 Paola Coppola
Paola Coppola Essam Kerwash
Essam Kerwash Janet Nooney
Janet Nooney Amro Omran
Amro Omran Susan Cole
Susan Cole- Medicines and Healthcare Regulatory Agency, London, United Kingdom
Pregnancy-related physiological changes can alter the absorption, distribution, metabolism and excretion of medicines which may affect the safety and efficacy of the medicines administered in pregnancy. Pharmacokinetic data can thus be instrumental in supporting dose adjustments required in this population. This review considers the availability of published pharmacokinetic data for over 200 medicines of interest for use in pregnancy in the UK, to identify whether sufficient data currently exists, in principle, for any medicine or group of medicines to support dose adjustments to maintain maternal health through pregnancy. Very limited data was found for many of the medicines of interest. Nevertheless, well documented, large changes of exposure for some drugs, where data is available, highlights the urgent need to collect more data of good quality to inform appropriate doses, when needed, in this population. In addition, clinical study methodology can have an impact on the usefulness of the data and key clinical design aspects are highlighted for consideration in future clinical study design.
Introduction
The benefit-risk decision to inform medicine use in pregnancy relies on the available information about the effect of the medicine on pregnancy outcomes and the effect of pregnancy on the medicine, i.e., the medicine's ability to maintain maternal health during pregnancy. There is, however, limited scientific information to support the safe and optimal use of medicines in pregnancy, and the data available to inform pregnant women about drug safety and efficacy are considered inadequate (1). The lack of information appears to encompass every aspect of pharmaceutics, including limited pharmacokinetic (PK) and pharmacodynamic (PD) information during and after pregnancy to ensure proper dosing.
Several physiological changes occurring during pregnancy may affect the way the body normally deals with administered medicines, leading to possible changes in the PK and PD of medicines as pregnancy advances. Possible changes in the absorption, distribution, metabolism and excretion processes of medicines may result either in increased or decreased blood levels of medicines during pregnancy compared to non-pregnant individuals (2), with consequences for both the mother and baby. Particularly, decreased drug blood levels may lead to loss of efficacy and reduced control of maternal health which may affect fetal health and/or development, whereas increased levels could result in exposing the mother and the fetus to unnecessarily high doses, increasing the risk of adverse effects. To avoid this, some dosing regimens may need adjustments to achieve efficacious levels to maintain maternal health in pregnancy. However, optimal dosages are not usually investigated in pregnant women, PK changes are unknown or poorly characterized for most medications, and it is unknown whether a single dosing regimen will be sufficient throughout pregnancy or whether one or several dose changes would be required as pregnancy advances (3).
The European Medicines Agency (EMA) and US Food and Drug Administration (FDA) recommend that, when possible, PK and PD studies should be conducted in pregnant women to understand better how pregnancy affects the blood levels of medicines commonly used, particularly during the first trimester, and to develop evidence-based guidance for dose and frequency of administration for use in pregnancy (4, 5). Nevertheless, at the time of licensing, this data is not available and most new medicines in the European Union advise avoiding use in pregnancy because of a lack of sufficient data in pregnant women.
Where there is a lack of efficacy and safety data in a population of interest, pharmacokinetic data can be used to extrapolate efficacy from a population which has been studied, as has been proposed and is increasingly being used for pediatrics (6). Pharmacokinetic data can therefore be very useful to support medicine use in a new population. In light of the lack of efficacy data in pregnancy, we collected pharmacokinetic data for a number of medicines important for use during pregnancy, in order to consider whether this could be used to support posology, or possible dose adjustments, when use is clinically essential in this population.
The aim of this work was to determine the extent of literature information that was available for each medicine and to determine which medicines had rich data sets. This review evaluates the availability of published PK data in pregnancy for medicines commonly used by pregnant women and highlights some common issues that limit usefulness of existing data in this population.
Methods
A number of therapeutic indications and particular medicines of interest for pregnancy were identified, with input from clinical assessors at the MHRA. Thereafter, further advice on the medicines list was sought from Independent Experts on the Commission on Human Medicines (CHM) and its Advisory committees, including the Medicines for Women's Health Expert Advisory Group (MWHEAG) and other relevant therapeutic area Expert Advisory Groups (EAGs).
The list (Table 1) comprised 214 drug, or drug combinations, and was used to drive literature searches which were aimed at collecting literature pharmacokinetic data in pregnancy for each medicine of interest. More detailed analysis of the PK data was conducted for a subset of the drugs and will be the focus of future publications.
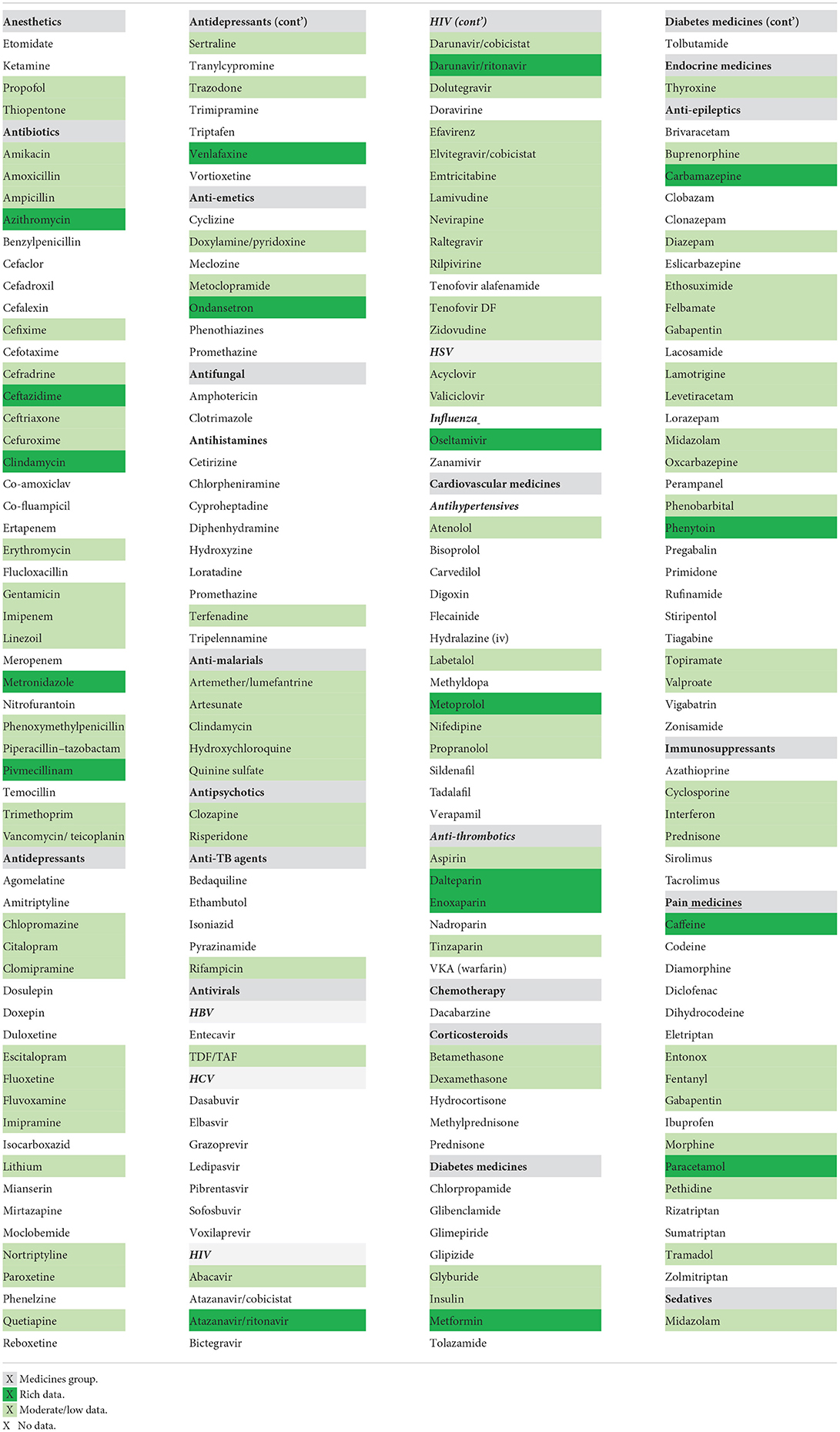
Table 1. List of medicines commonly used in pregnancy in the UK.
The focus was on maternal exposure, and publicly available pregnancy data up to 2019 have been collected from the literature, clinical trial databases or published assessment reports for Regulatory submissions. PubMed and SciFinder searches were conducted using the search terms “pharmacokinetics of medicine name in pregnant women” within the abstract or title of the published papers. In addition, the clinical trial databases i.e., www.clinicaltrials.gov and were searched using the terms “medicine name AND pregnancy.” Papers were selected for review if they were published in English and focused on the evaluation and/or collection of PK data in at least one gestational trimester in women. Published studies with data on PD, safety and efficacy in pregnant population and data on fetal exposure or transfer into breastmilk were noted but not included here as these were out of scope for this review.
Information was captured on whether the study included pre- or postpartum data in the same individuals or if comparisons were made to data from non-pregnant individuals in other clinical studies. Likewise, information was collected on whether or not protein binding was considered, where this was known from non-pregnant individuals to influence PK significantly.
Generally, PK parameters are reported using standard compartmental analysis however in some cases, where data is sparse, population PK (PopPK) modeling was used. This modeling approach is, in general, used to characterize PK in a typical individual and to study the variability in drug concentrations between subjects within the population and considering individual characteristics such as age, sex, weight or disease state (7). For the purposes of this review, studies which collected data for PopPK modeling were included as available data. However, details on the PK models used to describe the data, e.g., PopPK and Physiologically Based Pharmacokinetic Modeling (PBPK) are not included here.
To obtain an overall picture of the availability of pregnancy PK data, each medicine was classified according to the quantity of available data, taking into account the abundance of information, the availability of data across the three trimesters and the richness and quality of the data. Table 2 outlines the aspects that were considered when evaluating the richness and quality of the data for each medicine.

Table 2. Study design methodology factors which may affect reliability of PK studies.
Medicines with high quality data in all gestational trimesters were generally considered to have rich data and classified as high category, while medicines with high quality data in two trimesters, or with sparse samples in all trimesters, were classified as medium category. Medicines for which data was limited, usually referring to only one trimester and/or with only sparse sampling from a single trimester, were classified as low category.
Results
The literature searches were performed for the 214 drugs from the therapeutic areas identified to be of interest in pregnancy. Relevant PK data were identified for 109 out of 214 medicines, with no PK data identified for the remaining 105 drug searches. Therefore, available published PK data were found and collected for only approximately half of 214 medicines included in the search. Figure 1 shows the availability of published PK data, as percentage, for the medicines reported in Table 1.
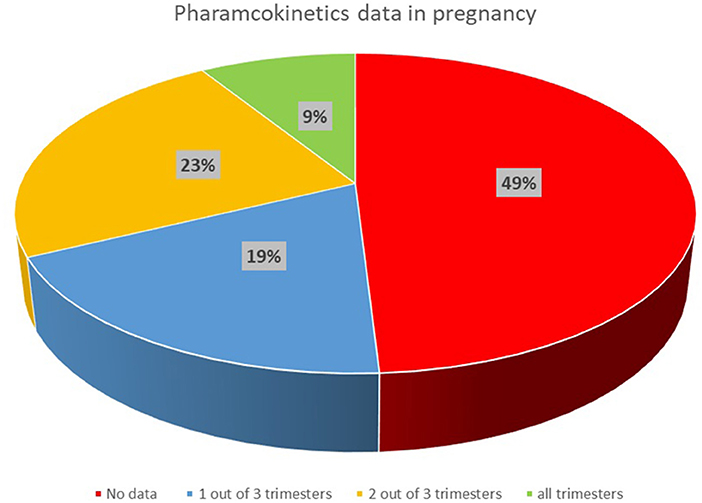
Figure 1. Availability of published clinical PK data in pregnancy for medicines of interest in the UK.
Clinical PK data with relatively rich sampling were available for 19 out of 109 medicines in all gestational trimesters (high category), while data were available for 50 medicines in two trimesters or where only sparse samples were available in all of the trimesters, and only very limited data were collected for 40 out of 109 medicines.
In some cases, it was difficult to classify the availability of PK data when considering the balance of data quantity vs. quality. For example, the effect of pregnancy on the PK of clozapine and risperidone was investigated in one study only with sparse samples, so met the criteria for low PK data availability. However, the study appeared well designed and allowed the characterization of the expected PK changes in all gestational trimesters (8).
In total over 420 publications with relevant data were identified. The amount of quality data available is very dependent on the therapeutic area, with some areas quite well studied in this population (e.g., antiviral (HIV), malaria, and antibiotics), whereas in other areas (e.g., antiemetics, TB, cardiovascular and diabetes), data is very limited, with most drugs having no data despite there being a need for treatment in this population. Figure 2 shows the availability of published PK data in pregnancy grouped by the investigated therapeutic areas.
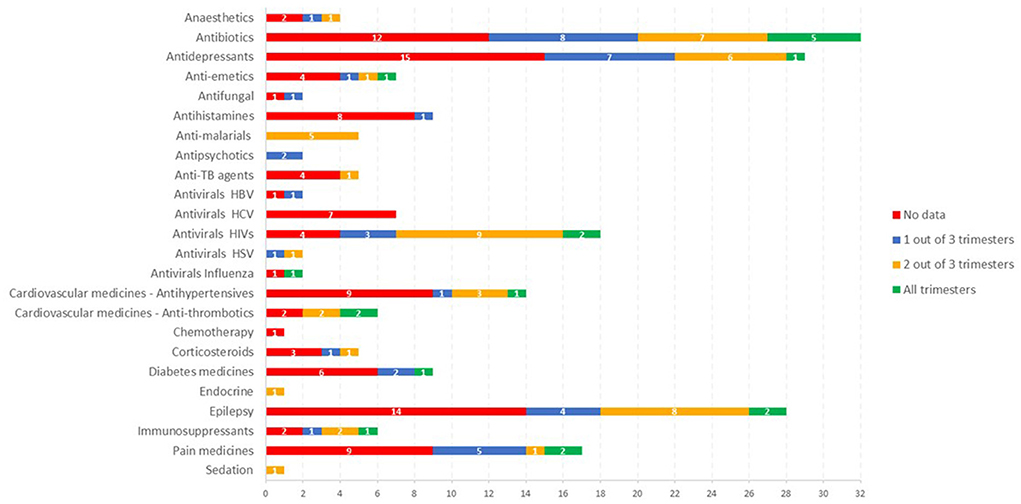
Figure 2. The availability of published PK data in pregnancy grouped by the investigated therapeutic areas.
In the antiviral area, HIV drugs are relatively well studied in pregnancy, with some data for most drugs and data is available across all trimesters for some medicines, e.g., atazanavir (9, 10). The availability of data in this area has been greatly facilitated by the IMPAACT study, which has studied at least 12 antiviral medicines in pregnancy using a platform study design.
For medicines in the antimalarial area, some data was available for all the drugs of interest (Figure 2) but in none of the cases were comprehensive data available across all trimesters. In some cases, the sparse data available was analyzed using a population PK analysis to show changes in pregnancy (12, 13).
For some antihypertensive medicines, some data was available for all trimesters, but full PK profiles were not available. Nevertheless, this allowed some conclusions on the impact of pregnancy compared with postpartum, e.g., with findings of up to 5 fold decreased exposure during pregnancy documented for metoprolol (14–16).
For medicines indicated for pain, there is currently reasonable data on the drugs blood levels in pregnancy for some drugs including, e.g., paracetamol (17–23) while limited PK data are available for other compounds in this therapeutic area (e.g., aspirin, morphine, tramadol, gabapentin, entonox, buprenorphine and opioids such as fentanyl and pethidine). However no PK data were identified in the pregnant population for some medicines, e.g., ibuprofen, triptans, codeine.
Corticosteroids as well as anti-hypertensives are commonly used during pregnancy. However, our search showed that the PK of those medicines' groups are not extensively studied in pregnancy i.e., PK data are not found for all pregnancy trimesters for medicines such as labetalol, nifedipine, dexamethasone or betamethasone. In other cases, e.g., methyldopa, PK data were not available in literature at the time of this review.
The PK of about half of the antidepressant drugs included in the search has been evaluated in pregnant women, although often a complete characterization of the PK profile is not available or data are not available for all gestational trimesters (e.g., citalopram, fluoxetine, sertraline, trazodone, lithium). Venlafaxine has been investigated in all trimesters of pregnancy, and decreased blood venlafaxine levels were observed in pregnancy compared to postpartum, although not all studies included postpartum measures or comparative data in non-pregnant individuals (24–26).
Amongst the immunosuppressant medicines, the PK of tacrolimus (11, 27, 28) has been investigated most extensively in pregnancy although the available data are not available in all gestational trimesters or consists of sparse samples. Of note, the available data suggest higher concentrations of free tacrolimus which may be clinically significant (11).
The impact of pregnancy on the PK of a number of antiepileptic drugs has been investigated by several authors. For some drugs (e.g., carbamazepine, phenytoin, levetiracetam, lamotrigine, valproate), PK data are available in all gestational trimesters, or at least two of them, although in general only sparse data have been collected (29–33). Significant decreases in exposure have been reported for lamotrigine (33, 34), whereas, although some authors reported a small reduction in total carbamazepine (11), the pharmacologically active free carbamazepine and its clearance did not, in general, significantly change during pregnancy (30–32).
Anesthetics (e.g., propofol, thiopentone) as well as sedative medicines (e.g., midazolam) do not appear to have been investigated during pregnancy, although some limited PK data are available in women undergoing cesarean section (35–38).
A limited number of studies were found which investigated the PK of antipsychotics (8, 9) in pregnancy, although in these studies data were collected in each gestational trimesters as well as at postpartum.
For diabetic medicines, the PK of metformin (39, 40) has been investigated during pregnancy, however limited data are available for other medicines commonly used, e.g., insulin and glyburide.
Discussion
More than 80% of women take at least one medicine during pregnancy (41, 42) and recent estimates suggest that one in five UK pregnancies were reported to be affected by polypharmacy (taking two or more prescription medicines at the same time1. It is therefore important to understand the impact of pregnancy-related physiological changes on the systemic exposure of medicines. The collection of PK data to evaluate this can support the benefit-risk evaluations of medicines used in pregnancy and support requirements for dose selection. This review focusses on PK. Dose adjustments based solely on PK assume that there are no changes in PD in this population, which is not always the case, e.g., glycemia control, or blood pressure. This needs to be considered on an individual therapeutic basis.
Our review of the literature for pharmacokinetic data on a number of medicines of interest for use in pregnancy has highlighted significant shortfalls in the availability of data. However, where data is available, significant changes in exposure are seen with decreased exposure of up to 5 fold documented, e.g., metoprolol (14, 15) and lamotrigine (33, 34), while for other medicines no changes are seen, e.g., carbamazepine (30–32). Higher concentrations of free drug than total drug can be observed in pregnancy, and these changes may be clinically significant for medicines such as tacrolimus (11). For other medicines, a decrease in total drug levels may not be related to a decrease in unbound concentrations. For instance, some authors reported a small reduction in total carbamazepine (29), but the pharmacologically active free carbamazepine and its clearance did not, in general, significantly change during pregnancy (30–32). This highlights the importance of study design in evaluating PK. A well designed PK study with a rich sampling set allows a good understanding of the PK profile and changes in important parameters like Volume of distribution, Clearance and elimination half-life. Whereas studies with sparse opportunistic samples may only allow an understanding of the overall change in concentration. However even in the latter case, if used in a well characterized population PK model, this data can also be used to inform changes in parameters.
The differential changes in exposure levels underline why more studies are urgently needed to support dosing of medicinal products in this population. There is also generally a dearth of new medications to treat obstetrical and lactation disorders (1). Although much needed, treatments for obstetric complications appear underdeveloped compared with other fields of medicine, and the appetite of pharmaceutical companies to invest in research for obstetric syndromes is generally reduced by concerns for maternal, fetal, and infant safety, poor definition, and regulatory paths toward product approval (43). In addition to ethical considerations, extra logistical challenges exist for clinical studies in pregnant women that need to be addressed. Therefore, it is important to maximize the usefulness of studies in this population.
A number of limitations in existing study methodology were noted in the PK data collected and are summarized (Table 2), consideration of these can be used to influence the design and maximize the output from future studies in this population.
Variability in exposure levels and other PK parameters made it difficult to determine if and when changes related to pregnancy occur. In some studies, this was likely due to inclusion of too few participants, so the studies were underpowered to detect statistically significant changes. Studies should include a sufficient number of subjects to allow significant changes in exposure to be detected. Consideration of the variability of the drug in non-pregnant individuals can be used to inform the number of subjects, similar to the approach taken for pediatric studies (44, 45).
Information on time of sampling since dose was often poorly reported, especially for studies with sparse sampling. Inaccuracies in this can underestimate peak plasma levels and affect estimates of elimination time. Ideally, the drugs levels should be measured at several time points to allow an accurate characterization of the PK changes in pregnancy. If plasma samples cannot be collected at the exact scheduled time, it is important to record accurately information about the time of the sample collection in relation to when the drug was administered. Consideration should be given to the dose route and formulation when considering appropriateness of sample points, especially around time of expected peak plasma levels, since intravenous or oral dosing can give quite different profiles, and immediate or modified release formulations will influence the shape of the plasma concentration profile (e.g., venlafaxine).
A combination of sparse sampling and population PK modeling such as in the study by Karanam (46) can allow the characterization of PK changes in different gestational trimesters and allows the integration of other data to better inform changes in pregnancy. This suggests that more use could be made of opportunistic samples, particularly if it is difficult to obtain plasma samples so a rich PK dataset is not feasible. Since medical visits are usually frequent in pregnancy, opportunistic samples from routine blood collection for laboratory tests could be used for PK evaluation, provided good bioanalytical support is available. Such an approach should ideally collect at least some samples around expected Tmax and during the elimination phase to support the understanding of possible impact of pregnancy on the medicine absorption and elimination.
It is worth considering aspects specific to this population that are important when interpreting data from these studies. Changes in exposure with time as pregnancy progresses can be rapid whereas changes that occur with age are usually slow. Where large changes in exposure are expected or seen during pregnancy it is important to have data at early stages of pregnancy, e.g., first trimester and to have earlier time points post-partum, to determine the time away from, and to return, of normal physiology. Knowledge of expected changes due to physiological changes or PBPK models would support the design of studies. Few studies also considered the impact of disease and/or polymorphisms in drug metabolizing enzymes on the exposure, although these could differentially affect pregnancy-related changes in drug blood levels as was seen for metoprolol (16).
Several studies evaluated PK changes in pregnancy by the comparison of the drug systemic exposure to the systemic exposure at postpartum or pre-pregnancy. However, time since delivery varied widely in some studies from several days to months in different studies (or even within the same study) and was poorly reported in others. For true post-partum, for most medicines, data should be collected at least 6 weeks after delivery as the pregnancy-related physiological changes are expected to revert to the non-pregnant status (47, 48). Moreover, if possible, data should be collected in the same woman during both pregnancy and postpartum in order to reduce the possible effect of intersubject variability. Data from the same woman in multiple trimesters may only be feasible for chronic treatments. However, sampling during pregnancy and postpartum from the same individuals and collected from women at different trimesters should be feasible for acute or short term treatments. Population PK analysis could be used to characterize pregnancy related changes for medicines use in non-chronic conditions.
Alternative approaches include platform type trials, as used in Impaact which can be used to efficiently investigate a number of medicines in a particular therapeutic area, the use of innovative designs and complex trial designs has also been proposed by other publications (49), Alternatively it can be efficient to use existing data, e.g., from therapeutic monitoring programmes, to examine changes in exposure in this population.
The importance of a study with rich sampling over a study with sparse samples would depend on how well characterized the PK profile is in the non-pregnant population and the level of PD understanding. If a trough concentration is recognized as the important determination of efficacy, and there are limited safety concerns, then determination of the trough concentration could be sufficient, in other cases, where PKPD is less well understood, it would be important to characterize the whole profile (AUC, Cmax and T1/2) with a dedicated PK study with rich sampling or informed time points analyzed in conjunction with a PopPK model.
Generally, the integrated approach to support dosing in pregnancy, as has been advocated by other publications (50, 51), utilizing all available methodologies and supported by modeling is supported and would be considered to add great value to inform dosing in this population.
It is recognized that this study has some limitations. Papers without references to PK pregnancy data in either the title or abstract may not be included in the results of this search. Moreover, the search terms “blood/serum concentrations” were not used.
Cited references were selected as good example of the currently available information on the impact of pregnancy on the PK, or of the clinical study methodology. A full list of papers will be provided on the MHRA pregnancy website.
The data identified in this review spans some 35+ years of PK studies in pregnancy (30). Some of the limitations in reporting of key information may be related to age of the studies. Over this time period the CONSORT (http://www.consort-statement.org/) and STROBE (https://www.strobe-statement.org/) statements have improved reporting for clinical trials and observational studies respectively. There is currently no agreed standard for reporting of PK studies akin to these, likewise there is no regulatory guidance for the design of PK studies in pregnancy however guidance does exist for the reporting of PK studies in pediatrics (6, 45) and of population pharmacokinetics (52) for regulatory purposes. Whilst these provide useful guidance on considerations on the design of clinical studies, many studies are conducted by independent academics, so a statement on standards for publication of PK studies in scientific journals would help the design of efficient studies to provide valuable data.
In conclusion, pregnancy-related physiological changes are expected to alter the pharmacokinetics of medicines commonly used by pregnant women. Despite this, our review shows that very limited data are currently available to characterize the impact of those changes on the maternal blood levels of most medicines commonly used during pregnancy. An improved understanding of the pharmacokinetic changes in all gestational trimesters is urgently needed to aid the benefit-risk evaluations of medicines used by pregnant women and to support safe and effective doses in this population. When designing clinical studies there are a number of important considerations on the study methodology to maximize the value of the data and minimize the impact on subjects in the trials.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The MHRA received a grant from the Bill & Melinda Gates Foundation, Seattle, Washington, United States, for a project to investigate the utilization of PK and PBPK to improve drug use in pregnancy.
Conflict of interest
PC was an employee of the MHRA at the time of the research study and manuscript writing. She is currently an employee of AstraZeneca UK Ltd.
The authors declare that this study received funding from the Bill and Melinda Gates Foundation. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, and the decision to submit it for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agencies or other organizations with which the authors are affiliated.
Footnotes
References
1. Caritis SN, Venkataramanan R. Obstetrical, fetal, and lactation pharmacology—a crisis that can no longer be ignored. Am J Obstetr Gynaecol. (2021) 12:10–20. doi: 10.1016/j.ajog.2021.02.002
2. Feghali M, Venkataramanan R, Caritis S. Pharmacokinetics of drugs in pregnancy. Semin Perinatol. (2015) 39:512–9. doi: 10.1053/j.semperi.2015.08.003
3. Cole S, Coppola P, Kerwash E, Nooney J, Lam SP. Pharmacokinetic characterization to enable medicine use in pregnancy, the potential role of physiologically-based pharmacokinetic modeling: a regulatory perspective CPT. Pharmacometrics Syst Pharmacol. (2020) 9:547–9. doi: 10.1002/psp4.12551
4. European Medicines Agency. EMA Guideline on the Exposure to Medicinal Products During Pregnancy: Need for Post-Authorisation Data (2005).
5. US FDA. Pregnant Women: Scientific and Ethical Considerations for Inclusion in Clinical Trials. Guidance for Industry (2018).
6. European Medicines Agency. Reflection Paper on the Use of Extrapolation in the Development of Medicines for Paediatrics (2018).
7. Aarons L. Population pharmacokinetics: theory and practice. Br J Clin Pharmacol. (1991) 32:669–70.
8. Westin AA, Brekke M, Molden E, Skogvoll E, Castberg I, Spigset O. Treatment with antipsychotics in pregnancy: changes in drug disposition. Clin Pharmacol Ther. (2018) 103:477–84. doi: 10.1002/cpt.770
9. Else LJ, Jackson V, Brennan M, Back DJ, Khoo SH, Coulter-Smith S et al. Therapeutic drug monitoring of atazanavir/ritonavir in pregnancy. HIV Med. (2014) 15:604–10. doi: 10.1111/hiv.12164
10. Lê MP, Mandelbrot L, Descamps D, Soulié C, Ichou H, Bourgeois-Moine A, et al. Pharmacokinetics, safety and efficacy of ritonavir-boosted atazanavir (300/100 mg once daily) in HIV-1-infected pregnant women. Antivir Ther. (2015) 20:507–13. doi: 10.3851/IMP2936
11. Zheng S, Easterling TR, Umans JG, Miodovnik M, Calamia JC, Thummel KE, et al. Pharmacokinetics of tacrolimus during pregnancy. Ther Drug Monit. (2012) 34:660–70. doi: 10.1097/FTD.0b013e3182708edf
12. Mosha D, Guidi M, Mwingira F, Abdulla S, Mercier T, Decoster LA, et al. Population pharmacokinetics and clinical response for artemether lumefantrine in pregnant and non-pregnant women with uncomplicated plasmodium falciparum malaria in Tanzania Dominic. Antimicrob Agents Chemother. (2014) 24:4583–92. doi: 10.1128/AAC.02595-14
13. Kloprogge K, Jullien V, Piola P, Dhorda M, Muwanga S, Nosten F, et al. Population pharmacokinetics of quinine in pregnant women with uncomplicated Plasmodium falciparum malaria in Uganda. J Antimicrob Chemother. (2014) 69:3033–40. doi: 10.1093/jac/dku228
14. Högstedt S, Lindberg B, Rane A. Increased oral clearance of metoprolol in pregnancy. Eur J Clin Pharmacol. (1983) 24:217–20. doi: 10.1007/BF00613820
15. Högstedt S, Lindberg B, Peng DR, Regårdh CG, Rane A. Pregnancy-induced increase in metoprolol metabolism. Clin Pharmacol Ther. (1985) 37:688–92. doi: 10.1038/clpt.1985.114
16. Ryu RJ, Eyal S, Easterling TR, Caritis SN, Venkataraman R, Hankins G, et al. Pharmacokinetics of metoprolol during pregnancy and lactation. J Clin Pharmacol. (2016) 56:581–9. doi: 10.1002/jcph.631
17. Rayburn W, Shukla U, Stetson P, Piehl E. Acetaminophen pharmacokinetics: comparison between pregnant and non-pregnant women. Am J Obstet Gynecol. (1986) 155:1353–6. doi: 10.1016/0002-9378(86)90173-0
18. Beaulac-Baillargeon L, Rocheleau S. Paracetamol pharmacokinetics during the first trimester of human pregnancy. Eur J Clin Pharmacol. (1994) 46:451-4. Drug Invest. 6 (3):176–179, 1993 01 I 4–2402/93/0003–01 76j$02.00/0 © Adis International Limited. All rights reserved. DRll333 Paracetamol Pharmacokinetics in Pregnancy doi: 10.1007/BF00191910
19. Beaulac-Baillargeon L. Rocheleau paracetamol pharmacokinetics in pregnancy. S Drug Invest. (1993) 6:176–9. doi: 10.1007/BF03259739
20. Kulo A, van de Velde M, de Hoon J, Verbesselt R, Devlieger R, Deprest J, et al. Pharmacokinetics of a loading dose of intravenous paracetamol post caesarean delivery. Int J Obstet Anesth. (2012) 21:125–8. doi: 10.1016/j.ijoa.2011.12.007
21. Kulo A, Peeters MY, Allegaert K, Smits A, de Hoon J, Verbesselt R, et al. Pharmacokinetics of paracetamol and its metabolites in women at delivery and post-partum. Br J Clin Pharmacol. (2013) 75:850–60. doi: 10.1111/j.1365-2125.2012.04402.x
22. Kulo A, Van Calsteren K, van de Velde M, Mulabegovic N, Verbesselt R, de Hoon JN, et al. Weight, pregnancy and oral contraceptives affect intravenous paracetamol clearance in young women. Eur Rev Med Pharmacol Sci. (2014) 18:599–604. Avalable online at: https://www.europeanreview.org/article/7016
23. Nitsche JF, Patil AS, Langman LJ, Penn HJ, Derleth D, Watson WJ, et al. Transplacental passage of acetaminophen in term pregnancy. Am J Perinatol. (2017) 34:541–3. doi: 10.1055/s-0036-1593845
24. Horst PG, Larmené-Beld KH, Bosman J, van der Veen EL, Wieringa A, Smit JP. Concentrations of venlafaxine and its main metabolite O-desmethylvenlafaxine during pregnancy. J Clin Pharm Ther. (2014) 39:541–4. doi: 10.1111/jcpt.12188
25. Rampono J, Simmer K, Ilett KF, Hackett LP, Doherty DA, Elliot R, et al. Placental transfer of SSRI and SNRI antidepressants and effects on the neonate. Pharmacopsychiatry. (2009) 42:95–100. doi: 10.1055/s-0028-1103296
26. Klier CM, Mossaheb N, Saria A, Schloegelhofer M, Zernig G. Pharmacokinetics and elimination of quetiapine, venlafaxine, and trazodone during pregnancy and postpartum. J Clin Psychopharmacol. (2007) 27:720–2. doi: 10.1097/JCP.0b013e31815a57d8
27. Zheng S, Easterling TR, Hays K, Umans JG, Miodovnik M, Clark S et al. Tacrolimus placental transfer at delivery and neonatal exposure through breast milk. Br J Clin Pharmacol. (2013) 76:988–96. doi: 10.1111/bcp.12122
28. Aktürk S, Çelebi ZK, Erdogmuş S, Kanmaz AG, Yüce T, Sengül S, et al. Pregnancy after kidney transplantation: outcomes, tacrolimus doses, and trough levels. Transplant Proc. (2015) 47:1442–4. doi: 10.1016/j.transproceed.2015.04.041
29. Lander CM, Eadie MJ. Plasma antiepileptic drug concentrations during pregnancy. Epilepsia. (1991) 32:257–66. doi: 10.1111/j.1528-1157.1991.tb05253.x
30. Yerby MS. Problems and management of the pregnant woman with epilepsy. Epilepsia. (1987) 28:S29–36. doi: 10.1111/j.1528-1157.1987.tb05775.x
31. Yerby MS, Friel PN, McCormick K, Koerner M, Van Allen M, Leavitt AM, et al. Pharmacokinetics of anticonvulsants in pregnancy: alterations in plasma protein binding. Epilepsy Res. (1990) 5:223–8. doi: 10.1016/0920-1211(90)90042-T
32. Tomson T, Lindbom U, Ekqvist B, Sundqvist A. Disposition of carbamazepine and phenytoin in pregnancy. Epilepsia. (1994) 35:131–5. doi: 10.1111/j.1528-1157.1994.tb02922.x
33. Reisinger TL, Newman M, Loring DW, Pennell PB, Meador KJ. Antiepileptic drug clearance and seizure frequency during pregnancy in women with epilepsy. Epilepsy Behav. (2013) 29:13–8. doi: 10.1016/j.yebeh.2013.06.026
34. Pennell PB, Newport DJ, Stowe ZN, Helmers SL, Montgomery JQ. Henry TR. The impact of pregnancy and childbirth on the metabolism of lamotrigine. Neurology. (2004) 62:282–95. doi: 10.1212/01.WNL.0000103286.47129.F8
35. Gin T, Gregory MA, Chan K, Buckley T, Oh TE. Pharmacokinetics of propofol in women undergoing elective caesarean section. Br J Anaesth. (1990) 64:148–53. doi: 10.1093/bja/64.2.148
36. Kanto J, Rosenberg P. Propofol in cesarean section: a pharmacokinetic and pharmacodynamic study. Methods Find Exp Clin Pharmacol. (1990) 12:707–11.
37. Morgan DJ, Blackman GL, Paull JD, Wolf LJ. Pharmacokinetics and plasma binding of thiopental. II: Stud Cesarean Sect Anesthesiol. (1981) 54:474–80. doi: 10.1097/00000542-198106000-00006
38. Wilson CM, Dundee JW, Moore J, Howard PJ, Collier PS, A. comparison of the early pharmacokinetics of midazolam in pregnant and non-pregnant women. Anaesthesia. (1987) 42:1057–62. doi: 10.1111/j.1365-2044.1987.tb05168.x
39. De Oliveira Baraldi C, Moisés EC, de Jesus Ponte Carvalho TM, de Jesus Antunes N, Lanchote VL, Duarte G, et al. Effect of type 2 diabetes mellitus on the pharmacokinetics of metformin in obese pregnant women. Clin Pharmacokinet. (2012) 51:743–9. doi: 10.1007/s40262-012-0008-7
40. Hughes RC, Gardiner SJ, Begg EJ, Zhang M. Effect of pregnancy on the pharmacokinetics of metformin. Diabet Med. (2006) 23:323–6. doi: 10.1111/j.1464-5491.2005.01769.x
41. Lupattelli A, Spigset O, Twigg MJ, Zagorodnikova K, Mårdby AC, Moretti ME, et al. Medication use in pregnancy: a cross-sectional, multinational web-based study. BMJ Open. (2014) 4:e004365. doi: 10.1136/bmjopen-2013-004365
42. Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S, National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol. (2011) 205:51. doi: 10.1016/j.ajog.2011.02.029
43. Bahmanyar ER, Out HJ, van Duin M. Women and babies are dying from inertia: a collaborative framework for obstetrical drug development is urgently needed. Am J obstetr Gynaecol. (2021) 21:44–50. doi: 10.1016/j.ajog.2021.03.024
44. Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clinic Pharmacol. (2012) 52:1601–6. doi: 10.1177/0091270011422812
45. European Medicines Agency. Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. (2006). EMEA/CHMP/EWP/147013/2004 Corrigendum.
46. Karanam A, Pennell P. Lamotrigine clearance increases by 5 weeks gestational age: relationship to estradiol concentrations and gestational age. Ann Neurol. (2018) 84:556–63. doi: 10.1002/ana.25321
47. Mulligan N, Best BM, Wang J, Capparelli EV, Stek A, Barr E et al. Dolutegravir pharmacokinetics in pregnant and postpartum women living with HIV. AIDS. (2018) 32:729–37. doi: 10.1097/QAD.0000000000001755
49. Hoffman RM, Mngqibisa R, Averitt D, Currier JS. Accelerating drug discovery for pregnant and lactating women living with HIV. J Int AIDS Soc. (2021) 24:e25680. doi: 10.1002/jia2.25680
50. Eke AC, Olagunju A, Best BM, Mirochnick M, Momper JD, Abrams E et al. Innovative approaches for pharmacology studies in pregnant and lactating women: a viewpoint and lessons from HIV. Clin Pharmacokinet. (2020) 59:1185–94. doi: 10.1007/s40262-020-00915-w
51. Eke AC, Olagunju A, Momper J, Penazzato M, Abrams EJ, Best BM et al. Optimizing pharmacology studies in pregnant and lactating women using lessons from HIV: a consensus statement. Clin Pharmacol Ther. (2021) 110:36–48. doi: 10.1002/cpt.2048
Keywords: pregnancy, pharmacokinetics, clinical trials, regulatory, ADME
Citation: Coppola P, Kerwash E, Nooney J, Omran A and Cole S (2022) Pharmacokinetic data in pregnancy: A review of available literature data and important considerations in collecting clinical data. Front. Med. 9:940644. doi: 10.3389/fmed.2022.940644
Received: 10 May 2022; Accepted: 14 September 2022;
Published: 04 October 2022.
Edited by:
Steffen Thirstrup, European Medicines Agency, NetherlandsReviewed by:
Karel Allegaert, University Hospitals Leuven, BelgiumAngela Colbers, Radboud University, Netherlands
Copyright © 2022 Coppola, Kerwash, Nooney, Omran and Cole. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Cole, c3VzYW4uY29sZUBtaHJhLmdvdi51aw==