 Rajalingham Sakthiswary1*
Rajalingham Sakthiswary1* Rajeswaran Uma Veshaaliini1
Rajeswaran Uma Veshaaliini1 Kok-Yong Chin2
Kok-Yong Chin2 Srijit Das3
Srijit Das3 Srinivasa Rao Sirasanagandla3
Srinivasa Rao Sirasanagandla3- 1Department of Medicine, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
- 2Department of Pharmacology, Universiti Kebangsaan Malaysia Medical Centre, Kuala Lumpur, Malaysia
- 3Department of Human and Clinical Anatomy College of Medicine and Health Sciences Sultan Qaboos University, Muscat, Oman
Rheumatoid arthritis (RA) is an autoimmune disease, in which the inflammatory processes involve the skeletal system and there is marked destruction of the bones and the surrounding structures. In this review, we discuss the current concepts of osteoimmunology in RA, which represent the molecular crosstalk between the immune and skeletal systems, resulting in the disruption of bone remodeling. Bone loss in RA can be focal or generalized, leading to secondary osteoporosis. We have summarized the recent studies of bone loss in RA, which focused on the molecular aspects, such as cytokines, autoantibodies, receptor activator of nuclear kappa-β ligand (RANKL) and osteoprotegerin (OPG). Apart from the above molecules, the role of aryl hydrocarbon receptor (Ahr), which is a potential key mediator in this process through the generation of the Th17 cells, is discussed. Hence, this review highlights the key insights into molecular mechanisms of bone loss in RA.
Introduction
Bone remodeling mechanisms in RA
Physiological bone remodeling occurs through cell-mediated processes in response to biomechanical signals. The initial phase of bone resorption which is mediated by osteoclasts is followed by the recruitment of osteoblasts which mediate bone formation (1). Under normal circumstances, there is a tight coupling of osteoclastic and osteoblastic activities. Transforming growth factor-β (TGF-β) and bone morphogenetic proteins (BMPs) are bone growth factors which are sequestered into the bone matrix and have been strongly implicated in linking bone resorption and formation (2, 3). Besides, osteoclasts may independently modulate osteoblast differentiation and activity. There are other factors that influence physiologic bone remodeling include spingosine-1-phosphate, Wnt 10b and BMP6 that enhance bone formation (4) and semaphorin 4D that inhibits bone formation (5).
Rheumatoid arthritis (RA) is a form of chronic inflammatory arthritis with negative effects on skeletal remodeling. Periarticular bone erosions represent the radiographic hallmark of RA (6). Matrix metalloproteinases (MMPs) which are produced by synovial fibroblasts have been implicated in the degradation of extracellular matrix; mainly the proteoglycans and collagens of the articular cartilage (7). MMP-1 and MMP-3 levels were found to correlate strongly with joint erosions but there is a paucity of published data on the effects of MMPs on osteoclastogenesis (8–10). In active RA, disruptions in physiologic bone remodeling occur, whereby a mismatch in osteoclastic-osteoblastic activity results in enhanced bone resorption with a lack of compensatory bone formation. the processes that regulate the coupling of bone resorption and formation which usually occur under physiologic conditions. There is a mismatch in osteoclastic-osteoblastic activity resulting in enhanced bone resorption with a lack of compensatory bone formation. Beyond the disordered focal bone remodeling associated with the active synovitis of the affected joints, there is generalized axial and appendicular bone loss at distant sites which are not inflamed. Hence, there are 3 types of bone loss i.e., local, juxta-articular and systemic. While all these forms are related to inflammation, the underlying mechanisms for each type of bone loss vary. Systemic bone loss or osteoporosis in RA is caused by systemic inflammation, the use of glucocorticoid therapy and physical impairment while local bone loss in the form of joint erosions due to the effects of the synovial immune cells and cytokines (11). In RA, studies have consistently pointed out that biochemical markers of bone turnover have a significant positive correlation with the disease activity. The systemic bone loss is mediated by synovial cytokines with osteoclastogenic activity that enter the blood circulation and adversely affect the generalized bone remodeling (12).
The bone pannus interface in patients with RA exhibits a heterogeneous population of cells including osteoclasts. Animal models have implicated osteoclasts for the marginal joint erosions in RA. The rheumatoid synovium contains abundant osteoclast precursors and factors with potent pro-osteoclast differentiation and activation activity. Receptor activator of nuclear kappa-β ligand (RANKL), a member of the tumor necrosis factor ligand family, plays a vital role in osteoclastogenesis. RANKL is expressed by synovial fibroblasts and activated T cells in the joints of RA patients. Hence, treatment with osteoprotegerin (the soluble receptor that inhibits RANKL activity) results in marked inhibition of joint erosions (Figure 1).

Figure 1. RANKL-dependent osteoclastogenesis.
In RA, an additional mechanism of bone adaptation involves the process of endochondral bone formation whereby there is replacement of cartilaginous matrix with mineralised bone. Local production of bone growth factors, including TGF-β and BMPs have been implicated in this process (13). Multiple cytokines, lipid mediators and growth factors have been shown to function as anabolic mediators of bone formation either via effects on osteoblasts and their precursors or via chondrogenesis. Of these mediators, particular attention has focused more recently on the wingless (WNT) signaling pathway (14). The Wnt ligands regulate bone formation via several distinct pathways, including the WNT/b-catenin pathway, the non-canonical WNT/planar cell polarity pathway and the WNT/calcium pathway. Signaling via the canonical pathway is initiated by the binding of WNT ligands to a dual receptor complex comprising low-density lipoprotein receptor-related protein (LRP) 5 or LRP6. This leads to the inactivation of a multiprotein β-catenin complex that targets β-catenin to proteosomal degradation. β-Catenin then accumulates in the cytoplasm and translocates to the nucleus, where it stimulates osteoblast differentiation. Activation of the canonical β-catenin pathway results in transcriptional activation of the osteoprotegerin (OPG) gene (15). OPG is a potent inhibitor of RANK ligand (RANKL), which is the key regulator of osteoclast differentiation and activation. The WNT signaling pathway is regulated by families of both activators and inhibitors. Sclerostin, is one of the most potent inhibitors of this pathway (16). The Dickkopf (DKK) proteins and sclerostin, as well as soluble frizzled related protein play a role in modulating the pattern of bone repair and regeneration in RA (17).
Receptor activator of nuclear kappa-β ligand (RANKL)/osteoprotegerin (OPG)
The bone remodeling process is delicately orchestrated by osteoclasts and osteoblasts through multiple signaling pathways. The RANKL/OPG is the most recognized axis in mediating the formation of osteoclasts (18). Osteoblasts secrete RANKL that binds with RANK on the osteoclasts precursors to promote their differentiation into mature osteoclasts with bone-resorption ability. Apart from osteoblasts, hypertrophic chondrocytes and osteocytes are the major sources of RANKL during growth and maturation (19). In patients with RA and animals with experimentally induced inflammatory arthritis, fibroblast-like synoviocytes (FLS) represent a major source of RANKL (20, 21). Briefly, the binding of RANKL to RANK recruits adaptor protein tumor necrosis factor receptor and activates mitogen-activated protein kinases, nuclear factor-kappa β and activator protein-1. Coupled with immunoreceptor tyrosine-based activation motif-mediated calcium ion signaling, this eventually leads to activation and amplification nuclear factor of activated T-cells cytoplasmic 1, which transcribes genes that stimulate osteoclast differentiation and bone resorption (22, 23). OPG acts as a decoy receptor to bind with RANKL, thus preventing the activation of RANK-RANKL signaling (23, 24) (Figure 2).
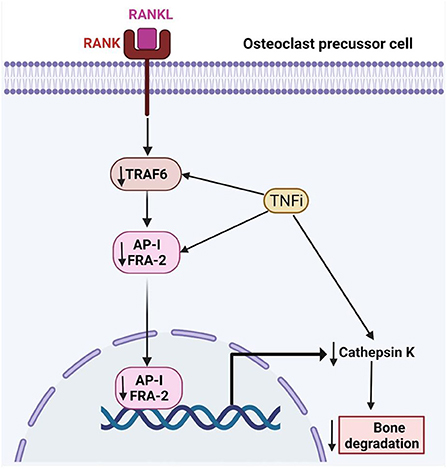
Figure 2. The effects of tumor necrosis factor inhibitors on RA-related bone loss. TRAF6, TNF-receptor associated factor-6; FRA-2, Fos-related antigen-2; AP-1, Activator protein 1; RANK, receptor activator of nuclear factor kappa-B; RANKL, Receptor activator of nuclear factor kappa-B ligand.
The expression of RANKL and OPG is triggered by various factors, including inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, IL-17 present in high levels among patients with RA (25). Of note, IL-6 and soluble IL-6 receptor induce RANKL expression in FLS and they are vital for the TNF-α and IL-17 mediated RANKL expression (26). A recent study revealed that the level of lymphotoxin-α, a close homolog of TNF-α, is higher in patients with RA compared to those with OA. It can induce RANKL expression in chondrocytes in a manner like TNF-α, which subsequently promotes the differentiation of peripheral mononuclear cells into osteoclast-like cells in vitro (27). Thus, these factors could contribute to increased RANKL levels leading to bone erosions; observed in RA patients. In tissues samples of patients with RA, RANKL and OPG are co-expressed at the sites of articular bone erosion, with RANK-positive multinucleated cells (28). In addition, RANKL expression by FLS is shown to play a more significant role in RA-associated bone erosion based on a study using cell-specific RANKL knockout animals (29). Conventional anti-RA agents like methotrexate have been shown to prevent bone resorption by reducing serum soluble RANKL levels (30).
Epidemiological studies have reported that RANKL level is higher in patients with RA compared to normal controls (31), and correlates with Larsen score and radiological progression (31). A meta-analysis showed that RANKL gene rs2277438 polymorphism increases RA risk, while RANK gene rs1805034, OPG gene rs3102735, rs2073618, rs3134069 polymorphism were not related to RA risk (32). A Polish study revealed that RANK rs8086340 single nucleotide polymorphism affects disease susceptibility while RANK rs8086340 and RANKL rs7325635 GG were associated with lower C-reactive protein levels. RANKL rs8086340 and rs1805034 CC homozygotes were associated with higher alkaline phosphatase levels compared to other phenotypes whereas RANKL rs7988338 GG homozygotes were associated with a lower number of swollen joints compared to the A allele (33). A meta-analysis of three French cohorts found that the OPG gene G allele of rs2073618 is associated with bone erosion defined by the Sharp score, but not RANKL rs7325635 gene allele (34).
Given the important role of RANKL in RA, denosumab, an antibody against RANKL, could ameliorate the effects of RANKL on bone damage. Individual randomized controlled trials have demonstrated the efficacy of denosumab in reducing new bone erosion and promoting bone erosion healing in patients with RA (35, 36). Discontinuation of denosumab among patients with RA receiving glucocorticoids leads to a gradually increased bone remodeling and decreased bone mineral density (BMD) to baseline level after 12 months (37). These data show that inhibition of RANKL activity is a promising approach to reduce bone damage in patients with RA. A study on Fc-osteoprotegerin infusion in animals with collagen-induced arthritis showed that the treatment can prevent bone erosion by inhibiting osteoclast formation, but not cartilage erosion (38). Table 1 summarizes the recent studies on RANKL/OPG in RA.
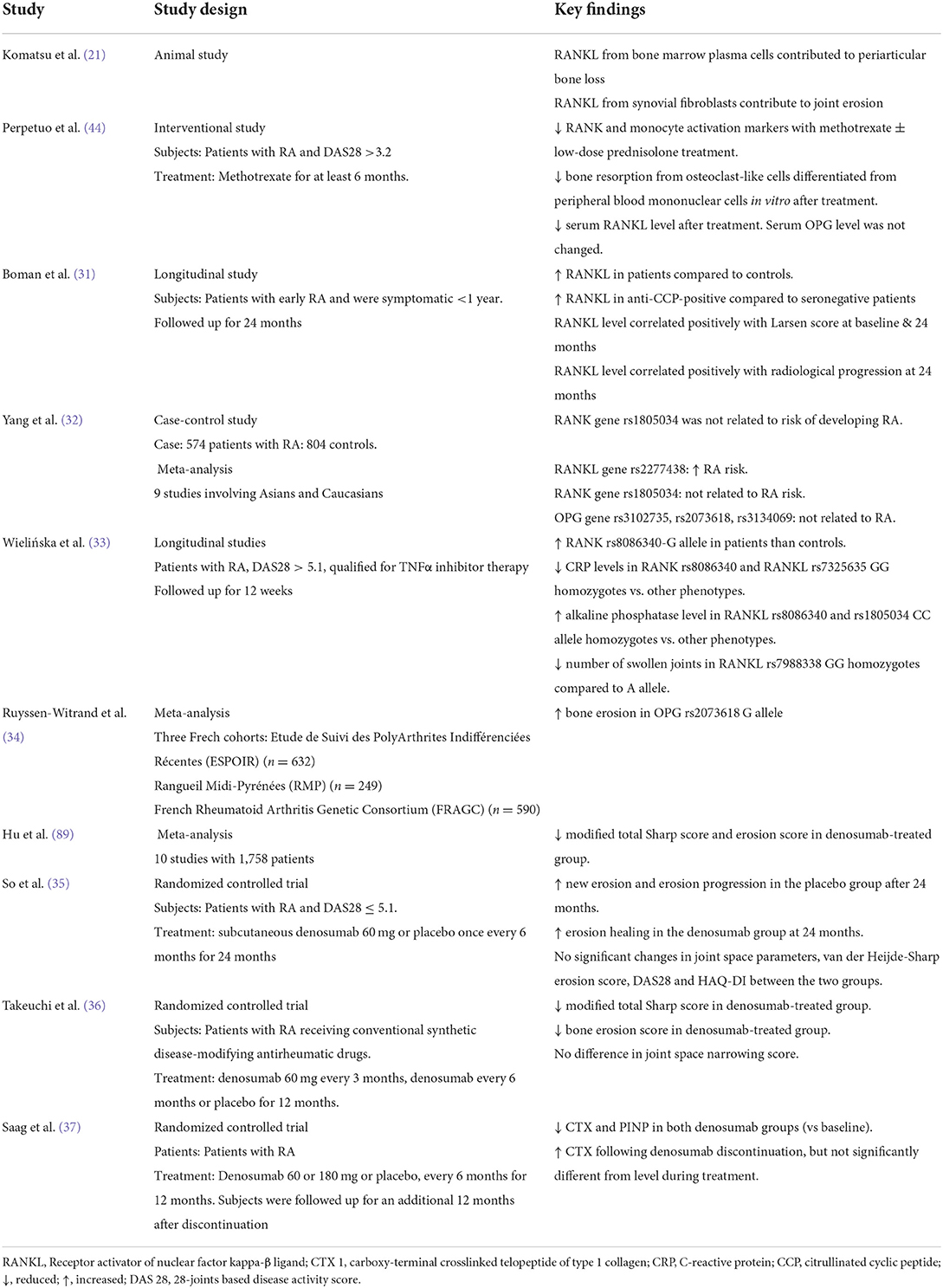
Table 1. Recent evidence on the association between RANKL/OPG and RA.
The central role of cytokines
The RANKL/OPG ratio determines the physiological balance of bone formation and resorption, with a higher ratio favoring increased bone resorption. A higher RANKL/OPG ratio is associated with increased radiographic damage in RA patients (39). Various proinflammatory cytokines regulate the expression of RANKL and OPG including TNFα, IL-6 and IL-1 (40). Table 2 summarizes the findings of recent studies on cytokine related bone loss in RA.
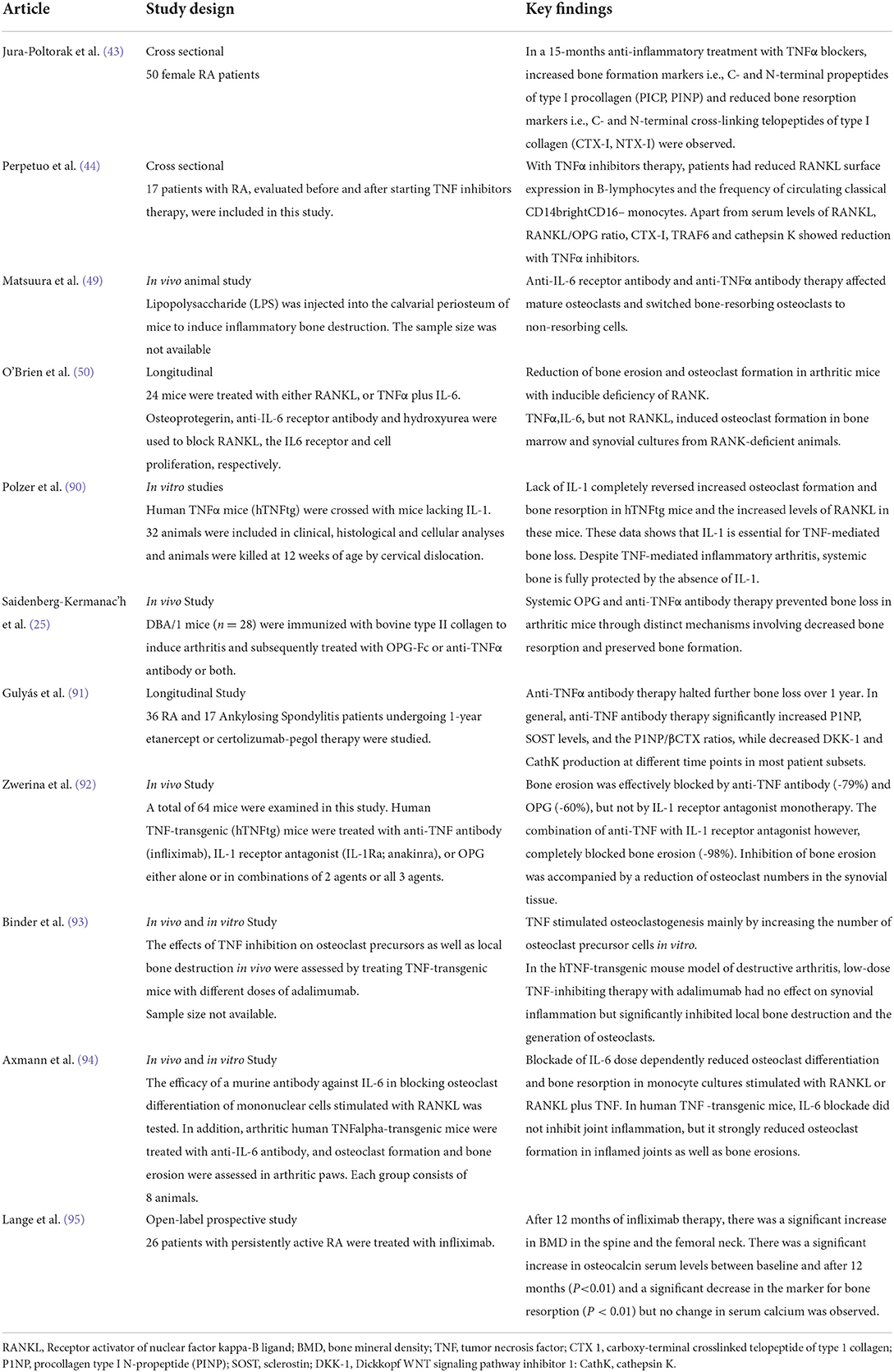
Table 2. Recent studies on cytokines-related bone loss in RA.
TNFα
TNFα promotes osteoclastogenesis by promoting the expression of the essential osteoclast differentiation factor, RANKL, and/or OPG, by bone marrow stromal cells (41, 42). Jura-Półtorak et al. demonstrated that the 15-month anti-inflammatory treatment with TNFα inhibitors was associated with increased bone formation markers i.e., C- and N-terminal propeptides of type I procollagen (PICP, PINP) and reduced bone resorption markers i.e., C- and N-terminal cross-linking telopeptides of type I collagen (CTX-I, NTX-I) (43). In keeping with these findings, Perpetuo et al. disclosed that after TNFα inhibitor therapy, patients had reduced RANKL surface expression in B-lymphocytes and the frequency of circulating classical CD14brightCD16− monocytes. Apart from the serum levels of RANKL, RANKL/OPG ratio, CTX-I, TRAF6 and cathepsin K showed a reduction with TNFα inhibitors (44).
IL-6
IL-6 has a wide variety of biological effects in RA such as induction of proliferation of B cells and plasma cells, enhancement of production of acute-phase protein by hepatocytes and stimulation of differentiation of T helper cells. IL-6 is one of the culprit cytokines responsible for joint destruction in RA.
In murine bone marrow cells, IL-6 suppresses osteoclast differentiation through inhibition of the nuclear factor of activated T cells cytoplasmic 1 (NFATc1) (45). Besides, animal studies have suggested that IL-6 is essential for the differentiation from naïve T cells to Th17 cells (46, 47). Th17 cells, a subset of T-helper cells, are also known to express RANKL (48). Following treatment with anti-IL-6 receptor antibodies in murine models of arthritis, osteoclasts tend to transform into non-resorbing cells (49). Recent studies have also reported that co-stimulation of IL-6 and TNFα may trigger osteoclast differentiation from bone marrow-derived macrophages. This form of osteoclastogenesis is considered to occur in a RANKL-independent manner, given that it is not inhibited by OPG (50).
IL-1
The IL-1 family of ligands includes 11 members, with IL-1β as the main culprit in many inflammatory conditions. The inactive IL-1β precursor is cleaved by caspase-1 into an active cytokine (51), which binds to type I (IL-1RI) and type II (IL-1RII) specific receptors. IL-1β is a strong stimulator of osteoclastogenesis (52) through upregulation of the production of RANKL and downregulation of OPG (53). Besides, IL-1β increases prostaglandin synthesis (54), which is a potent resorption stimulus (10). Prostaglandins, such as prostaglandin E2 (PGE2), may mediate the upregulation of RANKL by activating cell-surface receptors (55). Furthermore, IL-1β also stimulates osteoclast activity by increasing the production of macrophage colony-stimulating factor (M-CSF) and inhibiting osteoclast apoptosis. IL-1β has an intimate relationship with TNF-α and experimental evidence suggests that blocking IL1β and TNF-α may completely halt bone resorption (56).
Apart from enhancing osteoclastogenesis, IL-1β strongly inhibits osteoblastogenesis. The inhibition of osteoblast activity is modulated via mitogen-activated protein kinase (MAPK), by activated signal transducers and activators of transcription (STATs). IL-1β also upregulates DKK1- and sclerostin, which may inhibit the production of osteoblasts (57). In a Chinese population of patients with postmenopausal osteoporosis, there was a strong association between the Taq I IL-1β exon 5 gene polymorphism and a reduced BMD (58) (Figure 3).
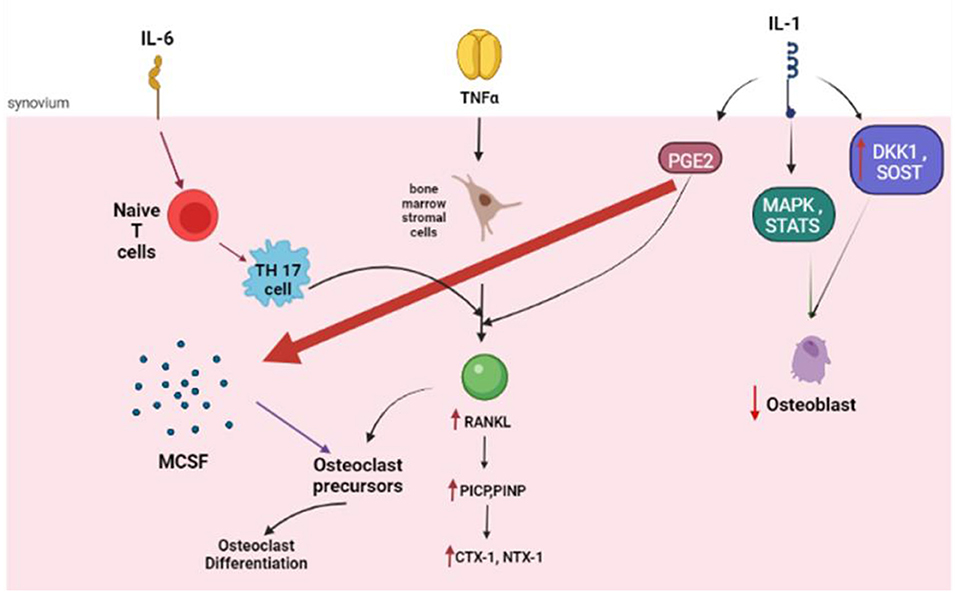
Figure 3. The role of the key cytokines in osteoclastogenesis. IL, Interleukin; NF, Tumor Necrosis factor; RANKL, Receptor activator of nuclear factor kappa-B ligand; PICP, propeptides of type I procollagen; PINP, N-terminal propeptides of type I procollagen; CTX 1, carboxy-terminal crosslinked telopeptide of type 1 collagen; NTX 1, amino-terminal crosslinked telopeptide of type 1 collagen; PGE, prostaglandin; MAPK, mitogen-activated protein kinase; DKK1, Dickkopf WNT signaling pathway inhibitor 1; SOST, Sclerostin.
Other cytokines
Many other cytokines such as IL-22, IL-9, and IL-17A have been implicated to a lesser extent in RA-related bone loss (59–61). IL-17A which has a well-established role in the pathogenesis of enthesitis in spondyloarthritides. It exerts its pathogenic effects on FLS via IL-17/IL-17RA/STAT-3 signaling. IL-17 was found to have osteoclastogenic potential through the upregulation of RANKL (59). In a similar manner, IL-9 stimulation was reported to significantly enhance M-CSF -RANKL-mediated osteoclast formation and differentiation. IL-9 regulates the expression of genes in the metabolic pathways and the expression of matrix metalloproteinases (MMPs), which are responsible for bone degradation (60).
Autoantibodies and bone resorption
Autoantibodies, namely rheumatoid factor (RF) and anti-citrullinated cyclic peptide (anti-CCP), play pivotal roles in RA for diagnostic and prognostic purposes. Seropositive disease is associated with a more severe and aggressive disease course (62). Emerging evidence suggests an association between anti-CCP and bone resorption. Clinical studies have consistently revealed that anti-CCP positivity was related to an increased risk of major fracture, lower BMD and osteopenia (63, 64). Bugatti et al. reported that anti-CCP positivity negatively affected the Z-scores of the spine and hip. The above association was observed even at low levels of RF [adjusted OR (95 % CI) 2.65 (1.01 to 7.24)], but was further increased by concomitant high RF [adjusted OR (95 % CI) 3.38 (1.11 to 10.34)] (64). This finding lends credence to the notion that autoantibodies may have a direct causative role in bone remodeling. In a Spanish cohort of patients with early arthritis and a median disease duration of 5.1 months, anti-CCP positivity remained significantly associated with lower BMD at the lumbar spine, femoral neck, and hip but not at the metacarpophalangeal joints despite adjustment for gender, age and body mass index (65). Taken together, these results may suggest that autoantibodies tend to induce systemic bone loss earlier than local or articular bone loss. Table 3 summarizes the findings of recent studies on anti-CCP and bone loss. However, there is a lack of data on the relationship between RF and bone loss.

Table 3. Recent studies on the association between anti-CCP and bone mineral density.
The levels of the active TRAP5b isoform, which represent the number and activity of osteoclasts (35, 36), and cathepsin K, which degrades the bone matrix proteins (37, 38), were both found to be significantly higher in the bone marrow samples with anti-CCP. The anti-CCP-positive bone marrow samples also contained increased concentrations of CTX-I, which is produced as a result of the enzymatic activity of cathepsin K (66, 67).
The key orchestrator of anti-CCP-dependent osteoclastogenesis is IL-8 (68). The concentration of this cytokine increases during osteoclast differentiation and in the presence of anti-CCP. Bone marrow samples from RA patients revealed increased IL-8 levels in anti-CCP positive bone marrow. TRAP5b and cathepsin K levels were found to correlate positively with IL-8 concentration (63).
Apart from RF and anti-CCP, anti-carbamylated protein antibodies, which are not routinely tested in clinical practice, were found to have an independent association with lower BMD in the spine and hip but not the metacarpophalangeal joint (69) (Figure 4).
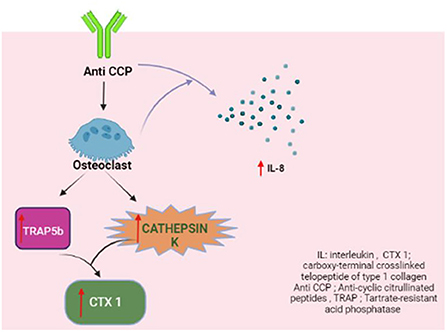
Figure 4. Anti-CCP and osteoclastogenesis.
Role of aryl hydrocarbon receptor in RA-associated bone erosion
Aryl hydrocarbon receptor (Ahr) is a ligand-activated transcription factor that belongs to the Per-Arnt-Sim protein superfamily. It is a nuclear receptor and usually stays inactive in the cytosol due to its binding with cochaperones. When it binds to a specific ligand, its structure transforms and it is translocated to the nucleus, where it forms heterodimerization with aromatic hydrocarbon receptor nuclear transfer protein (ARNT). Subsequently, the Ahr/ARNT complex regulates biological functions, including toxicity, biological evolution, bone remodeling, and immune response by promoting gene transcription of various prototypic genes (70). Increasing evidence confirms its close association with the pathogenesis of immune diseases, including RA. Ahr activation significantly contributes to the development of RA (71). In experimental studies, Ahr activation with its agonists contributed to RA disease progression, bone damage and osteoclast differentiation (72). Ahr expression was found to be 2-fold higher in RA patients when compared to controls. Furthermore, a positive association between cigarette smoking and Ahr activation in RA patients has been demonstrated (73) (Figure 5).
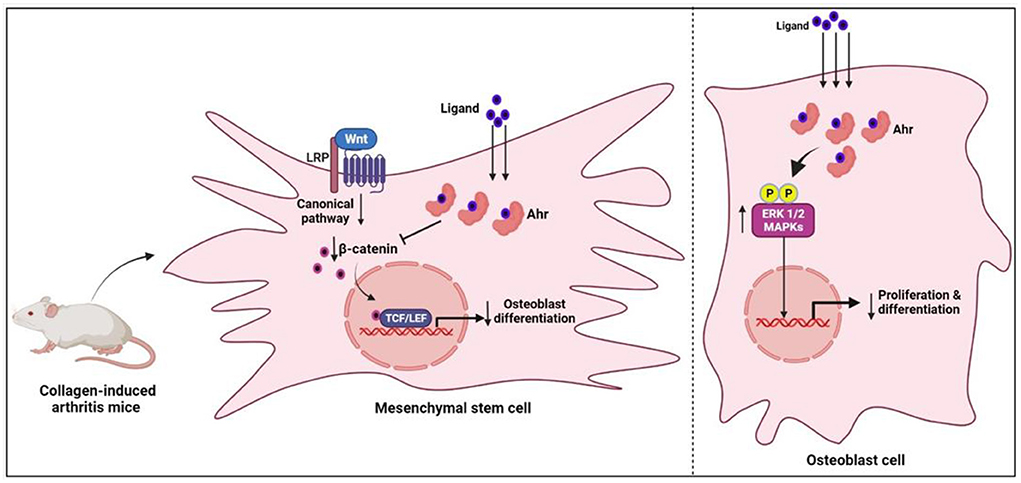
Figure 5. The mechanism of osteogenesis inhibition by Ahr-associated mesenchymal stem cell via the Wnt/-catenin pathway. Ahr, aryl hydrocarbon receptor; ERK1/2, extracellular signal_regulated protein kinase; MAPK, mitogen-activated protein kinase.
Maintaining homeostasis in bone remodeling requires a balance between the relative activity of bone-forming osteoblasts and bone-resorbing osteoclasts. The modulation of AhR signaling via the NF-B, Wnt, and MAP kinase pathways has been linked to changes in bone remodeling (74–78). The activated Ahr may affect bone remodeling by interfering with the functional differentiation of osteoblasts and osteoclasts (79). The influence of Ahr signaling on osteoblast function is well–established in recent studies. The bone erosion in arthritic mice was found to be positively associated with high Ahr expression, and these highly expressed Ahr levels were relatively localized to the osteoblast cells (80). Additionally, in in vitro conditions, the activated Ahr inhibited the proliferation and differentiation of osteoblast cells by upregulating the ERK/MAPK signaling pathway (80). In further investigations, it was concluded that the activated Ahr in mesenchymal stem cells, derived from collagen-induced arthritis mice, inhibited osteogenesis by downregulating β-catenin (81). On the other hand, the influence of Ahr signaling on osteoclastogenesis is not clearly understood. In in vivo and in vitro studies of rodents, there were inconsistent results regarding the effects of Ahr on osteoclast differentiation (82). However, findings from a recent study on human osteoclast cells revealed that Ahr activation through kynurenine inhibited osteoclast differentiation by downregulating NFATc1 protein expression (83). The same study concluded that Ahr activation could be a potential target to treat the bone loss in RA. In support of this, natural products derived Ahr ligands/agonists have been shown to prevent bone erosions in RA (84, 85). Overall, the influence of Ahr signaling on RA-associated bone erosion is still controversial as it inhibits the osteoblast cells and may either inhibit or promote osteoclast differentiation.
Disease-modifying anti-rheumatic drugs (DMARDs) and bone loss
Disease-modifying anti-rheumatic drugs (DMARDs) are the mainstay of treatment in RA. There are conventional (methotrexate, leflunomide, sulfasalazine), biologic (adalimumab, etanercept, golimumab, tocilizumab etc) and targeted synthetic DMARDs (tofacitinib, baricitinib, upadacitib). Apart from suppression of inflammation, there is ample evidence that all of these classes of drugs prevent articular erosions and radiographic progression of joint damage. Chen et al. reported that after 3 years of biologic or targeted synthetic DMARD therapy, BMD remained stable at the femoral neck, hip and lumbar vertebra (86). These findings were echoed by other studies (87, 88).
Conclusions
There is compelling evidence that the interplay of various pathways and different mechanisms contribute to bone loss and secondary osteoporosis in RA. Osteoclastogenesis may occur via RANKL-dependent and RANKL-independent processes. TNFα, IL-1, IL-6 anti-CCP which are the key orchestrators of inflammation tend to affect bone remodeling adversely.
Author contributions
RS, RU, K-YC, and SD contributed to the literature search and manuscript composition. SS contributed to creation of the figures and manuscript composition. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by a grant by the Ministry of Higher Education of Malaysia (FRGS/1/2021/SKK08/UKM/02/1) and the National University of Malaysia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eriksen EF. Cellular mechanisms of bone remodeling. Rev Endocr Metab Disord. (2010) 11:219–27. doi: 10.1007/s11154-010-9153-1
2. Nistala H, Lee-Arteaga S, Siciliano G, Smaldone S, Ramirez F. Extracellular regulation of transforming growth factor beta and bone morphogenetic protein signaling in bone. Ann N Y Acad Sci. (2010) 1192:253–6. doi: 10.1111/j.1749-6632.2009.05350.x
3. Wu X, Shi W, Cao X. Multiplicity of BMP signaling in skeletal development. Ann N Y Acad Sci. (2007) 1116:29–49. doi: 10.1196/annals.1402.053
4. Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A. (2008) 105:20764–9. doi: 10.1073/pnas.0805133106
5. Negishi-Koga T, Shinohara M, Komatsu N, Bito H, Kodama T, Friedel RH, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med. (2011) 17:1473–80. doi: 10.1038/nm.2489
6. Sargin G, Kose R, Senturk T. Relationship between bone mineral density and anti-citrullinated protein antibody and rheumatoid factor in patients with rheumatoid arthritis. Eur J Rheumatol. (2019) 6:29–33. doi: 10.5152/eurjrheum.2018.18099
7. Araki Y, Mimura T. Matrix metalloproteinase gene activation resulting from disordred epigenetic mechanisms in rheumatoid arthritis. Int J Mol Sci. (2017) 18:905. doi: 10.3390/ijms18050905
8. Green MJ, Gough AK, Devlin J, Smith J, Astin P, Taylor D, et al. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology (Oxford). (2003) 42:83–8. doi: 10.1093/rheumatology/keg037
9. Di Spigna G, Rossi FW, Mormile I, Ladogana P, Buonavolonta L, Covelli B, et al. Serum Metalloprotease 3 (MMP-3) biomarker of therapeutic efficacy during treatment of rheumatoid arthritis. J Biol Regul Homeost Agents. (2021) 35:1041–5. doi: 10.23812/21–86–L
10. Sun Y, Huang Y, Chen T, Li X, Chen J, Wang Z, et al. Effect of downregulation of serum MMP-3 levels by traditional Chinese medicine ingredients combined with methotrexate on the progression of bone injury in patients with rheumatoid arthritis: A protocol for a systematic review and meta-analysis. Medicine (Baltimore). (2020) 99:e22841. doi: 10.1097/MD.0000000000022841
11. Llorente I, García-Castañeda N, Valero C, González-Álvaro I, Castañeda S. Osteoporosis in rheumatoid arthritis: dangerous liaisons. Front Med (Lausanne). (2020) 7:601618. doi: 10.3389/fmed.2020.601618
12. Goldring SR. The effects of inflammatory arthritis on bone remodeling. Arthritis Res Ther. (2005) 7:S12. doi: 10.1186/ar1518
13. Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, et al. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. (2004) 104:4029–37. doi: 10.1182/blood-2004-01-0041
14. O'Brien CA. Control of RANKL gene expression. Bone. (2010) 46:911–9. doi: 10.1016/j.bone.2009.08.050
15. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. (2007) 109:3839–48. doi: 10.1182/blood-2006-07-037994
16. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. doi: 10.1038/16852
17. Braun T, Zwerina J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res Ther. (2011) 13:235. doi: 10.1186/ar3380
18. Ono T, Hayashi M, Sasaki F, Nakashima T. RANKL biology: bone metabolism, the immune system, and beyond. Inflamm Regen. (2020) 40:2. doi: 10.1186/s41232-019-0111-3
19. Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE. (2015) 10:e0138189. doi: 10.1371/journal.pone.0138189
20. Kim KW, Cho ML, Oh HJ, Kim HR, Kang CM, Heo YM, et al. TLR-3 enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Immunol Lett. (2009) 124:9–17. doi: 10.1016/j.imlet.2009.02.006
21. Komatsu N, Win S, Yan M, Huynh NC, Sawa S, Tsukasaki M, et al. Plasma cells promote osteoclastogenesis and periarticular bone loss in autoimmune arthritis. J Clin Invest. (2021) 131. doi: 10.1172/JCI143060
22. Park JH, Lee NK, Lee SY. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol Cells. (2017) 40:706–13. doi: 10.14348/molcells.2017.0225
23. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. (2014) 5:511. doi: 10.3389/fimmu.2014.00511
24. Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. (2007) 9 (Suppl. 1):S1. doi: 10.1186/ar2165
25. Saidenberg-Kermanac'h N, Cohen-Solal M, Bessis N, De Vernejoul MC, Boissier MC. Role for osteoprotegerin in rheumatoid inflammation. Joint Bone Spine. (2004) 71:9–13. doi: 10.1016/S1297-319X(03)00131-3
26. Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford). (2008) 47:1635–40. doi: 10.1093/rheumatology/ken363
27. Takeshita A, Nishida K, Yoshida A, Nasu Y, Nakahara R, Kaneda D, et al. RANKL expression in chondrocytes and its promotion by lymphotoxin-α in the course of cartilage destruction during rheumatoid arthritis. PLoS ONE. (2021) 16:e0254268. doi: 10.1371/journal.pone.0254268
28. Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM. RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology (Oxford). (2006) 45:1068–76. doi: 10.1093/rheumatology/kel045
29. Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis. (2016) 75:1187–95. doi: 10.1136/annrheumdis-2014-207137
30. Perpétuo IP, Caetano-Lopes J, Rodrigues AM, Campanilho-Marques R, Ponte C, Canhão H, et al. Methotrexate and low-dose prednisolone downregulate osteoclast function by decreasing receptor activator of nuclear factor-κβ expression in monocytes from patients with early rheumatoid arthritis. RMD Open. (2017) 3:e000365. doi: 10.1136/rmdopen-2016-000365
31. Boman A, Kokkonen H, Ärlestig L, Berglin E, Rantapää-Dahlqvist S. Receptor activator of nuclear factor kappa-B ligand (RANKL) but not sclerostin or gene polymorphisms is related to joint destruction in early rheumatoid arthritis. Clin Rheumatol. (2017) 36:1005–12. doi: 10.1007/s10067-017-3570-4
32. Yang H, Liu W, Zhou X, Rui H, Zhang H, Liu R. The association between RANK, RANKL and OPG gene polymorphisms and the risk of rheumatoid arthritis: a case-controlled study and meta-analysis. Biosci Rep. (2019) 39:BSR20182356. doi: 10.1042/BSR20182356
33. Wielińska J, Kolossa K, Swierkot J, Dratwa M, Iwaszko M, Bugaj B, et al. Polymorphisms within the RANK and RANKL encoding genes in patients with rheumatoid arthritis: association with disease progression and effectiveness of the biological treatment. Arch Immunol Ther Exp (Warsz). (2020) 68:24. doi: 10.1007/s00005-020-00590-6
34. Ruyssen-Witrand A, Degboé Y, Cantagrel A, Nigon D, Lukas C, Scaramuzzino S, et al. Association between RANK, RANKL and OPG polymorphisms with ACPA and erosions in rheumatoid arthritis: results from a meta-analysis involving three French cohorts. RMD Open. (2016) 2:e000226-e. doi: 10.1136/rmdopen-2015-000226
35. So H, Cheng IT, Lau SL, Chow E, Lam T, Hung VW, et al. Effects of RANKL inhibition on promoting healing of bone erosion in rheumatoid arthritis using HR-pQCT: a 2-year, randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2021) 80:981–8. doi: 10.1136/annrheumdis-2021-219846
36. Takeuchi T, Tanaka Y, Soen S, Yamanaka H, Yoneda T, Tanaka S, et al. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase 3 trial. Ann Rheum Dis. (2019) 78:899–907. doi: 10.1136/annrheumdis-2018-214827
37. Saag KG, McDermott MT, Adachi J, Lems W, Lane NE, Geusens P, et al. The effect of discontinuing denosumab in patients with rheumatoid arthritis treated with glucocorticoids. Arthritis Rheumatol. (2022) 74:604–11. doi: 10.1002/art.41981
38. Romas E, Sims NA, Hards DK, Lindsay M, Quinn JW, Ryan PF, et al. Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol. (2002) 161:1419–27. doi: 10.1016/S0002-9440(10)64417-3
39. van Tuyl LH, Voskuyl AE, Boers M, Geusens P, Landewé RB, Dijkmans BA, et al. Baseline RANKL:OPG ratio and markers of bone and cartilage degradation predict annual radiological progression over 11 years in rheumatoid arthritis. Ann Rheum Dis. (2010) 69:1623–8. doi: 10.1136/ard.2009.121764
40. Xu S, Wang Y, Lu J, Xu J. Osteoprotegerin and RANKL in the pathogenesis of rheumatoid arthritis-induced osteoporosis. Rheumatol Int. (2012) 32:3397–403. doi: 10.1007/s00296-011-2175-5
41. Zhao B. TNF and bone remodeling. Curr Osteoporos Rep. (2017) 15:126–34. doi: 10.1007/s11914-017-0358-z
42. Zerbini CAF, Clark P, Mendez-Sanchez L, Pereira RMR, Messina OD, Uña CR, et al. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos Int. (2017) 28:429–46. doi: 10.1007/s00198-016-3769-2
43. Jura-Poltorak A, Szeremeta A, Olczyk K, Zon-Giebel A, Komosinska-Vassev K. Bone metabolism and RANKL/OPG ratio in rheumatoid arthritis women treated with TNF-alpha inhibitors. J Clin Med. (2021) 10:2905. doi: 10.3390/jcm10132905
44. Perpetuo IP, Caetano-Lopes J, Rodrigues AM, Campanilho-Marques R, Ponte C, Canhao H, et al. Effect of tumor necrosis factor inhibitor therapy on osteoclasts precursors in rheumatoid arthritis. Biomed Res Int. (2017) 2017:2690402. doi: 10.1155/2017/2690402
45. Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J Biol Chem. (2008) 283:11535–40. doi: 10.1074/jbc.M607999200
46. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
47. Moser T, Akgun K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: therapeutic implications. Autoimmun Rev. (2020) 19:102647. doi: 10.1016/j.autrev.2020.102647
48. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. (2006) 203:2673–82. doi: 10.1084/jem.20061775
49. Matsuura Y, Kikuta J, Kishi Y, Hasegawa T, Okuzaki D, Hirano T, et al. In vivo visualisation of different modes of action of biological DMARDs inhibiting osteoclastic bone resorption. Ann Rheum Dis. (2018) 77:1219–25. doi: 10.1136/annrheumdis-2017-212880
50. O'Brien W, Fissel BM, Maeda Y, Yan J, Ge X, Gravallese EM, et al. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. (2016) 68:2889–900. doi: 10.1002/art.39837
51. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
52. Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol. (2013) 25:389–93. doi: 10.1016/j.smim.2013.10.001
53. Nakamura I, Jimi E. Regulation of osteoclast differentiation and function by interleukin-1. Vitam Horm. (2006) 74:357–70. doi: 10.1016/S0083-6729(06)74015-8
54. Ruscitti P, Cipriani P, Carubbi F, Liakouli V, Zazzeroni F, Di Benedetto P, et al. The role of IL-1β in the bone loss during rheumatic diseases. Mediators Inflamm. (2015) 2015:782382. doi: 10.1155/2015/782382
55. Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. (2008) 29:403–40. doi: 10.1210/er.2007-0038
56. Lacativa PG, Farias ML. Osteoporosis and inflammation. Arq Bras Endocrinol Metabol. (2010) 54:123–32. doi: 10.1590/S0004-27302010000200007
57. Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. (2012) 11:234–50. doi: 10.1038/nrd3669
58. Chen HY, Chen WC, Wu MC, Tsai FJ, Lin CC. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphism in postmenopausal women: correlation to bone mineral density and susceptibility to osteoporosis. Maturitas. (2003) 44:49–54. doi: 10.1016/S0378-5122(02)00313-4
59. Ganesan R, Rasool M. Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol Immunol. (2017) 91:134–44. doi: 10.1016/j.molimm.2017.09.003
60. Kar S, Gupta R, Malhotra R, Sharma V, Farooque K, Kumar V, et al. Interleukin-9 facilitates osteoclastogenesis in rheumatoid arthritis. Int J Mol Sci. (2021) 22:10397. doi: 10.3390/ijms221910397
61. Min HK, Won JY, Kim BM, Lee KA, Lee SJ, Lee SH, et al. Interleukin (IL)-25 suppresses IL-22-induced osteoclastogenesis in rheumatoid arthritis via STAT3 and p38 MAPK/IkappaBalpha pathway. Arthritis Res Ther. (2020) 22:222. doi: 10.1186/s13075-020-02315-8
62. Choi S, Lee KH. Clinical management of seronegative and seropositive rheumatoid arthritis: a comparative study. PLoS ONE. (2018) 13:e0195550. doi: 10.1371/journal.pone.0195550
63. Kurowska W, Slowinska I, Krogulec Z, Syrowka P, Maslinski W. Antibodies to citrullinated proteins (ACPA) associate with markers of osteoclast activation and bone destruction in the bone marrow of patients with rheumatoid arthritis. J Clin Med. (2021) 10:1778. doi: 10.3390/jcm10081778
64. Bugatti S, Bogliolo L, Vitolo B, Manzo A, Montecucco C, Caporali R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res Ther. (2016) 18:226. doi: 10.1186/s13075-016-1116-9
65. Llorente I, Merino L, Ortiz AM, Escolano E, González-Ortega S, García-Vicuña R, et al. Anti-citrullinated protein antibodies are associated with decreased bone mineral density: baseline data from a register of early arthritis patients. Rheumatol Int. (2017) 37:799–806. doi: 10.1007/s00296-017-3674-9
66. Lv Y, Wang G, Xu W, Tao P, Lv X. Wang Y. Tartrate-resistant acid phosphatase 5b is a marker of osteoclast number and volume in RAW 2647 cells treated with receptor-activated nuclear κB ligand. Exp Ther Med. (2015) 9:143–6. doi: 10.3892/etm.2014.2071
67. Goto T, Yamaza T, Tanaka T. Cathepsins in the osteoclast. J Electron Microsc (Tokyo). (2003) 52:551–8. doi: 10.1093/jmicro/52.6.551
68. Krishnamurthy A, Joshua V, Haj Hensvold A, Jin T, Sun M, Vivar N, et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
69. Regueiro C, Ortiz AM, Boveda MD, Castañeda S, Gonzalez-Alvaro I, Gonzalez A. Association of high titers of anti-carbamylated protein antibodies with decreased bone mineral density in early arthritis patients. PLoS ONE. (2018) 13:e0202583. doi: 10.1371/journal.pone.0202583
70. Yu H, Jiang L, Wan B, Zhang W, Yao L, Che T, et al. The role of aryl hydrocarbon receptor in bone remodeling. Prog Biophys Mol Biol. (2018) 134:44–9. doi: 10.1016/j.pbiomolbio.2017.12.005
71. Nguyen NT, Nakahama T, Kishimoto T. Aryl hydrocarbon receptor and experimental autoimmune arthritis. Semin Immunopathol. (2013) 35:637–44. doi: 10.1007/s00281-013-0392-6
72. Fu J, Nogueira SV, Drongelen VV, Coit P, Ling S, Rosloniec EF, et al. Shared epitope-aryl hydrocarbon receptor crosstalk underlies the mechanism of gene-environment interaction in autoimmune arthritis. Proc Natl Acad Sci U S A. (2018) 115:4755–60. doi: 10.1073/pnas.1722124115
73. Kazantseva MG, Highton J, Stamp LK, Hessian PA. Dendritic cells provide a potential link between smoking and inflammation in rheumatoid arthritis. Arthritis Res Ther. (2012) 14:R208. doi: 10.1186/ar4046
74. Øvrevik J, Låg M, Lecureur V, Gilot D, Lagadic-Gossmann D, Refsnes M, et al. AhR and Arnt differentially regulate NF-κB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun Signal. (2014) 12:48. doi: 10.1186/s12964-014-0048-8
75. Schneider AJ, Branam AM, Peterson RE. Intersection of AHR and Wnt signaling in development, health, and disease. Int J Mol Sci. (2014) 15:17852–85. doi: 10.3390/ijms151017852
76. Wang Q, Kurita H, Carreira V, Ko CI, Fan Y, Zhang X, et al. Ah receptor activation by dioxin disrupts activin, BMP, and WNT signals during the early differentiation of mouse embryonic stem cells and inhibits cardiomyocyte functions. Toxicol Sci. (2016) 149:346–57. doi: 10.1093/toxsci/kfv246
77. Wincent E, Stegeman JJ, Jönsson ME. Combination effects of AHR agonists and Wnt/β-catenin modulators in zebrafish embryos: Implications for physiological and toxicological AHR functions. Toxicol Appl Pharmacol. (2015) 284:163–79. doi: 10.1016/j.taap.2015.02.014
78. Occhi G, Barollo S, Regazzo D, Bertazza L, Galuppini F, Guzzardo V, et al. A constitutive active MAPK/ERK pathway due to BRAFV600E positively regulates AHR pathway in PTC. Oncotarget. (2015) 6:32104–14. doi: 10.18632/oncotarget.5194
79. Korkalainen M, Kallio E, Olkku A, Nelo K, Ilvesaro J, Tuukkanen J, et al. Dioxins interfere with differentiation of osteoblasts and osteoclasts. Bone. (2009) 44:1134–42. doi: 10.1016/j.bone.2009.02.019
80. Yu H, Du Y, Zhang X, Sun Y, Li S, Dou Y, et al. The aryl hydrocarbon receptor suppresses osteoblast proliferation and differentiation through the activation of the ERK signaling pathway. Toxicol Appl Pharmacol. (2014) 280:502–10. doi: 10.1016/j.taap.2014.08.025
81. Tong Y, Niu M, Du Y, Mei W, Cao W, Dou Y, et al. Aryl hydrocarbon receptor suppresses the osteogenesis of mesenchymal stem cells in collagen-induced arthritic mice through the inhibition of β-catenin. Exp Cell Res. (2017) 350:349–57. doi: 10.1016/j.yexcr.2016.12.009
82. Park R, Madhavaram S, Ji JD. The role of aryl-hydrocarbon receptor (AhR) in osteoclast differentiation and function. Cells. (2020) 9:2294. doi: 10.3390/cells9102294
83. Kim SY, Oh Y, Jo S, Ji JD, Kim TH. Inhibition of human osteoclast differentiation by kynurenine through the aryl-hydrocarbon receptor pathway. Cells. (2021) 10:3498. doi: 10.3390/cells10123498
84. Tong B, Yuan X, Dou Y, Wu X, Wang Y, Xia Y, et al. Sinomenine induces the generation of intestinal Treg cells and attenuates arthritis via activation of aryl hydrocarbon receptor. Lab Invest. (2016) 96:1076–86. doi: 10.1038/labinvest.2016.86
85. Tong B, Yuan X, Dou Y, Wu X, Chou G, Wang Z, et al. Norisoboldine, an isoquinoline alkaloid, acts as an aryl hydrocarbon receptor ligand to induce intestinal Treg cells and thereby attenuate arthritis. Int J Biochem Cell Biol. (2016) 75:63–73. doi: 10.1016/j.biocel.2016.03.014
86. Chen JF, Hsu CY Yu SF, Ko CH, Chiu WC, Lai HM, et al. The impact of long-term biologics/target therapy on bone mineral density in rheumatoid arthritis: a propensity score-matched analysis. Rheumatology (Oxford). (2020) 59:2471–80. doi: 10.1093/rheumatology/kez655
87. Chen MH Yu SF, Chen JF, Chen WS, Liou TL, Chou CT, et al. Different effects of biologics on systemic bone loss protection in rheumatoid arthritis: an interim analysis of a three-year longitudinal cohort study. Front Immunol. (2021) 12:783030. doi: 10.3389/fimmu.2021.783030
88. Chen YM, Chen HH, Huang WN, Liao TL, Chen JP, Chao WC, et al. Tocilizumab potentially prevents bone loss in patients with anticitrullinated protein antibody-positive rheumatoid arthritis. PLoS ONE. (2017) 12:e0188454. doi: 10.1371/journal.pone.0188454
89. Hu Q, Zhong X, Tian H, Liao P. The efficacy of denosumab in patients with rheumatoid arthritis: a systematic review and pooled analysis of randomized or matched data. Front Immunol. (2021) 12:799575. doi: 10.3389/fimmu.2021.799575
90. Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, et al. Interleukin-1 is essential for systemic inflammatory bone loss. Ann Rheum Dis. (2010) 69:284–90. doi: 10.1136/ard.2008.104786
91. Gulyás K, Horváth Á, Végh E, Pusztai A, Szentpétery Á, Pethö Z, et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin Rheumatol. (2020) 39:167–75. doi: 10.1007/s10067-019-04771-3
92. Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, et al. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. (2004) 50:277–90. doi: 10.1002/art.11487
93. Binder NB, Puchner A, Niederreiter B, Hayer S, Leiss H, Blüml S, et al. Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis Rheum. (2013) 65:608–17. doi: 10.1002/art.37797
94. Axmann R, Böhm C, Krönke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. (2009) 60:2747–56. doi: 10.1002/art.24781
95. Lange U, Teichmann J, Müller-Ladner U, Strunk J. Increase in bone mineral densit y of patients with rheumatoid arthritis treated with anti-TNF-alpha antibody: a prospective open-label pilot study. Rheumatology (Oxford). (2005) 44:1546–8. doi: 10.1093/rheumatology/kei082
96. Cheng TT Yu SF, Su FM, Chen YC, Su BY, Chiu WC, et al. Anti-CCP-positive patients with RA have a higher 10-year probability of fracture evaluated by FRAX®: a registry study of RA with osteoporosis/fracture. Arthritis Res Ther. (2018) 20:16. doi: 10.1186/s13075-018-1515-1
97. Hafström I, Ajeganova S, Andersson ML, Bala SV, Bergman S, Bremander A, et al. A Swedish register-based, long-term inception cohort study of patients with rheumatoid arthritis - results of clinical relevance. Open Access Rheumatol. (2019) 11:207–17. doi: 10.2147/OARRR.S218448
98. Wysham KD, Shoback DM, Imboden JB, Katz PP. Association of high anti-cyclic citrullinated peptide seropositivity and lean mass index with low bone mineral density in rheumatoid arthritis. Arthritis Care Res (Hoboken). (2018) 70:961–9. doi: 10.1002/acr.23440
99. Amkreutz J, de Moel EC, Theander L, Willim M, Heimans L, Nilsson J, et al. Association between bone mineral density and autoantibodies in patients with rheumatoid arthritis. Arthritis Rheumatol. (2021) 73:921–30. doi: 10.1002/art.41623
100. Ahmad HA, Alemao E, Guo Z, Iannaccone CK, Frits ML, Weinblatt M, et al. Association of low bone mineral density with anti-citrullinated protein antibody positivity and disease activity in established rheumatoid arthritis: findings from a US observational cohort. Adv Ther. (2018) 35:232–42. doi: 10.1007/s12325-017-0657-x
Keywords: rheumatoid arthritis, bone, osteoporosis, cytokines, ligands, autoantibodies
Citation: Sakthiswary R, Uma Veshaaliini R, Chin K-Y, Das S and Sirasanagandla SR (2022) Pathomechanisms of bone loss in rheumatoid arthritis. Front. Med. 9:962969. doi: 10.3389/fmed.2022.962969
Received: 07 June 2022; Accepted: 01 August 2022;
Published: 17 August 2022.
Edited by:
Kayo Masuko, Sanno Medical Center, JapanReviewed by:
Lan Xiao, Queensland University of Technology, AustraliaTing Zheng, Hospital for Special Surgery, United States
Santos Castañeda, Hospital de La Princesa, Spain
Copyright © 2022 Sakthiswary, Uma Veshaaliini, Chin, Das and Sirasanagandla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajalingham Sakthiswary, c2FrdGhpczVAaG90bWFpbC5jb20=