regulation of mitochondrial biogenesis and its intersection with inflammatory responses. Antioxid Redox Signal. (2015) 22:965–76. doi: 10.1089/ars.2014.6200
143. Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, et al. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury–associated coagulopathy in mice. Blood. (2016) 127:2763–72. doi: 10.1182/blood-2015-12-688838
144. Cap AP, Hunt B. Acute traumatic coagulopathy. Curr Opin Crit Care. (2014) 20:638–45. doi: 10.1097/MCC.0000000000000158
145. Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. (2014) 124:2173–83. doi: 10.1182/blood-2014-05-573543
146. Riedel B, Rafat N, Browne K, Burbury K, Schier R. Perioperative implications of vascular endothelial dysfunction: current understanding of this critical sensor-effector organ. Curr Anesthesiol Rep. (2013) 3:151–61. doi: 10.1007/s40140-013-0024-7
147. Johansson PI, Stensballe J, Ostrowski SR. Shock induced endotheliopathy (SHINE) in acute critical illness - a unifying pathophysiologic mechanism. Crit Care. (2017) 21:25. doi: 10.1186/s13054-017-1605-5
148. Henriksen HH, Mcgarrity S, Sigurethardottir RS, Nemkov T, D’alessandro A, Palsson BO, et al. Metabolic systems analysis of shock-induced endotheliopathy (SHINE) in trauma: a new research paradigm. Ann Surg. (2020) 272:1140–8. doi: 10.1097/SLA.0000000000003307
149. Hart DA. Human heterogeneity and survival of the species: how did it arise and being sustained?—the conundrum facing researchers. J Biomed Sci Eng. (2021) 14:212–21. doi: 10.4236/jbise.2021.145018
150. Tsukamoto T, Pepe HC. Animal models for trauma research: what are the options? Shock. (2009) 31:3–10. doi: 10.1097/SHK.0b013e31817fdabf
151. Dobson GP, Letson HL, Biros E, Morris JL. Specific pathogen-free (SPF) animal status as a variable in biomedical research: have we come full circle? EBioMedicine (Lancet). (2019) 41:42–3. doi: 10.1016/j.ebiom.2019.02.038
152. Letson HL, Morris JL, Biros E, Dobson GP. Conventional and specific-pathogen free rats respond differently to anesthesia and surgical trauma. Sci Rep. (2019) 9:9399. doi: 10.1038/s41598-019-45871-z
153. Dobson GP, Morris J, Biros E, Letson HL. Specific pathogen-free animals for civilian and military trauma: a cautionary note in the translation of new drug therapies. Shock. (2020) 54:232–6. doi: 10.1097/SHK.0000000000001495
154. Foster HL. Housing of disease-free vertebrates. Ann N Y Acad Sci. (1959) 78:80–8. doi: 10.1111/j.1749-6632.1959.tb53096.x
155. Masopust D, Sivula CP, Jameson SC. Of mice, dirty mice and men: using mice to understand human immunology. J Immunol. (2017) 199:383–8. doi: 10.4049/jimmunol.1700453
156. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. (2016) 532:512–6. doi: 10.1038/nature17655
157. Schubert KA, Boerema AS, Vaanholt LM, De Boer SF, Strijkstra AM, Daan S. Daily torpor in mice: high foraging costs trigger energy-saving hypothermia. Biol Lett. (2010) 6:132–5. doi: 10.1098/rsbl.2009.0569
158. Dobson GP. The August Krogh principle: seeking unity in diversity. Shock. (2014) 42:480. doi: 10.1097/SHK.0000000000000229
159. Bouma HR, Carey HV, Kroese GM. Hibernation: the immune system at rest? J Leukocyte Biol. (2010) 88:619–24. doi: 10.1189/jlb.0310174
160. Franconi F, Campesi I, Colombo D, Antonini P. Sex-gender variable: methodological recommendations for increasing scientific value of clinical studies. Cells. (2019) 8:476. doi: 10.3390/cells8050476
161. Gölz C, Kirchhoff FP, Westerhorstmann J, Schmidt M, Hirnet T, Rune GM, et al. Sex hormones modulate pathogenic processes in experimental traumatic brain injury. J Neurochem. (2019). [Epub ahead of print]. doi: 10.1111/jnc.14678
162. Gupte R, Brooks WM, Vukas RR, Pierce JD, Harris JL. Sex differences in traumatic brain injury: what we know and what we should know. J Neurotrauma. (2019). [Epub ahead of print]. doi: 10.1089/neu.2018.6171
163. Choudhry MA, Bland KI, Chaudry IH. Trauma and immune response–effect of gender differences. Injury. (2007) 38:1382–91. doi: 10.1016/j.injury.2007.09.027
164. Chaudry IH, Bland KI. Cellular mechanisms of injury after major trauma. Br J Surg. (2009) 96:1097–8. doi: 10.1002/bjs.6697
165. Shoemaker WC, Beez M. Pathophysiology, monitoring, and therapy of shock with organ failure. Appl Cardiopul Pathophysiol. (2010) 14: 5–15.
166. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
167. Mehta P, Mcauley DF, Brown M, Sanchez E, Tattersall RS, Mansen JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
168. Moore TJ, Zhang H, Anderson G, Alexander GC. Estimated costs of pivotal trials for novel therapeutic agents approved by the US food and drug administration, 2015-2016. JAMA Intern Med. (2018) 178:1451–7. doi: 10.1001/jamainternmed.2018.3931
169. Moore FD. The growth of surgical biology. Ann Surg. (1953) 138:807–22. doi: 10.1097/00000658-195311000-00023
170. Letson HL, Granfeldt A, Jensen TH, Mattson TH, Dobson GP. ALM supports a high flow, hypotensive, vasodilatory state with improved O2 delivery and cerebral protection in a pig model of non-compressible hemorrhage. J Surg Res. (2020) 253:127–38. doi: 10.1016/j.jss.2020.03.048
171. Letson HL, Dobson GP. 3.0% NaCl adenosine, lidocaine, Mg2+ (ALM) bolus and 4 hours ‘drip’ infusion reduces non-compressible hemorrhage by 60% in a rat model. J Trauma Acute Care Surg. (2017) 82:1063–72. doi: 10.1097/TA.0000000000001454
172. Letson HL, Dobson GP. Adenosine, lidocaine and Mg2+ (ALM) fluid therapy attenuates systemic inflammation, platelet dysfunction and coagulopathy after non-compressible truncal hemorrhage. PLos One. (2017) 12:e0188144. doi: 10.1371/journal.pone.0188144
173. Letson HL, Dobson GP. Adenosine, lidocaine and Mg2+ (ALM) resuscitation fluid protects against experimental traumatic brain injury. J Trauma Acute Care Surg. (2018) 84:908–16. doi: 10.1097/TA.0000000000001874
174. Letson HL, Morris JL, Biros E, Dobson GP. ALM fluid therapy leads to 72 hr survival after hemorrhagic shock: a model for studying differential gene expression and extending biological time. J Trauma Acute Care Surg. (2019) 87:606–13. doi: 10.1097/TA.0000000000002397
175. Griffin MJ, Letson HL, Dobson GP. Adenosine, lidocaine and Mg2+ (ALM) induces a reversible hypotensive state, reduces lung edema and prevents coagulopathy in the rat model of polymicrobial sepsis. J Trauma Acute Care Surg. (2014) 77:471–8. doi: 10.1097/TA.0000000000000361
176. Griffin MJ, Letson HL, Dobson GP. Small-volume adenosine, lidocaine and Mg2+ (ALM) 4 hour infusion leads to 88% survival after 6 days of experimental sepsis in the rat without antibiotics. Clin Vaccine Immunol. (2016) 23:863–72. doi: 10.1128/CVI.00390-16
177. Granfeldt A, Letson HL, Dobson GP, Shi W, Vinten-Johansen J, Tonnesen E. Cardioprotective and anti-inflammatory effects of treatment with adenosine, lidocaine and Mg2+ in a porcine model of endotoxemia. Circulation. (2013) 18:682–70.
178. Letson HL, Biros E, Morris JL, Dobson GP. ALM fluid therapy shifts sympathetic hyperactivity to parasympathetic dominance in the rat model of non-compressible hemorrhagic shock. Shock. (2022) 57:264–73. doi: 10.1097/SHK.0000000000001886
179. Letson HL, Dobson GP. Differential contributions of platelets and fibrinogen to early coagulopathy in a rat model of hemorrhagic shock. Thromb Res. (2016) 141:58–61. doi: 10.1016/j.thromres.2016.03.007
180. Torres Filho IP, Torres LN, Salgado C, Dubick MA. Novel adjunct drugs reverse endothelial glycocalyx damage after hemorrhagic shock in rats. Shock. (2017) 48:583–9. doi: 10.1097/SHK.0000000000000895
181. Seyhan AA. Lost in translation: the valley of death across preclinical and clinical divide – identification of problems and overcoming obstacles. Transl Med Commun. (2019) 4:18. doi: 10.1186/s41231-019-0050-7
182. Downing NS, Shah ND, Aminawung JA, Pease AM, Zeitoun JD, Krumholz HM, et al. Postmarket safety events among novel therapeutics approved by the US food and drug administration between 2001 and 2010. JAMA. (2017) 317:1854–63. doi: 10.1001/jama.2017.5150
183. Huston JM, Tracey KJ. The pulse of inflammation: heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J Intern Med. (2011) 269:45–53. doi: 10.1111/j.1365-2796.2010.02321.x
184. Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. (2013) 62:1214–22. doi: 10.1136/gutjnl-2012-302550
185. Dobson GP. Addressing the global burden of sepsis: importance of a systems-based approach. Crit Care Med. (2014) 42:e797–8. doi: 10.1097/CCM.0000000000000595
186. Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, et al. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology. (2007) 107:776–84. doi: 10.1097/01.anes.0000286984.39328.96
187. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflügers Arch. (2000) 440:653–66. doi: 10.1007/s004240000307
188. Pillinger NL, Kam PCA. Endothelial glycocalyx: basic science and clinical implications. Anaesth Intensive Care. (2017) 45:3. doi: 10.1177/0310057X1704500305
189. Vink H, Duling BR. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res. (1996) 79:581–9. doi: 10.1161/01.RES.79.3.581
190. Squire JM, Chew M, Nneji G, Neal C, Barry J, Michel C. Quasi-periodic substructure in the microvessel endothelial glycocalyx: a possible explanation for molecular filtering? J Struct Biol. (2001) 136:239–55. doi: 10.1006/jsbi.2002.4441
191. Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proc Natl Acad Sci USA. (2003) 100:7988–95. doi: 10.1073/pnas.1332808100
192. Arkill KP, Knupp C, Michel CC, Neal CR, Qvortrup K, Rostgaard J, et al. Similar endothelial glycocalyx structures in microvessels from a range of mammalian tissues: evidence for a common filtering mechanism? Biophys J. (2011) 101:1046–56. doi: 10.1016/j.bpj.2011.07.036
Keywords: trauma, hemorrhage, immune, inflammation, mitochondria, system, ALM, cytokines
Citation: Dobson GP, Morris JL and Letson HL (2022) Immune dysfunction following severe trauma: A systems failure from the central nervous system to mitochondria. Front. Med. 9:968453. doi: 10.3389/fmed.2022.968453
Received: 14 June 2022; Accepted: 01 August 2022;
Published: 30 August 2022.
Edited by:
Klemens Horst, University Hospital RWTH Aachen, GermanyReviewed by:
Birte Weber, University Hospital Frankfurt, GermanyCopyright © 2022 Dobson, Morris and Letson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geoffrey P. Dobson, Z2VvZmZyZXkuZG9ic29uQGpjdS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.