 Jinlin Cheng1
Jinlin Cheng1 Ling Gong
Ling Gong Wenjun Yang
Wenjun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 08 December 2022
Sec. Hepatology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.962408
This article is part of the Research TopicEditors' Showcase: HepatologyView all 10 articles
Objective: To improve the accuracy of the diagnosis of familial progressive intrahepatic cholestasis type 3 (PFIC3, https://www.omim.org/entry/602347).
Materials and methods: Between September 2019 and March 2021, we recruited four patients with PFIC3 from two liver centers in East China. Molecular genetic findings of ATP-binding cassette subfamily B member 4 [ATP binding cassette transporter A4 (ABCB4), https://www.omim.org/entry/171060] were prospectively examined, and clinical records, laboratory readouts, and macroscopic and microscopic appearances of the liver were analyzed.
Results: Four patients experienced cholestasis, mild jaundice, and elevated levels of serum direct bilirubin, γ-glutamyltransferase, or total bile acids. All patients had moderate-to-severe liver fibrosis or biliary cirrhosis, and their liver biopsy specimens stained positive with rhodamine. Molecular immunohistochemistry revealed reduced or absent MDR3 expression in all liver specimens. A novel mutation of ABCB4 (c.1560 + 2T > A) was identified in patients with PFIC3, which is of high clinical significance and may help understand mutant ABCB4 pathogenesis.
Conclusion: MDR3 immunohistochemistry and molecular genetic analyses of ABCB4 are essential for the accurate diagnosis of PFIC3.
Progressive familial intrahepatic cholestasis (PFIC) is a genetic disorder with autosomal recessive inheritance. It primarily manifests as jaundice and itchy skin and then progresses from persistent cholestasis to cirrhosis and liver failure before late childhood (1). Thus far, PFIC has been reported in Caucasian, Asian, and African patients. The estimated incidence of PFIC ranges from 1 per 50,000 to 1 per 100,000 worldwide (2). Based on the gene carrying the mutation, PFIC is currently identified to result from mutations in six genes, namely ATP8B1, ATP-binding cassette subfamily B member 11 (ABCB11), ATP binding cassette transporter A4 (ABCB4), TJP2, NR1H4, and MYO5B. There are three classic types of PFIC, referred to as PFIC1, PFIC2, and PFIC3, which result from a defect in a biliary protein involved in bile formation and circulation in the liver (3). PFIC1 and PFIC2 represent two-thirds of PFIC cases, whereas PFIC3 represents one-third of PFIC cases (4–8).
PFIC1 and PFIC2 show clinical symptoms in the neonatal period and rapidly progress to liver failure but with normal or reduced GGT levels. Conversely, PFIC3 is clinically present in adolescence or adulthood, progresses slowly, and GGT levels are elevated. PFIC3 is caused by mutations in the ABCB4 gene (The ATP binding cassette subfamily B member 4), which result in multidrug resistance-associated protein 3 (MDR3) to be truncated, unstable, misfolded, or impaired during its transport (9, 10). ABCB4 gene is located on human chromosome 7q21, with a total of 75504 bases and 28 exons, of which 27 exons have coding sequences and the first exon has a 26-bp non-translated region (11).
MDR3 protein is a protein encoded by the ABCB4 gene. It plays an important role in normal bile formation and is distributed in the surface of cholangiole of hepatocytes. It has 12 transmembrane regions; the N-terminus and C-terminus are on the inner side of the hepatocyte membrane, which is actually phosphatidyl transferase enzyme, and mediates the transport of phosphatidylcholine (PC) to bile from the inside of the hepatocyte membrane to the outside. The phospholipids in normal bile can form stable mixed particles (mixed micelles) in appropriate proportions with bile salts. They emulsify bile salts and avoid them from precipitating to form crystals, thus protecting bile duct epithelial cells and promoting cholesterol secretion (12). Disease-specific ABCB4 gene polymorphisms have been described in some intrahepatic cholestasis diseases, suggesting decreased MDR3 expression and function (12).
Recent studies have shown that the ABCB4 gene mutation types include truncated mutations, missense mutations, frameshift mutations, nonsense mutations, and splicing site mutations. Some of these may clinically manifest as various chronic progressive lesions (13). Some mutant heterozygotes usually have normal clinical manifestations; however, they can still exhibit various symptoms caused by low MDR3 function when induced by certain factors, such as drugs.
Variations introducing premature stop codons cause a defect in protein expression; however, more than 70% of disease-causing ABCB4 variations are missense variations, and the ways in which such variations affect ABCB4 are generally unknown (14). Theoretically, missense mutations could affect the protein in several ways, either by modulating its expression at the canalicular membrane or by impairing its activity.
At present, clinical examination can identify ABCB4 gene deficiency-related diseases based on the patient’s medical history, blood biochemical examination, liver tissue biopsy, liver imaging, and other auxiliary tests; however, the method commonly used to confirm ABCB4 deficiency is genetic sequence detection and MDR3 immunohistochemical staining of liver tissue slices. Notably, gene polymorphisms of ABCB4 may affect the function of MDR3 proteins. MDR3 immunohistochemical staining methods are simple, but whether the staining results are consistent with genetic mutations requires further exploration.
This study was aimed at improving the diagnosis of PFIC3 by observing whether the expression of MDR3 staining in liver puncture tissue sections was consistent with the gene mutation and analyzing clinical, pathologic, genetic, and clinical features of four cases of Chinese patients.
Between September 2019 and March 2021, four patients with PFIC3 were recruited from two liver centers in East China, including three patients from the Affiliated Hospital of Hangzhou Normal University and one patient from the First Affiliated Hospital of Zhejiang University. Cholestasis was defined as follows: (1) γ-glutamyl transpeptidase (GGT) levels higher than three times the upper limit of normal (ULN) and serum alkaline phosphatase levels higher than 1.5 times the upper limit of normal (ULN); (2) direct bilirubin (DBil) levels higher than 17. 1 μmol/L and serum total bilirubin (TBil) levels lower than 85.0 mol/L; or (3) DBil accounting for > 20% of TBil when TBil is ≥ 85.0 mol/L (15).
In this study, PFIC3 presented as (1) the appearance of clinical symptoms of cholestasis, including mild yellow dye in the skin and sclera, (2) elevated serum DBil, GGT, or total bile acid (TBA) levels, (3) the presence of the ABCB4 gene and its mutants, and (4) cholestasis identified as not having any cause besides the aforementioned ones through ultrasound B, isotope liver scan, and genetic metabolic disease screening. Clinical courses and molecular genetic results for ABCB4 were examined retrospectively and prospectively for patients with PFIC3 for laboratory values, macroscopic and microscopic liver characteristics, and molecular genetic characteristics of the liver. Our study protocol was compliant with ethical guidelines of the Declaration of Helsinki of 1975 (2004 revision), the Ethical Committee of the Affiliated Hospital of Hangzhou Normal University (approval number: 20211223090600282150), and the First Affiliated Hospital of Zhejiang University (approval number: 2021IIT0286). Considering the retrospective design of the study and anonymity of the patients, the need for obtaining written informed consent from the patients was waived by the ethics committees.
Patients with PFIC3 had liver biopsy specimens collected during causal evaluations of their cholestasis. To determine basic histologic characteristics and check for collagen and copper granule deposits in hepatocytes, 4-μm sections of formalin-fixed, paraffin-embedded blocks were cut and stained with hematoxylin and eosin, Masson’s trichrome, and rhodamine. Epithelial cells of the bile duct were immunohistochemically stained for CK7. Immunohistochemical staining was also performed for MDR3 (clone EPR23697-35, dilution 1:3000; Abcam, London, United Kingdom). The immunohistochemistry of MDR3 was considered positive only if it stained in a canalicular pattern. Two pathologists evaluated all biopsy specimens.
Whole-exome sequencing (WES) and bioinformatics were used to detect and analyze genetic variations in ABCB4 using the Illumina NovaSeq 6000 sequencing system. Although exons account for only 1–2% of the human genome, they contain 85% of the pathogenic variants. Our sequencing was based on a standardized target area capture sequence platform. The sequencing quality control indicators are the average coverage rate of 99.11% and an average sequencing depth of 158.81 ×.
To describe the nucleotide substitutions, we used the NCBI transcript sequence of ABCB4: NM_000443.3, which contains all candidates for single nucleotide variants and small indels. We used a public database (gnomAD database) to estimate the prevalence of mutations in the ABCB4 gene in the East Asian populations and used the ClinVar database and Human Gene Mutation Database of 2021 to predict the correlation between ABCB4 gene mutation and disease occurrence. Then, we predicted the function of proteins (harmful or harmless) coded by genes with missense mutation of the novel variants using the Sorting Intolerant From Tolerant (SIFT) program and Polymorphism Phenotyping v2 (PolyPhen-2). The pathogenicity of the detected mutations was evaluated principally on the basis of the relevant guidelines of the American College of Medical Genetics and Genomics (16).
The study included four patients (one man and three women) with a median age at referral of 26 years (range, 17–45 years; Table 1). Cholestasis presented with jaundice as the first clinical sign in all patients, except for patient 2, who presented with thrombocytopenia as the first clinical sign, which was identified in routine medical examination findings. By appropriately investigating these causes, other common causes of cholestasis, including biliary atresia, congenital biliary dilation, viral hepatitis, and other metabolic disorders, such as citrin deficiency, benign recurrent intrahepatic cholestasis, and bile acid synthesis defects, were excluded. Currently, no commercial tests are available in China for testing urinary coproporphyrin isomers. In all patients who had fasted for 3 h, abdominal ultrasound revealed liver cirrhosis and different manifestations of portal hypertension, including splenomegaly, esophageal varicose veins, thrombocytopenia, and ascites (Table 1).
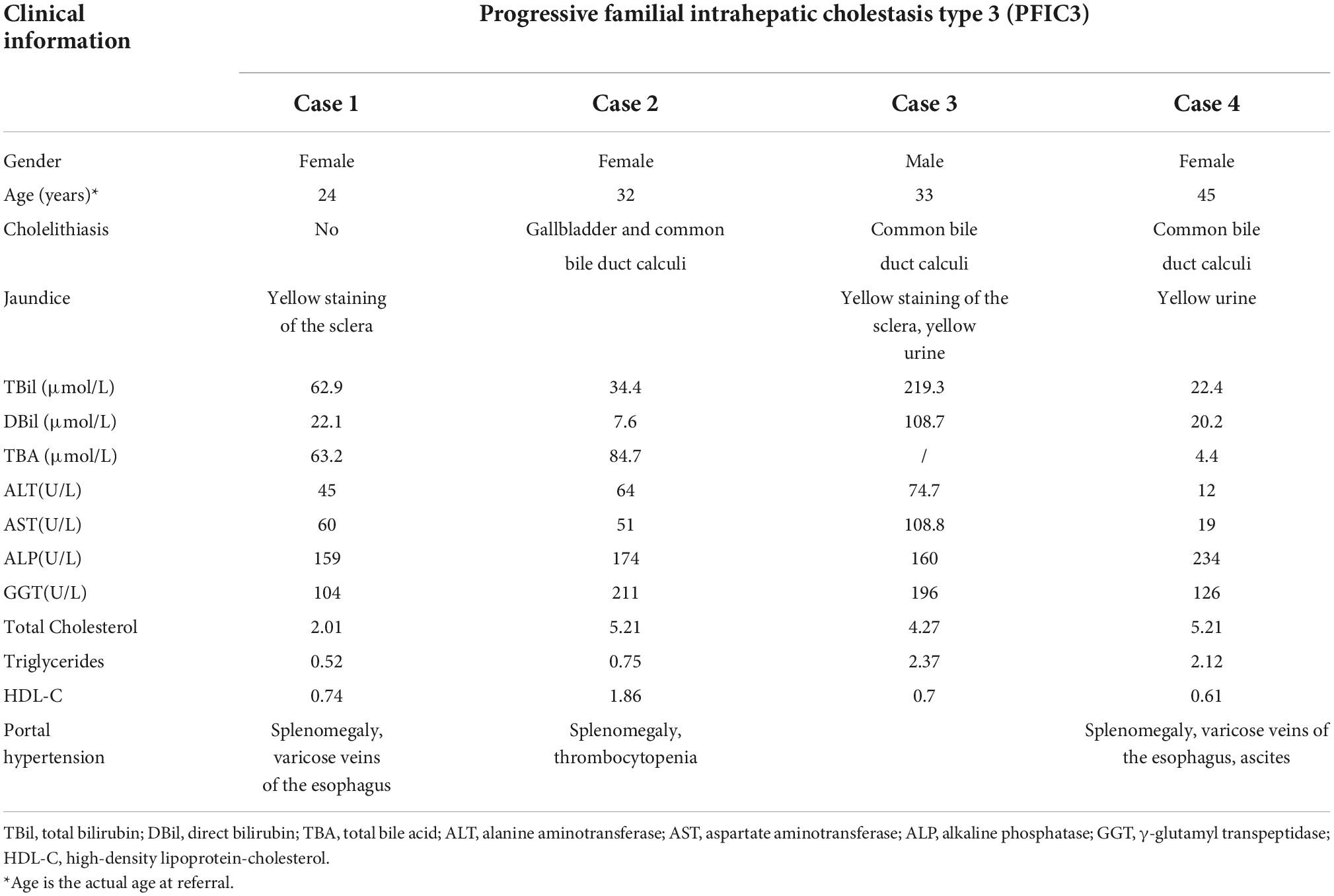
Table 1. Clinical characteristics and laboratory findings.
All patients had abnormal liver functions suggestive of cholestasis (Table 1).
Hepatopathological analyses revealed that all patients had extensive portal fibrosis and biliary cirrhosis (Figure 1) classified by Ishak stages 3–4 and Knodell stage 3 (17), and a missing bile duct or biliary epithelium injury was not noted. Histopathological approaches revealed an air halo in the plasma of the hepatocytes around the nodules in patient 1 (Figure 1A), an incomplete liver cirrhosis nodule and expansion of the hepatic sinuses in patient 2 (Figure 1B), inflammatory infiltration accompanied by ductular proliferation in patient 3 (Figure 1C), and liver fibrosis in patient 4 (Figure 1D). PFIC3 patients showed some pathologic features of cholestatic cirrhosis (Figure 2). Bile plugs were observed in some ductules and lobules with hepatocyte cholestasis (Figure 2A). Rhodamine staining was positive for copper particles which appeared deposited in hepatocyte plasma (Figure 2B). CK7 immunostaining within the portal tract revealed extensive proliferation of ducts (Figure 2C) and CK7 expression in some intermediate hepatocytes (Figure 2D). Bile duct reaction was found in 3/4 patients, cholestasis in all patients, portal area inflammation in 3/4 patients, and cirrhosis in all patients. Decreased expression of MDR3 was found in 3/4 patients (Table 2). In this study, hepatology analysis revealed two different patterns: one involving portal fibrosis and ductular proliferation and the other involving extensive fibrosis and biliary cirrhosis. Four of the liver specimens were examined immunohistochemically. One had absent expression of MDR3; the other three had decreased expression of MDR3 (Figure 3).
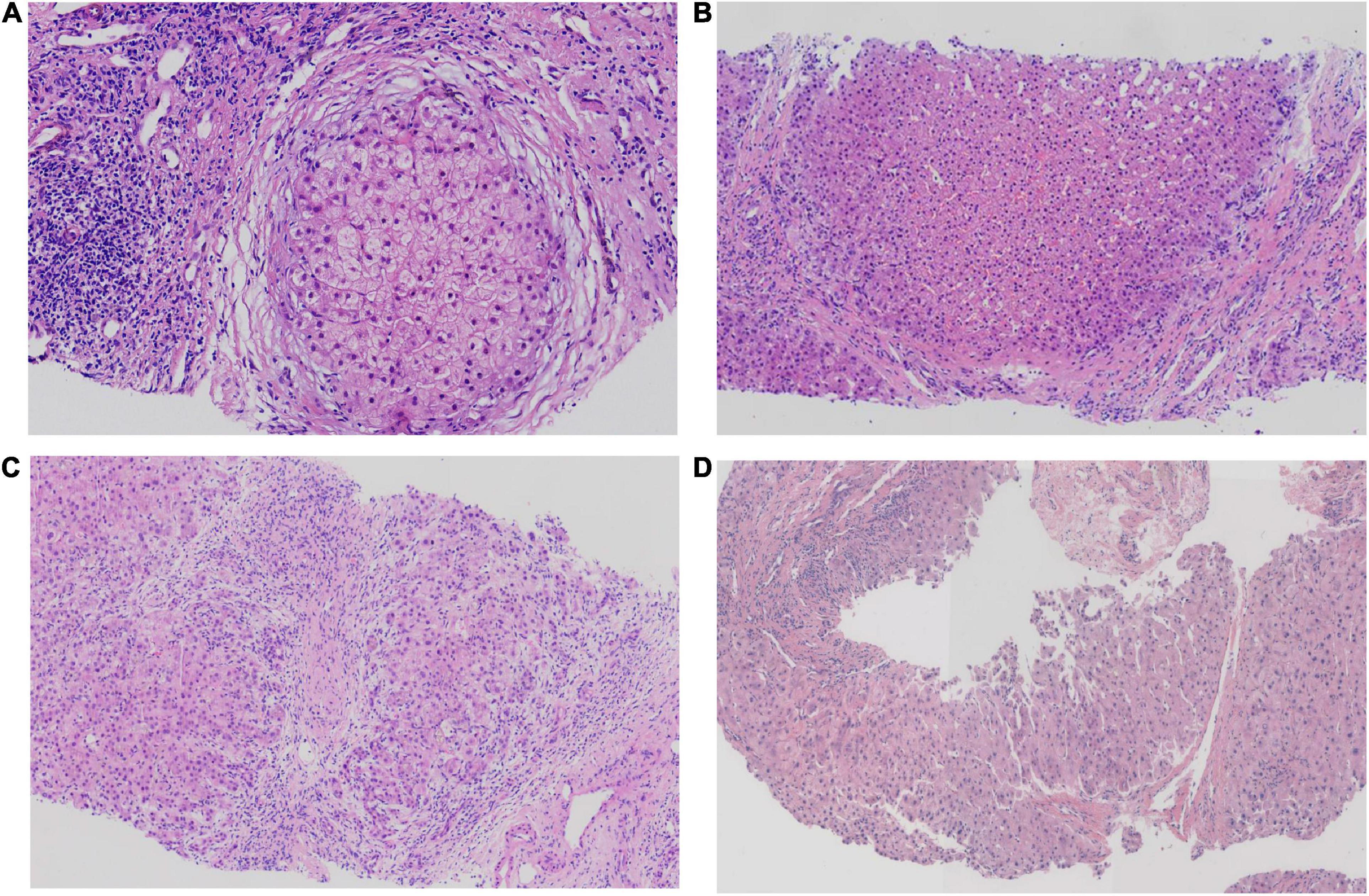
Figure 1. Pathologic Features of Patients with PFIC3 in liver biopsy specimens. All patients with severe liver fibrosis and even cirrhosis (A–D). Panel (A) shows a cirrhosis nodule and an air halo in the plasma of the hepatocytes around the nodules in patient 1. Panel (B) shows an incomplete cirrhosis nodule and the expansion of the hepatic sinuses in patient 2. Panel (C) shows severe liver fibrosis and mild inflammatory infiltration accompanied by ductular proliferation in patient 3. Panel (D) shows liver cirrhosis in patient 4. Panels (A–D) show H&E-stained samples; magnification: 200× in (A–C) and 100× in (D).
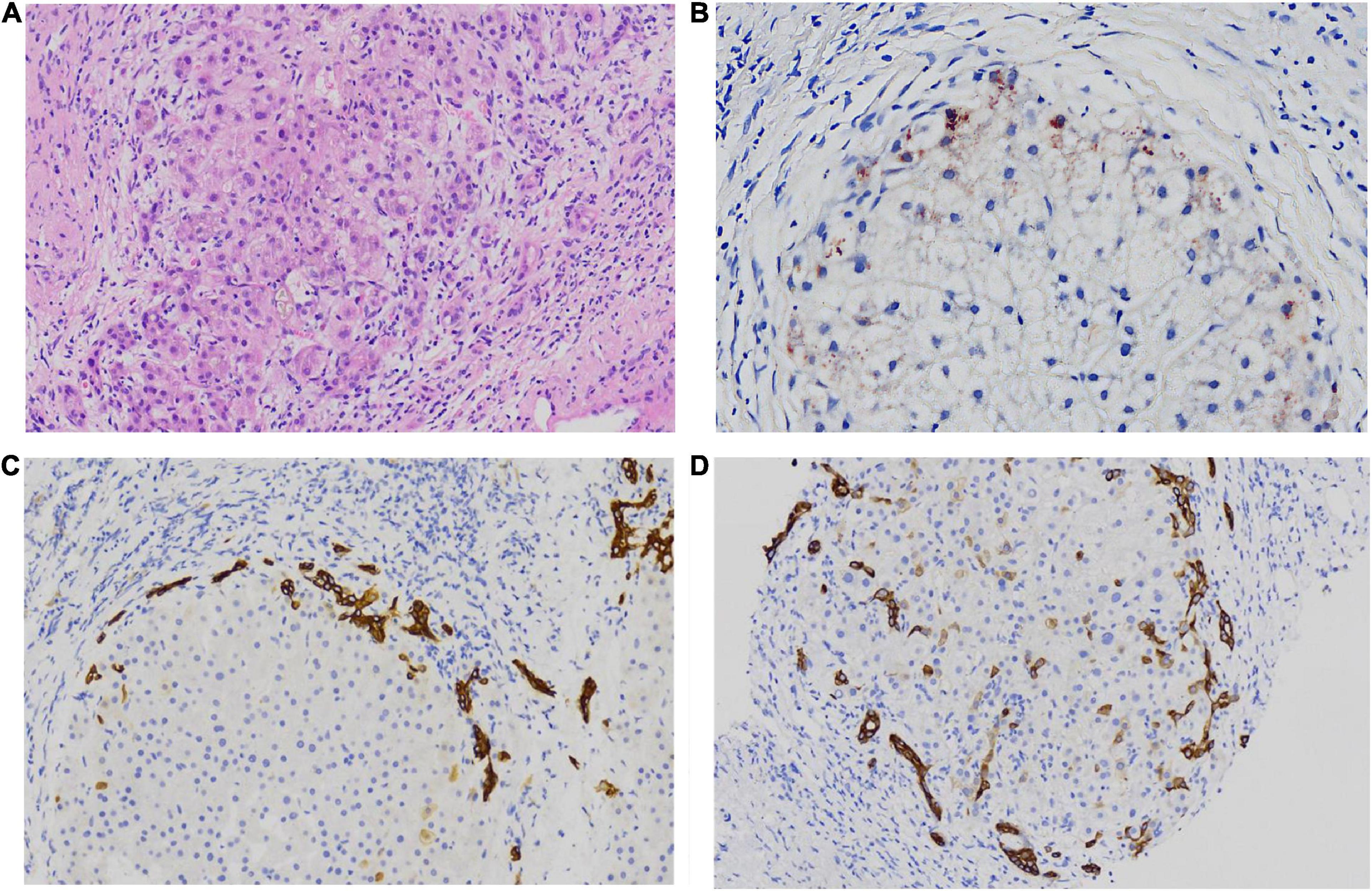
Figure 2. some special pathologic features of patients with PFIC3 in liver biopsy specimens. Panel (A) shows the presence of bile plugs in some ductules and lobules with hepatocyte cholestasis; Panel (B) shows orange granule deposits of copper in periportal hepatocytes (Rhodamine staining). Panel (C) shows immunohistochemical staining for cytokeratin 7 (ck7), which confirms the presence of interlobular duct and ductular reaction. Panel (D) shows the heterotopic expression of ck7 on periportal hepatocytes, indicating chronic cholestasis. Panels (A,B) show H&E-stained samples; Panels (C,D) show immunohistochemically stained samples; magnification: 200× in (A–D).

Table 2. Summary of histologic characteristics.
Figure 3. Biopsy specimens of liver samples expressing Multidrug-Resistant Protein 3. (A) Normal MDR3 expression is visible in liver biopsy samples from a healthy individual. (B) Liver biopsy specimens from Case 2: loss of MDR3 expression. (C–E) Liver biopsy specimens from Case 1, 3, and 4: The MDR3 staining was significantly reduced. Images showing immunohistochemical staining at a magnification of 200×.
The four patients were identified as having two types of pathogenic variants of ABCB4: splice site (patient 1) and missense mutations (patients 2, 3, and 4) (Table 3). ABCB4_ex13 (c.1560 + 2T > A) was a hybrid splicing mutation, whereas ABCB4_ex17 c.2177C > T (p. Pro726Leu), ABCB4_ex15 c.1756G > T (p. Val586Leu), and ABCB4_ex15 c.247IT > C (p. Val824Ala) were hybrid missense mutations.
Table 3. Patients with pathogenic ABCB4 variants.
One novel variant was identified (c.1560 + 2T > A) in patient 1; this was a hybrid splicing mutation that occurred at the classical shear site. This variation is currently not mentioned in the ClinVar database, and mutations at this site have not been reported in East Asian populations. A comprehensive judgment for suspected pathogenic mutation was made using SIFT and PolyPhen-2.
Notably, a hybrid missense mutation, c.2177C > T (p. Pro726Leu), was found in patients 2 and 3; this is a suspected pathogenic mutation according to the ClinVar database. Mutations at this site have a frequency of 0 in healthy East Asian populations. SIFT and PolyPhen-2 predicted that the function of the resultant truncated protein would be harmful.
Furthermore, c.1756G > T (p. Val586Leu) and c.2471 T > C (p. Val824Ala) were found in patients 2 and 3, respectively. These are hybrid missense mutations that have not yet been mentioned in the ClinVar database. These mutations are not known to frequently occur in East Asian populations. The function of the resultant truncated protein was predicted to be harmful by SIFT and suspected to be harmful by PolyPhen-2.
Every variation in ABCB4 genes in our patients was verified by Sanger sequencing, and a validation diagram was prepared (Figure 4). Furthermore, we traced the source of patients’ variations. Interestingly, all female patients inherited the genetic variants from their fathers; conversely, patient 3 (the only male patient) inherited one variation of the ABCB4 gene from each parent. This also indicates that the variation in the ABCB4 gene is dominantly inherited.
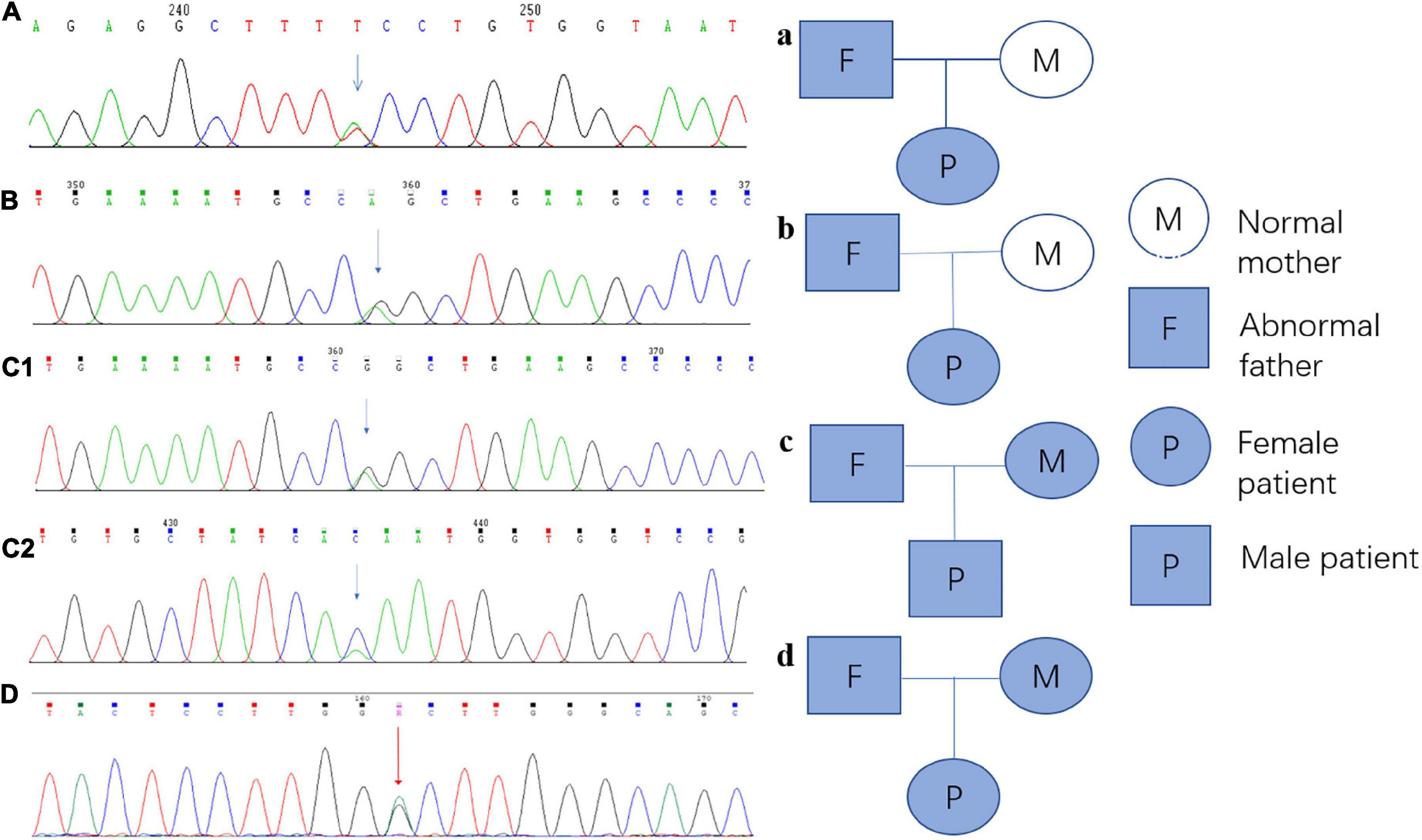
Figure 4. Sanger sequencing verification diagram and pedigree of the ABCB4 variations in each patient and their parents. (A) Sanger sequencing verification of ABCB4_ex13 c.1560 + 2T > A in patient 1; a: the pedigree of patient 1 and her parents, and this variation originated from her father. (B) Sanger sequencing verification of ABCB4_ex17 c.2177C > T (p. Pro726Leu) in patient 2; b: the pedigree of patient 2 and her parents, and this variation originated from her father. (C1) Sanger sequencing verification of ABCB4_ex17 c.2177C > T (p. Pro726Leu) in patient 3, c1: the pedigree of patient 3 and his parents, and this variation originated from his mother. (C2) Sanger sequencing verification of ABCB4_ex15 c.1756G > T (p. Val586Leu) in patient 3, c2: the pedigree of patient 3 and his parents, and this variation originated from his father. (D) Sanger sequencing verification of ABCB4_ex17 c.2171T > C (p. Vol824Ala) in patient 4; d: the pedigree of patient 4, and this variation originated from her father.
Clinical, pathologic, and genetic aspects of PFIC3 were studied in four Chinese patients. All patients had severe portal fibrosis, even biliary cirrhosis. All patients had similar clinical courses, with mild yellow dye in the skin and sclera and yellow urine, culminating into severe cholestasis. The ABCB4 genetic test was conducted for all patients.
The biochemical indices of liver functions indicated that serum alanine aminotransferase (ALT), GGT, and TBA levels were continuously high at more than three times the normal values. The changes in serum GGT levels were consistent with those of ALT, TBil, and TBA levels. In the presence of high serum TBA concentration, primary bile acid synthesis disorders can be excluded. In patients with PFIC3, serum GGT activity is high, whereas in patients with PFIC1 and PFIC2, serum GGT activity is normal. A distinguishing feature of patients with PFIC3 over patients with PFIC1/PFIC2 is that the former patients are more likely to present with cholestatic jaundice later in life rather than early in infancy or childhood (18). In patients with elevated GGT levels who were suspected as having cholestasis without an obvious cause, PFIC3 should be suspected after excluding other possible causes of cholestasis (19).
Four patients underwent liver biopsies because of their prolonged jaundice, severe portal fibrosis, and abnormal laboratory values, such as increased serum DBil, GGT, and TBA; the findings and values convincingly suggested biliary cholestasis. All patients had extensive fibrosis, ductular proliferation, and biliary cirrhosis. Holostasis (bile plugs) was found in all patients. The plasma of the hepatocytes around the nodules appearing as an air halo, copper particle deposition, and positive CK7 expression of intermediate liver cells are morphological characteristics indicative of biliary cirrhosis, and all patients had these findings. In previous research, these findings have been reported to be consistent with histological changes in the liver of adult patients with PFIC3 (20). Furthermore, liver biopsies revealed ductular proliferation with mild inflammation, which is indicative of early stages of fibrosis in the portal tract. In other studies, PFIC3 has been described as having two histological patterns: the first is slight portal fibrosis and ductular proliferation and the second is extensive fibrosis and biliary cirrhosis that may present at as early as 5 years of age (21). This suggests that patients with PFIC3 can develop extensive fibrosis or bile siltation cirrhosis during adolescence or later, and the diagnosis of PFIC3 needs to be distinguished from that of other diseases, such as Wilson’s disease and primary biliary cirrhosis, on the basis of its morphological characteristics. Therefore, immunohistochemical staining was also applied in this study to assist the diagnosis.
Immunohistochemical analyses discovered absence or decreased expression of MDR3 in all patients. Because alterations in MDR3 expression are infrequent in other types of cholestatic disease, absence of or a decrease in MDR3 expression could serve as a diagnostic indicator of PFIC3. PC, a phospholipid, is transported by MDR3 through the canalicular membrane of the liver (22). Hepatocytes use MDR3 to transfer phospholipid molecules from their inner canalicular membrane to their outer canalicular membrane via the flippase activity. Notably, PC protects hepatocyte membranes from bile salts’ detergent activity (23–25). The activity of MDR3 is essential for bile salts to attach to PC in the canaliculus, as demonstrated in in vitro studies (26). Inflammation and tissue death result from the detergent action of free bile salts that can solubilize the apical membrane and hepatobiliary epithelium, leading to biliary canaliculi injury, cirrhosis, and ultimately liver failure (27). Moreover, bile with low phospholipid levels can cause cholesterol to crystallize, thus resulting in bile duct obstruction. Compared to weak or normal staining of MDR3 in patients with a reciprocal missense mutation of ABCB4, Jacquemin et al. observed completely negative staining of MDR3 in patients harboring a missense mutation of ABCB4, which results in a truncated MDR3; this is consistent with the findings of the present study. These results suggest that a diagnosis of PFIC3 requires a combination of histopathologic examination and immunohistochemical assessment of MDR3 expression. Despite this, normal immunostaining of MDR3 does not exclude genetic defects because mutations may lead to loss of function while maintaining normal synthesis. Therefore, genetic testing of ABCB4 is essential (28).
In patients with PFIC3, genetic mutations associated with ABCB4 occur homozygously, heterozygously, or compound heterozygously. The biallelic mutation of the ABCB4 gene, which encodes the phospholipid flippase MDR3 on chromosome 7q21.1, contributes to the development of PFIC3 (29–31).
More than 40 different ABCB4 gene mutations have been reported, including missense mutations, senseless mutations, deletion mutations, and insertion of small fragments of bases (32). Different types of pathogenic mutations in ABCB4 (≥ 70% missense) lead to distinct clinical outcomes and are associated with different cumulative risks (33). Delaunay et al. devised a classification system for the various forms of these mutations. Class I includes mutations that result in defective synthesis, class II covers variations that prevent protein maturation, class III includes mutations that result in mature but defective proteins, class IV includes unstable variations, and class V includes mutations associated with unknown pathogenicity. These classifications are useful in determining potential therapies for patients depending on their genotype (14).
According to the human gene mutation database,1 ABCB4 has been associated with more than 202 biliary diseases, 50 of which are related to PFIC3 (34). Besides truncated and missense mutations, nonsense mutations, deletions, and frameshift mutations occur as well. ABCB4 gene mutation can lead to defective MDR3 expression, bile phospholipid deficiency, and persistent inflammation and fibrosis in the liver and bile tube cells caused by continuous exposure to bile salt toxicity, thus resulting in bile siltation. A nonsense mutation or deletion often manifests with severe clinical symptoms due to the severity of the mutation.
In this study, four pathogenic variants of the ABCB4 gene (c.1560 + 2 T > A, c.2177 C > T, c.1756 G > T, and c.2471 T > C) were identified in the patients. The human gene mutation database listed the c.2177 C > T (p. Pro726Leu) hybrid missense mutation in ex17 of ABCB4 as one that has been detected in patients with PFIC3; this mutation was found in two of our patients.2 Adolescent and adult patients with PFIC3 reportedly have the c.2177 C > T mutation (35–37). Patients with PFIC can also develop cholelithiasis and intrahepatic cholestasis because of mutations causing cholelithiasis and pregnancy-induced liver disease. Functional experiments indicate that the expression of mutant ABCB4 in the bile duct has little or no effect on MDR3 protein expression, but completely impaired secretion of phosphatidyl choline results in loss of MDR3 protein expression. Mutations in the ABCB4 gene are indicated by the absence of or a significant reduction in the expression of the MDR3 protein in liver tissues.
In addition, we identified a novel mutant of ABCB4 in a hybrid splicing environment, namely ABCB4_ex13 (c.1560 + 2T > A). In accordance with the Exome Aggregation Consortium, Cambridge, Massachusetts,3 c. 1560 + 2T > A is a hybrid splicing mutation, which occurs at the classical shear site. This mutation may change the cutting pattern of the RNA precursor and produce abnormal splicing variants later in transcription, which subsequently affects the expression or function of the protein. The frequency of a mutation in this site in healthy East Asian population remains unknown. There is no record for this variant in ClinVar as well. It was predicted to be a pathogenic variation based on comprehensive judgment. Although the mutation was predicted to be or suspected as being harmful according to the predictive software, knowledge of the patients’ familial history and a larger sample size are needed to further investigate the correlation between the genotype and clinical phenotype.
Notably, the effect that the hybrid missense mutations c.1756 G > T and c.2471 T > C will have according to the American College of Medical Genetics and Genomics standard remains unknown. PFIC3 is caused by the autosomal recessive inheritance of the pathogenic mutation of the ABCB4 gene (see Appendix OMIM). However, the two mutations were detected in the exon region of a single allele, which makes the patient only a carrier of the mutation. PFIC3 in this patient may be associated with this mutation; however, we did not exclude the possibility that PFIC3 in this patient could also have been caused by pathogenic mutations in non-exon regions or by other types of mutations.
A liver biopsy revealed no evidence of canalicular MDR3 expression in patient 1; this patient had a homozygous missense mutation that caused MDR3 to be defectively trafficked to the apical membrane. Regarding the other patients who exhibited canalicular expression of MDR3, albeit at a reduced level, they carried mutations that did not completely alter where the protein localized in the membrane. Treatment with ursodeoxycholic acid is normally effective for patients with reduced MDR3 expression.
Our study has some limitations, including its retrospective design, the small number of patients, and the inclusion of only East Asian patients. There is a need for larger prospective studies in Asia and globally. Patients should be recruited from different age groups, with a particular focus on the neonatal population. Furthermore, we acknowledge that family lineage testing of patients should have been conducted; during follow-up visits, we tried our best to persuade the parents to take the ABCB4 gene test to effectively verify the pathogenicity of genetic mutations, particularly that of newly discovered mutations; however, any further information in this regard depends on the willingness of the patient’s parents to be tested. Molecular methods should be improved to elucidate the role of and mechanisms behind the new genetic mutations reported in our study.
A diagnosis of PFIC3 should be considered in patients with long-term liver dysfunction and elevated phospholipid levels presenting with severe fibrosis or bile cirrhosis. We believe that for an accurate diagnosis of PFIC3 and exclusion of other causes of cholestasis, immunohistochemistry staining of liver specimens for MDR3 and molecular genetic analysis of ABCB4 are essential.
The data presented in this study are deposited in the NCBI BioProject repository, accession number: PRJNA876380.
The studies involving human participants were reviewed and approved by Ethical Committee of The Affiliated Hospital of Hangzhou Normal University. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
JC wrote the manuscript. LG acquired clinical information of the patient. XM analyzed the genetic results of the patients. XW and JZ performed the pathological sections and histochemical staining. WY designed and revised the manuscript. All authors read and approved the final manuscript.
Thus study was funded by Zhejiang Province Public Welfare Technology Application Research Project (No. GF19H160052).
The authors thank the patients and their relatives for agreeing to report her case and for providing a detailed medical history.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
PFIC3, progressive familial intrahepatic cholestasis type 3; MDR3, multidrug resistance-associated protein 3; AKP, alkaline phosphatase; GGT, γ-glutamyle transpeptidase; DBil, direct bilirubin; TBil, total bilirubin; TBA, total bile acids; HDL-C, high-density lipoprotein cholesterol; CK7, cytokeratin 7; CK19, cytokeratin 19.
1. Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. (2011) 31:3–10. doi: 10.1055/s-0031-1272831
2. Vitale G, Gitto S, Vukotic R, Raimondi F, Andreone P. Familial intrahepatic cholestasis: new and wide perspectives. Dig Liver Dis. (2019) 51:922–33. doi: 10.1016/j.dld.2019.04.013
3. Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. (2014) 4:25–36. doi: 10.1016/j.jceh.2013.10.005
4. Lang T, Haberl M, Jung D, Drescher A, Schlagenhaufer R, Keil A, et al. Genetic variability, haplotype structures, and ethnic diversity of hepatic transporters MDR3 (ABCB4) and bile salt export pump (ABCB11). Drug Metab Dispos. (2006) 34:1582–99. doi: 10.1124/dmd.105.008854
5. Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. (2010) 51:1645–55. doi: 10.1002/hep.23539
6. Mullenbach R, Lammert F. An update on genetic analysis of cholestatic liver diseases: digging deeper. Dig Dis. (2011) 29:72–7. doi: 10.1159/000324137
7. Sharma A, Poddar U, Agnihotry S, Phadke SR, Yachha SK, Aggarwal R. Spectrum of genomic variations in Indian patients with progressive familial intrahepatic cholestasis. BMC Gastroenterol. (2018) 18:107. doi: 10.1186/s12876-018-0835-6
8. Amirneni S, Haep N, Gad MA, Soto-Gutierrez A, Squires JE, Florentino RM. Molecular overview of progressive familial intrahepatic cholestasis. World J Gastroenterol. (2020) 26:7470–84. doi: 10.3748/wjg.v26.i47.7470
9. Ganne-Carrié N, Baussan C, Grando V, Gaudelus J, Cresteil D, Jacquemin E. Progressive familial intrahepatic cholestasis type 3 revealed by oral contraceptive pills. J Hepatol. (2003) 38:693–4. doi: 10.1016/s0168-8278(03)00049-7
10. Degiorgio D, Crosignani A, Colombo C, Bordo D, Zuin M, Vassallo E, et al. ABCB4 mutations in adult patients with cholestatic liver disease: impact and phenotypic expression. J Gastroenterol. (2016) 51:271–80. doi: 10.1007/s00535-015-1110-z
11. Reichert MC, Lammert F. ABCB4 gene aberrations in human liver disease: an evolving spectrum. Semin Liver Dis. (2018) 38:299–307. doi: 10.1055/s-0038-1667299
12. Wei G, Cao J, Huang P, An P, Badlani D, Vaid KA, et al. Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4(-/-) mouse model of PFIC3. J Hepatol. (2021) 74:1416–28. doi: 10.1016/j.jhep.2020.12.010
13. Zhang W, Lin R, Lu Z, Sheng H, Xu Y, Li X, et al. Phenotypic and molecular characteristics of children with progressive familial intrahepatic cholestasis in South China. Pediatr Gastroenterol Hepatol Nutr. (2020) 23:558–66. doi: 10.5223/pghn.2020.23.6.558
14. Delaunay JL, Durand-Schneider AM, Dossier C, Falguières T, Gautherot J, Davit-Spraul A, et al. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology. (2016) 63:1620–31. doi: 10.1002/hep.28300
15. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. (2009) 51:237–67. doi: 10.1016/j.jhep.2009.04.009
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Goodman ZD. Grading and staging systems for inflammation and fibrosis in chronic liver diseases. J Hepatol. (2007) 47:598–607. doi: 10.1016/j.jhep.2007.07.006
18. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. (2010) 30:134–46. doi: 10.1055/s-0030-1253223
19. Poupon R, Rosmorduc O, Boelle PY, Chretien Y, Corpechot C, Chazouilleres O, et al. Genotype-phenotype relationships in the low-phospholipid-associated cholelithiasis syndrome: a study of 156 consecutive patients. Hepatology. (2013) 58:1105–10. doi: 10.1002/hep.26424
20. Elderman JH, Ter Borg PC, Dees J, Dees A. Pregnancy and ABCB4 gene mutation: risk of recurrent cholelithiasis. BMJ Case Rep. (2015) 2015:bcr2014206919. doi: 10.1136/bcr-2014-206919
21. Olsen JA, Alam A, Kowal J, Stieger B, Locher KP. Structure of the human lipid exporter ABCB4 in a lipid environment. Nat Struct Mol Biol. (2020) 27:62–70. doi: 10.1038/s41594-019-0354-3
22. Reichert MC, Hall RA, Krawczyk M, Lammert F. Genetic determinants of cholangiopathies: Molecular and systems genetics. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1484–90. doi: 10.1016/j.bbadis.2017.07.029
23. Dixon PH, Williamson C. The pathophysiology of intrahepatic cholestasis of pregnancy. Clin Res Hepatol Gastroenterol. (2016) 40:141–53. doi: 10.1016/j.clinre.2015.12.008
24. Khabou B, Durand-Schneider AM, Delaunay JL, Ait-Slimane T, Barbu V, Fakhfakh F, et al. Comparison of in silico prediction and experimental assessment of ABCB4 variants identified in patients with biliary diseases. Int J Biochem Cell Biol. (2017) 89:101–9. doi: 10.1016/j.biocel.2017.05.028
25. Gunaydin M, Bozkurter Cil AT. Progressive familial intrahepatic cholestasis: diagnosis, management, and treatment. Hepat Med. (2018) 10:95–104. doi: 10.2147/HMER.S137209
26. Prescher M, Smits SHJ, Schmitt L. Stimulation of ABCB4/MDR3 ATPase activity requires an intact phosphatidylcholine lipid. J Lipid Res. (2020) 61:1605–16. doi: 10.1194/jlr.RA120000889
27. Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. (2001) 120:1448–58. doi: 10.1053/gast.2001.23984
28. Keitel V, Burdelski M, Warskulat U, Kuhlkamp T, Keppler D, Haussinger D, et al. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. (2005) 41:1160–72. doi: 10.1002/hep.20682
29. Groen A, Romero MR, Kunne C, Hoosdally SJ, Dixon PH, Wooding C, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. (2011) 141:e1921–4. doi: 10.1053/j.gastro.2011.07.042
30. Guyot C, Stieger B. Interaction of bile salts with rat canalicular membrane vesicles: evidence for bile salt resistant microdomains. J Hepatol. (2011) 55:1368–76. doi: 10.1016/j.jhep.2011.04.014
31. Park HJ, Kim TH, Kim SW, Noh SH, Cho KJ, Choi C, et al. Functional characterization of ABCB4 mutations found in progressive familial intrahepatic cholestasis type 3. Sci Rep. (2016) 6:26872. doi: 10.1038/srep26872
32. Hochrath K, Ehnert S, Ackert-Bicknell CL, Lau Y, Schmid A, Krawczyk M, et al. Modeling hepatic osteodystrophy in Abcb4 deficient mice. Bone. (2013) 55:501–11. doi: 10.1016/j.bone.2013.03.012
33. Saleem K, Cui Q, Zaib T, Zhu S, Qin Q, Wang Y, et al. Evaluation of a novel missense mutation in ABCB4 gene causing progressive familial intrahepatic cholestasis type 3. Dis Markers. (2020) 2020:6292818. doi: 10.1155/2020/6292818
34. Gautherot J, Delautier D, Maubert MA, Ait-Slimane T, Bolbach G, Delaunay JL, et al. Phosphorylation of ABCB4 impacts its function: insights from disease-causing mutations. Hepatology. (2014) 60:610–21. doi: 10.1002/hep.27170
35. Lincke CR, Smit JJ, van der Velde-Koerts T, Borst P. Structure of the human MDR3 gene and physical mapping of the human MDR locus. J Biol Chem. (1991) 266:5303–10. doi: 10.1016/s0021-9258(19)67788-4
36. Degiorgio D, Colombo C, Seia M, Porcaro L, Costantino L, Zazzeron L, et al. Molecular characterization and structural implications of 25 new ABCB4 mutations in progressive familial intrahepatic cholestasis type 3 (PFIC3). Eur J Hum Genet. (2007) 15:1230–8. doi: 10.1038/sj.ejhg.5201908
Keywords: PFIC, PFIC3, ABCB4, cholestasis, MDR3
Citation: Cheng J, Gong L, Mi X, Wu X, Zheng J and Yang W (2022) Case series of progressive familial intrahepatic cholestasis type 3: Characterization of variants in ABCB4 in China. Front. Med. 9:962408. doi: 10.3389/fmed.2022.962408
Received: 27 June 2022; Accepted: 09 November 2022;
Published: 08 December 2022.
Edited by:
Luigi Tornillo, University of Basel, SwitzerlandReviewed by:
Patryk Lipiński, Children’s Memorial Health Institute (IPCZD), PolandCopyright © 2022 Cheng, Gong, Mi, Wu, Zheng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenjun Yang, eWFuZ3dlbmp1bjM0NjNAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.