 Selami Kocak Toprak
Selami Kocak Toprak
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 15 July 2022
Sec. Hematology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.967900
This article is part of the Research TopicConstructing New Motifs in HematologyView all 10 articles
Myelodysplastic syndromes (MDS) is a heterogeneous group of disorders characterized by increased risk of acute myeloid leukemia transformation and cytopenia. The prognosis of MDS patients can be evaluated with various scoring systems, the most commonly used are IPSS (International Prognostic Scoring System), revised-IPSS, and WPSS (WHO classification-based prognostic scoring system). MDS treatment is decided according to the risk classification. The goal of treatment in low-risk MDS is to improve cytopenia, reduce transfusion needs, improve quality of life, prolong overall survival, and maybe reduce the risk of progression to leukemia. In the near future, combining both genomics-based, ex vivo functional based and molecular stratification analysis will lead the way to a personalized and targeted approach.
Myelodysplastic Syndrome (MDS) can be defined as a common name given to a heterogeneous group of diseases in which ineffective hematopoiesis and cytopenia are predominate. There are specific characteristic changes in blood and bone marrow, mainly dysplasia (dyserythropoiesis, dysgranulopoiesis and monocytosis, dysmegakaryopoiesis) in all three series of bone marrow.
Although it can be observed in almost any age group, MDS, which is known to affect mostly the elderly, poses an important problem for hematologists especially in terms of diagnosis and ability to determine and administer an appropriate treatment in limited time (1). The median age at diagnosis is 71, and the annual incidence of disease is 0.1/100,000 for the population under the age of 40, while this rate is 30.2/100,000 for the 70–79 age group and 59.8/100,000 for those over 80 years old, according to the data of the United States (US) National Cancer Institute (NCI) (2, 3).
Cytopenia, bone marrow dysplasia, and some characteristic chromosomal anomalies define this disease according to the World Health Organization (WHO); however, understanding the pathogenesis, classification, and prognosis of the disease has actually become much easier as a result of the integration of next-generation DNA sequencing (NGS) technique into daily practice and its integration with morphology, cytogenetic and molecular genetic techniques (2).
The highly heterogeneous nature of MDS complicates the treatment of the disease and requires individualization as well. The only “curative” approach among many treatment options actually comes with allogeneic hematopoietic stem cell transplantation (AHSCT), but this form of treatment can unfortunately be applied to a limited number of “fit” patients. For the majority of patients, the preferred treatment methods are generally “non-intensive” options due to age, comorbidities, etc. of these patients, and the ideal choice is to use risk-based approaches/treatments. Almost all of a wide range of treatment options ranging from growth factor to lenalidomide and hypomethylating drugs are the preferred approaches to correct cytopenia, improve quality of life, and if possible, prevent disease progression, rather than radical treatment of the disease.
In this review, the past, present and possible future view of low-risk MDS management will be discussed including general definition, classification, clinical presentation, risk stratification, prognostic evaluation.
Myelodysplastic syndrome is a wide spectrum of heterogeneous diseases in which different sizes of cytopenia and morphological dysplasia that have the risk of developing into acute myeloblastic leukemia (AML) (2). It is clear that the most important determinant for MDS is dysplasia, which can be detected in early and mature cells of the bone marrow, rather than the presence of cytopenia.
Dyserythropoiesis is identified by various anomalies in the nucleus and cytoplasm of erythroid cells. In the nucleus, budding, bridging, karyorexia, presence of multiple nuclei and megaloblastoid changes are present; cytoplasmic ones are classified as ring sideroblast, vacuolization and Periodic acid–Schiff (PAS) positivity (2). Dysgranulopoiesis, on the other hand, is characterized by very small or abnormally large myeloid lineage cells, nuclear hyposegmentation (pseudo-Pelger-Huet), nuclear hypersegmentation, reduction or absence in granulation, the presence of Pseudo-Chédiak-Higashi granules, Döhle bodies, Auer rods, and Barr bodies (2). Dysmegakaryopesis is classified as the presence of micromegakaryocytes, nuclear hypolobation, and multiple nuclei (2).
It was first named as preleukemia in 1953, and as a result of various definitions such as chronic erythremic myelosis, hypoplastic acute myeloid leukemia, and dysmyelopoietic syndrome, its first morphological classification (FAB, French–American–British) was made in 1982 using the name MDS (2, 4). WHO updated this classification in 2001 and 2008, and finally, in 2016, made a morphological and cytogenetic-based classification and defined 6 disease types in general, including subgroups (5). It should be emphasized, however, that the issue of exactly where MDS and AML diverge continues to be debated. The only difference between the two diseases is not only the number of blasts, but also the clinical progression rate, as well as the biological and morphological characteristics of the diseases. Especially, the US National Comprehensive Cancer Network (NCCN) panel team and also WHO state that a disease with a blast rate of between 20 and 29% and a stable clinical course for at least 2 months can be defined as “high-risk MDS” (3). However, in this situation, it would be more accurate to define patients with FLT3 or NPM1 mutations as AML rather than MDS (2, 6).
Myelodysplastic syndrome is known to be a hematopoietic stem cell disease that develops as a result of the interaction of bone marrow microenvironment and immune system with various genetic and epigenetic factors for many years (2). In MDS, it was reported that structural genetic defects, mostly caused by various “unbalanced changes” due to chromosomal losses and excesses, such as deletion (del) 5q, monosomy 7, trisomy 8, and del 20q and were identified in almost more than 50% of cases (7). With the adaptation of the NGS technique to clinical practice, recurrent mutations of more than 50 genes involved in DNA methylation, chromatin modification, RNA “splicing”, “cohesion” formation, transcription control, and DNA repair and signaling processes have been detected (2, 8). It has been suggested that there are various immunological imbalances, especially in the T lymphocyte series; for example, cytotoxic T cells increase in low-risk MDS and regulatory T cells dominate the presentation together with the immune “escape” mechanism in the high-risk subgroup (2, 9). Its shown that low risk MDS patients also have chronic inflammation (10).
Another MDS developmental process for which the underlying mechanism is largely unknown is “therapy related” disease, which is cytotoxic drug or radiotherapy related. The remarkable feature here is that the disease develops at a higher rate in individuals carrying a CHIP (clonal hematopoiesis of indeterminate prognosis) clone (11).
Long-standing macrocytic anemia, mild thrombocytopenia, and neutropenia may be identified during the development of MDS before the disease clinically develops. In fact, the diagnosis can be made by identifying cytopenia in routine blood tests by chance even when the patient is not more symptomatic and at relatively earlier times.
Complaints such as weakness, fatigue, decrease in effort capacity, dizziness, and cognitive dysfunction, which are triggered by tissue hypoxia related to anemia and caused by the fact that almost the whole organism, mainly the musculoskeletal system, cardiovascular system and central nervous system are affected, are remarkable. Although mild at first, skin/mucosal bleeding caused by overt/deep thrombocytopenia is not surprising in cases in whom the diagnosis is delayed. Thrombocytopenia can mistakenly be diagnosed as immune thrombocytopenia if dysplastic changes are ignored and not recognized. Neutropenia accompanied by functional disorders may be the cause of life-threatening bacterial and fungal infections. There is a substantial number of MDS cases diagnosed with fever, cough, dysuria, and even septic shock.
In the historical process, many approaches have been developed that try to determine the prognostic classification through defining a MDS patient and consider various criteria such as clinical features, bone marrow blast rate, cytopenia, age, lactate dehydrogenase level, and cytogenetic features (2). The first of these was the International Prognostic Scoring System for MDS-IPSS, which divided patients into four subgroups in 1997 according to their cytopenia, bone marrow blast rate and cytogenetic features, and their median survival of 0.4–5.7 years (12). In the last 15 years, the need for a new classification has arisen as a result of the addition of ferritin, beta 2 microglobulin, bone marrow fibrosis, comorbidity, and performance status of patients, morphological re-classification of MDS, and finally the new cytogenetic features that can be detected with the developing technology (2). With the evaluation of 7,012 patients belonging to the database of the MDS study group (International Working Group for Prognosis in MDS-IWG-PM), which was formed with international multicentre participation, a more detailed scoring system -revised (R) IPSS- was developed which defines 5 prognostic categories with five different cytogenetic features, and classifies the depth of cytopenia and bone marrow blast ratio in more detail (13) (Table 1). Moreover, a WHO classification-based prognostic scoring system (WPSS) was developed with the participation of two centers from Italy and Germany in 2007, and five different subgroups were defined with a median survival of 12–103 months (14) (Table 2). Although anemia is a poor prognostic subgroup in this classification, the depth of anemia, in other words Hb level, was also included in the prognostic classification in a recent analysis as its depth and its clinical reflection were not sufficiently correlated (15). IWG-PM reported in its analysis comparing WPSS with R-IPSS that WPSS was also a very effective scoring method in the prognostic classification of untreated MDS cases and AML transformation (16).

Table 1. R-IPSS scoring system (13).

Table 2. WHO classification-based prognostic scoring system (WPSS) scoring system (14).
Although there are many different approaches today, it is clear that the R-IPSS risk classification system is generally used in making treatment decisions by considering the age and performance status of a patient (2, 3).
The presence of various somatic mutations which are not in these systems, but have been identified with NGS in recent years, such as TP53, ASXL1, EZH2, ETV6, and RUNX1 that lead to poor clinical course, and SF3B1 that leads to a good clinical outcome, are tried to be integrated into scoring systems by some centers (17–19).
International prognostic scoring systems that divide MDS into two large groups as low or high risk have actually revealed two disease subtypes with completely different treatment goals (20). While treatment policies in low-risk patients focus mostly on the correction of cytopenia which is reducing blood product support, especially erythrocyte suspension support, improving the quality of life, and maintaining it, if possible; those in the high-risk subgroup are correction of cytopenia, as well as delaying leukemic progression, and if possible, ensuring the survival of patients (Table 3) (21).

Table 3. Treatment goals in low- and high-risk myelodysplastic syndromes (MDS) patients (20).
There is no conflict regarding the inclusion to low-risk MDS of patients who belong to “low” and “intermediate-1” risk groups according to the IPSS, and “very low” and “low” risk groups according to the R-IPSS. However, there is a conflict as to whether patients in “intermediate” risk group in R-IPSS have low or high risk. While some researchers place those in the “intermediate” risk group directly into the high-risk MDS group, others consider those in the “intermediate” risk group above 3.5 points only as high-risk MDS (20, 21). Nevertheless, it is obvious that the first things to consider when making a decision are the worsening of disease or side effects might be caused with the treatment.
In fact, there is no need to move beyond “supportive” therapy due to the presence of mild and asymptomatic cytopenia in a substantial number of low-risk MDS patients. This approach can be considered applicable to all low- or high-risk MDS patients who do not have a long-life expectancy due to age and comorbidities. Patients who are fit, asymptomatic, without blast increase, and who do not have a high-risk cytogenetic/molecular profile do not require any special treatment other than regular controls (21, 22). Results from a recent real-world cohort study (n = 125) indicate that over a third of patients with low-MDS have been managed using watchful waiting only, with no systemic treatment or transfusions received (23). However, it is recommended that asymptomatic low-risk MDS patients should be followed up more closely if they have various mutations that are not currently included in the risk classification and show a genotypically high-risk prognostic profile, such as ASXL1 mutation (21). The most important markers in these patients are rapid worsening cytopenia, an increase in the number of blasts in the peripheral blood or bone marrow, and the change of findings in cytogenetic/molecular studies (21).
Current treatment approach in low-risk MDS patients focuses on combating cytopenia, especially anemia, and the poor results of transfusion load.
Symptoms reflecting anemia-related tissue hypoxia such as weakness, fatigue, and decreased exercise capacity are the most frequently reported complaints in MDS patients. These patients also need regular erythrocyte suspension (ES) support. Regular and frequent ES support, on the other hand, means a very problematic complication such as transfusional hemosiderosis, as well as a financial load and the need for social support.
The use of “erythropoiesis-stimulating agents” such as recombinant erythropoietin (EPO) or darbepoetin (DAR) as a single drug has been the mainstay of treatment for many years in low-risk patients who are at the forefront of anemia and need frequent transfusion support. On the other hand, phase three randomized studies on both drugs were conducted more recently and the use of EPO-alpha was eventually approved (20, 21). In a multicenter, randomized phase 3 study from European countries, 147 low-risk MDS patients with hemoglobin (Hb) level of ≤10 g/dL, EPO level of ≤500 mU/mL, and low transfusion load were randomized to receive DAR-alpha or placebo at the rate of 2/1 (24). Following placebo or subcutaneous administration of 500 μg DAR-alpha every 3 weeks for 24 weeks, the frequency of transfusion was significantly higher in the placebo arm (59.2 vs 36.1% and p = 0.008), while the recovery of anemia was significantly higher in the DAR arm (14.7 vs 0% and p = 0.016). In another multicenter, double-blind, placebo-controlled phase 3 study from European countries, 130 low-risk (IPSS low and medium-1) MDS patients with similar characteristics to the other study were randomized to EPO-alpha or placebo arms at the rate of 2/1 (25). After subcutaneous administration of 450 IU/kg/w EPO-alpha or placebo for 24 weeks, the recovery of anemia was significantly higher in the EPO arm (31.8 vs. 4.4% and p = 0.001).
Some features in low-risk MDS patients have become important in predicting the response to “erythropoiesis stimulating agents.” These features are low endogenous EPO level (<500, <100 IU/L) and having a total ES transfusion load of less than 4 units in 2 months (26, 27).
In European Union countries, endogenous EPO level should be <200 IU/L for reimbursement approval of EPO-alpha, whereas in our country this value is determined as <500 IU/L. In many low-risk MDS patients, the effect of “erythropoiesis stimulating agents” becomes apparent in about 3 weeks, while it is also stated that this effect can be sustained for a median of 15–18 months (21).
In patients whose expected response cannot be obtained with “erythropoiesis stimulating agents” and especially in the subgroup with ring sideroblasts, recovery can be achieved in approximately 20% of patients with subcutaneous application of granulocyte colony stimulating factors (G-CSF) of 1–2 μg/kg/w (28).
In del(5q) positive low-risk MDS patients with low transfusion load and symptomatic anemia, the first-line treatment of anemia is again “erythropoiesis stimulating agents” (26). However, it is noteworthy that most of these patients have high endogenous EPO levels, usually associated with low and short-term response rates and correlated with high clonality rate in myeloid precursor cells (23). Lenalidomide seems to be a good choice with a treatment success of up to 70% in the treatment of anemia, especially for patients with high EPO levels and a constant need for ES transfusion (29, 30). The most common side effects of the drug are diarrhea, rash, nausea, constipation, fever, itching, shortness of breath, recurrent arterial thrombotic events, and hematological side effects such as neutropenia and/or thrombocytopenia, and it is obvious that these complications should be considered very important considering the average age and fitness of MDS patients (20).
In an international multicenter, randomized, phase 3 study published in 2016, 239 low-risk and del(5q) negative patients were randomized 2/1 to lenalidomide or placebo arm (31). ES transfusion independence was clearly superior in the lenalidomide arm, while no significant deterioration in quality of life was detected in the drug arm. In another recent randomized study, 131 patients resistant to “erythropoiesis stimulating agents”, who were del(5q) negative low-risk and ES transfusion-dependent, were randomized to either lenalidomide alone or lenalidomide + EPO arms (32). The use of lenalidomide in combination with “erythropoiesis stimulating agents” provided a significant superiority in anemia response compared to the arm in which it was given alone (39.4 vs 23.1% and p = 0.044). In a recent study, the combination of lenalidomide and erythropoiesis stimulating agents yields 38.9% of major erythroid responses who relapsed or unresponsive to erythropoiesis stimulating agents (33).
On the other hand, patients with TP53 mutations, which constitute approximately 20% of all patients, did not have the expected benefit from lenalidomide with high leukemic transformation rates (21). In the LEMON5 study, the overall response rates and survival in patients with TP53 mutation who were followed up with lenalidomide monotherapy were significantly lower than those in patients without TP53 mutation (34). Moreover, TET2 and RUNX1 mutations are associated with poor outcome and lenalidomide unresponsiveness (21). Even if there is no disease progression or leukemic transformation in patients who do not respond adequately to lenalidomide in the presence of TP53 or other mutations, it is recommended to administer hypomethylating (HMI) agents or HSCT in the absence of an ongoing clinical trial (21).
Many MDS patients live dependent on regular ES transfusions as part of supportive therapy. In these patients, “non-transferrin bound iron” and labile plasma iron fraction increase, and ultimately and stored especially in the heart, liver and endocrine glands. In a phase 3 study comparing placebo and deferasirox, there was a significant risk reduction of 36.4% in event-free survival (event: worsening cardiac function, hospitalization with heart failure, liver dysfunction, liver cirrhosis, and AML transformation) in the iron chelation group (35).
Many international guidelines recommend iron chelation based on the ferritin level; the opinion is that this amount should be at least 1,000 ng/mL (21). It is known that the most commonly used chelator is deferasirox, and the patient compliance problem is also improved with the film-coated tablets released in recent years. Iron chelation is an application that must be included in the algorithm before HSCT, and it positively affects the transplant outcome.
Immune dysregulation is better understood in the etiopathogenesis of MDS and is considered to be responsible for ineffective hematopoiesis. In this context, it was reported that very satisfactory response rates (16–67%), including all three series, were obtained with antithymocyte globulin (ATG) and/or cyclosporine treatment (31). Some features become important in predicting patients who will benefit from immunosuppressive therapy. Presence of dysplasia which is the subtype previously classified as refractory anemia, absence of ring sideroblast subtype, especially hypoplastic/hypocellular bone marrow, having HLA DR15 typing, young age (<60 years), female gender, presence of trisomy 8, and relatively short duration of transfusion need are among these characteristics (21). Interestingly, in a retrospective study involving a large number of patients, in which the presence of SF3B1, a somatic mutation known to be associated with good clinical outcome in MDS, adversely affected the overall response, it was reported that a total response rate of up to 45% was obtained with the use of horse-derived ATG (32). A meta-analysis was unable to associate specific biomarkers predictive of response given the overall lack of prospective, randomized controlled studies for the use of immunosuppressive treatment in low risk MDS (33).
Hypomethylating agents (HMIs), used at standard or reduced doses, are included in the treatment of low-risk MDS. Although it is generally approved by the US Food and Drug Administration (FDA), it has a more limited use in European countries. The reason may be disappointing results obtained in the studies. In a phase 2 study of the Nordic group, which included patients with low-risk MDS who were resistant to the EPO + G-CSF combination or were not suitable for transfusion, a response of only 20% was achieved with azacitidine (AZA) at a dose of 75 mg/m2/day administered for 5 days every 28 days. Moreover, this response was both short-term and more toxic than expected (36). Similarly, in prospective studies of the French group, in which they randomized 98 low-risk patients with a median age of 72 years to AZA vs AZA + EPO treatment arms, transfusion independence at the end of six courses was only achieved in 16.3% of patients (14.3% in the AZA-EPO arm) (37). In a study which 113 low-risk MDS patients were included, a total response rate of 70% was achieved with 3 days of 20 mg/m2/day decitabine (DEC) every 28 days, while transfusion independence was achieved in 32% of patients (38). Although the oral formulation of AZA, which has been developed in recent years, promises ease of administration and longer lasting efficacy at lower doses, the international multicenter phase 3 study which compared CC-486 (oral AZA) with placebo and included low-risk MDS patients with ES transfusion dependent and thrombocytopenia was terminated earlier than expected due to toxicity (21, 39).
In 2020, the FDA and Canadian authorities approved DEC/cedazuridine (ASTX727 or DEC-C, oral decitabine) for the treatment of all subtypes of adult MDS and CMML in any stage of disease based on the 60% of overall response rate (40). In the Ascertain trial, the low grade MDS patients had 57% overall response rate, 48% became ES independent and 67% platelet transfusion independent (41).
“Luspatercept” and “sotatercept”, also named as “erythropoiesis maturation agents”, are specific activin receptor fusion proteins that act as ligand traps to neutralize negative regulators of late-stage erythropoiesis (2, 20, 21). In the PACE-MDS study which included 57 MDS patients with low transfusion load and in IPSS low and intermediate-1 risk groups, hematological recovery and transfusion independence were reported as 63 and 38%, respectively, in the group using relatively higher dose (0.75–1.75 mg/kg) luspatercept (42). In an international multicenter, double-blind, placebo-controlled, randomized, phase 3 study (MEDALIST) published in January 2020, 229 MDS patients with ring sideroblasts who were in very low, low, and intermediate risk groups according to R-IPSS were randomized 2/1 to luspatercept and placebo arms (43). Transfusion independence (38 vs 13%) and hematological recovery (58 vs 17%) were significantly superior to placebo in the luspatercept arm, while AML transformation in both arms was not different. In April 2020, the FDA approved luspatercept for use in MDS patients with ring sideroblasts who are with very low, low, and intermediate risk and who require two units or more of ES for 8 weeks and have not benefited from “erythropoiesis stimulating agents”.
Roxadustat, on the other hand, is a small molecule that can be used orally and is an inhibitor of the “hypoxia-inducible factor” “prolyl hydroxylase” (20). There are ongoing studies on the use of this drug, which stimulates endogenous EPO production, eliminates the negative effects of inflammation in endogenous EPO production, triggers erythropoiesis and Hb production, and regulates iron regulation through hepcidin metabolism, in anemia in MDS and chronic kidney disease (21). The data for the low burden transfusion dependent low risk MDS are promising which 38% of the patient achieved transfusion independency over 8 weeks in weeks 1–28 of treatment while 42% achieved this during 52 weeks of treatment (44). The results of a recent phase 3, randomized, double-blind, placebo-controlled study showed that transfusion independence was achieved in nine patients (37.5%) at 28 and 52 weeks; seven of the patients patients received 2.5 mg/kg dose (NCT03263091) (45).
Imetelstat is a drug that help maintaining normal hematopoiesis by acting as a telomerase inhibitor in cells with short telomere length and hyper telomerase activity (21). Short telomeres and high telomerase activity are associated with shorter overall survival in MDS (2, 20). In a study published at the annual meeting of the American Society of Hematology in 2017, transfusion independence was found in 34% and erythroid-hematological improvement in 63% of patients with imetelstat in MDS patients with low and intermediate-1 risk who were resistant/unresponsive to “erythropoiesis stimulating agents” (46). When administered intravenously every 4 weeks, transfusion independence was reported in 42% of patients and response duration may be over 1 year in 30% of patients up to 2.8 years (47). Furthermore, specific inhibitors of IDH1 or IDH2 genes, ivosidenib and enasidenib showed promising responses (50%) in patients that carry the somatic mutations (48, 49). KER-050, a modified activin receptor type IIA inhibitor, is designed to target transforming growth factor-β ligands, including activin A. In an open label phase II study in very low to intermediate risk MDS patients, overall erythroid response rate was 60% (n = 6/10). 33% (n = 1/3) non-transfused participants had a hemoglobin increase of ≥1.5 g/dL sustained ≥8 weeks. Increases in platelets were also observed (50). The new treatment approaches are given in Table 4.

Table 4. New treatment strategies in low grade myelodysplastic syndromes (MDS).
In low-risk MDS with thrombocytopenia, platelet suspension transfusion and thrombopoietin (TPO) receptor agonists, in addition to HMIs, constitute a remarkable treatment option (21). In a randomized study in which “romiplostim” was compared with placebo in low-risk MDS patients, it was found that the platelet counts increased, bleeding episodes and the need for thrombocyte suspensions decreased significantly in patients who received romiplostim compared to the placebo group (51). In the 5-year long-term analysis of the same study, transformation to AML and death rates were not different in the drug group than in the placebo group (52). In a prospective, multicenter EUROPE phase II trial, mutated SRSF2 occurred more often in responders of romiplostim compared with non-responders (41 vs 16%, p = 0.018) (53).
In a phase 2 study in which 90 low-risk thrombocytopenic MDS patients were randomized to “eltrombopag” and placebo at a rate of 2/1, decrease in thrombocyte response and bleeding episodes were found to be significantly superior in the eltrombopag arm (47 vs. 3%, p = 0.0017 and 14 vs 42%, p = 0.0025, respectively) (54). AML transformation was also reported to be same between the two groups. A similar response rate of 44% was observed in a second phase 2 dose escalation study. The predictors of response were the presence of a PNH clone, marrow hypocellularity, thrombocytopenia, and baseline elevated plasma TPO levels (55). The combination of eltrombopag and lenalidomide in low and intermediate risk MDS demonstrated good efficacy with ORR of 40.9%, response durability and an acceptable safety profile (56). The mechanisms of potential treatment alternatives for low grade MDS are summarized in Figure 1.
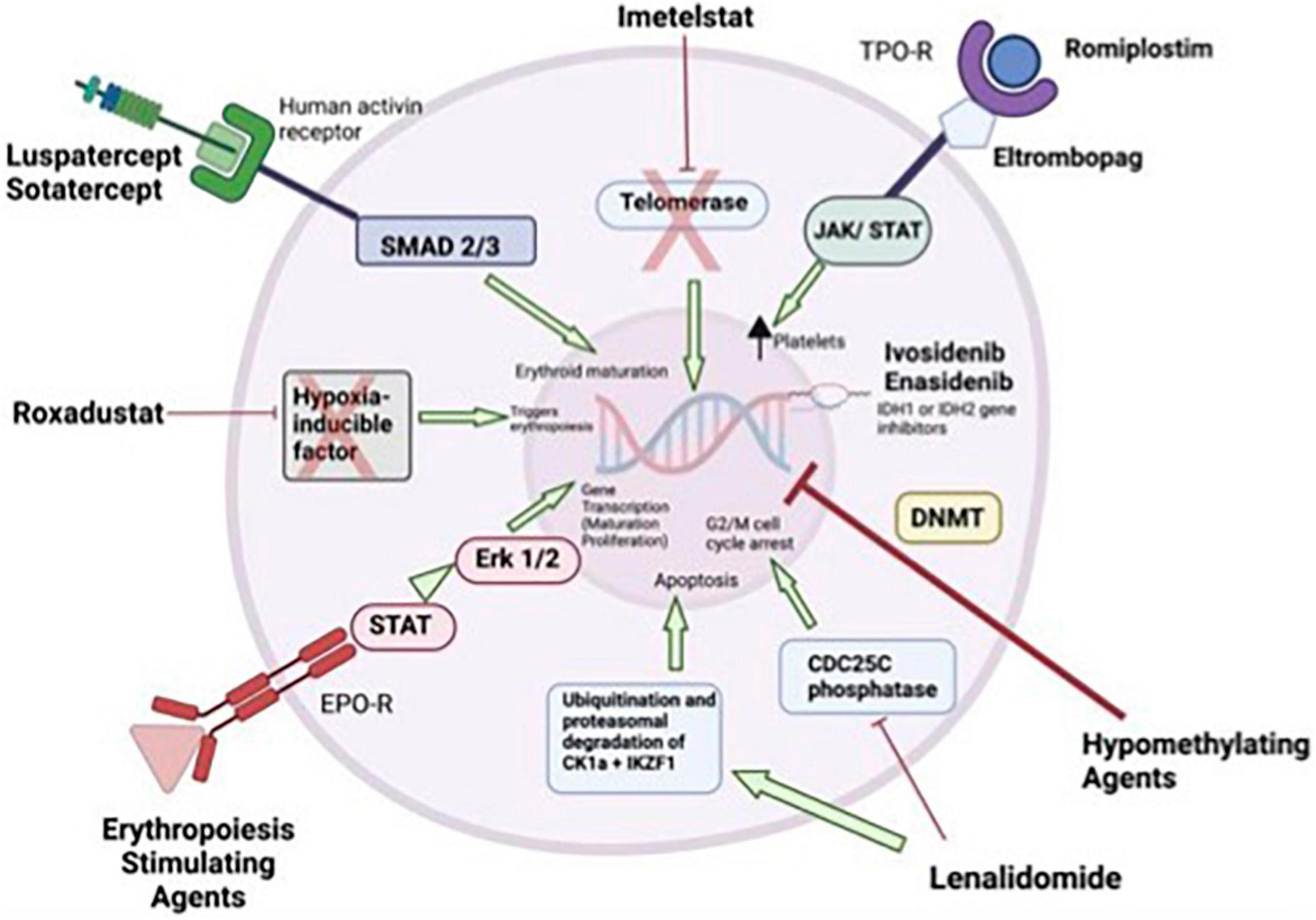
Figure 1. Mechanism of actions for treatments in low-risk myelodysplastic syndromes (MDS). Erythropoiesis stimulating agents stimulate gene transcription of maturation and proliferation of erythrocytes through JAK/STAT and Erk1/2. Lenalidomide inhibits the CDC25C phosphatase and by decrease in CK1α levels. Inhibition of the CDC25C phosphatase leads to a stoppage of proliferation by induction of an arrest in the cell cycle at the transition between G2 and M phase. Hypomethylating agents induce DNA hypomethylation. Ivosidenib, and enasidenib inhibits IDH1 or 1DH2 genes. Thrombopoietic receptor agonists activate signaling leads to increased platelet production. Imetelstat is a drug that help maintaining normal hematopoiesis by acting as a telomerase inhibitor in cells with short telomere length and hyper telomerase activity. “Luspatercept” and “sotatercept” specific activin receptor fusion proteins that act as ligand traps to neutralize negative regulators of late-stage erythropoiesis. Roxadustat is a small molecule that can be used orally and is an inhibitor of the “hypoxia-inducible factor” (20, 21, 61–64). EPO-R, erythropoietin receptor; JAK, janus kinase; STAT, signal transducer and activator of transcription; TPO-R, thrombopoietin receptor.
It is clear that performing AHSCT earlier in the course of the disease will provide a more favorable outcome in the long term. On the other hand, it is necessary to avoid a treatment process that carries a substantial risk of death in low-risk patients who have a high chance of obtaining a response with standard first-line “soft” treatment options (57). General opinion is that IPSS low and moderate-1 risk subgroup patients do not have any indications for AHSCT at the time of diagnosis, except IPSS intermediate-1 risk subgroup with cytopenia and/or poor cytogenetic karyotype (58). In a multicenter biologic assignment trial, reduced intensity AHSCT showed advantage to hypomethylating therapy or best supportive care on 3-year OS (47.9 vs 26.6%, P = 0.0001) and 3-year leukemia-free survival (35.8 vs 20.6%, P = 0.003) in subjects 50–75 years of age with intermediate-2 or high risk MDS (59). In the retrospective analysis of the European Society for Blood and Marrow Transplantation (EBMT), which included 246 MDS patients with low and moderate-1 risk according to IPSS, a 3-year overall survival rate of 58% was achieved with AHSCT and the non-relapse mortality rate was reported to be 30% (60). Therefore, in patients with unresponsive disease to first-line therapies and having poor prognostic characteristics such as life-threatening infection, grade 2 or greater bone marrow fibrosis, severe thrombocytopenia, severe neutropenia, severe anemia, ES transfusion dependency, high-risk molecular anomalies, and treatment-related MDS, AHSCT should be considered an option as an individualized treatment approach (21, 57).
In conclusion, for the majority of MDS patients, the therapeutic approach is based on IPSS (or IPSS-R/WPSS) stratification, with some non-curative options, except AHSCT. However, it should be known that the heterogeneity and complexity of low risk MDS requires a personalized management that, unfortunately, does not yet exist (20).
It should be kept in mind that there are a substantial number of asymptomatic patients who can only be kept under close follow-up with the option of “watchful waiting”. It is known that “erythropoiesis stimulating agents” in low-risk MDS where anemia is at the forefront and lenalidomide in those with del (5q) positive are beneficial. The combination of lenalidomide + G-CSF may be a good alternative for patients who are del (5q) negative and are resistant to “erythropoiesis stimulating agents”. Immunosuppressive treatment options should not be disregarded, especially in subtypes with low ring sideroblasts, high endogenous EPO levels and additionally HLA DR15 positivity. Although thrombocytopenia and neutropenia are encountered less frequently, they are two characteristics that are more difficult to treat (21). HMIs can be used at adjusted doses and with profit/loss calculations in low-risk MDS patients with both anemia and thrombocytopenia/neutropenia, those who do not respond to the first-line treatments that are specified, and those who have unfavorable somatic mutations. Although mortality rates are high, in patients who are resistant to first-line therapy, are transfusion-dependent, and have additionally high-risk molecular anomalies, the option of AHSCT should be considered without delay, by explaining the possible complications to the patient and family at the appropriate time and in the appropriate order. Recommendations are summarized in Figure 2 (20, 21).

Figure 2. Current treatment approaches in low-risk myelodysplastic syndromes (MDS) (19, 20). MDS, myclodysplastic syndrome; IPSS, International Prognostic Scoring System; R-IPSS, Revised International Prognostic Scoring System; TPO, thrombopoietin; EPO, erythropoietin; ESA, erythropoiesis stimulating agents; G-CSF, granulocyte colony stimulating factor; HMA, hypomethylating agents; AHSCT, allogeneic hematopoietic stem cell transplantation; ATG, antithymocyte globulin; CsA, cyclosporine.
It seems impossible to consider a single gold standard treatment option that will be successful in a large proportion of patients because of underlying stem cell disease and the combination of many different mechanisms in its etiopathogenesis (2). However, with the analysis of the results of ongoing ex vivo functional studies and genomic-based studies, it may be possible in the non-distant future to create more specific treatment options that will work in low risk MDS.
ST conceived and designed the review, collected the data, wrote the manuscript, and approved the submitted version.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The author thank Pinar Ataca Atilla for the assistance in drawing the Figure 1. The figure is created with Biorender.com.
1. Löwenberg B. Introduction to a review series on myelodysplastic syndromes. Blood. (2019) 133:1001. doi: 10.1182/blood-2018-12-886549
2. Hellström-Lindberg E, Tobiasson M, Greenberg P. Myelodysplastic syndromes: moving towards personalized management. Haematologica. (2020) 105:1765–79. doi: 10.3324/haematol.2020.248955
3. Greenberg PL, Stone RM, Al-Kali A, Barta SK, Bejar R, Bennett JM, et al. Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2017) 15:60–87. doi: 10.6004/jnccn.2017.0007
4. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. (1982) 51:189–99. doi: 10.1111/j.1365-2141.1982.tb08475.x
5. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World health organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
6. Bains A, Luthra R, Medeiros LJ, Zuo Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: frequency and potential value for predicting progression to acute myeloid leukemia. Am J Clin Pathol. (2011) 135:62–9. doi: 10.1309/AJCPEI9XU8PYBCIO
7. Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. (2007) 110:4385–95. doi: 10.1182/blood-2007-03-082404
8. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. (2014) 28:241–7. doi: 10.1038/leu.2013.336
9. Chamuleau ME, Westers TM, van Dreunen L, Groenland J, Zevenbergen A, Eeltink CM, et al. Immune mediated autologous cytotoxicity against hematopoietic precursor cells in patients with myelodysplastic syndrome. Haematologica. (2009) 94:496–506. doi: 10.3324/haematol.13612
10. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med. (2021) 218:e20201544. doi: 10.1084/jem.20201544
11. Takahashi K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. (2017) 18:100–11. doi: 10.1016/S1470-2045(16)30626-X
12. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. (1997) 89:2079–88. doi: 10.1182/blood.V89.6.2079
13. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. (2012) 120:2454–65. doi: 10.1182/blood-2012-03-420489
14. Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. (2007) 25:3503–10. doi: 10.1200/JCO.2006.08.5696
15. Malcovati L, Della Porta MG, Strupp C, Ambaglio I, Kuendgen A, Nachtkamp K, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based prognostic scoring system (WPSS). Haematologica. (2011) 96:1433–40. doi: 10.3324/haematol.2011.044602
16. Della Porta MG, Tuechler H, Malcovati L, Schanz J, Sanz G, Garcia-Manero G, et al. Validation of WHO classification-based prognostic scoring system (WPSS) for myelodysplastic syndromes and comparison with the revised international prognostic scoring system (IPSS-R). A study of the international working group for prognosis in myelodysplasia (IWG-PM). Leukemia. (2015) 29:1502–13. doi: 10.1038/leu.2015.55
17. Bejar R, Papaemmanuil E, Haferlach T, Garcia-Manero G, Maciejewski JP, Sekeres MA, et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS-R: analysis of combined datasets from the international working group for prognosis in MDS-molecular committee. Blood. (2015) 126:907. doi: 10.1182/blood.V126.23.907.907
18. Nazha A, Al-Issa K, Hamilton BK, Radivoyevitch T, Gerds AT, Mukherjee S, et al. Adding molecular data to prognostic models can improve predictive power in treated patients with myelodysplastic syndromes. Leukemia. (2017) 31:2848–50. doi: 10.1038/leu.2017.266
19. Tefferi A, Gangat N, Mudireddy M, Lasho TL, Finke C, Begna KH, et al. Mayo alliance prognostic model for myelodysplastic syndromes: integration of genetic and clinical information. Mayo Clin Proc. (2018) 93:1363–74. doi: 10.1016/j.mayocp.2018.04.013
20. Scalzulli E, Pepe S, Colafigli G, Breccia M. Therapeutic strategies in low and high-risk MDS: what does the future have to offer? Blood Rev. (2020) 45:100689. doi: 10.1016/j.blre.2020.100689
22. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. (2011) 364:2496–506. doi: 10.1056/NEJMoa1013343
23. Zimmerman Savill KM, Gajra A, Price K, Kish JK, Brown-Bickerstaff C, Falkenstein A, et al. Lower-risk myelodysplastic syndromes: erythropoiesis-stimulating agent treatment approaches and outcomes in the United States. Blood. (2021) 138(Suppl. 1):4657. doi: 10.1182/blood-2021-146461
24. Platzbecker U, Symeonidis A, Oliva EN, Goede JS, Delforge M, Mayer J, et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia. (2017) 31:1944–50. doi: 10.1038/leu.2017.192
25. Fenaux P, Santini V, Spiriti MAA, Giagounidis A, Schlag R, Radinoff A, et al. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-α in anemic patients with low-risk MDS. Leukemia. (2018) 32:2648–58. doi: 10.1038/s41375-018-0118-9
26. Hellström-Lindberg E, Gulbrandsen N, Lindberg G, Ahlgren T, Dahl IM, Dybedal I, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol. (2003) 120:1037–46. doi: 10.1046/j.1365-2141.2003.04153.x
27. Buckstein R, Balleari E, Wells R, Santini V, Sanna A, Salvetti C, et al. ITACA: a new validated international erythropoietic stimulating agent-response score that further refines the predictive power of previous scoring systems. Am J Hematol. (2017) 92:1037–46. doi: 10.1002/ajh.24842
28. Kelaidi C, Park S, Sapena R, Beyne-Rauzy O, Coiteux V, Vey N, et al. Long-term outcome of anemic lower-risk myelodysplastic syndromes without 5q deletion refractory to or relapsing after erythropoiesis-stimulating agents. Leukemia. (2013) 27:1283–90. doi: 10.1038/leu.2013.16
29. Fenaux P, Giagounidis A, Selleslag D, Beyne-Rauzy O, Mufti G, Mittelman M, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with del5q. Blood. (2011) 118:3765–76. doi: 10.1182/blood-2011-01-330126
30. Giagounidis A, Mufti GJ, Mittelman M, Sanz G, Platzbecker U, Muus P, et al. Outcomes in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with isolated deletion 5q treated with lenalidomide: a subset analysis from the MDS-004 study. Eur J Haematol. (2014) 93:429–38. doi: 10.1111/ejh.12380
31. Santini V, Almeida A, Giagounidis A, Gröpper S, Jonasova A, Vey N, et al. Randomized phase III study of lenalidomide versus placebo in RBC transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes and ineligible for or refractory to erythropoiesis-stimulating agents. J Clin Oncol. (2016) 34:2988–96. doi: 10.1200/JCO.2015.66.0118
32. Toma A, Kosmider O, Chevret S, Delaunay J, Stamatoullas A, Rose C, et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia. (2016) 30:897–905. doi: 10.1038/leu.2015.296
33. List AF, Sun Z, Verma A, Bennett JM, Komrokji RS, McGraw K, et al. Lenalidomide-epoetin alfa versus lenalidomide monotherapy in myelodysplastic syndromes re-fractory to recombinant erythropoietin. J Clin Oncol. (2021) 39:1001–9. doi: 10.1200/JCO.20.01691
34. Mossner M, Jann JC, Nowak D, Platzbecker U, Giagounidis A, Götze K, et al. Prevalence, clonal dynamics and clinical impact of TP53 mutations in patients with myelodysplastic syndrome with isolated deletion (5q) treated with lenalidomide: results from a prospective multicenter study of the german MDS study group (GMDS). Leukemia. (2016) 30:1956–9. doi: 10.1038/leu.2016.111
35. Angelucci E, Li J, Greenberg PL, Depei W, Hou M, Montaño Figueroa E, et al. Safety and efficacy, including event-free survival, of deferasirox versus placebo in iron-overloaded patients with low- and int-1-risk myelodysplastic syndromes (MDS): outcomes from the randomized, double-blind Telesto study. Abstract #234. In: Paper Presented at the 2018 ASH Annual Meeting. San Diego, CA (2018).
36. Parikh AR, Olnes MJ, Barrett AJ. Immunomodulatory treatment of myelodysplastic syndromes: antithymocyte globulin, cyclosporine, and alemtuzumab. Semin Hematol. (2012) 49:304–11. doi: 10.1053/j.seminhematol.2012.07.004
37. Stahl M, DeVeaux M, de Witte T, Neukirchen J, Sekeres MA, Brunner AM, et al. The use of immunosuppressive therapy in MDS: clinical outcomes and their predictors in a large international patient cohort. Blood Adv. (2018) 2:1765–72. doi: 10.1182/bloodadvances.2018019414
38. Maximilian S, Jan Philipp B, Smith G, Wang R, Zeidan AM. Use of immunosuppressive therapy for management of myelodysplastic syndromes: a systematic review and meta-analysis. Haematologica. (2020) 105:102–11. doi: 10.3324/haematol.2019.219345
39. Tobiasson M, Dybedahl I, Holm MS, Karimi M, Brandefors L, Garelius H, et al. Limited clinical efficacy of azacitidine in transfusion-dependent, growth factor-resistant, low- and int-1-risk MDS: results from the nordic NMDSG08A phase II trial. Blood Cancer J. (2014) 4:e189. doi: 10.1038/bcj.2014.8
40. Thépot S, Ben Abdelali R, Chevret S, Renneville A, Beyne-Rauzy O, Prébet T, et al. A randomized phase II trial of azacitidine +/- epoetin-β in lower-risk myelodysplastic syndromes resistant to erythropoietic stimulating agents. Haematologica. (2016) 101:918–25. doi: 10.3324/haematol.2015.140988
41. Jabbour E, Short NJ, Montalban-Bravo G, Huang X, Bueso-Ramos C, Qiao W, et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood. (2017) 130:1514–22. doi: 10.1182/blood-2017-06-788497
42. Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. (2017) 18:1338–47. doi: 10.1016/S1470-2045(17)30615-0
43. Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. (2020) 382:140–51.
44. Henry DH, Glaspy J, Harrup R, Mittelman M, Zhou A, Carraway HE, et al. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: open-label, dose-selection, lead-in stage of a phase 3 study. Am J Hematol. (2021) 92:174–84. doi: 10.1002/ajh.26397
45. Henry DH, Glaspy J, Harrup R, Mittelman M, Zhou A, Carraway HE, et al. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: open-label, dose-selection, lead-in stage of a phase 3 study. Am J Hematol. (2022) 97:174–84.
46. Fenaux P, Raza A, Vellenga E, Platzbecker U, Santini V, Samarina I, et al. Efficacy and safety of imetelstat in RBC transfusion-dependent (TD) IPSS low/int-1 MDS relapsed/refractory to erythropoiesis-stimulating agents (ESA) (IMerge). Blood. (2017) 130(Suppl. 1):4256.
47. Steensma DP, Fenaux P, Van Eygen K, Raza A, Santini V, Germing U, et al. Imetelstat achieves meaningful and durable transfusion independence in high transfusion–burden patients with lower-risk myelodysplastic syndromes in a phase II study. J. Clin. Oncol. (2021) 39:48–56. doi: 10.1200/JCO.20.01895
48. Sebert M, Cluzeau T, Beyne Rauzy O, Bastard AS, Dimicoli-Salazar S, Thepot S, et al. Ivosidenib monotherapy is effective in patients with IDH1 mutated myelodysplastic syndrome (MDS): the idiome phase 2 study by the GFM group. Blood. (2021) 138:62. doi: 10.1182/blood-2021-146932
49. Ades L, Dimicoli-Salazar S, Sebert M, Cluzeau T, Bastard AS, Laribi K, et al. Enasidenib (ENA) is effective in patients with IDH2 mutated myelodysplastic syndrome (MDS): the ideal phase 2 study by the GFM group. Blood. (2021) 138:63. doi: 10.1182/blood-2021-147898
50. Ross DM, Arbelaez A, Chee LCY, Fong CY, Hiwase D, Kannourakis G, et al. A phase 2, open-label, ascending dose study of ker-050 for the treatment of anemia in patients with very low, low, or intermediate risk myelodysplastic syndromes. Blood. (2021) 138(Suppl. 1):3675.
51. Giagounidis A, Mufti GJ, Fenaux P, Sekeres MA, Szer J, Platzbecker U, et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1-risk myelodysplastic syndrome and thrombocytopenia. Cancer. (2014) 120:1838–46. doi: 10.1002/cncr.28663
52. Kantarjian HM, Fenaux P, Sekeres MA, Szer J, Platzbecker U, Kuendgen A, et al. Long-term follow-up for up to 5 years on the risk of leukaemic progression in thrombocytopenic patients with lower-risk myelodysplastic syndromes treated with romiplostim or placebo in a randomised double-blind trial. Lancet Haematol. (2018) 5:e117–26. doi: 10.1016/S2352-3026(18)30016-4
53. Kubasch AS, Giagounidis A, Metzgeroth G, Jonasova A, Herbst R, Diaz JMT, et al. Clinical and molecular markers for predicting response to romiplostim treatment in lower-risk myelodysplastic syndromes. Abstract S169. In: Paper Presented at the European Hematology Association (EHA) 2022 Congress. Vienna (2022).
54. Oliva EN, Alati C, Santini V, Poloni A, Molteni A, Niscola P, et al. Eltrombopag versus placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): phase 1 results of a single-blind, randomised, controlled, phase 2 superiority trial. Lancet Haematol. (2017) 4:e127–36. doi: 10.1016/S2352-3026(17)30012-1
55. Vicente A, Patel BA, Gutierrez-Rodrigues F, Groarke EM, Giudice V, Lotter J, et al. Eltrombopag monotherapy can improve hematopoiesis in patients with low to intermediate risk-1 myelodysplastic syndrome. Haematologica. (2020) 105:2785–94. doi: 10.3324/haematol.2020.249995
56. Gonzalez-Lugo JD, Kambhampati S, Yacoub A, Donnellan W, Fehn P, Remy C, et al. Lenalidomide and eltrombopag for treatment in low or intermediate risk myelodysplastic syndrome: result of a phase 2 study combination clinical trial. Blood. (2021) 138(Suppl. 1):65.
57. de Witte T, Bowen D, Robin M, Malcovati L, Niederwieser D, Yakoub-Agha I, et al. Allogeneic hematopoietic stem cell transplantation for MDS and C recommendations from an international expert panel. Blood. (2017) 129:1753–62. doi: 10.1182/blood-2016-06-724500
58. Robin M, Fenaux P. Which lower risk myelodysplastic syndromes should be treated with allogeneic hematopoietic stem cell transplantation? Leukemia. (2020) 34:2552–60.
59. Nakamura R, Saber W, Martens MJ, Ramirez A, Scott B, Oran B, et al. Biologic assignment trial of reduced-intensity hematopoietic cell transplantation based on donor availability in patients 50-75 years of age with advanced myelodysplastic syndrome. J Clin Oncol. (2021) 39:3328–39. doi: 10.1200/JCO.20.03380
60. Robin M, Porcher R, Zinke-Cerwenka W, van Biezen A, Volin L, Mufti G, et al. Allogeneic haematopoietic stem cell transplant in patients with lower risk myelodysplastic syndrome: a retrospective analysis on behalf of the chronic malignancy working party of the EBMT. Bone Marrow Transplant. (2017) 52:209–15. doi: 10.1038/bmt.2016.266
61. Kubasch AS, Platzbecker U. Setting fire to ESA and EMA resistance: new targeted treatment options in lower risk myelodysplastic syndromes. Int J Mol Sci. (2019) 20:3853. doi: 10.3390/ijms20163853
62. Stomper J, Rotondo JC, Greve G, Lübbert M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: mechanisms of resistance and novel HMA-based therapies. Leukemia. (2021) 35:1873–89. doi: 10.1038/s41375-021-01218-0
63. Stahl M, Zeidan AM. Lenalidomide use in myelodysplastic syndromes: insights into the biologic mechanisms and clinical applications. Cancer. (2017) 123:1703–13. doi: 10.1002/cncr.30585
Keywords: myelodysplastic syndrome, low risk, treatment, anemia, thrombocytopenia
Citation: Toprak SK (2022) Past, present and future in low-risk myelodysplastic syndrome. Front. Med. 9:967900. doi: 10.3389/fmed.2022.967900
Received: 13 June 2022; Accepted: 28 June 2022;
Published: 15 July 2022.
Edited by:
Erden Atilla, Mersin State Hospital, TurkeyReviewed by:
Ender Soydan, Güven Hospital, TurkeyCopyright © 2022 Toprak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Selami Kocak Toprak, c2t0b3ByYWtAeWFob28uY29t, orcid.org/orcid.org//0000-0001-7717-5827
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.