 Henry W. Chen
Henry W. Chen Grant Barber
Grant Barber Benjamin F. Chong
Benjamin F. Chong- Department of Dermatology, University of Texas Southwestern Medical Center, Dallas, TX, United States
Cutaneous lupus erythematosus (CLE) is an autoimmune connective tissue disease that can exist as a disease entity or within the context of systemic lupus erythematosus (SLE). Over the years, efforts to elucidate the genetic underpinnings of CLE and SLE have yielded a wealth of information. This review examines prior studies investigating the genetics of CLE at the DNA and RNA level and identifies future research areas. In this literature review, we examined the English language literature captured within the MEDLINE and Embase databases using pre-defined search terms. First, we surveyed studies investigating various DNA studies of CLE. We identified three predominant areas of focus in HLA profiling, complement deficiencies, and genetic polymorphisms. An increased frequency of HLA-B8 has been strongly linked to CLE. In addition, multiple genes responsible for mediating innate immune response, cell growth, apoptosis, and interferon response confer a higher risk of developing CLE, specifically TREX1 and SAMHD1. There was a strong association between C2 complement deficiency and CLE. Second, we reviewed literature studying aberrations in the transcriptomes of patients with CLE. We reviewed genetic aberrations initiated by environmental insults, and we examined the interplay of dysregulated inflammatory, apoptotic, and fibrotic pathways in the context of the pathomechanism of CLE. These current learnings will serve as the foundation for further advances in integrating personalized medicine into the care of patients with CLE.
Introduction
Cutaneous lupus erythematosus (CLE) is a heterogeneous autoimmune disease that can be skin-limited or exist within the context of systemic lupus erythematosus (SLE). With the advancement of genetic sequencing technology at the DNA and RNA level, more dysregulated pathways and gene networks that contribute to the development of CLE have been identified. Specifically, differential expression of key genes involved in various pathways, such as inflammation, apoptosis, and immunity has revealed a complex, heterogeneous picture. These new gene expression profiles offer the opportunity to further delineate classification subsets of CLE and potentially predict prognosis, such as response to treatments and progression to systemic disease (1, 2).
Up until recently, the development of new therapies for CLE has been stymied by an incomplete understanding of the underlying pathophysiology of CLE. Given the importance of understanding the genetic landscape of CLE, we performed a literature review to summarize studies examining DNA and RNA genetic aberrations in CLE.
Methods
This was a review of the English-language literature captured within the MEDLINE and Embase databases using pre-defined search terms (Supplementary Table 1) from inception through 7 February 2022. Two independent reviewers (H.W.C. and G.B.) reviewed all studies, and a third reviewer (B.F.C.) resolved any discrepancies. Inclusion criteria were original studies, case series, and case reports related to CLE in humans in the English language. Articles underwent title and abstract screening with a subsequent full-text review. Articles were included if their findings were pertinent to DNA or RNA in the context of CLE. Reviews, conference abstracts, editorials, and all non-peer-reviewed findings were excluded from this review. In total, 1,253 studies were identified for screening, and 105 studies were ultimately included for final review after applying inclusion and exclusion criteria (Supplementary Figure 1).
Results
DNA
Studies examining DNA have long been performed to better our understanding of cutaneous lupus. Three major themes emerged from our review of these studies, such as HLA profiling, complement deficiencies, and genetic polymorphisms.
Human Leukocyte Antigen Genes Have Been Associated With CLE and Its Subtypes
Human leukocyte antigen (HLA) is quintessential in the differentiation of self and non-self and plays a strong role in autoimmunity. HLA profiling studies have been performed in small groups of patients with CLE and controls to better understand genetic variations. Fowler et al. (3) found an increased frequency of HLA-DRw6 among both White and Black patients with CLE. In further studies, HLA-B8 has repeatedly been found to be increased among patients with discoid lupus erythematosus (DLE) and subacute cutaneous lupus erythematosus (SCLE) (4–7). Bielsa et al. (4) found an increased frequency of HLA-B8 and HLA-DR3 along with a decreased frequency of HLA-DR5 in patients with annular SCLE when compared with controls. Another study of 11 Finnish patients with SCLE and 23 controls showed that HLA-DR3, HLA-B8, and HLA-DR2 were higher in patients with SCLE vs. controls (7). Fischer et al. (8) found the HLA-DQA1*OI02 allele was significantly increased among 26 patients with chronic CLE (CCLE) vs. healthy controls. The HLA-DQA1 alleles have also been studied in neonatal lupus erythematosus (NLE), with mothers of seven NLE children all carrying at least one DQA1 allele with glutamine at position 34 of the first domain compared with just 44% of controls (9). Another study of 28 patients with DLE showed a higher frequency of HLA-DRB1*04 (10). These studies highlight the inherent importance of HLA variations in CLE, though many are limited by small sample sizes. Larger studies, such as genome-wide association studies, in diverse populations, would help elucidate these variations.
Deficiencies in the Complement Cascade Contribute to Cutaneous Lupus Erythematosus Pathogenesis
The complement cascade is indispensable in mediating phagocytosis and inflammation. C1q is a subcomponent of C1 comprised of three heterotrimeric subunits (C1qA, C1qB, and C1qC). In a cohort of 19 White patients with SCLE, homozygous C1qA A > G transition mutation in exon 2, which results in a synonymous mutation, was found to occur more frequently in patients with SCLE relative with healthy controls (11). Despite no alterations in the protein sequence, decreased C1q protein was still observed. Another case study identified a homozygous G > C transversion mutation in C1qC exon 1, resulting in a Gly61Arg mutation (12). Multiple case studies have identified C2 deficiency in patients with SCLE and DLE (13–15). The gene encoding C2 lies within the major histocompatibility complex and is thus linked with HLA-A10, -A25, -B18, -DR2, and -Dw2. Agnello et al. (16) examined the pedigrees of four patients with DLE and found a partial genetic deficiency of C4 in patients carrying the null C4 allele B*QO. Given the important function of the complement cascade in mediating phagocytosis and inflammation, further studies on the complement system’s role in the pathogenesis of CLE would be beneficial.
Genetic Polymorphisms Have Been Featured in Familial Chilblain Lupus and Other Cutaneous Lupus Erythematosus Subtypes
Genetic polymorphisms have also been a frequent focus of study for lupus. Table 1 summarizes prominent ones that have been identified, such as TREX1, SAMHD1, and tumor necrosis factor (TNF). TREX1 has been identified as a significant factor in familial chilblain lupus. Günther et al. (17) identified a potential mutation hotspot for TREX1 where 4 of 6 families affected by familial chilblain lupus all present the same mutation. Günther et al. (18) also linked TREX1 with the upregulation of type I interferon (IFN) activity in familial chilblain lupus. Another case report of a family with familial chilblain lupus revealed that three affected individuals all carried the same heterozygous mutation (19). Heterozygous mutations in SAMHD1 have also been identified in patients with familial chilblain lupus independently of TREX1 mutations, as reported by Ravenscroft et al. (20). Further, Linggonegoro et al. (21) found a deletion of the SAMHD1 gene’s initiator region in a child with familial chilblain lupus, who did not show an increased IFN signature. TNF is another gene investigated extensively in lupus. Mutations involving this gene seem to be distinctly associated with SCLE rather than DLE (6, 22, 23). Millard et al. (6) reported a significantly higher frequency of TNF-α (−308 G/A) single nucleotide polymorphism (SNP) among 36 patients with SCLE compared with 49 patients with DLE and 102 healthy relatives of patients with lupus. Similarly, TNF-α (−308 G/A) SNP was found to be a significant risk factor among 192 patients with SLE, but not among 56 patients with DLE (23). Studies on ITGAM polymorphisms compared in patients with DLE and SLE have shown conflicting results. One study of 21 patients with DLE and 35 patients with SLE showed polymorphisms of ITGAM among patients with SLE but not among patients with DLE (24). However, another study of 177 patients with DLE and 85 patients with SLE found ITGAM polymorphisms to be 3-fold greater in DLE compared to controls and five times greater than in SLE (25).
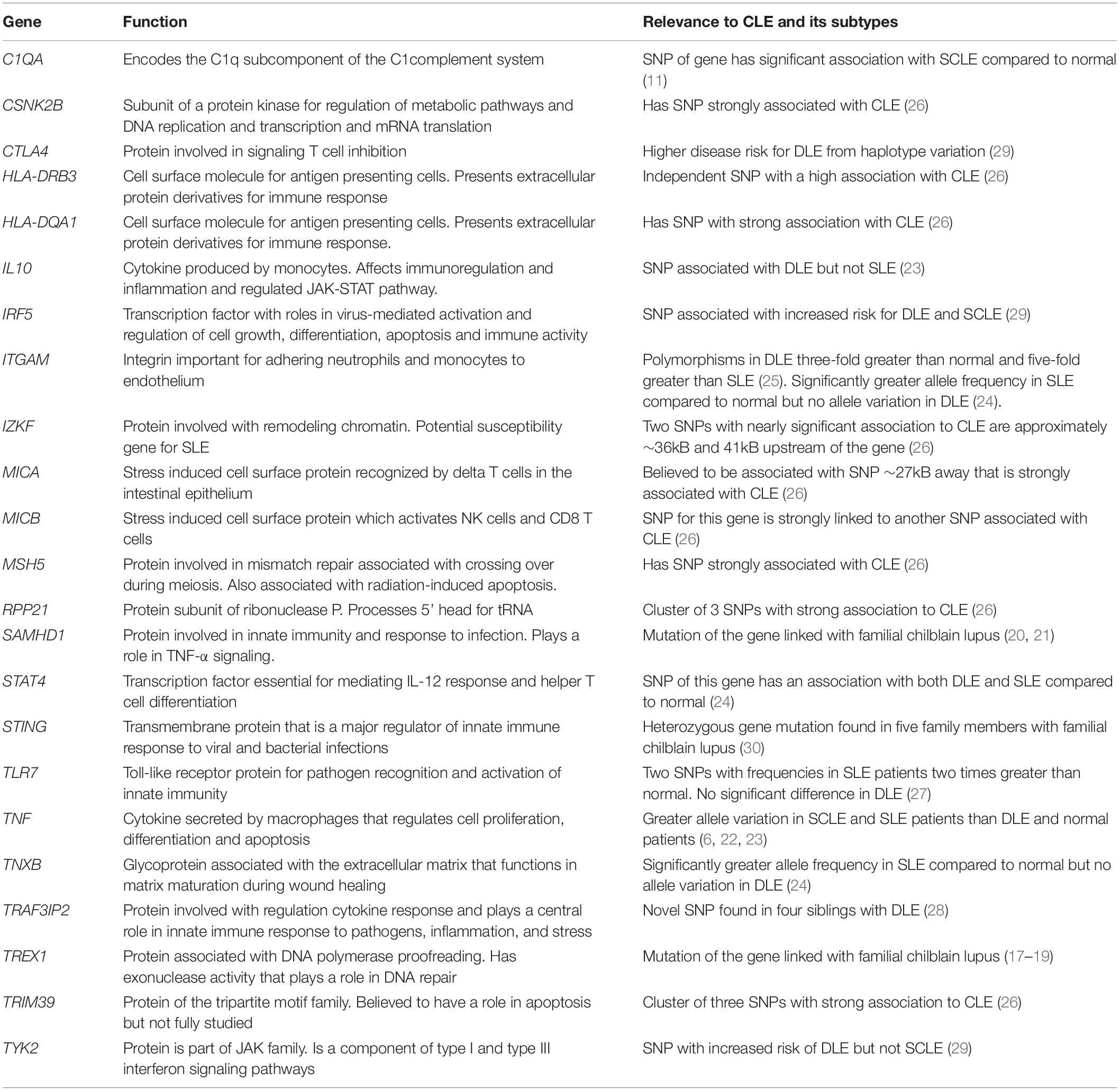
Table 1. Genetic polymorphisms investigated in cutaneous lupus studies and their functions.
To date, only Kunz et al. (26) have performed a genome-wide association study specifically examining patients with CLE. A comparison of 183 German patients with CLE, including DLE, SCLE, and lupus erythematosus tumidus (LET) subtypes, against healthy controls with a validation set of Finnish patients with DLE (n = 177) and SCLE (n = 42), revealed 62 SNPs predominantly on chromosome 6 in the major histocompatibility complex region. The presence of SNPs associated with apoptosis and inflammation (i.e., TRIM39/RPP21) and previously described in SLE (i.e., HLA-DQA1, MICA/B, and IZKF) suggests unique and overlapping genetic underpinnings of CLE and SLE.
In summary, numerous genes were found to have SNPs that were associated with a greater risk of CLE and SLE, such as TLR7, TRAF3IP2, TYK2, IRF5, IL10, C1QA, and STAT4 (11, 23, 24, 27–29). Table 1 summarizes other additional genes whose polymorphisms are distinctly different in CLE and SLE groups (23, 24, 28–30).
RNA
Understanding the genetic aberrations at the DNA level serves as a foundation for examining the changes in the CLE transcriptome. The interplay of multiple pathways, namely, inflammation, apoptosis, and fibrosis, lays the framework for the pathomechanisms behind CLE (Figure 1). Herein, we describe the major contributors to CLE pathogenesis identified in gene expression analyses.
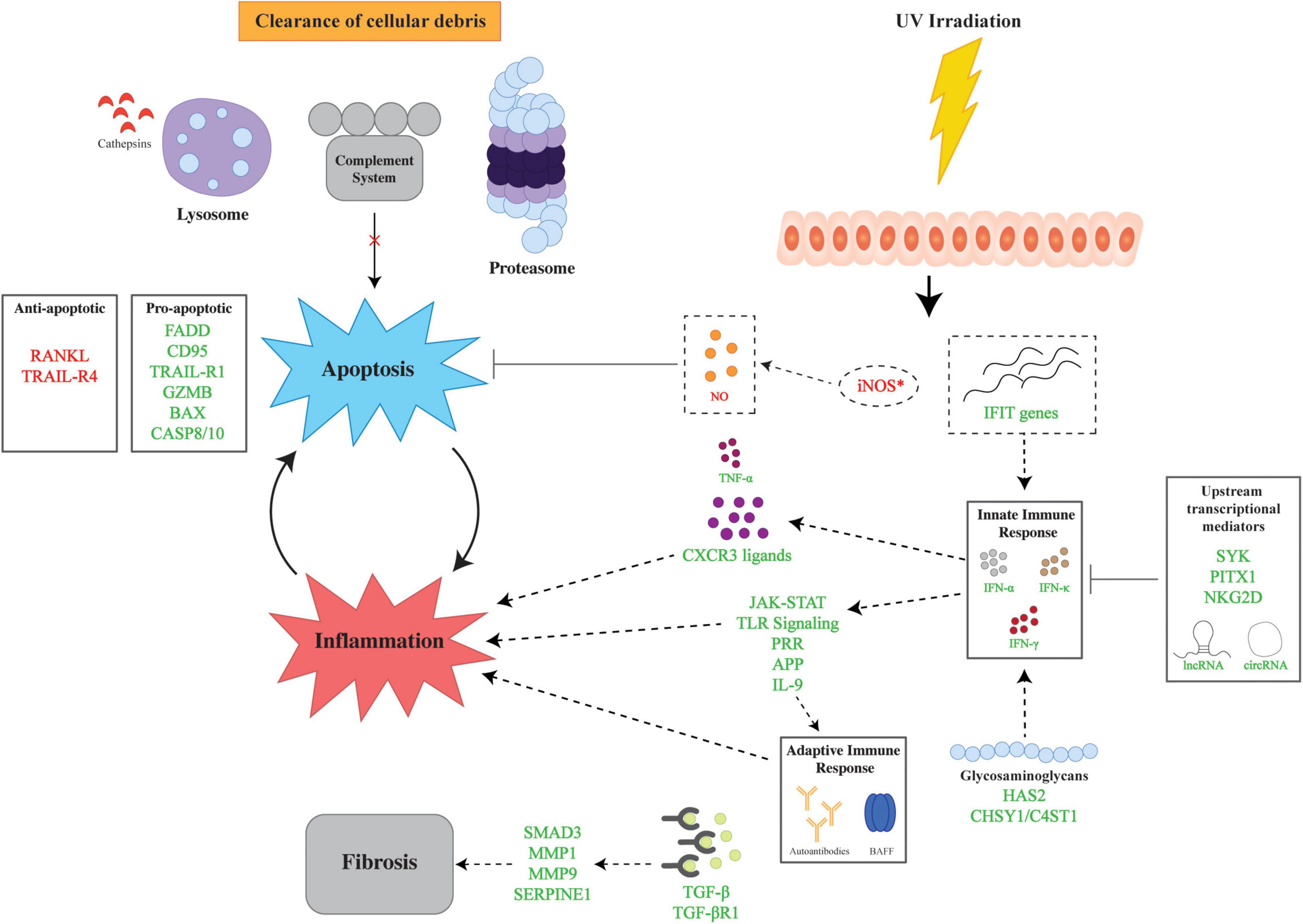
Figure 1. Overview of major pathways in the genetic pathophysiology of cutaneous lupus erythematosus (CLE). The genetic pathophysiology of CLE encompasses three main pathways: inflammation, apoptosis, and fibrosis. UV irradiation is a major environmental insult that serves to initiate CLE pathogenesis by aberrant expression of iNOS and IFIT genes which lead to apoptosis and inflammation, respectively. The inflammatory response is mediated by the innate and adaptive immune response via IFNs and autoantibodies. Upstream transcriptional mediators regulate the innate immune response via transcriptional factors and non-coding RNAs. Glycosaminoglycans also modulate the inflammatory response via the upregulation of HAS2 and CHSY1/C4ST1. Apoptosis is facilitated by dysregulation of pro- and anti-apoptotic genes in tandem with impaired cellular clearance via aberrations in the lysosome, proteasome, and complement. Finally, fibrosis is mediated by TGF-β and downstream upregulation of effector molecules. Lines with bars indicate inhibition. Arrows depict subsequent effect of molecule or process. Green text signifies upregulation and red text signifies downregulation. APP, antigen processing and presentation; BAX, Bcl-2 Associated X-protein; GZMB, Granzyme B; C4ST1, carbohydrate sulfotransferase 11; CASP8/10, caspase 8/10; CD95, Fas; CHSY1, chondroitin sulfate synthase 1; circRNAs, circular RNAs; CXCR3, C-X-C motif chemokine receptor 3; FADD, Fas Associated via Death Domain; HAS2, hyaluronan synthase 2; IFIT, interferon induced proteins with tetratricopeptide repeats; IFN-α, interferon alpha, IFN-γ, interferon gamma; IFN-κ, interferon kappa; JAK-STAT, Janus kinase signal transducer and activator of transcription; lncRNAs, long non-coding RNAs; MMP1, matrix metallopeptidase 1; MMP9, matrix metallopeptidase 9, NKG2D, natural killer group 2D; PITX1, paired like homeodomain 1; RANKL, receptor activator of nuclear factor kappa-beta ligand, SERPINE1, Serpin Family E Member 1; SMAD3, SMAD Family Member 3, SYK, spleen tyrosine kinase; TGF-β, transforming growth factor beta; TGF-βR1, transforming growth factor beta receptor 1; TNF-α, tumor necrosis factor alpha; TLR, toll-like receptor; TRAIL-R1, TRAIL Receptor 1; TRAIL-R4, TRAIL Receptor 4; UV, ultraviolet.
UV Irradiation Is a Major Initiator in the Pathogenesis of Cutaneous Lupus Erythematosus
UV irradiation has been thought to play a key role in the development of CLE, specifically due to the induction of autoantigens. After UV irradiation, nitric oxide is synthesized by nitric oxide synthases, such as inducible nitric oxide synthase (iNOS), and functions to protect cells, such as keratinocytes, from apoptosis (31, 32). Early work showed abnormal iNOS gene expression in the skin of patients with CLE patients who demonstrated delayed kinetics of iNOS induction by 72 h relative to controls (33). UVB irradiation induces chemokines, such as CXCR3 ligands CXCL9, CXCL10, and CXCL11, necessary to orchestrate the innate and adaptive response central to the immunopathogenesis of CLE (34). More recently, Katayama et al. (35) showed upregulation of the IFIT gene family, HLA-DPA1, and normal UV response genes (i.e., nucleic acid binding and erythematous reactions) in CLE skin relative to healthy skin. The IFIT gene family has subsequently been shown to be the top hub genes in bioinformatics analysis of DLE skin (36). This inflammatory response is mediated by IFNs with greater concordant elevations in IFN-α levels in SCLE relative to DLE.
Innate and Adaptive Immune Responses Drive Inflammation in Cutaneous Lupus Erythematosus Pathogenesis
For many years, unfettered inflammation secondary to dysregulated Th1 axis has been understood to be at the heart of CLE pathogenesis. Early studies using reverse-transcriptase PCR identified the potential role of type 1 cytokines and inflammation pathways. Patients with DLE without SLE were found to have increased expression of IFN-γ and IL-2 (37). An examination of the T-cell cytokine profile in CLE showed an upregulation of IFN-γ but also IL-5, indicating a possible role for Th2 cells (38). In the context of the B7-CD28 pathway, the importance of T-cells in the pathogenesis of CLE is underscored by findings of B7-1 and B7-2 RNA expression primarily in the dermis of patients with DLE, SCLE, and SLE (39). Microarray experiments comparing DLE to psoriasis confirmed a predominant Th1 signature and no Th17 signature, which is a hallmark of psoriasis (40). Bioinformatics analysis of gene networks notes an overlap of Th1 skewing of DLE with sarcoidosis (41). When CLE subtypes are compared, DLE and SCLE gene expression predominantly had a type I IFN signature, but DLE had a relatively increased expression of Th1-related cytokines (42).
Innate immune response functions upregulated by the Th1 phenotype include JAK/STAT signaling, toll-like receptor signaling, pattern recognition receptors, and antigen processing and presentation. Microarray and RNA-sequencing experiments consistently demonstrate upregulation of these inflammatory pathways in CLE skin (43–46). Recently, JAK/STAT upregulation in CLE has been the focus of targeted therapies, with JAK1-specific inhibition being explored as a promising approach for the treatment of CLE (47, 48). Enhanced toll-like receptor-dependent and pattern recognition receptor pathways contribute to both innate and adaptive immune responses in CLE (45). While often overlooked, only one study has examined the glycome of CLE given the function of glycosaminoglycans in mediating inflammation by acting as pathogen-associated molecular patterns (49, 50). Upregulation of hyaluronan and chondroitin sulfate via HAS2 and CHSY1/C4ST1, respectively, provides some evidence by which glycosaminoglycans participate in the characteristic inflammatory response of CLE. Finally, in a study by Zhu et al. (1), a unique machine learning approach leveraging modular analysis uncovered large heterogeneity in CLE, but central themes of Th1 dysregulation and interferon activation were largely preserved in identified clusters.
The adaptive immune response is also important in the pathogenesis of CLE via dysregulation in antigen presentation, activation of B-cells, and autoantibody production (36). Moreover, key players, such as IL-9 and B-cell activating factor (BAFF) may be important in CLE progression to SLE and the distinction of CLE from SLE. Elevated IL9 expression has been linked to production of autoantibodies in lupus-prone mice (51) and in skin of CLE patients who progressed to SLE versus those who did not (52). Similarly, our group found that higher BAFF mRNA and protein levels in patients with DLE with SLE than in those without SLE (53). Taken together, these findings suggest intricate interactions between the innate and adaptive immune response in mediating CLE pathogenesis and facilitating progression to SLE.
Upstream Regulation Impacts the Degree of Inflammatory Signatures in Cutaneous Lupus Erythematosus
Upstream mediators of inflammation have also been investigated. Spleen tyrosine kinase (SYK) is known to be a mediator of multiple innate and adaptive immune responses (54). Gene expression analysis via microarray of DLE (n = 7) and SCLE (n = 5) skin revealed upregulation of SYK and multiple SYK-regulated innate immune-related genes relative to healthy skin (n = 5) (55). Vorwerk et al. (56) postulate NKG2D, an immune receptor on NK cells and a subset of CD8 + T cells, contributes to CLE, giving upregulation on whole transcriptome RNA sequencing. However, NKG2D plays a selective role in autoimmune disease, and further mechanistic studies are required to understand its role in CLE (57). Recent RNA-sequencing data of SLE skin have identified another transcription factor, PITX1, which facilitates hypersensitive responses to type I IFNs in lupus keratinocytes (58). Finally, RNA-sequencing analysis of long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) was differentially expressed in patients with DLE and correlated with inflammatory immune response-related genes on coding and non-coding gene network analysis (59). Further functional studies of lncRNAs and circRNAs are required to fully understand their biological function.
Upregulated Cytokines and Chemokines Enhance Inflammatory Response in Cutaneous Lupus Erythematosus
One member of the type I IFN family, IFN-α, has had a well-established signature within SLE patients with skin involvement (60). In CLE, IFN-α is upregulated in both lesional and non-lesional skin (61). IFN-α recently has been shown to promote the adherence of Staphylococcus aureus with CLE and SLE keratinocytes (62). Thus, in concert with dysregulation of barrier proteins, such as filaggrin, IFN-α has been mechanistically implicated in the colonization of CLE lesions. In addition to IFN-α, IFN-κ has also been important in the dysregulation of CLE keratinocytes (58, 63, 64). Responsiveness to hydroxychloroquine therapy has been associated with an increased type I IFN signature, while high TNF-α was associated with response to adjunct quinacrine (2).
Upregulated expression of chemokines CXCL9, CXCL10, and CXCL11 and its receptor CXCR3 is a hallmark of CLE (65). These chemokines exert their effect on CXCR3-expressing cells and orchestrate the Th1 immune response by promoting Th1 cell migration (34, 48, 66). These chemokines have been repeatedly shown to facilitate interface dermatitis (66). CXCL9 and CXCL10 expression were strongly correlated with IFN-γ expression in DLE (n = 15), SCLE (n = 11), and LET (n = 21) skin. (67). Novel bioinformatics approaches have shown that these chemokines as key genes are involved in CLE (68).
Apoptosis Perpetuates Inflammation
Apoptosis is broadly comprised of the extrinsic and intrinsic pathways. Current evidence suggests increased activity of the extrinsic apoptotic pathway via the upregulation of the TRAIL receptor system and CD95 (69, 70). Specifically, an increase of apoptotic keratinocytes has been observed in CLE with concomitant increased epidermal expression of TRAIL-R1, CD95, and FADD (64, 69). Apoptotic keratinocytes contribute to the pathogenesis of CLE via the release of cellular debris, which results in a positive feedback loop of inflammation. Recent work by Kingsmore et al. (71) noted increased apoptotic mitochondrial gene signatures in DLE and lupus nephritis, suggesting a role for the intrinsic apoptotic pathway, and positive correlation with inflammatory cell signatures supports the intrinsic link of apoptosis with inflammation. Gene set expression analysis with microarray and RNA-sequencing of CLE skin and blood identified other genes, such as GZMB, BAX, and various caspases (CASP8/10) among others (1, 64, 72–75). Differential expression analysis of CLE lesional skin and blood skewed toward lesional skin, though apoptosis signatures were noted in both environments. Apoptosis and necroptosis pathways via RIP3 are activated in interface dermatitis characteristic in CLE (76).
Downregulation of anti-apoptotic genes has also been identified in CLE. RANKL, a regulator of apoptosis, is notably absent from CLE skin (77). While TRAIL-R1 has been shown to be pro-apoptotic, TRAIL-R4 serves as a decoy receptor, blocking TRAIL-induced apoptosis, and has been shown to be downregulated in CLE relative to psoriasis and lichen planus (69, 78).
Complements, Lysosomes, and Proteosome Contribute to Impaired Clearance of Cell Debris in Cutaneous Lupus Erythematosus
The complement cascade plays a pivotal role in the opsonization of cells undergoing apoptosis to facilitate phagocytosis, and the timely clearance of cellular debris is important to prevent the generation of autoantibodies. Similarly, lysosomal and proteasomal clearance of cellular debris via proteolysis plays an important role and has been shown to be dysregulated in CLE. Skin gene expression of complement has been shown to be more dysregulated relative to blood gene expression in CLE (74, 79, 80). This is contrasted with the upregulation of cathepsins associated with lysosomes and proteasome-related genes in CLE peripheral blood relative to lesional skin (35, 74). The complex dysregulation of the systems involved in the clearance of cellular debris at a localized and systemic level highlights the complexity of the pathogenesis of CLE.
Fibrosis Is Likely Driven by TGF-β in Cutaneous Lupus Erythematosus
Of the CLE subtypes, DLE has been most associated with scarring lesions with associated fibrosis. Comparison of patients with DLE and SCLE using Ingenuity Pathway Analysis revealed pathways associated with fibrotic processes, and longitudinal microarray analysis of patients with DLE and SCLE revealed sustained elevations of TGF-B1, TGF-BR1, SMAD3, MMP1, MMP9, and SERPINE1 (42). Interestingly, while TGF-β, a M2 macrophage-related protein, was noted to be overexpressed in DLE skin relative to normal skin in an independent experiment, other M2 macrophage-related genes, such as CD206, CD209, FOLR2, IL10, and arginase-1, were not differentially expressed (65). Taken together, TGF-β likely plays a key role in fibrogenesis in scarring DLE lesions, though the exact downstream mechanisms have yet to be fully defined.
Conclusion
Our understanding of the underlying genetics governing the pathophysiology of CLE has greatly increased, thanks to advances in gene expression technology. Insights into the underlying genetic polymorphisms that predispose patients to CLE and knowledge of key dysregulated pathways in CLE afford the opportunity to develop targeted therapies for patients with CLE. Most recently, pathogenesis-directed therapy has focused on blockade of IFN receptors, such as anifrolumab (81). Other approaches, such as targeting the JAK/STAT pathway, are under investigation (82–84). The limitations of reviewed studies include small sample size, specific, non-generalizable cohorts, and technical limitations of gene expression profiling approaches, such as low resolution in microarray studies. Further studies using newer technologies, such as single-cell RNA sequencing, are warranted to define the genetic pathophysiology of CLE at greater resolution. Greater understanding of the underlying genetics of CLE can lead to further development of targeted therapies for CLE.
Author Contributions
HC and BC conceived and designed the study. HC and GB acquired, analyzed, interpreted the data, and drafted the original manuscript. All authors contributed to critical revision of the manuscript for important intellectual content, read, and approved the submitted version.
Conflict of Interest
BC was an investigator for Daavlin Corporation, Biogen Incorporated, and Pfizer Incorporated and consultant for Bristol Meyers Squibb, EMD Serono, Horizon Therapeutics, and Biogen Incorporated.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Sojung L. Chen for her invaluable assistance with figure generation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.916011/full#supplementary-material
Supplementary Figure 1 | Flow diagram of workflow for the literature review.
Abbreviations
BAFF, B-cell activating factor; CCLE, chronic cutaneous lupus erythematosus; circRNAs, circular RNAs; CLE, cutaneous lupus erythematosus; DLE, discoid lupus erythematosus; HLA, human leukocyte antigen; IFN, interferon; IL, interleukin; iNOS, inducible nitric oxide synthase; LET, lupus erythematosus tumidus; lncRNAs, long non-coding RNAs; NLE, neonatal lupus erythematosus SLE, systemic lupus erythematosus; SNP, single nucleotide polymorphism; SYK, spleen tyrosine kinase.
References
1. Zhu JL, Tran LT, Smith M, Zheng F, Cai L, James JA, et al. Modular Gene analysis reveals distinct molecular signatures for subsets of patients with cutaneous lupus erythematosus. Brit J Dermatol. (2021) 185:563–72. doi: 10.1111/bjd.19800
2. Zeidi M, Kim HJ, Werth VP. Increased myeloid dendritic cells and tnf-alpha expression predicts poor response to hydroxychloroquine in cutaneous lupus erythematosus. J Investig Dermatol. (2019) 139:324–32. doi: 10.1016/j.jid.2018.07.041
3. Fowler JF, Callen JP, Stelzer GT, Cotter PK. Human histocompatibility antigen associations in patients with chronic cutaneous lupus erythematosus. J Am Acad Dermatol. (1985) 12:73–7.
4. Bielsa I, Herrero C, Ercilla G, Collado A, Font J, Ingelmo M, et al. Immunogenetic findings in cutaneous lupus erythematosus. J Am Acad Dermatol. (1991) 25:251–7.
5. Günther C, Meurer M, Stein A, Viehweg A, Lee-Kirsch M-A. Familial chilblain lupus–a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in trex1. Dermatology. (2009) 219:162–6.
6. Millard T, Kondeatis E, Cox A, Wilson A, Grabczynska S, Carey B, et al. A candidate gene analysis of three related photosensitivity disorders: cutaneous lupus erythematosus, polymorphic light eruption and actinic prurigo. Br J Dermatol. (2001) 145:229–36.
7. Partanen J, Koskimies S, Johansson E. C4 null phenotypes among lupus erythematosus patients are predominantly the result of deletions covering C4 and closely linked 21-hydroxylase a genes. J Med Genet. (1988) 25:387–91.
8. Fischer GF, Pickl WF, Faé I, Anegg B, Milota S, Volc-Platzer B. Association between chronic cutaneous lupus erythematosus and Hla class II alleles. Hum Immunol. (1994) 41:280–4.
9. Miyagawa S, Shinohara K, Fujita T, Kidoguchi K-I, Fukumoto T, Hashimoto K, et al. Neonatal lupus erythematosus: analysis of Hla class II Alleles in mothers and siblings from seven Japanese families. J Am Acad Dermatol. (1997) 36:186–90.
10. Lopez-Tello A, Rodríguez-Carreón A, Jurado F, Yamamoto-Furusho J, Castillo-Vázquez M, Chávez-Muñoz C, et al. Association of Hla-Drb1* 16 with chronic discoid lupus erythematosus in Mexican mestizo patients. Clin Exp Dermatol Exp Dermatol. (2007) 32:435–8.
11. Racila DM, Sontheimer CJ, Sheffield A, Wisnieski JJ, Racila E, Sontheimer RD. Homozygous single nucleotide polymorphism of the complement C1qa gene is associated with decreased levels of C1q in patients with subacute cutaneous lupus erythematosus. Lupus. (2003) 12:124–32. doi: 10.1191/0961203303lu329oa
12. Jlajla H, Sellami MK, Sfar I, Laadhar L, Zerzeri Y, Abdelmoula MS, et al. New C1q mutation in a tunisian family. Immunobiology. (2014) 219:241–6. doi: 10.1016/j.imbio.2013.10.010
13. Callen JP, Hodge SJ, Kulick KB, Stelzer G, Buchino JJ. Subacute cutaneous lupus erythematosus in multiple members of a family with C2 deficiency. Arch Dermatol. (1987) 123:66–70.
14. Belin DC, Bordwell BJ, Einarson ME, McLean RH, Weinstein A, Yunis EJ, et al. Familial discoid lupus erythematosus associated with heterozygote C2 deficiency. Arthritis Rheum. (1980) 23:898–903. doi: 10.1002/art.1780230804
15. Levy SB, Pinnell SR, Meadows L, Snyderman R, Ward FE. Hereditary C2 deficiency associated with cutaneous lupus erythematosus: clinical, laboratory, and genetic studies. Arch Dermatol. (1979) 115:57–61.
16. Agnello V, Gell J, Tye MJ. Partial genetic deficiency of the C4 component of complement in discoid lupus erythematosus and Urticaria/Angioedema. J Am Acad Dermatol. (1983) 9:894–8. doi: 10.1016/s0190-9622(83)70205-7
17. Günther C, Hillebrand M, Brunk J, Lee-Kirsch MA. Systemic involvement in trex1-associated familial chilblain lupus. J Am Acad Dermatol. (2013) 69:e179–81.
18. Günther C, Berndt N, Wolf C, Lee-Kirsch MA. Familial chilblain lupus due to a novel mutation in the exonuclease III domain of 3’ repair exonuclease 1 (Trex1). JAMA Dermatol. (2015) 151:426–31.
19. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in Trex1 cause familial chilblain lupus and dominant aicardi-goutieres syndrome. Am J Hum Genet. (2007) 80:811–5.
20. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in samhd1 causing familial chilblain lupus. Am J Med Genet Part A. (2011) 155:235–7.
21. Linggonegoro D, Song H, Jones K, Lee P, Schmidt B, Vleugels R, et al. Familial chilblains lupus in a child with heterozygous mutation in samhd1 and normal interferon signature. Br J Dermatol. (2021) 185:650–2.
22. Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M, et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheumat. (2005) 52:3639–45.
23. Suárez A, López P, Mozo L, Gutiérrez C. Differential effect of IL10 and Tnfα genotypes on determining susceptibility to discoid and systemic lupus erythematosus. Ann Rheumat Dis. (2005) 64:1605–10.
24. Skonieczna K, Czajkowski R, Kaszewski S, Gawrych M, Jakubowska A, Grzybowski T. Genetic similarities and differences between discoid and systemic lupus erythematosus patients within the polish population. Adv Dermatol Allergol Post?py Dermatol Alergol. (2017) 34:228.
25. Järvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Panelius J, Hasan T, et al. Polymorphisms of the itgam gene confer higher risk of discoid cutaneous than of systemic lupus erythematosus. PLoS One. (2010) 5:e14212. doi: 10.1371/journal.pone.0014212
26. Kunz M, Konig IR, Schillert A, Kruppa J, Ziegler A, Grallert H, et al. Genome-wide association study identifies new susceptibility loci for cutaneous lupus erythematosus. Exp Dermatol. (2015) 24:510–5. doi: 10.1111/exd.12708
27. Skonieczna K, Woźniacka A, Czajkowski R, Styczyñski J, Krenska A, Robak E, et al. X-linked Tlr7 gene polymorphisms are associated with diverse immunological conditions but not with discoid lupus erythematosus in Polish patients. Adv Dermatol Allergol. (2018) 35:26.
28. Nemer G, El-Hachem N, Eid E, Hamie L, Bardawil T, Khalil S, et al. A novel Traf3ip2 variant causing familial scarring alopecia with mixed features of discoid lupus erythematosus and folliculitis decalvans. Clin Genet. (2020) 98:116–25.
29. Järvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Koskinen LL, Jeskanen L, et al. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp Dermatol. (2010) 19:123–31.
30. König N, Fiehn C, Wolf C, Schuster M, Costa EC, Tüngler V, et al. Familial chilblain lupus due to a gain-of-function mutation in sting. Ann Rheumat Dis. (2017) 76:468–72.
31. Weller R, Schwentker A, Billiar TR, Vodovotz Y. Autologous nitric oxide protects mouse and human keratinocytes from ultraviolet B radiation-induced apoptosis. Am J Physiol Cell Physiol. (2003) 284:C1140–8. doi: 10.1152/ajpcell.00462.2002
32. Suschek CV, Krischel V, Bruch-Gerharz D, Berendji D, Krutmann J, Kroncke KD, et al. Nitric oxide fully protects against Uva-induced apoptosis in tight correlation with Bcl-2 up-regulation. J Biol Chem. (1999) 274:6130–7. doi: 10.1074/jbc.274.10.6130
33. Kuhn A, Fehsel K, Lehmann P, Krutmann J, Ruzicka T, Kolb-Bachofen V. Aberrant timing in epidermal expression of inducible nitric oxide synthase after Uv irradiation in cutaneous lupus erythematosus. J Invest Dermatol. (1998) 1:149–53.
34. Meller S, Winterberg F, Gilliet M, Muller A, Lauceviciute I, Rieker J, et al. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment - an amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum US. (2005) 52:1504–16. doi: 10.1002/art.21034
35. Katayama S, Panelius J, Koskenmies S, Skoog T, Mahonen K, Kisand K, et al. Delineating the healthy human skin Uv response and early induction of interferon pathway in cutaneous lupus erythematosus. J Invest Dermatol. (2019) 139:2058–61e4. doi: 10.1016/j.jid.2019.02.035
36. Xiang MM, Chen Q, Feng Y, Wang YL, Wang J, Liang J, et al. Bioinformatic analysis of key biomarkers and immune filtration of skin biopsy in discoid lupus erythematosus. Lupus. (2021) 30:807–17. doi: 10.1177/0961203321992434
37. Toro JR, Finlay D, Dou X, Zheng SC, LeBoit PE, Connolly MK. Detection of type 1 cytokines in discoid lupus erythematosus. Arch Dermatol. (2000) 136:1497–501. doi: 10.1001/archderm.136.12.1497
38. Stein LF, Saed GM, Fivenson DP. T-cell cytokine network in cutaneous lupus erythematosus. J Am Acad Dermatol. (1997) 36:191–6. doi: 10.1016/s0190-9622(97)70279-2
39. Denfeld RW, Kind P, Sontheimer RD, Schopf E, Simon JC. In Situ expression of B7 and Cd28 receptor families in skin lesions of patients with lupus erythematosus. Arthritis Rheum US. (1997) 40:814–21. doi: 10.1002/art.1780400507
40. Jabbari A, Suarez-Farinas M, Fuentes-Duculan J, Gonzalez J, Cueto I, Franks AG Jr., et al. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J Invest Dermatol. (2014) 134:87–95. doi: 10.1038/jid.2013.269
41. Nickles MA, Huang K, Chang YS, Tsoukas MM, Sweiss NJ, Perkins DL, et al. Gene co-expression networks identifies common hub genes between cutaneous sarcoidosis and discoid lupus erythematosus. Front Med. (2020) 7:606461. doi: 10.3389/fmed.2020.606461
42. Sole C, Gimenez-Barcons M, Ferrer B, Ordi-Ros J, Cortes-Hernandez J. Microarray study reveals a transforming growth factor-beta-dependent mechanism of fibrosis in discoid lupus erythematosus. Br J Dermatol. (2016) 175:302–13. doi: 10.1111/bjd.14539
43. Tsoi LC, Gharaee-Kermani M, Berthier CC, Nault T, Hile GA, Estadt SN, et al. Il18-containing 5-gene signature distinguishes histologically identical dermatomyositis and lupus erythematosus skin lesions. JCI Insight. (2020) 5:20. doi: 10.1172/jci.insight.139558
44. Dong Q, Chen K, Xie J, Han H, Feng Y, Lu J, et al. Identification of key genes and pathways in discoid lupus skin via bioinformatics analysis. Medicine. (2021) 100:e25433. doi: 10.1097/MD.0000000000025433
45. Scholtissek B, Zahn S, Maier J, Klaeschen S, Braegelmann C, Hoelzel M, et al. Immunostimulatory endogenous nucleic acids drive the lesional inflammation in cutaneous lupus erythematosus. J Invest Dermatol. (2017) 137:1484–92. doi: 10.1016/j.jid.2017.03.018
46. Merola JF, Wang W, Wager CG, Hamann S, Zhang X, Thai A, et al. Rna tape sampling in cutaneous lupus erythematosus discriminates affected from unaffected and healthy volunteer skin. Lupus Sci Med. (2021) 8:428. doi: 10.1136/lupus-2020-000428
47. Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective janus kinase 1 inhibition is a promising therapeutic approach for lupus erythematosus skin lesions. Front. (2020) 11:344. doi: 10.3389/fimmu.2020.00344
48. Calugareanu A, Grolleau C, Le Buanec H, Chasset F, Jachiet M, Battistella M, et al. clinical efficacy of selective Jak1 inhibition and transcriptome analysis of chronic discoid lupus erythematosus. J Eur Acad Dermatol Venereol. (2022) 36:e308–10. doi: 10.1111/jdv.17839
49. Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. (2006) 20:9–22. doi: 10.1096/fj.05-4682rev
50. Chang LM, Maheshwari P, Werth S, Schaffer L, Head SR, Kovarik C, et al. Identification and molecular analysis of glycosaminoglycans in cutaneous lupus erythematosus and dermatomyositis. J Histochem Cytochem. (2011) 59:336–45. doi: 10.1369/0022155410398000
51. Yang J, Li Q, Yang X, Li M. Interleukin-9 is associated with elevated anti-double-stranded DNA antibodies in lupus-prone mice. Mol Med. (2015) 21:364–70. doi: 10.2119/molmed.2014.00237
52. Xie Y, Liu B, Wu Z. Increased interleukin-9 levels in skin lesions from cutaneous lupus erythematosus patients may predict the progression to systemic lupus erythematosus. J Dermatol Sci. (2021) 101:78–80. doi: 10.1016/j.jdermsci.2020.10.016
53. Chong BF, Tseng LC, Kim A, Miller RT, Yancey KB, Hosler GA. Differential expression of baff and its receptors in discoid lupus erythematosus patients. J Dermatol Sci. (2014) 73:216–24. doi: 10.1016/j.jdermsci.2013.11.007
54. Mocsai A, Ruland J, Tybulewicz VL. The syk tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. (2010) 10:387–402. doi: 10.1038/nri2765
55. Braegelmann C, Holzel M, Ludbrook V, Dickson M, Turan N, Ferring-Schmitt S, et al. Spleen tyrosine kinase (Syk) is a potential target for the treatment of cutaneous lupus erythematosus patients. Exp Dermatol. (2016) 25:375–9. doi: 10.1111/exd.12986
56. Vorwerk G, Zahn S, Bieber T, Wenzel J. Nkg2d and its ligands as cytotoxic factors in cutaneous lupus erythematosus. Exp Dermatol. (2021) 30:847–52. doi: 10.1111/exd.14311
57. Guerra N, Pestal K, Juarez T, Beck J, Tkach K, Wang L, et al. A selective role of Nkg2d in inflammatory and autoimmune diseases. Clin Immunol. (2013) 149:432–9. doi: 10.1016/j.clim.2013.09.003
58. Tsoi LC, Hile GA, Berthier CC, Sarkar MK, Reed TJ, Liu J, et al. Hypersensitive Ifn responses in lupus keratinocytes reveal key mechanistic determinants in cutaneous lupus. J Immunol. (2019) 202:2121–30. doi: 10.4049/jimmunol.1800650
59. Xuan J, Xiong YY, Shi LJ, Aramini B, Wang HY. Do lncrnas and circrnas expression profiles influence discoid lupus erythematosus progression?-a comprehensive analysis. Ann Translat Med. (2019) 7:10. doi: 10.21037/atm.2019.12.10
60. Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, Ronnblom L. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. (2001) 10:484–90. doi: 10.1191/096120301678416042
61. Wongpiyabovorn J, Ruchusatsawat K, Onganantapong Y, Sintupak W, Hirankarn N. Interferon alpha Mrna level and subtypes in lesion and non-lesion from discoid lupus erythematosus patients without systemic lupus erythematosus. Asian Biomed. (2011) 5:643–7. doi: 10.5372/1905-7415.0505.085
62. Sirobhushanam S, Parsa N, Reed TJ, Berthier CC, Sarkar MK, Hile GA, et al. Staphylococcus aureus colonization is increased on lupus skin lesions and is promoted by Ifn-mediated barrier disruption. J Investig Dermatol. (2020) 140:1066.e–74.e. doi: 10.1016/j.jid.2019.11.016
63. Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing XY, et al. Lupus skin is primed for IL-6 inflammatory responses through a keratinocyte-mediated autocrine type I interferon loop. J Investig Dermatol. (2017) 137:115–22. doi: 10.1016/j.jid.2016.09.008
64. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I Ifn responses in cutaneous lupus are driven by epidermal-derived interferon Kappa. Ann Rheum Dis. (2018) 77:1653–64. doi: 10.1136/annrheumdis-2018-213197
65. Chong BF, Tseng LC, Hosler GA, Teske NM, Zhang S, Karp DR, et al. A subset of Cd163+ macrophages displays mixed polarizations in discoid lupus skin. Arthritis Res Ther. (2015) 17:324. doi: 10.1186/s13075-015-0839-3
66. Flier J, Boorsma DM, van Beek PJ, Nieboer C, Stoof TJ, Willemze R, et al. Differential expression of Cxcr3 targeting chemokines Cxcl10, Cxcl9, and Cxcl11 in different types of skin inflammation. J Pathol. (2001) 194:398–405. doi: 10.1002/1096-9896(200108)194:43.0.co;2-s
67. Gambichler T, Genc Z, Skrygan M, Scola N, Tigges C, Terras S, et al. Cytokine and chemokine ligand expression in cutaneous lupus erythematosus. Eur J Dermatol. (2012) 22:319–23. doi: 10.1684/ejd.2012.1725
68. Gao ZY, Su LC, Wu QC, Sheng JE, Wang YL, Dai YF, et al. Bioinformatics analyses of gene expression profile identify key genes and functional pathways involved in cutaneous lupus erythematosus. Clin Rheumatol. (2022) 41:437–52. doi: 10.1007/s10067-021-05913-2
69. Toberer F, Sykora J, Gottel D, Hartschuh W, Werchau S, Enk A, et al. Apoptotic signal molecules in skin biopsies of cutaneous lupus erythematosus: analysis using tissue microarray. Exp Dermatol. (2013) 22:656–9. doi: 10.1111/exd.12216
70. Zahn S, Rehkamper C, Ferring-Schmitt S, Bieber T, Tuting T, Wenzel J. Interferon-alpha stimulates trail expression in human keratinocytes and peripheral blood mononuclear cells: implications for the pathogenesis of cutaneous lupus erythematosus. Br J Dermatol. (2011) 165:1118–23. doi: 10.1111/j.1365-2133.2011.10479.x
71. Kingsmore KM, Bachali P, Catalina MD, Daamen AR, Heuer SE, Robl RD, et al. Altered expression of genes controlling metabolism characterizes the tissue response to immune injury in lupus. Sci. (2021) 11:14789. doi: 10.1038/s41598-021-93034-w
72. Ko WC, Li L, Young TR, McLean-Mandell RE, Deng AC, Vanguri VK, et al. Gene expression profiling in the skin reveals strong similarities between subacute and chronic cutaneous lupus that are distinct from lupus nephritis. J Invest Dermatol. (2021) 141:2808–19. doi: 10.1016/j.jid.2021.04.030
73. Dey-Rao R, Sinha AA. Genome-wide transcriptional profiling of chronic cutaneous lupus erythematosus (Ccle) peripheral blood identifies systemic alterations relevant to the skin manifestation. Genomics. (2015) 105:90–100. doi: 10.1016/j.ygeno.2014.11.004
74. Dey-Rao R, Sinha AA. In silico analyses of skin and peripheral blood transcriptional data in cutaneous lupus reveals Ccr2-a novel potential therapeutic target. Front Immunol. (2019) 10:640. doi: 10.3389/fimmu.2019.00640
75. Dey-Rao R, Smith JR, Chow S, Sinha AA. Differential gene expression analysis in Ccle lesions provides new insights regarding the genetics basis of skin Vs. systemic disease. Genomics. (2014) 104:144–55. doi: 10.1016/j.ygeno.2014.06.003
76. Lauffer F, Jargosch M, Krause L, Garzorz-Stark N, Franz R, Roenneberg S, et al. Type I immune response induces keratinocyte necroptosis and is associated with interface dermatitis. J Invest Dermatol. (2018) 138:1785–94. doi: 10.1016/j.jid.2018.02.034
77. Toberer F, Sykora J, Gottel D, Ruland V, Hartschuh W, Enk A, et al. Tissue microarray analysis of rankl in cutaneous lupus erythematosus and psoriasis. Exp Dermatol. (2011) 20:600–2. doi: 10.1111/j.1600-0625.2011.01303.x
78. Nguyen V, Cudrici C, Zernetkina V, Niculescu F, Rus H, Drachenberg C, et al. Trail, Dr4 and Dr5 are upregulated in kidneys from patients with lupus nephritis and exert proliferative and proinflammatory effects. Clin Immunol. (2009) 132:32–42. doi: 10.1016/j.clim.2009.02.011
79. Alase A, Shalbaf M, Berekmeri A, Yusof MYM, Botchkareva N, Goodfield M, et al. Analysis of noninvasively collected hair follicles may be sufficient for the diagnosis of chronic discoid lupus erythematosus of the scalp. Br J Dermatol. (2019) 180:E205–6.
80. Shalbaf M, Alase AA, Berekmeri A, Md Yusof MY, Pistolic J, Goodfield MJ, et al. Plucked hair follicles from patients with chronic discoid lupus erythematosus show a disease-specific molecular signature. Lupus Sci Med. (2019) 6:e000328. doi: 10.1136/lupus-2019-000328
81. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
82. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
83. Werth VP, Fleischmann R, Robern M, Touma Z, Tiamiyu I, Gurtovaya O, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomised, double-blind, placebo-controlled study. Rheumatology. (2021) 2021:keab685. doi: 10.1093/rheumatology/keab685
Keywords: cutaneous lupus erythematosus, DNA, RNA, genetic polymorphism, microarray, inflammation, apoptosis, fibrosis
Citation: Chen HW, Barber G and Chong BF (2022) The Genetic Landscape of Cutaneous Lupus Erythematosus. Front. Med. 9:916011. doi: 10.3389/fmed.2022.916011
Received: 08 April 2022; Accepted: 04 May 2022;
Published: 02 June 2022.
Edited by:
Filippa Nyberg, Karolinska University Hospital, SwedenReviewed by:
Kumutnart Chanprapaph, Mahidol University, ThailandCopyright © 2022 Chen, Barber and Chong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin F. Chong, QmVuLkNob25nQFVUU291dGh3ZXN0ZXJuLmVkdQ==