 Yina Wang1
Yina Wang1 Yu Yan
Yu Yan Li Zuo
Li Zuo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 25 August 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.911998
This article is part of the Research TopicIntegrated Management of Chronic Kidney Disease PatientsView all 24 articles
Combination of monoclonal immunoglobulin deposition disease (MIDD) and immunotactoid glomerulopathy (ITG) is a rare form of monoclonal immunoglobulin (MIg)-associated renal disease. We retrospectively reviewed the native kidney biopsy specimens at Peking University People’s Hospital from 2011 to 2020. Five patients were diagnosed as MIDD + ITG. Their clinical and pathological characteristics were studied. The typical clinical features were nephritic syndrome and renal dysfunction with prominent anemia, but hematuria was mild. Unlike single MIDD and single ITG, on light microscopy, segmentally distributed mesangial nodular sclerosis on the basis of mesangial matrix hyperplasia was the major lesion. Others including membranoproliferative glomerulonephritis (MPGN)-like lesion, glomerular basement membrane thickness, and mild to moderate mesangial and endothelial proliferations might presented at the same time and in the same glomeruli. On immunofluorescence, MIg, usually monoclonal light chains, deposited along glomerular basement membranes and tubular basement membranes, while the intact MIg or monoclonal heavy chain deposited in the mesangial regions. Corresponding to the depositions on immunofluorescence, punctate “powdery” deposits along glomerular basement membranes and tubular basement membranes under electronic microscopy indicated the presence of MIDD. Microtubular substructures (diameters of 20–50 nm) exhibiting hollow cores arranged in parallel arrays in mesangial regions indicated the presence of ITG. Patients treated with bortezomib-based regimen seemed to have better outcomes. In conclusion, MIDD + ITG is a rare combination form of MIg-associated renal disease. Accurate diagnosis requires the comprehensive pathological investigations.
Monoclonal immunoglobulin (MIg)-associated renal disease has heterogeneous morphologic forms (1). It usually presented in one form (2). Some of literatures reported combinations with two or more different pathologic forms (3–5). The most common pathologic form is monoclonal immunoglobulin deposition disease (MIDD) coexisting with light chain cast nephropathy (LCCN) (4, 6–9). However, the combination of two forms of glomerular diseases, especially the co-deposition of organized and non-organized structures was rare (3, 10). The presentation of this combination was not simply the add-on, but had unique characteristics. Up to date, no case of MIDD + ITG was reported. Here, we report the clinicopathological features, treatments and outcomes of 5 patients with MIDD + ITG, and help to understand the characteristics of this pattern.
All 11,767 native kidney biopsy specimens from 2011 to 2020 at Peking University People’s Hospital were reviewed from patients’ medical records. Patients fulfilled the diagnostic criteria of both MIDD and immunotactoid glomerulopathy (ITG) were enrolled in the study. MIDD and ITG were diagnosed according to previous literatures (2, 11, 12). Briefly, the patients were diagnosed as MIDD and ITG when there were typical depositions on EM (“powdery” deposits along basement membrane for MIDD and microtubular substructure exhibiting hollow cores arranged in parallel arrays for ITG) with IF proved MIg deposition.
Demographic and clinical information including age, gender, clinical symptoms, past histories of hypertension and diabetes, blood pressure, hemoglobin, urinalysis, urine protein output, serum albumin, serum creatinine were collected at the time of biopsy. MIg was detected by serum and/or urine immunofixation electrophoresis (IEF) and free light chains (FLCs) (Freelite, Binding Site, United Kingdom) test. Treatment and follow-up data were also obtained from the patients’ medical records.
All kidney biopsy samples were processed for light microscopy (LM), immunofluorescence (IF) and electronic microscopy (EM) examination using standard techniques. IF was performed on cryosections (5 μm) using polyclonal fluorescein isothiocyanate (FITC)-conjugated antibodies against IgG, IgM, IgA, C3, C1q, κ and λ light chains (Dako, Denmark), respectively. Determination of the IgG subclasses was performed using monoclonal FITC-conjugated antibodies to IgG1, IgG2, IgG3, and IgG4 (SouthernBiotech, United States). For LM, kidney biopsy specimens were stained with hematoxylin and eosin, periodic acid-Schiff (PAS), Masson’s trichrome, periodic acid-silver methenamine, respectively. Also, Congo red and immunohistochemical (IHC) staining (CD38, CD 138, CD3, and CD20) were performed. Ultrastructural evaluation was performed using a transmission electron microscope (Thermo Scientific, TECNAI SPIRIT, United States).
This research was carried out in accordance with International ethical guidelines for biomedical research involving human subjects (CIOMS) and Helsinki Declaration. This research was approved by the Ethics Committee of Peking University People’s Hospital (2121PHB-84-001). Informed consent was obtained from all participants.
Continuous variables with normal distribution were expressed as mean ± SD and variables with non-normal distribution were expressed as median (Q25, Q75). Categorical variables were expressed as numbers or percentages.
The incidence of MIDD + ITG was quite low, accounting for only 0.04% of biopsied patients in our center. Five patients were identified. The demographic characteristics are shown in Table 1. There were three males (60%) and two females with an average age of 61.0 ± 4.6 years at kidney biopsy.
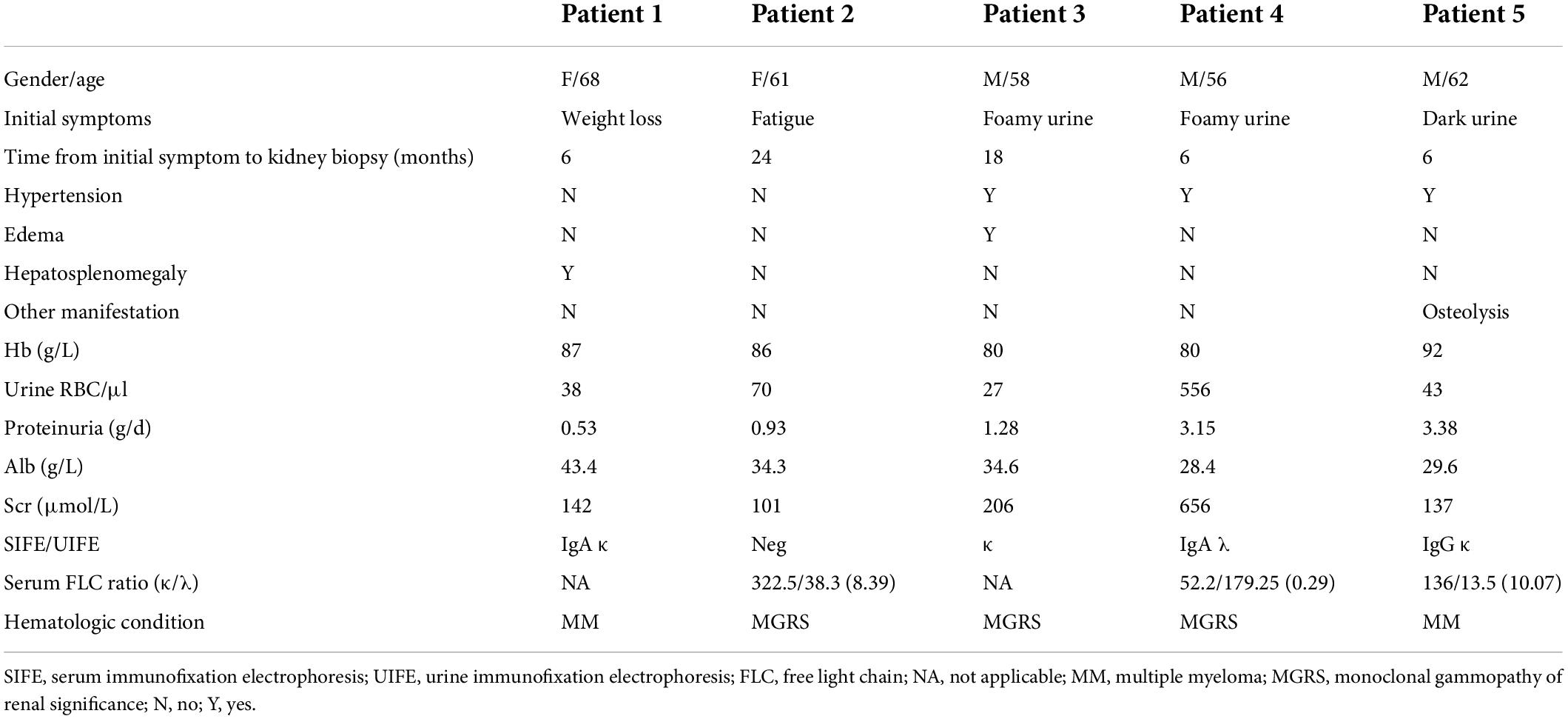
Table 1. Demographics and clinical characteristics.
The clinical characteristics of the five patients are shown in Table 1. Foamy urine, dark urine, fatigue and weight loss were the main initial symptoms. The median duration from onset to diagnosis was 6 (6, 21) months (range 6–24 months). Microscopic hematuria was seen in all patients. Proteinuria level was 1.85 ± 1.32 g/d. Serum albumin was 34.1 ± 5.9 g/L. Four patients suffered renal insufficiency as chronic kidney disease (CKD) in three and acute kidney injury (AKI) in one. Patient 4 was dialysis-dependent before renal biopsy. Only one patient (patient 2) had normal renal function. The median serum creatinine level was 142 (range 101–656) μmol/L. Three patients (60%) had hypertension, and none had diabetes.
As shown in Table 1, two patients fulfilled the established diagnostic criteria for MM and three were monoclonal gammopathy of renal significance (MGRS). All patients had prominent anemia with hemoglobin level of 85.0 ± 5.1 g/L. Serum and/or urine IEF confirmed the presence of MIg in four patients except patient 2. She was proved to have MIg for the high κ/λ ratio of 8.39, despite the negative results of serum and urine IEF. The complement levels (C3 and C4) were normal in all the five patients. The results of autoantibodies, hepatitis B and C, and serum cryoglobulin were all negative.
The pathologic findings are shown in Table 2 and Figure 1. Mesangial nodular sclerosis was the major lesion on LM usually distributing segmentally, formed on the basis of mesangial matrix hyperplasia lesions. There were also mesangial and segmental endocapillary proliferation. Some manifested membranoproliferative glomerulonephritis (MPGN)-like lesions due to mesangial interposition resulted in double-contour or multicontour. However, the lesion only distributed focally and segmentally and the GBMs were thickened mainly at the site of double-contour or multicontour. The GBMs in the non-sclerotic area were not thickened. Congo red staining were negative.
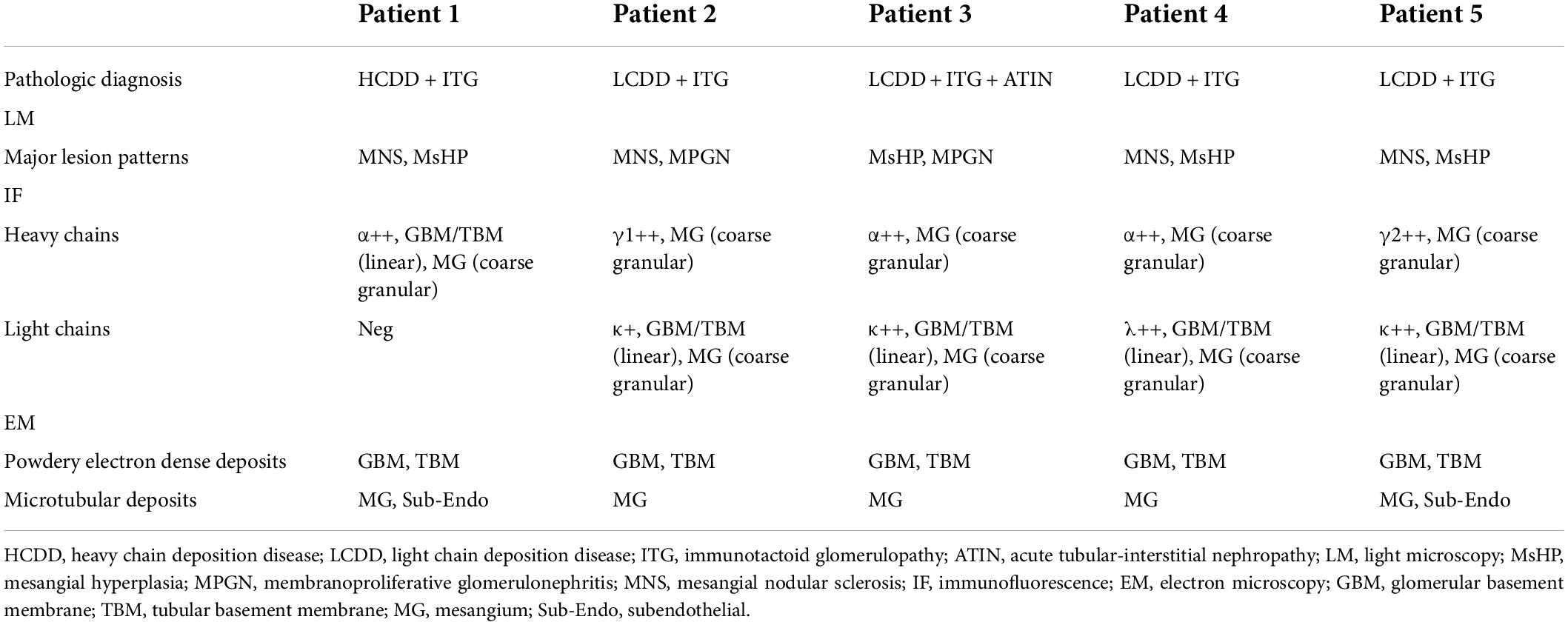
Table 2. Renal pathological findings.
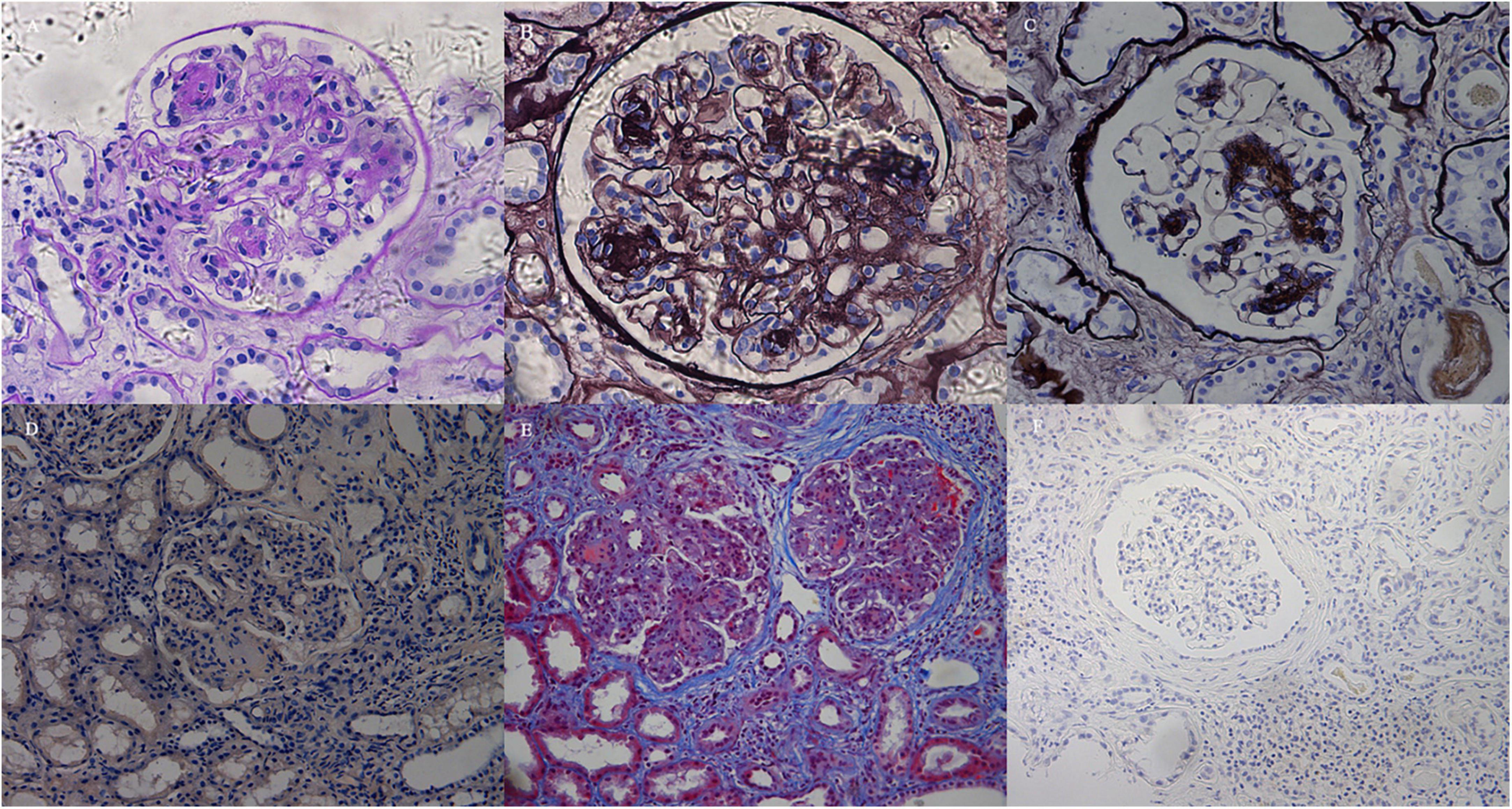
Figure 1. Light microscopy findings. (A) The glomerulus exhibited mesangial expansion and extensive proliferation of mesangial cells and matrix, with mesangial nodular sclerosis. The nodules and tubular basement membranes were periodic acid–Schiff (PAS) positive (Patient 3, PAS; × 200). (B) Diffuse membranoproliferative-like features revealed GBM duplication and mesangial interposition with double-contour or multicontour appearances (Patient 3, PASM; × 400). (C) The glomerulus exhibited segmental nodular sclerosis with mild mesangial hypercellularity. The GBMs in non-sclerotic areas were not thickened (Patient 4, PASM; × 200). (D) Congo red staining was negative (Patient 2, Congo red staining; × 100). (E) The mesangium expanded with diffuse hypercellularity and mesangial matrix proliferation. There was tubular atrophy and multifocal infiltration of lymphocytes and monocytes in the interstitium with fibrosis (Patient 2, Masson; × 100). (F) Lymphocytes and monocytes infiltrating the interstitium were CD38-negative by immunohistochemical staining (Patient 3, CD38; × 100).
Except for the glomerular lesions, there were various degrees of tubulointerstitial injury, with tubular atrophy and multifocal inflammatory infiltration. Lymphocytes (CD3+ or CD20+) and monocytes were seen without eosinophils according to the HE staining. In IHC staining, CD38+ or CD138+ cells were rarely seen indicating no plasma cells infiltration (shown in Figure 1). No PAS-negative casts were found in the tubules.
On IF, immune depositions with monoclonal light chain and/or heavy chain were consistent with the type of serum/urine MIg (Table 2). Monoclonal heavy chain without light chain was found in one patient, while the deposits of monoclonal light chain + heavy chain were found in other four patients. κ chain was the major type of light chain, while α and γ chains were the major types of heavy chains. In all the patients, light chains or heavy chain α deposited in mesangial region, along the GBMs and segmentally along the TBMs. Heavy chains were found mainly in mesangial region without deposition along GBMs and TBMs in four patients besides patient 1 (shown in Figure 2).
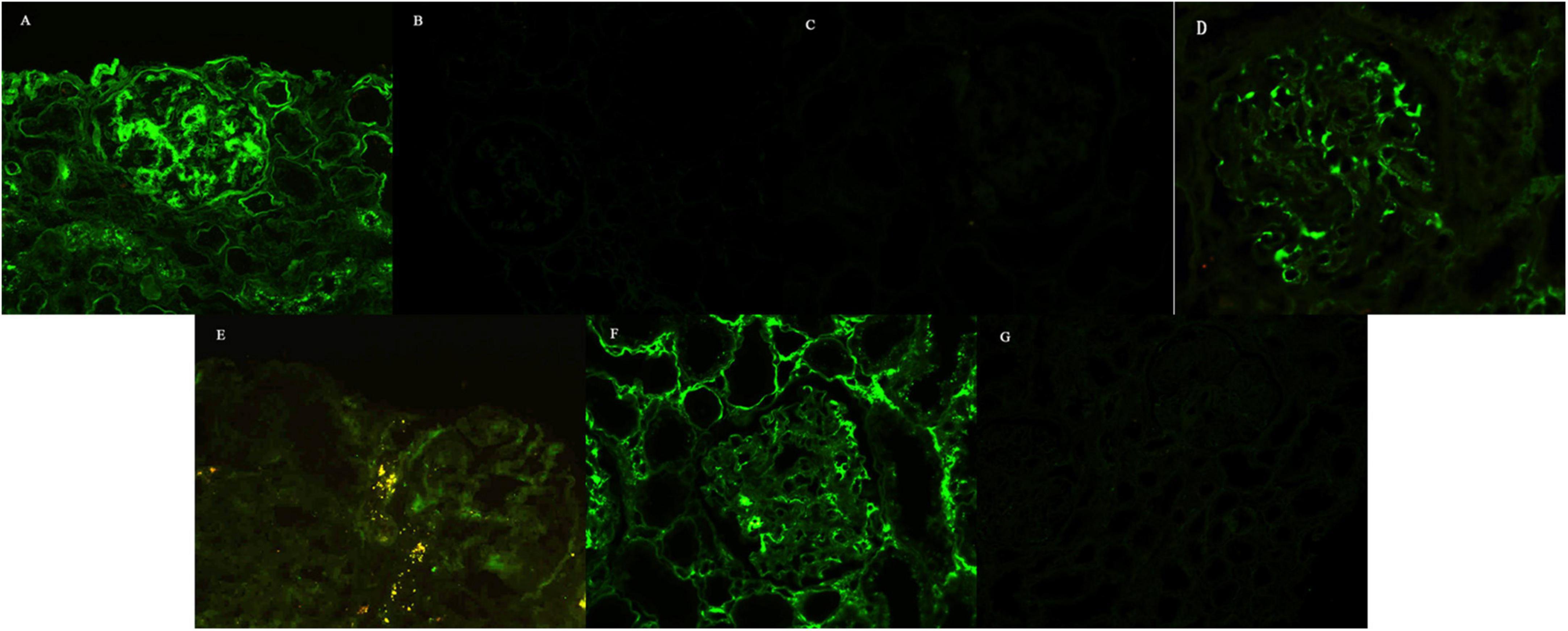
Figure 2. Immunofluorescence findings. (A) IgA linearly deposited along the GBMs and segmentally along the TBMs, while it was coarsely granularly deposited in the mesangial regions, with 3 + intensity (Patient 1, ×100). Staining for κ-chain (B) and λ-chain (C) was negative (Patient 1, ×100). Staining for IgG (D) and IgG2 (E) revealed coarse granular deposition only in the mesangial regions, without deposition along GBMs and TBMs (Patient 5, × 200). Restrictive κ-chain (F) was linearly deposited along the GBMs, segmentally deposited along the TBMs, and granularly deposited in the mesangial regions, while staining for λ-chain (G) was negative (Patient 5, × 100).
Punctate “powdery” electron-dense deposits were seen in mesangial regions and along the GBMs and/or TBMs on EM. Concurrently, coarse granular deposits were found in the mesangial regions with a microtubular substructure (diameters of 20–50 nm) exhibiting hollow cores arranged in parallel arrays (shown in Table 2 and Figures 2, 3).
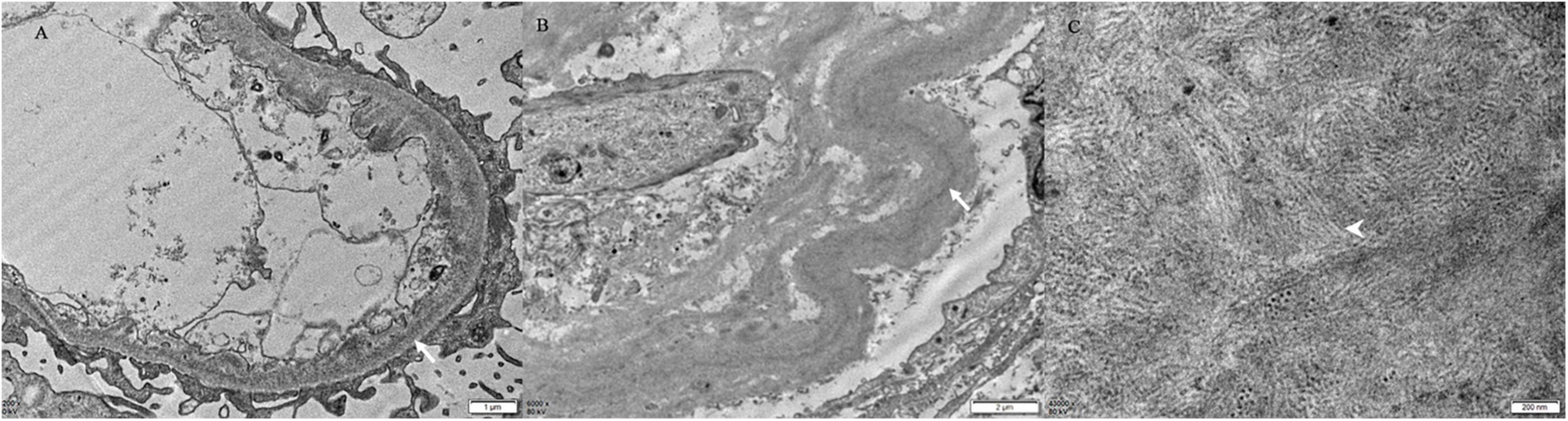
Figure 3. Electron microscopy findings. There are finely punctate “powdery” electron-dense deposits (white arrow) involving the inner aspect of the GBM (A) and the outer aspect of the TBM [white arrow, (B)] [Patient 4, (A) × 200, (B) × 6,000]. (C) The mesangial deposits comprised microtubular substructures (diameters of 20–50 nm) with hollow cores (arrowhead) arranged at least focally in parallel arrays (Patient 4 × 43,000).
Follow-up data were available for four patients (except patient 1) with a median duration of 31 (20, 54) months (range 18–60 months). Patients 2 and 5 received bortezomib-based therapy with or without autologous stem cell transplantation. They achieved complete remission of hematological and renal symptoms with improved renal function. Patient 3 was treated with steroids combined with cyclophosphamide and lenalidomide. Four months later, he advanced to end stage renal failure albeit hematological complete remission was achieved. After 60 months of follow-up, he remained dialysis-dependent. Patient 4 refused any chemotherapy and was treated with hemodialysis. After 18 months of follow-up, he also remained dialysis-dependent.
Although the incidence of single MIDD or ITG patients was relatively low (6, 13, 14), here we described a rarer series of patients with MIDD + ITG. Their characteristic pathological features had great diagnostic value, despite the non-specific clinical presentations. Segmental mesangial nodular sclerosis on the basis of mesangial matrix hyperplasia was the main lesion. MPGN-like lesion, GBM thickness and mild to moderate mesangial and endothelial proliferations presented at the same time and in the same glomeruli. EM manifestations of “powdery” deposits along GBM and/or TBM, and microtubular substructures with hollow cores arranged in parallel arrays in mesangial regions indicated the presence of both MIDD and ITG. At the same time, IF proved MIg depositions.
The patients in this study were all in the middle aged or older. It was similar to that previously reported in single MIDD or ITG patients (6, 13, 14). However, due to the small sample size in this study, we could not determine whether there was a gender difference in MIDD + ITG. The duration from onset to diagnosis was relatively long due to atypical early symptoms and late renal biopsy. Nephritic syndrome with chronic renal dysfunction was the prominent presentation of MIDD + ITG, in accordance with most MIg-associated renal diseases. The remarkable anemia incompatible with the patients’ renal function indicated the possible existence of hematologic diseases. The results of serum/urine IEF and/or FLCs implied the possible diagnosis of MIg associated disease. Our study was consistent with the reports in the literature that not all the patients have positive M-spike on serum/urine IEF, and serum FLCs assay can make up for this deficiency (6, 12, 15, 16).
The prognosis of these patients was heterogeneous. Similar with previous studies (17, 18), patients treated with bortezomib-based regimen seemed to have better outcomes. Further study containing more patients with longer follow up is needed to confirm this hypothesis.
As the non-specificity of the clinical manifestations, the accurate diagnosis relied on renal pathology. The manifestations on LM displayed the heterogeneity of lesions including mesangial nodular sclerosis forming on the basis of mesangial matrix hyperplasia, MPGN-like lesions, GBM thickness and etc. This was a unique feature of MIDD + ITG. Although mesangial nodular sclerosis could be seen in both single MIDD and single ITG, there were prominent differences compared with that in MIDD + ITG (6, 11, 19). Mesangial nodular sclerosis distributed focally and segmentally in MIDD + ITG in contrast to the diffusely and globally distribution in single MIDD. This was consistent with the literature reports that the combination of LCDD with other forms of disease had lower presence of nodular sclerosis and was only diagnosed through the powdery deposits on EM (20). Considering ITG, although mesangial nodular sclerosis might present in the late stage, mesangial and endothelial proliferation leading to MPGN-like lesions were the typical pathological features (12, 14, 21–23). However, in MIDD + ITG, less proliferation and only segmental MPGN-like lesions were seen. At the same time, the partial thickened GBMs resulted from mesangial interposition with a double-contour or multicontour appearance was more comparable with the feature of ITG, rather than that of MIDD mainly presenting as diffuse thickness of GBMs. So, we speculated that ITG might play more roles in the formation of glomerular lesions in our patients. At the same time, due to the non-homogeneous distribution of mesangial nodular sclerosis lesions and negative result of Congo red staining, amyloidosis could be excluded.
On EM, there were characteristic features to differentiate the components of MIDD + ITG. The punctate “powdery” deposits along the GBMs and/or TBMs was the feature of MIDD, while the microtubular deposits with hollow cores in mesangial region indicated the presence of ITG (6, 15, 23). Although cryoglobulinemic glomerulonephritis (Cryo GN) also produce microtubular structures, there were several differences to ITG. In ITG, the microtubules were relatively thicker than Cryo GN. On cross-section, microtubules of Cryo GN have 8–12 spokes emanating from the perimeter, making the cross-sectional outer diameter about 33 nm (24). No EM features relating to Cryo GN with the negative results of serum cryoglobulin have excluded the diagnosis. Thus, the coexistence of both “powdery” deposits along the GBMs and/or TBMs and microtubular deposits with hollow cores in mesangial region confirmed the diagnosis when there was evidence of MIg deposition in the kidney.
On IF, κ was the major light chain in patients with MIDD + ITG, consisting with the immune type of single MIDD and single ITG (2, 6, 13–15). γ and α heavy chains were both commonly seen, while γ1 and γ2 were the main γ subclasses. This was in accordance with the immune deposition types of ITG in the literature (13). The features of MIg deposition indicated the existence of different forms of the disease. In this cohort, heavy chains mainly deposited in mesangial regions, and light chains deposited along GBMs and TBMs, as well as in mesangial regions. This suggested that intact MIg might be the component of ITG deposits while restricted light chains were the component of MIDD. Thus, the four patients except patient 1 were diagnosed as LCDD + ITG. For patient 1, α heavy chain participated in both MIDD and ITG deposits indicating the diagnosis of HCDD + ITG.
Various degrees of tubular interstitial lesions incompatible with glomerular lesions were also visible in four patients except patient 2 in this study. These might explain the renal dysfunction presented in the patients. Pathologically, according to IHC results, the infiltrated inflammatory cells were mainly CD3+ T cells and CD20+ B cells. Together with manifestations of routine staining, there was no evidence of allergic nephritis, LCCN, myeloma infiltration, or any tubulointerstitial damage caused by MM or MGRS. Considering the significant immune deposition along TBMs, renal dysfunction presented either chronically or acutely in patients with MIDD + ITG, may be related to the involvement of renal tubules and interstitium of MIDD. Taken together, careful and thorough investigations with the combined application of IF, EM, LM and IHC could provide an accurate diagnosis of MIDD + ITG.
We reported a rare MIg-associated renal disease in which MIg deposited in both organized and non-organized ultrastructures. According to the pathogenesis reported (10), MIDD and ITG might resulted from immunoglobulins (MIgs) of different origins with unusual or abnormal structures. The deposition may be the consequence of the acquired defects in podocyte functions relating the clearance of the filtrated and retained immunoglobulin, which created the unique environment for deposition (19, 25). According to the immune phenotype, we hypothesize that in MIDD + ITG, intact MIgs with abnormal structures might result in organized deposits. At the same time, light chains or heavy chains were more likely to deposit on GBM, due to the abnormal physicochemical properties of MIg. We presumed that this might be one of the reasons to cause the acquired defects in podocyte functions. These defects promoted the retaining and deposition of organized intact MIgs with abnormal structures, leading to the formation of MIDD + ITG.
At the same time, whether the organized and non-organized deposits resulted from the same MIg of different conformations remained to be elucidated. In the series of LCDD + AL reported by Said, through the method of laser microdissection-assisted liquid chromatography-tandem mass spectrometry (LC-MS/MS), the combination was thought to be caused by pathological light chains produced by subclones stemming from one immunoglobulin light chain rearrangement, with a distinct mutated complementary determining region. Despite the lack of LC-MS/MS exam, we speculated that MIDD + ITG in this study was also caused by the same MIg for the following reasons. Firstly, MIDD and ITG deposits on IF had the same light chain or heavy chain isotype. Secondly, none of the patients had two different MIgs in the serum or urine. However, the accurate evidence needs to be obtained from LC-MS/MS.
In this study, the extraordinary combination of MIDD + ITG were described. The major limitation of the current study was that because of the limited number of cases, it was difficult to accurately summarize the prognostic factors for the outcomes. More cases are needed to further confirm our findings and speculations.
MIDD + ITG is a rare form of MIg-associated renal disease. Clinically it mainly presents as nephritic syndrome and renal dysfunction with prominent anemia. Serum/urine IEF or serum FLCs may prove the existence of MIg. The accurate diagnosis relies on the comprehensive pathologic investigations. Larger cohort would help to determine the best choice of treatment of MIDD + ITG. The accurate evidence of whether the coexisted organized and non-organized deposits came from the same MIg, needs to be obtained from LC-MS/MS.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of Peking University People’s Hospital. The patients/participants provided their written informed consent to participate in this study.
YW, YY, and BD concepted and designed the study. WZ, XL, CS, and LJ analyzed and interpreted the data. YW and YY drafted the article and revised the article. MW and LZ provided intellectual content of critical importance to the work described. YY final approved the version to be published. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China (81000296) and the Specialized Research Fund for the Doctoral Program of Higher Education (20090001120098).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Motwani SS, Herlitz L, Monga D, Jhaveri KD, Lam AQ. Paraprotein-related kidney disease: glomerular diseases associated with paraproteinemias. Clin J Am Soc Nephrol. (2016) 11:2260–72. doi: 10.2215/CJN.02980316
2. Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D’Agati VD, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. (2019) 15:45–59. doi: 10.1038/s41581-018-0077-4
3. Adapa S, Konala VM, Naramala S, Nast CC. Multiple morphological phenotypes of monoclonal immunoglobulin disease on renal biopsy: significance of treatment. Clin Nephrol Case Stud. (2020) 8:17–24. doi: 10.5414/CNCS110052
4. Qian Q, Leung N, Theis JD, Dogan A, Sethi S. Coexistence of myeloma cast nephropathy, light chain deposition disease, and nonamyloid fibrils in a patient with multiple myeloma. Am J Kidney Dis. (2010) 56:971–6. doi: 10.1053/j.ajkd.2010.06.018
5. Sundaram S, Mainali R, Norfolk ER, Shaw JHT, Zhang PL. Fibrillary glomerulopathy secondary to light chain deposition disease in a patient with monoclonal gammopathy. Ann Clin Lab Sci. (2007) 37:370–4.
6. Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, D’Agati VD, et al. Renal monoclonal immunoglobulin deposition disease: a report of 64 patients from a single institution. Clin J Am Soc Nephrol. (2012) 7:231–9. doi: 10.2215/CJN.08640811
7. Zand L, Nasr SH, Gertz MA, Dispenzieri A, Lacy MQ, Buadi FK, et al. Clinical and prognostic differences among patients with light chain deposition disease, myeloma cast nephropathy and both. Leuk Lymphoma. (2015) 56:3357–64. doi: 10.3109/10428194.2015.1040011
8. Lin J, Markowitz GS, Valeri AM, Kambham N, Sherman WH, Appel GB, et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol. (2001) 12:1482–92. doi: 10.1681/ASN.V1271482
9. Joly F, Cohen C, Javaugue V, Bender S, Belmouaz M, Arnulf B, et al. Randall-type monoclonal immunoglobulin deposition disease: novel insights from a nationwide cohort study. Blood. (2019) 133:576–87. doi: 10.1182/blood-2018-09-872028
10. Said SM, Best Rocha A, Valeri AM, Paueksakon P, Dasari S, Theis JD, et al. The characteristics of patients with kidney light chain deposition disease concurrent with light chain amyloidosis. Kidney Int. (2021) 101:152–63. doi: 10.1016/j.kint.2021.10.019
11. Charles JJ, Olson JL, Silva FG, D’Agati VD. Heptinstall’s Pathology of the Kidney. (2014). Available online at: https://shop.lww.com/Heptinstall-s-Pathology-of-the-Kidney/p/9781451144116# (accessed August 27, 2014).
12. Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, Fermand JP, et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. (2015) 87:698–711.
13. Nasr SH, Kudose SS, Said SM, Santoriello D, Fidler ME, Williamson SR, et al. Immunotactoid glomerulopathy is a rare entity with monoclonal and polyclonal variants. Kidney Int. (2021) 99:410–20. doi: 10.1016/j.kint.2020.07.037
14. Nasr SH, Fidler ME, Cornell LD, Leung N, Cosio FG, Sheikh SS, et al. Immunotactoid glomerulopathy: clinicopathologic and proteomic study. Nephrol Dial Transplant. (2012) 27:4137–46. doi: 10.1093/ndt/gfs348
15. Li XM, Rui HC, Liang DD, Xu F, Liang SS, Zhu XD, et al. Clinicopathological characteristics and outcomes of light chain deposition disease: an analysis of 48 patients in a single Chinese center. Ann Hematol. (2016) 95:901–9. doi: 10.1007/s00277-016-2659-1
16. Sethi S, Rajkumar SV, D’Agati VD. The Complexity and heterogeneity of monoclonal immunoglobulin-associated renal diseases. J Am Soc Nephrol. (2018) 29:1810–23. doi: 10.1681/ASN.2017121319
17. Cohen C, Royer B, Javaugue V, Szalat R, El Karoui K, Caulier A, et al. Bortezomib produces high hematological response rates with prolonged renal survival in monoclonal immunoglobulin deposition disease. Kidney Int. (2015) 88:1135–43. doi: 10.1038/ki.2015.201
18. Bridoux F, Javaugue V, Bender S, Leroy F, Aucouturier P, Debiais-Delpech C, et al. Unravelling the immunopathological mechanisms of heavy chain deposition disease with implications for clinical management. Kidney Int. (2017) 91:423–34. doi: 10.1016/j.kint.2016.09.004
19. Korbet SM, Schwartz MM, Lewis EJ. Immuotactoid glomerulopathy (fibrillary glomerulonephritis). Clin J Am Soc Nephrol. (2006) 1:1351–6. doi: 10.2215/CJN.01140406
20. Lin ZS, Zhang X, Li DY, Yu XJ, Qin AB, Dong Y, et al. Clinicopathological features and outcomes of coexistent light chain cast nephropathy and light chain deposition disease in patients with newly diagnosed multiple myeloma. J Clin Pathol. (2021) [Online ahead of print]. doi: 10.1136/jclinpath-2021-207449
21. Jain A, Haynes R, Kothari J, Khera A, Soares M, Ramasamy K. Pathophysiology and management of monoclonal gammopathy of renal significance. Blood Adv. (2019) 3:2409–23. doi: 10.1182/bloodadvances.2019031914
22. Batko K, Malyszko J, Jurczyszyn A, Vesole DH, Gertz MA, Leleu X, et al. The clinical implication of monoclonal gammopathies: monoclonal gammopathy of undetermined significance and of renal significance. Nephrol Dial Transplant. (2019) 34:1440–52. doi: 10.1093/ndt/gfy259
23. Kanzaki G, Okabayashi Y, Nagahama K, Ohashi R, Tsuboi N, Yokoo T, et al. Monoclonal immunoglobulin deposition disease and related diseases. J Nippon Med Sch. (2019) 86:2–9.
24. Cameron C. Diagnostic electron microscopy. A Text/Atlas (2nd edn). G. Richard Dickersin. Springer-Verlag, New York, 2000. No. of pages: 1005 (894 illustrations). ISBN: 0 387 98388 0. J Pathol. (2001) 194:137–8. doi: 10.1002/1096-9896(200105)194:1<137::AID-PATH841>3.0.CO;2-9
Keywords: monoclonal immunoglobulin deposition disease, immunotactoid glomerulopathy, pathology, glomerulonephritis, multiple myeloma, renal biopsy
Citation: Wang Y, Yan Y, Dong B, Zou W, Li X, Shao C, Jiang L, Wang M and Zuo L (2022) Clinicopathological manifestations of coexistent monoclonal immunoglobulin deposition disease and immunotactoid glomerulopathy. Front. Med. 9:911998. doi: 10.3389/fmed.2022.911998
Received: 03 April 2022; Accepted: 29 July 2022;
Published: 25 August 2022.
Edited by:
Gian Marco Ghiggeri, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Emanuele De Simone, Azienda Sanitaria Locale “Città di Torino”, ItalyCopyright © 2022 Wang, Yan, Dong, Zou, Li, Shao, Jiang, Wang and Zuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Yan, eWFubWFpbDEwMDRAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.