 Dongling Liu1†
Dongling Liu1† Linlin Wen2,3†
Linlin Wen2,3† Zhandong Wang2Yang Hai4Dan Yang2Yanying Zhang4,5Min Bai5Bing Song4,5Yongfeng Wang5*
Zhandong Wang2Yang Hai4Dan Yang2Yanying Zhang4,5Min Bai5Bing Song4,5Yongfeng Wang5*- 1School of Pharmacy, Gansu University of Chinese Medicine, Lanzhou, China
- 2School of Traditional Chinese and Western Medicine, Gansu University of Chinese Medicine, Lanzhou, China
- 3County People’s Hospital, Pingliang, China
- 4Gansu University of Chinese Medicine/Scientific Research and Experimental Center, Lanzhou, China
- 5Gansu Provincial Engineering Laboratory for Research and Promotion of Quality Standardization of Authentic Medicinal Materials in Gansu Province/Provincial Key Laboratory of Pharmaceutical Chemistry and Quality Research in Colleges and Universities in Gansu Province/Gansu Provincial Laboratory Animal Industry Technology Center, Lanzhou, China
Acute pancreatitis (AP), as a common cause of clinical acute abdomen, often leads to multi-organ damage. In the process of severe AP, the lungs and intestines are the most easily affected organs aside the pancreas. These organ damages occur in succession. Notably, lung and intestinal injuries are closely linked. Damage to ML, which transports immune cells, intestinal fluid, chyle, and toxic components (including toxins, trypsin, and activated cytokines to the systemic circulation in AP) may be connected to AP. This process can lead to the pathological changes of hyperosmotic edema of the lung, an increase in alveolar fluid level, destruction of the intestinal mucosal structure, and impairment of intestinal mucosal permeability. The underlying mechanisms of the correlation between lung and intestinal injuries are inflammatory response, oxidative stress, and endocrine hormone secretion disorders. The main signaling pathways of lung and intestinal injuries are TNF-α, HMGB1-mediated inflammation amplification effect of NF-κB signal pathway, Nrf2/ARE oxidative stress response signaling pathway, and IL-6-mediated JAK2/STAT3 signaling pathway. These pathways exert anti-inflammatory response and anti-oxidative stress, inhibit cell proliferation, and promote apoptosis. The interaction is consistent with the traditional Chinese medicine theory of the lung being connected with the large intestine (fei yu da chang xiang biao li in Chinese). This review sought to explore intersecting mechanisms of lung and intestinal injuries in AP to develop new treatment strategies.
Introduction
Acute pancreatitis (AP) is one of the common causes of acute abdomen in clinical practice (1, 2). It is closely caused by two predisposing factors – gallstones and alcohol misuse (3). These two factors stimulate the pancreas to secrete a large amount of digestive enzymes, leading to autodigestion, inflammation, edema, and bleeding, and even necrosis of the pancreas or surrounding fatty tissue. Some studies have demonstrated that AP is associated with fat necrosis around the peripancreatic and intra-pancreatic areas. The necrosis develops because adipocytes in the peritoneum are destroyed and digested by inappropriately activated pancreatic enzymes (4). Stored triglycerides in the adipocytes are released and split by pancreatic lipases into fatty acids and glycerol molecules, and the unsaturated free fatty acids increase damage to the surrounding tissues, and systemic inflammation develops (5). Necrosis of adipocytes serves as an important inflammatory source by the release of a huge number of inflammatory mediators (6). In AP, various inflammatory factors trigger the waterfall cascade of inflammatory mediators through the “trigger-like action.” Subsequently, which causes the systemic inflammatory response syndrome (SIRS), and eventually leads to multiple organ dysfunction syndrome (MODS), both of which contribute to the high mortality rate of AP (7).
The lungs and intestine are generally considered to be the most directly targeted organs of AP, and resulting in lung injury and intestinal injury are the main causes of early death in AP patients, with a mortality rate of 60% (8, 9). In AP, intestinal injury is an early event involving damage of intestinal mucosa barrier and altered intestinal mucosal permeability that occurs in AP (10). This injury increase intestinal permeability and cause massive cell death in the intestinal epithelium and basement membrane, leading to the activation of the local response and inducing bacterial translocation out of the gastrointestinal tract. These phenomena contribute substantially to MODS and systemic inflammatory response, being able to damage distant organs (11–13). Of these multiple organs, the lung appears to be the earliest remote organ affected by AP (14). Acute lung injury (ALI) is the dominant death-associated factor in patients who have diffuse alveolar damage on chest X-ray with early-stage AP (8). The generation of inflammatory mediators, including cytokines and chemokines, as well as reactive oxygen species (ROS), is a fundamental cause of AP-induced lung injury. These mediators contribute to the accumulation of macrophages and neutrophils and, later, trigger a cascade of pathological changes in pulmonary microcirculation of the lungs, leading to the occurrence and aggravation of AP-induced lung injury (15, 16). Noteworthy, lung injury is one of the most important components of the MODS triggered by intestinal injury (14). Both lung and intestinal injury can together worsen the severity of AP, eventually resulting in death. Interestingly, it is consistent with the traditional Chinese medicine theory of the lung being connected with the large intestine (17).
There are some close connections between the physiology of lungs and intestine and also between the pathological changes and molecular mechanism of intestinal and lung injury caused by AP. Thus, it is necessary to investigate a common intervention strategy for pulmonary and intestinal injuries. This review summarized the AP-related lung and intestinal injury and their mechanisms, and subsequently discussed the intersections of these mechanisms. This review also aimed to explore the underlying co-mechanisms of multiorgan AP injury and find the novel clinical treatment methods as well as provide the valuable insights into clinical practice.
The Mechanism of Acute Pancreatitis
Acute pancreatitis is a relatively common pancreatic inflammatory disorder, leading to local and systemic inflammation. The main pathological mechanism of AP is activated trypsin-induced self-digestive acute inflammatory response in the pancreas (18, 19). Once pancreatic acinar cells are damaged, acinar cells would release many pro-inflammatory mediators, such as cytokines and chemokines, and stimulate the recruitment and activation of immune cells, including innate immune cells (neutrophils, macrophages, dendritic cells, mast cells, and NK cells), and adaptive immune cells (T cells and B cells), which will consequently exacerbate the pancreatic injury, resulting in necrosis of the pancreas (18, 20, 21). The necrotized pancreatic acinar cells release various kinds of damage-associated molecular patterns (DAMPs), such as high-mobility group box protein 1 (HMGB1), and activate infiltrating immune-associated cells to generate more inflammatory mediators, which in turn accelerate more immune cell infiltration and aggravate inflammation, even contributing to systemic inflammation (20, 21).
Numerous studies have shown that acinar cell necrosis and inflammation are induced by oxidative stress, Ca2+ overload, a disorder of organelles, early activation of trypsin, activation of Toll-like receptors 4 (TLR4), nuclear factor kappa B (NF-κB), NOD-like receptor protein 3 (NLRP3), signal transducer and activator of transcription 3 (STAT3), activator protein-1 (AP-1), and mitogen-activated protein kinases (MAPK) signaling pathways, and accumulation of inflammatory cells (16, 22). Extensive studies have suggested that Ca2+ overload and oxidative stress are early events in AP. Toxic by-products of AP (such as bile acids or alcohol metabolites) promote the production of ROS and the release of Ca2+, affecting pancreatic cell structure and function (23). ROS can also increase the activation of inflammatory signals, such as AP-1, STAT3, and MAPKs (24).
Acute pancreatitis-related multiorgan injury by activating STAT3 pathway. HMGB1, released in stress, promotes pancreatic injury and pro-inflammatory cytokine release and induces the Janus kinase 2 (JAK2)/STAT3 signal pathway, further intensifying the inflammatory reaction (25). TLRs, especially TLR4, are the first factors to become activated. Activated TLR4 and the NF-κB pathway promote cell synthesis, secreting pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and IL-6, which can also stimulate NF-κB, leading to its further activation, thus, resulting in persistent and intensified inflammation (26). TLR9 is expressed in resident immune cells of the pancreas, which are predominantly represented by macrophages, and it is an important DAMP receptors upstream of inflammasome activation, and caspase-1, and NLRP3 inflammasome are required for the development of inflammation in AP (20, 27). A recent study demonstrated that NLRP3 inflammasome and gasdermin D (GSDMD) activation-mediated pyroptosis in acinar cells is closely linked to pancreatic necrosis and systemic inflammation in AP (28). Another study recently indicated a new mechanism in which the p53-apoptosis-inducing factor (AIFM2) pathway regulates AP with multiorgan injury via activating transcription factor 6 (ATF6)-mediated apoptosis, suggesting that endoplasmic reticulum (ER)-mitochondrial-nuclear crosstalk plays an important role in AP development (29). Additionally, the inflammatory process is amplified following further secretion and overexpression of adhesion molecules including intercellular adhesion molecule 1 (ICAM-1) and vascular adhesion molecule 1 (VCAM-1), which represent ligands for lymphocyte function-associated antigen 1 on leukocytes and lymphocytes, αLβ2 and CD11a-CD18 on monocytes and integrin macrophage 1 antigen (Mac-1) on neutrophils, while their secretion is promoted by ROS generation and TNF-α itself (30). The potential targeted pathways in AP are shown in Table 1.
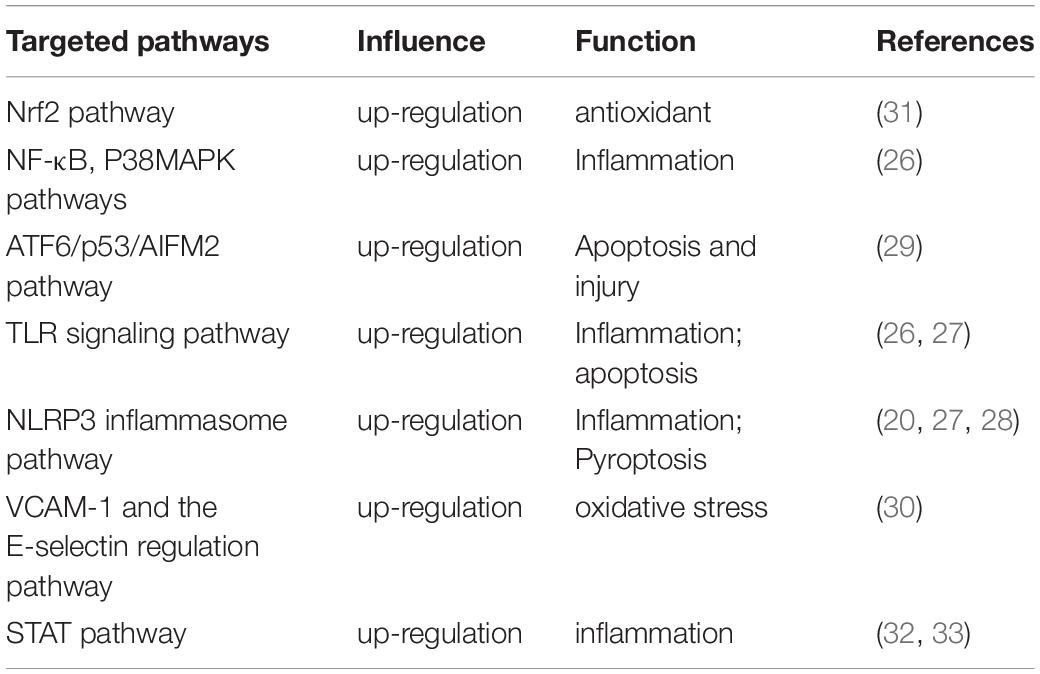
Table 1. Targeted pathways in AP.
Mechanisms of Lung Injury in Acute Pancreatitis
Lung injury is the most common feature and the main cause of early death in AP (8). The pathological changes are closely related to an increase in vascular endothelial cell space and permeability, aggregation of marginal concentration of leukocytes, and profuse expression of ICAM-1 (15, 16). During AP, the accumulation of inflammatory cells and inflammatory mediators in the lung destroys the blood-air barrier. Several studies have demonstrated that that the accumulation of a lot of neutrophils in the lungs enhanced generation of ROS and increased the production of pro-inflammatory cytokines, including HMGB1, endotoxin, TNF-α, IL-1β, and IL-6, which activate various intracellular signaling pathways (e.g., NF-κB, NLRP3, STAT3, MAPK, and AP-1) and adhesion molecules, thus, releasing inflammatory mediators, which exacerbate the damage and result in lung injury (16, 34). According to the relevant literature, the pathological reactions and their molecular mechanisms of AP-related lung injury can be summarized by mainly including inflammatory response outbreak, oxidative stress damage, and endocrine disorders (Figure 1).
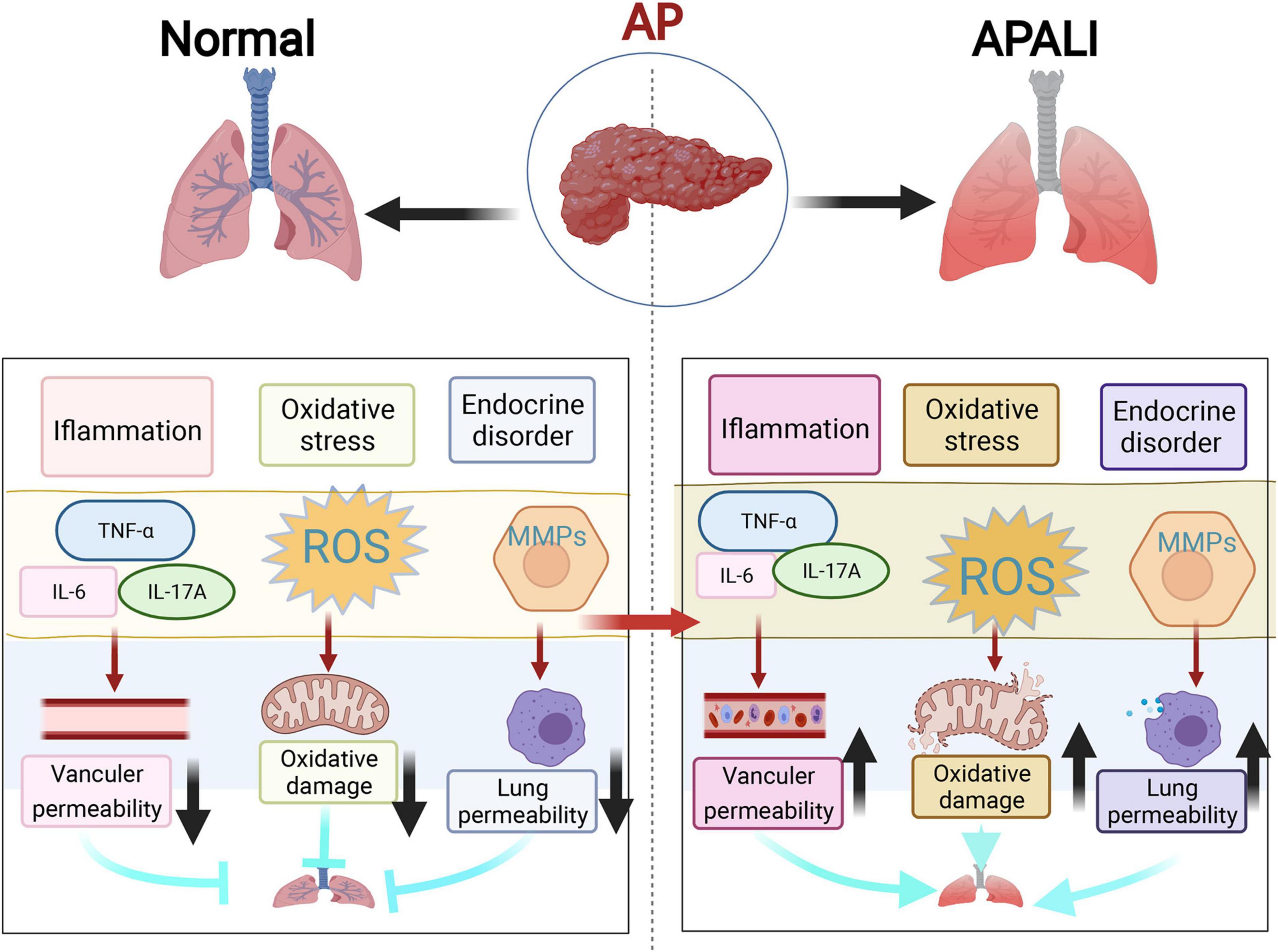
Figure 1. The three main mechanisms of lung injury in acute pancreatitis (AP). This figure shows three most highlighted mechanisms in the studies of lung injury in AP, which include inflammatory response outbreak, oxidative stress damage, and endocrine disorders represented by matrix metalloproteinases (MMPs).
Inflammatory Response of Lung Injury in Acute Pancreatitis
Inflammatory response plays a key role and has a major effect on the outcome of lung injury in AP (35). Pro-inflammatory factors play a key role in lung injury (36). Pro-inflammatory cytokines are considered to initiate and maintain lung injury (37), and they mainly influence human lung microvascular endothelial cells (HPMECs). Their effect leads to increased alveolar permeability, alveolar tissue fluid extravasation, pulmonary edema, and declined blood oxygen saturation (38). TNF-α, IL-17A, IL-6, IL-1β, and HMGB1 are some of the most important pro-inflammatory cytokines in AP-ALI (Figures 1, 2). The possible cytokines of lung injury in AP are shown in Table 2.
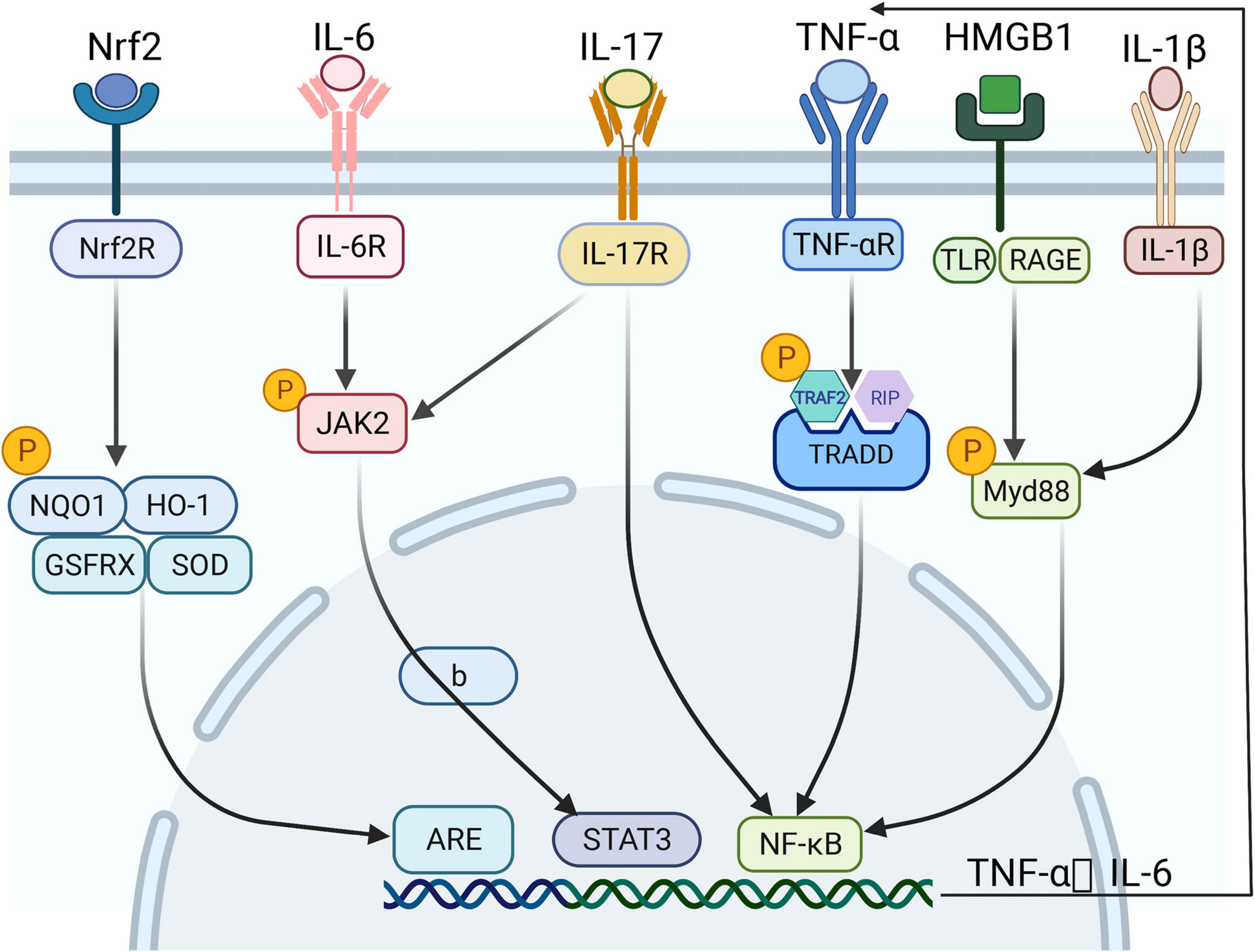
Figure 2. The signaling pathways of lung and intestinal injuries in acute pancreatitis (AP) in different environments and under certain physiological conditions. Several inflammatory activators, such as TNF-α, HMGB1, and proinflammatory factors, activate various intracellular signaling pathways – particularly NF-κB, STAT3, and ARE – by binding specific receptors, thereby releasing a range of inflammatory mediators and promoting each other, forming a vicious cycle.

Table 2. The cytokines of lung and intestinal injuries in AP.
TNF-α, which is released by macrophages and activated fixed circulating monocytes in activated fixed tissues, appears early and plays an important role in the occurrence and development of lung injury in AP (50). It first binds to TNF receptor 1 (TNF-R1) and interacts with adaptor proteins, such as TNF-R1-related death domain (TRADD) protein, TNF-R-related factor 2 (TRAF2), and receptor-interacting protein (RIP). Then, it docks and binds to the mentioned proteins, triggers the intracellular cascade reaction; and finally activates NF-κB (51, 52). NF-κB activation can enhance the transcription of TNF-α genes, thus, creating a vicious feedback loop that amplifies early inflammatory signals and exacerbates the initial inflammatory effects (53) (Figure 1).
IL-17A is closely associated with the pathogenesis of lung injury in AP, and it is mainly expressed by adaptive immune cells populations (39, 54). Study have shown that the expression level of IL-17A is elevated in serum and bronchoalveolar lavage fluid (BALF) in AP-related lung injury (55). Although IL-17A causes lung damage through neutrophil aggregation, which directly leads to pulmonary and pancreatic edema (40), it also acts through synergy with other inflammatory factors, including ligands for TNF-α, IL-1β, and TLRs, and can induce the production of other pro-inflammatory cytokines and chemokines (56). Studies have demonstrated that IL-17A stimulates IL-8 production through synergy with TNF-α and induces an accelerated secretion of IL-6 from human peripancreatic myofibroblasts, which can increase the production of CXCL family of chemokines (neutrophil recruitment) in alveolar type II epithelial cells, and exacerbate airway inflammation and lung ischemia-reperfusion injury (42, 57).
IL-6 is mostly produced by monocytes, macrophages, and dendritic cells (58). It is a multifunctional cytokine with clear pro-inflammatory and anti-inflammatory properties that inhibit proliferation and promote apoptosis and lung injury by activating the JAK2/STAT3 signaling pathway (59, 60). In the early stages of pancreatic lung injury, IL-6 is rapidly synthesized and plays a protective role in host defense. As the injury persists and leukocyte IL-6 production by leukocytes continues to increase, extravasation of alveolar tissue fluid and pulmonary edema occur, leading to impaired lung function, progression of AP, and increased mortality (61).
IL-1β is a member of the IL1 cytokine family. It is produced by activated macrophage preproteins and is hydrolyzed by cystathionine 1 (CASP1/ICE) protein. This cytokine is an important mediator of inflammatory response, and it is involved in various cellular activities, including cell proliferation, differentiation, and apoptosis (62). In lung injury, it is mainly produced upon activation of the NF-κB and JAK2/STAT3 signaling pathways, which intensifies the acute injury process by activating the aggregation of monocytes and macrophages (63, 64). It is expressed only on the surface of activated vascular endothelial cells and initiates the adhesion of leukocytes to vascular endothelial cells, playing an important role in the early development of lung injury in AP (65, 66).
High-mobility group box protein 1 has been proposed as a potent inflammatory mediator in ALI, and the blockade of HMGB1 has led to a significant reduction in lung inflammatory reaction. Luan et al. (34) and Ding et al. (67) demonstrated that the downregulation of HMGB1 can inhibit the activity of NF-κB, inhibit the expression of TNF-α, IL-1β, ICAM-1, and matrix metalloproteinase (MMP) 9 in the lung tissue, and tissue, and therefore decrease the severity of AP-associated ALI. Qu et al. (68) indicated that HMGB1 binds to certain receptors, such as receptor for advanced glycation end products (RAGE) and TLR4, activates the ROS and phosphatidylinositol-3 kinase (PI3K) pathways and myeloid differentiating factor 88 (MyD88), and results in the release of TNF-α, IL-1β, IL-6, and other cytokines contributing to ALI, suggesting that blocking HMGB1 may provide an effective treatment strategy for AP-associated ALI.
Oxidative Stress in Lung Injury in Acute Pancreatitis
Oxidative stress plays a crucial role in the development of lung injury in AP. An excessive amount of ROS can be generated by activating neutrophils, leading to severe oxidative stress damage (44, 69). Oxidative stress damage not only plays a role in the local damage of the pancreas but also plays a dominant role in damage to other organs, accelerating the occurrence and development of the SIRS and MODS (70, 71). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor of the alkaline locking chain and a major regulator of resistance to oxidative stress, and thus the Nrf2/antioxidant response element (ARE) pathway is the most important endogenous antioxidant signaling pathway in the body (72). Nrf2 can upregulate antioxidant genes and exert antioxidant effects by binding to the promoter sequence, ARE, and activation of Nrf2 can reduce neutrophilic airway inflammation by the upregulation of antioxidants and downregulation of inflammatory cytokines in the airways (73, 74). In AP, Nrf2 dysfunction and function abnormalities of key enzymes in the Nrf2/ARE pathway, such as heme oxygenase-1 (HO-1), quinone oxidoreductase-1 (NQO1), glutathione peroxidase (GSH-Px) and superoxide dismutase (SOD) occur, resulting in loss of antioxidant function and consequent lung tissue damage (75).
Endocrine Disorders Involving Matrix Metalloproteinases in Lung Injury in Acute Pancreatitis
HPMEs are damaged by inflammatory factors and inflammatory mediators of lung injury in AP (76). HPMECs can secrete MMPs, which are a type of zinc-dependent endopeptidases (77). MMP-2 and MMP-9 are the most extensively studied MPPs. They degrade extracellular basement membrane collagen and alter lung permeability, exacerbating lung injury (78, 79). MMP-2 and MMP-9 have been found to play an important role in ALI, acute respiratory distress syndrome (ARDS), and pulmonary fibrosis (80–82). MMP-9 is mainly involved in the ALI phase, whereas MMP-2 is mainly involved in the subsequent repair and fibrosis phase (83). MMP-9 levels in BALF have been reported to increase in patients with ARDS secondary to septic shock compared to normal controls, suggesting that MMP-9 plays an important role in the development of lung injury in AP (84).
Other Mechanisms of Lung Injury in Acute Pancreatitis
Abnormalities in the lysosomal degradation pathway, which underlie the emergence of differentiation disorders, can cause impairments in cell development, homeostasis, and survival (85). Studies have shown that abnormalities in the lysosomal degradation pathway, represented by impaired autophagy, play an important role in the evolution of lung injury in AP (86–88). Pulmonary intravascular macrophages (PIMs) damage lung endothelial cells, and they may play a role in another pathogenetic pathway of lung injury in AP (89). Water channel proteins (aquaporins) are a family of membrane proteins involved in the selective transport of water across cell membranes and play a key role in the development of acute pulmonary edema (47). Additional mechanisms, such as activation of ICAM-1 and alteration of circulating protease and phospholipase concentration, have been associated with AP-induced remote lung injury (90). Studies have indicated that ICAM-1 and E-selectin as important inflammatory cytokines are involved in the pathogenesis of AP-associated lung injury, which lead to increased leukocytic infiltration, permeability, proliferation, migration, pulmonary microcirculatory dysfunction and acute respiratory distress syndrome (91, 92).
The Experimental Animal Models of Acute Pancreatitis-Associated Acute Lung Injury
The experimental animal models of AP and AP-associated ALI have been relatively well-established. Sodium taurocholate, caerulein, or L-arginine are primarily used to create the models (Table 3).

Table 3. The experimental animal models of pancreatitis-associated ALI.
Where rats are were starved overnight and anesthetized with ketamine 40 mg/kg intraperitoneally. The hepatic portal of the bile duct is clamped, and 3.5% sodium taurocholate in a volume of 1 mL/kg is retrogradely injected into the biliopancreatic conduit at a steady pace (0.1 mL/min) (93). The animal models caused by caerulein and LPS have been made. Mice are treated intraperitoneally (i.p.) with 50 μg/kg body weight caerulein (10 mL/kg of body weight) twice every hour (seven injections in total) and 10 mg/kg of lipopolysaccharide (LPS) (10 mL/kg of body weight, i.p.) following the last dosage of caerulein immediately (74). The AP-associated ALI rat model has been established by administering two intraperitoneal injections (1 h apart) of L-arginine at a dose of 4 g/kg body weight (96).
Mechanisms of Intestinal Injury in Acute Pancreatitis
In intestinal injury caused by AP, the intestinal barrier becomes damaged the earliest. The pathological changes are closely related to the destruction of the intestinal mucosal structure and an increase in intestinal mucosal permeability (97, 98). The effects of intestinal barrier failure in AP manifest as intestinal immune deficiency, intestinal microflora disturbance, increased intestinal permeability, and excessive release of inflammatory mediators (like endotoxin, cytokines, chemokines, DAMPs, and mRNAs) (16, 97). Based on the relevant literature, this review summarizes the mechanisms of intestinal injury, including inflammatory response burst, oxidative stress injury, and endocrine disruption represented by angiopoietin (Ang) and mitochondrial injury (Figure 3).
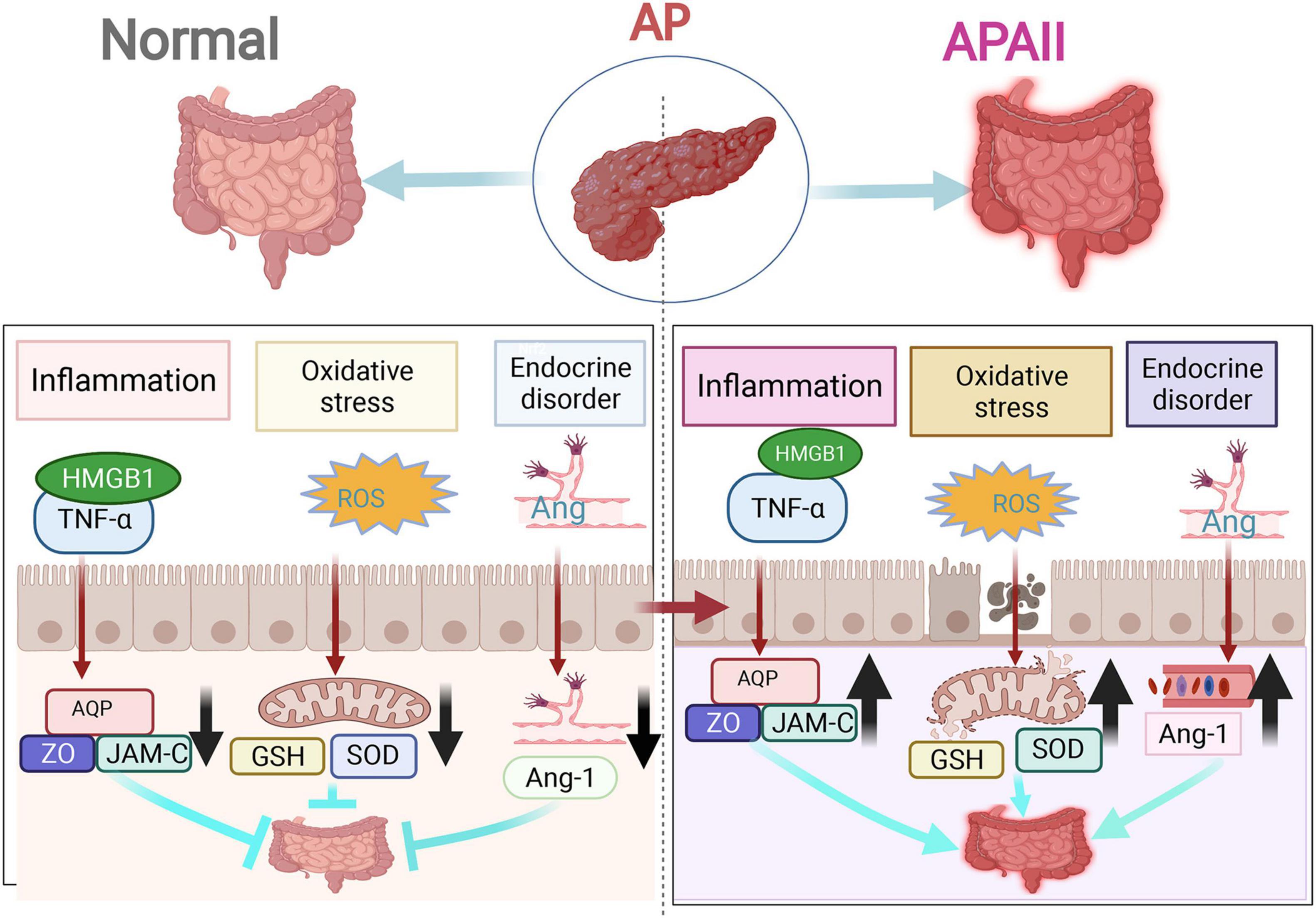
Figure 3. The three main mechanisms of intestinal injury in acute pancreatitis (AP). The three most highlighted mechanisms of intestinal injury in AP are inflammatory response outbreak, oxidative stress damage, and endocrine disorders.
Inflammatory Response to Intestinal Injury in Acute Pancreatitis
The inflammatory response plays a major role in the development and prognosis of intestinal injury in AP (16). Inflammatory cells and factors accumulate and infiltrate during the development of AP, leading to the disruption of the intestinal barrier and intestinal mucosal structure and an increase in intestinal mucosal permeability. The disruption of the intestinal barrier function is mainly manifested in two aspects: intestinal mucosal epithelial barrier dysfunction (mechanical, chemical, immunological, and biological barrier dysfunction) and intestinal capillary endothelial barrier dysfunction (99). In contrast, most of the inflammatory responses are caused by mechanical and chemical barrier dysfunction.
The intestinal mechanical barrier comprises intestinal epithelial cells (IECs) and tight intercellular junctions. IECs separate the intestinal lumen from the lamina propria. The tight junctions are the main determinants of paracellular permeability (100). IECs ensure proper digestion and barrier function with a high turnover rate of 4–5 days (101). Uncontrolled inflammatory response of the intestinal epithelium is a characteristic feature of intestinal barrier failure in severe AP. The caspase-3 pathway can be stimulated by an inflammatory factor, TNF-α, leading to severe apoptosis of intestinal mucosal cells and destruction of intestinal mucosal structures (102). Another inflammatory factor, HMGB1, activates RAGE and TLRs, which, in turn, activate the NF-κB signaling, release inflammatory mediators, and enhance the binding of ARE elements to downstream target mRNAs (such as those for TNF-α, IL-6, and IL-8), and maintain the stability of target mRNAs. As a result, they increase the translation of related proteins and induce a strong pro-inflammatory effect (16).
The intestinal chemical barrier consists of mucin (MUC), antimicrobial peptides (AMPs), and other digestive enzymes. AMPs have sterilizing and anti-inflammatory functions. Additionally, they promote tissue repair. In the intestine, AMPs, especially β-defensins, in the intestine are reduced due to the aggregation and infiltration of inflammatory cells in AP, increasing the rate of intestinal bacterial translocation and the possibility of retrograde infection (103). MUC2, one of the most common forms present of MUC, is the main component of the intestinal chemical barrier covering IECs and forms the intestinal mucus layer. It is the first line of defense of the intestinal mucosal barrier; therefore, it is the most vulnerable to inflammatory reactions and loss of barrier function (104) (Table 2).
The intestinal capillary endothelial barrier is a semi-selective barrier composed of a single layer of endothelial cells surrounding the vascular lumen and basement membrane. The occurrence of intestinal capillary endothelial barrier impairs capillary permeability, which affects intestinal mucosal permeability and causes intestinal edema. Capillary permeability is determined by three factors: endothelial cells, inter-endothelial connections, and basement membrane (49). Studies have shown that aquaporin 1 (AQP-1), MMP-9, and junctional adhesion molecule-C (JAM-C) play key roles in the regulation of capillary permeability in AP (105). Under the stimulation of inflammatory factors, intestinal permeability can increase when AQP-1 is downregulated, leading to an imbalance in water transfer and homeostasis between the cells and blood vessels. Microvascular permeability can increase when MMP-9 is decreased, leading to damage to the basement membrane. JAM-C is localized at cell junctions, increasing intercellular permeability when affected, which, in turn, increases intestinal mucosal permeability and causes intestinal edema (95).
Oxidative Stress of Intestinal Injury in Acute Pancreatitis
During oxidative stress, an imbalance between oxidative and antioxidant action occurs in the body. Inflammatory infiltration of neutrophils, increased secretion of proteases, and production of oxidative intermediates, such as oxygen radicals and lipid peroxides, lead to IEC damage in the early stages of AP (106). Oxygen radicals and lipid peroxides can be produced in damaged intestinal tissues by activated immune cells and alveolar cells, which can activate the Nrf2/ARE antioxidant signaling pathway (79), resulting in a decrease in total GSH and SOD levels (107, 108). As a result, intestinal barrier function, intestinal mucosal structure, and intestinal mucosal permeability become disrupted (109).
Endocrine Disorders of Intestinal Injury in Acute Pancreatitis
Ang has an important role in the pathology of intestinal capillary leakage syndrome in SAP. Normally, Ang-1 not only promotes endothelial cell chemotaxis and aggregation but also inhibits apoptosis, inflammation, exudation, and leukocyte adhesion (110). Additionally, it regulates the proliferation of endothelial cells and vascular smooth muscle cells, and promotes vascular maturation, which is important for maintaining vascular stability and integrity (111). In AP, a significant decrease in serum Ang-1 levels, a significant increase in the number of inflammatory factors in the serum, an increase in capillary permeability of the tissues, an increase in alveolar fluid, and the appearance of pulmonary edema are all observed. Related studies have shown that Ang1 not only effectively alleviated intestinal capillary leakage in a rat model of AP but also played a positive role in the protection of intestinal microcirculation. Additionally, it significantly upregulated the expression of AQP-1 and downregulated the expression of MMP-9 and JAM-C protein to provide intestinal protection (49).
Other Mechanisms of Intestinal Injury in Acute Pancreatitis
Mitochondria are central to cellular viability and function, controlling control many physiological metabolic processes (112, 113). Disorders of ROS and cytochrome C metabolism, which occur in mitochondria, can lead to abnormalities in intestinal metabolism (114). Significant mitochondrial dysfunction occurs in the jejunum in the early stages of severe AP in rat models (115). Additionally, swelling of mitochondria in the colonic mucosal epithelium occurs. In edematous pancreatitis, degeneration and rupture of colonic mitochondria are present in necrotizing pancreatitis, suggesting that the damage to the mitochondria is closely related to intestinal injury in AP (116).
Common Mechanisms of Lung and Intestinal Injuries in Acute Pancreatitis
The lungs and intestine are organs that are directly damaged organs in AP. Mesenteric lymph (ML) is the direct axial connection between the intestine and lungs (117). Physiologically, the lungs and large intestine can interact with each other to promote homeostasis, whereas pathologically, the balance and interaction between the two axes are disturbed. Lung-gut syndrome is a condition in which the lungs and large intestine become affected concurrently; respiratory and digestive tract disorders occur together (118). This interaction is consistent with the theory of the lung being connected with the large intestine in Chinese medicine. Inflammatory mediators reaching the lung and intestinal tissues cause an inflammatory response, oxidative stress, and endocrine hormone secretion disorders, impairing the balance axis between the lung and intestine.
Inflammatory Response in the Imbalance of the Lung-Gut Axis
Acute pancreatitis is an acute necrotic and inflammatory process that suddenly occurring around and inside the pancreas (119). Overactivation of the inflammatory response is important in the course of pancreatitis, including the production and activation of inflammatory factors and the accumulation of pro-inflammatory mediators. The main inflammatory factors, including TNF-α, IL-17, IL-1, and IL-8, are released into the bloodstream. They accumulate in the lungs and intestine, activating the NF-κB signaling pathway, amplifying the inflammatory effect, and inducing a systemic inflammatory response. These processes lead to inflammatory injury in the lungs and intestine (120). Thromboxane A2 (TXA2), platelet-activating factor (PAF), endothelin 1 (ET-1), phospholipase A2 (PLA2), and IL-1β can cause vasospasm, white blood cell and platelet aggregation, thrombosis, and vascular endothelial cell damage, leading to the loss of the intestinal barrier, which in turn causes lung injury (121). The main pro-inflammatory mediators include HMGB1 and TLR-4. HMGB1 is released by necrotic acinar cells and acts on its receptors, ARGE and TLR (122). Chen et al. (123) found that the expression of HMGB1 is positively related to intestinal barrier failure and inhibition of HMGB1 can significantly improve intestinal injury. Furthermore, HMGB1 activates the NF-κB signaling pathway and releases several downstream inflammatory factors by myd88 and appears during an early inflammatory response (124). Relevant studies have shown that the levels of HMGB1 and TLR-4 can be used as important indicators of AP progress (125) (Figure 2 and Table 2).
Oxidative Stress in the Imbalance of the Lung-Gut Axis
During AP, Ca2+ overload, inflammatory mediators, and zymogen activation can damage the arterial muscles and vascular endothelial cells in the pancreatic lobules, resulting in pancreatic vasoconstriction, shunting, and insufficient perfusion. Intestinal mucosal tissue is the most susceptible to insufficient perfusion (126). When the intestinal mucosa is signaled by ischemia and hypoxia, xanthine oxidase and hypoxanthine accumulate in the intestinal tissue, leading to adenosine triphosphate (ATP) depletion due to insufficient oxidative phosphorylation. In the subsequent reperfusion, the body converts hypoxanthine to xanthine, and then releases superoxide ions to generate more free oxygen radicals. The Nrf2/ARE antioxidant signal pathway is activated, causing lipid peroxidation and cell membrane damage, which accelerate intestinal barrier loss and lung function impairment (127). Liu et al. (43) have found that SAP can change the hemodynamics of rats, leading to severe microcirculation disturbances in the pancreas and other organs (especially the lungs) and causing redox imbalance. These changes result in hyperosmotic edema of the lungs and increased alveolar fluid level (10, 15), as well as other pathological changes, including the destruction of intestinal mucosal structure and changes in intestinal mucosal permeability (35) (Figure 2).
Endocrine Disorders in the Imbalance of the Lung-Gut Axis
The intestine is not simply a digestive organ: it has certain endocrine functions. The endocrine hormones secreted by the intestine play important roles in gastric content transport and digestion. Hormones, such as secreted cholecystokinin octapeptide (CCK-8) and blood vessel active intestinal peptide (VIP), can cause lung and intestinal damage in AP under pathological conditions (48). Blood CCK is mainly derived from intestinal secretory endothelial cells. Related studies have shown that it not only promotes gall bladder contraction but also relaxes the sphincter of Oddi and protects the gastric mucosa. Additionally, it can relax tracheal muscles, activate pulmonary interstitial macrophages (PIM), and reduce the occurrence of endotoxemia-related pneumonia. Although CCK is derived from enteroendocrine endothelial cells, it can cause lung and intestinal damage concurrently. VIP is a linear peptide widely distributed in the gastrointestinal tract and lungs. Its function is mainly to promote glandular secretion. It is negatively correlated with gastrointestinal motility. It can dilate systemic and pulmonary blood vessels, and inhibit the proliferation of pulmonary artery smooth muscle cells (128). Animal experiments have proven that VIP can reduce pulmonary hypertension by reducing pulmonary vascular resistance (45).
Other Mechanisms in the Imbalance of the Lung-Gut Axis
Water channel proteins play a vital role in maintaining the water homeostasis of the lungs and intestine. AQPs are a type of membrane water channels, consisting of six transmembrane spiral segments and two shorter spiral segments that do not span the entire membrane. Their main function is to promote water transport in passive transport, and AQPs are widely distributed in mammalian organs (46). Currently, 13 AQPs have been discovered, and at least eight AQPs have been shown to transport water in humans and rodents (47). It was previously shown that the expression levels of AQP-1 and AQP-5 were decreased in patients with pulmonary edema after a viral infection were decreased (129). Sakai et al. (130) that the genes of AQP4 and AQP8 are mainly expressed in the mouse colon. The regulation of transepithelial fluid transport in the gastrointestinal tract is based on ion and water transport by AQPs (131). Additionally, the appearance of pulmonary and bowel dysfunction is closely related to intra-abdominal hypertension (IAH) and/or abdominal cavity syndrome (ACS). Reduced IAH maintains the integrity of the intestinal barrier, promotes the recovery of intestinal function, inhibits inflammation, and increases blood oxygen saturation (SpO2) (132). To reduce tissue damage of the intestines and lungs (133), thus, other mechanisms of lung and bowel damage in AP can be targeted to reduce damage to the intestine and lungs (133).
Potential Targets for the Protection of Lung and Intestinal Injuries in Acute Pancreatitis
Studies have shown that lipoxin A4 inhibits the activation of TNF-α/TNF-R1 and downstream signals, such as TRADD, TRAF2, and RIP. The inhibition influences the activity of the NF-κB pathway, and consequently inhibits the production of inflammatory mediators and the impairment of pulmonary intestinal injury (94, 134). According to another study, TLR4, which is a key receptor for the recognition of pathogen-associated molecular patterns (PAMPs), may provide valuable guidance for the treatment of pulmonary intestinal injury in SAP (135).
BML-111, which is a commercially synthesized lipoxin receptor agonist, protects against emotion-induced LAI in mice by modulating the Nrf2/ARE signaling pathway (74). Isoglycyrrhizinate alleviates AP in mice by inhibiting oxidative stress and modulating the Nrf2/HO1 pathway (31). The protective effect of tanshinone IIA in AP in mice occurs by inhibiting oxidative stress through the Nrf2/ROS pathway (136). Protective effects of rhodopsin against AP-induced lung injury occur by the inhibition of NLRP3 inflammasome activation specifically activated by the Nrf2/HO-1 pathway (93). HMGB1 inhibitors reduce the severity of SAP by reducing intestinal permeability through the reduction in claudin-2 and occludin expression (25). Moreover, hydrostatin-SN10 significantly reduces IL-6 levels, reduces inflammatory response, and improves lung tissue injury by inhibiting JAK2/STAT3 phosphorylation (137).
Conclusion and Future Perspectives
This review described the three common mechanisms of lung and intestinal injury in AP. The common mechanisms of lung and intestinal injury in AP are inflammatory response, oxidative stress, and endocrine hormone secretion disorder. However, few studies have reported concurrent treatment options for lung and intestinal injury in AP. Therefore, future studies are required to investigate common treatment options for lung and intestinal injury in AP. The mechanism of inflammatory response is the most important element in lung and intestinal injury, because it is generally considered to be effective in the common prevention and treatment of lung and intestinal injury in AP. However, details regarding the involved pro-inflammatory mediators and cells are still lacking.
Mesenteric lymph can concurrently cause lung and intestinal injury in AP. Therefore, surgical diversion of ML represents a treatment option. An inflammatory response is an unclear mechanism of lung and intestinal injury in AP. Therefore, currently, the greatest challenge of studies on lung and intestinal injury in AP is the elucidation of the specific mechanisms and their precise targets. Accurate detection of these mechanisms and targets and demonstration of the remarkable effects of multitarget integration will be useful for lung and intestinal injury in AP in clinical practice. Additionally, immunotherapy is also a promising approach, and it could be an effective strategy targeting and altering the activities of immune cells in AP, which might have long-term therapeutic effects.
Author Contributions
YW and DL organized thoughts for the manuscript and revised the manuscript. LW drafted the manuscript and gathered information for the manuscript. DY revised the first manuscript. ZW, YH, and MB translated and revised the manuscript. YZ, DY, and BS offered opinions for the drawing of diagrams and charts in the manuscript. All authors read and approved the final version of the manuscript.
Funding
This project was supported by the National Natural Science Foundation of China (grant numbers 82160871, 81760828, and 82074419).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Gansu Provincial Engineering Laboratory for Research and Promotion of Quality Standardization of Authentic Medicinal Materials in Gansu Province, Provincial Key Laboratory of Pharmaceutical Chemistry and Quality Research in Colleges and Universities in Gansu Province, and Gansu Experimental Animal Industry Technology Center for providing support and assistance for this review article.
Abbreviations
AP, acute pancreatitis; ALI, acute lung injury; DAMPs, damage-associated molecular patterns; HMGB1, high-mobility group box protein 1; ML, mesenteric lymph; MMPs, matrix metalloproteinases; TNF- α, tumor necrosis factor- α; IL-17, interleukin-17; IL-6, interleukin-6; IL-1 β, interleukin-1 β; TNF-R1, tumor necrosis factor-R1; TRADD, tumor necrosis factor receptor 1 related death domain protein; TRAF2, tumor necrosis factor receptor related factor 2; TLR4, Toll-like receptors 4; NLRP3, NOD-like receptor protein 3; STAT3, signal transducer and activator of transcription 3; AP-1, activator protein-1; MAPK, mitogen-activated protein kinases; NF- κ B, nuclear factor kappa B; SIRS, systemic inflammatory response syndrome; MODS, multiple organ dysfunction syndrome; Nrf2, Related factor 2; ARE, antioxidant response element; AQPs, Water channel proteins; Ang, angiopoietin; IEC, intestinal epithelial cells; AMPs, antimicrobial peptides; JAM-C, junctional adhesion molecule-C; GSH, glutathione; SOD, superoxide dismutase; ROS, reactive oxygen species; VIP, blood vessel active intestinal peptide.
References
1. Gao E, Jiang Y, Li Z, Xue D, Zhang W. Association between high mobility group box-1 protein expression and cell death in acute pancreatitis. Mol Med Rep. (2017) 15:4021–6. doi: 10.3892/mmr.2017.6496
2. Yan TY, Li YX, Sun Y, Wang H, Wang J, Wang W, et al. Hospital acquired lower respiratory tract infections among high risk hospitalized patients in a tertiary care teaching hospital in China: an economic burden analysis. J Infect Public Health. (2018) 11:507–13. doi: 10.1016/j.jiph.2017.10.003
3. Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. (2015) 386:85–96. doi: 10.1016/S0140-6736(14)60649-8
4. Franco-Pons N, Gea-Sorlí S, Closa D, Chen T, Tian JY, Si HR. Release of inflammatory mediators by adipose tissue during acute pancreatitis. J Pathol. (2010) 221:175–82. doi: 10.1002/path.2691
5. Hussain N, Wu F, Zhu L, Thrall RS, Kresch MJ. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol. (1998) 19:867–74. doi: 10.1165/ajrcmb.19.6.3118
6. Dal M, Ulutas K. Assessment of visceral and subcutaneous obesity to understand the efficiency of adipose tissue in acute pancreatitis. Niger J Clin Pract. (2021) 24:993–993. doi: 10.4103/njcp.njcp_370_19
7. Qiao SF, Sun JB, Li F. Analysis of related factors and prevention measures of pancreatic infection in patients with severe acute pancreatitis. Clin Med Ch. (2006) 22:252–3.
8. Samanta J, Singh S, Arora S, Muktesh G, Aggarwal A, Dhaka N, et al. Cytokine profile in prediction of acute lung injury in patients with acute pancreatitis. Pancreatology. (2018) 18:878–84. doi: 10.1016/j.pan.2018.10.006
9. Chen H, Su H, Yuan l. Mechanism of lung and intestinal injury in rats with acute pancreatitis. Needle Res. (2018) 43:353–9. doi: 10.1186/s12906-017-1789-X
10. Ijaz M, Adrish M. Outcomes in patients with acute lung injury/ARDS vs cardiogenic pulmonary edema. Chest. (2015) 148:e194. doi: 10.1378/chest.14-3052
11. Tamura H, Reich J, Nagaoka I. Bacterial endotoxin assays relevant to host defense peptides. Juntendo Med J. (2016) 62:132–40. doi: 10.14789/jmj.62.132
12. Garg PK, Singh VP. Organ failure due to systemic injury in acute pancreatitis. Gastroenterology. (2019) 156:2008–23. doi: 10.1053/j.gastro.2018.12.041
13. Pasari LP, Khurana A, Anchi P, Saifi MA, Annaldas S, Godugu C. Visnagin attenuates acute pancreatitis via Nrf2/NFκB pathway and abrogates associated multiple organ dysfunction. Biomed Pharmacother. (2019) 112:108629. doi: 10.1016/j.biopha.2019.108629
14. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. (2005) 353:1685–93. doi: 10.1056/NEJMoa050333
15. Sarr MG, Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, et al. The new revised classification of acute pancreatitis. Surg Clin North Am. (2012) 93:549–62.
16. Ge P, Luo Y, Okoye CS, Chen H, Liu J, Zhang G, et al. Intestinal barrier damage, systemic inflammatory response syndrome, and acute lung injury: a troublesome trio for acute pancreatitis. Biomed Pharmacother. (2020) 132:110770. doi: 10.1016/j.biopha.2020.110770
17. Shao N. The theory and practice of “lung and large intestine being exterior-interior relationship”. Henan Prov Tradit Chin Med. (2020) 40:1768–72. doi: 10.16367/j.issn.1003-5028.2020.11.0446
18. Al Mofleh IA. Severe acute pancreatitis: pathogenetic aspects and prognostic factors. World J Gastroenterol. (2008) 14:675–84. doi: 10.3748/wjg.14.675
19. Chen C, Hu H, Li X, Zheng Z, Wang Z, Wang X, et al. Rapid detection of anti-SARS-CoV-2 antibody using a selenium nanoparticle-based lateral flow immunoassay. IEEE Trans Nanobioscience. (2022) 21:37–43. doi: 10.1109/TNB.2021.3105662
20. Lu G, Pan Y, Kayoumu A, Zhang L, Yin T, Tong Z, et al. Indomethacin inhabits the NLRP3 inflammasome pathway and protects severe acute pancreatitis in mice. Biochem Biophy Res Commun. (2017) 493:827–32. doi: 10.1016/j.bbrc.2017.08.060
21. Peng C, Li Z, Yu X. The role of pancreatic infiltrating innate immune cells in acute pancreatitis. Int J Med Sci. (2021) 18:534–45. doi: 10.7150/ijms.51618
22. Choudhury S, Ghosh S, Gupta P, Mukherjee S, Chattopadhyay S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic Res. (2015) 49:1371–83. doi: 10.3109/10715762.2015.1075016
23. Maléth J, Hegyi P. Ca2+ toxicity and mitochondrial damage in acute pancreatitis: translational overview. Philos T R Soc B. (2016) 371:20150425. doi: 10.1098/rstb.2015.0425
24. Yu JH, Kim H. Oxidative stress and inflammatory signaling in cerulein pancreatitis. World J Gastroenterol. (2014) 20:17324–9. doi: 10.3748/wjg.v20.i46.17324
25. Huang L, Zhang D, Han W, Guo C. High-mobility group box-1 inhibition stabilizes intestinal permeability through tight junctions in experimental acute necrotizing pancreatitis. Inflamm Res. (2019) 68:677–89. doi: 10.1007/s00011-019-01251-x
26. Chen ZP, Huang HP, He XY, Wu BZ, Liu Y. Early continuous blood purification affects TNF−α, IL-1β, and IL-6 in patients with severe acute pancreatitis via inhibiting TLR4 signaling pathway. Kaohsiung J Med Sci. (2022) 2022:1–7. doi: 10.1002/kjm2.12497
27. Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. (2011) 141:358–69. doi: 10.1053/j.gastro.2011.03.041
28. Gao L, Dong X, Gong W, Huang W, Xue J, Zhu Q, et al. Acinar cell NLRP3 inflammasome and gasdermin D (GSDMD) activation mediates pyroptosis and systemic inflammation in acute pancreatitis. Br J Pharmacol. (2021) 178:3533–52. doi: 10.1111/bph.15499
29. Tan JH, Cao RC, Zhou L, Zhou ZT, Chen HJ, Xu J, et al. ATF6 aggravates acinar cell apoptosis and injury by regulating p53/AIFM2 transcription in severe acute pancreatitis. Theranostics. (2020) 10:8298–314. doi: 10.7150/thno.46934
30. Gulla A, Gulbinas A, Dambrauskas Z, Strupas K. Heme oxygenase-1 polymorphism is associated with the development of necrotic acute pancreatitis via vascular cell adhesion molecule-1 and the E-selectin expression regulation pathway. Pancreas. (2019) 48:787–91. doi: 10.1097/MPA.0000000000001328
31. Liu XN, Zhu QT, Zhang M, Yin T, Xu R, Xiao W, et al. Isoliquiritigenin ameliorates acute pancreatitis in mice via inhibition of oidative stress and modulation of the Nrf2/HO-1 pathway. Oxid Med Cell Longev. (2018) 2018:7161592. doi: 10.1155/2018/7161592
32. Liu Z, Qi M, Tian S, Yang Q, Liu J, Wang S, et al. Ubiquitin-Specific Protease 25 Aggravates Acute Pancreatitis and Acute Pancreatitis-Related Multiple Organ Injury by Destroying Tight Junctions Through Activation of The STAT3 Pathway. Front Cell Dev Biol. (2021) 9:806850. doi: 10.3389/fcell.2021.806850
33. Qiu Z, Xu F, Wang Z, Yang P, Bu Z, Cheng F, et al. Blockade of JAK2 signaling produces immunomodulatory effect to preserve pancreatic homeostasis in severe acute pancreatitis. Biochem Biophys Rep. (2021) 28:101133. doi: 10.1016/j.bbrep.2021.101133
34. Luan ZG, Zhang XJ, Yin XH, Ma XC, Zhang H, Zhang C, et al. Downregulation of HMGB1 protects against the development of acute lung injury after severe acute pancreatitis. Immunobiology. (2013) 218:1261–70. doi: 10.1016/j.imbio.2013.04.013
35. Guo H, Suo DW, Zhu HP, Sun XM, Chen J. Early blood purification therapy of severe acute pancreatitis complicated by acute lung injury. Eur Rev Med Pharmacol Sci. (2016) 20:873–8.
36. Zhang Y, Yan L, Han W. Elevated level of miR-551b-5p is associated with inflammation and disease progression in patients with severe acute pancreatitis. Ther Apher Dial. (2018) 22:649–55. doi: 10.1111/1744-9987.12720
37. Pastor CM, Matthay MA, Frossard JL. Pancreatitis associated acute lung injury: new insights. Chest. (2003) 124:2341–51. doi: 10.1378/chest.124.6.2341
38. Yi L, Huang X, Guo F, Zhou Z, Chang M, Jingning H. GSK-3 beta-dependent activation of GEF-H1/ROCK signaling promotes LPS-induced lung vascular endothelial barrier dysfunction and acute lung injury. Front Cell Infect Microbiol. (2017) 7:357. doi: 10.3389/fcimb.2017.00357
39. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
40. Tang RM, Qiu L, Sun R, Sun R, Cheng L, Ma X, et al. Increased interleukin-23/17axis and C-reactive protein are associated with severity of acute pancreatitis in patients. Pancreas. (2015) 44:321–5.
41. Ahmed N, Naif OA, Sheikh FA, Mohammed MA, Abdullah SA, Ali SA, et al. Blockade of interleukin-2-inducible T-cell kinase signaling attenuates acute lung injury in mice through adjustment of pulmonary Th17/Treg immune responses and reduction of oxidative stress. Int Immunopharmacol. (2020) 83:106369. doi: 10.1016/j.intimp.2020.106369
42. Liu Y, Mei J, Gonzales L, Yang G, Dai N, Wang P, et al. IL-17A and TNF-α exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J Immunol. (2011) 186:3197–205. doi: 10.4049/jimmunol.1002016
43. Liu XM, Liu QG, Xu J, Pan CE. Microcirculation disturbance affects rats with acute severe pancreatitis following lung injury. World J Gastroenterol. (2005) 11:6208–11. doi: 10.3748/wjg.v11.i39.6208
44. Sacco P, Decleva E, Tentor F, Menegazzi R, Borgogna M, Paoletti S, et al. A suitable tool for sustained inhibition of ROS release by activated neutrophils. Macromol Biosci. (2017) 17:1700214. doi: 10.1002/mabi.201700214
45. Newman JH. Pulmonary hypertension. Dtsch Arztebl Int. (2005) 114:73–84. doi: 10.3238/arztebl.2017.0073
46. Sogami M, Era S, Murakami M, Seo Y, Watari H, Uyesaka N. Application of the transition state theory to water transport across cell membranes. Biochim Biophys Acta. (2001) 1511:42–8. doi: 10.1016/s0005-2736(00)00384-9
47. Towne JE, Harrod KS, Krane CM, Menon AG. Decreased expression of aquaporin AQP1 and AQP5 in mouse lung after acute viral infection. Respir Cell Mol Biol. (2000) 22:34–44. doi: 10.1165/ajrcmb.22.1.3818
48. Zhang X, Han D, Ding D, Yao Y. Cholecystokinin octapeptide inhibits the in vitro expression of CD14 in rat pulmonary interstitial macrophages induced by lipopolysaccharide. Chin Med J (Engl). (2002) 115:276–9.
49. Pana LY, Chena YF, Lia HC, Bi LM, Sun WJ, Sun GF, et al. Dachengqi decoction attenuates intestinal vascular endothelial injury in severe acute pancreatitis in vitro and in vivo. Cell Physiol Biochem. (2017) 44:2395–406. doi: 10.1159/000486155
50. Bhatia M. Novel therapeutic targets for acute pancreatitis and associated multiple organ dysfunction syndrome. Curr Drug Targets Inflamm Allergy. (2002) 1:343–51. doi: 10.2174/1568010023344517
51. Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. (2002) 296:1634–5. doi: 10.1126/science.1071924
52. Rangamani P, Sirovich L. Survival and apoptotic pathways initiated by TNF-alpha: modeling and predictions. Biotechnol Bioeng. (2007) 97:1216–29. doi: 10.1002/bit.21307
53. Zhang X, Wu D, Jiang X. ICAM-1 and acute pancreatis complicated by acute lung injury. J Pancreas. (2009) 10:8–14.
55. Dai SR, Li Z, Zhang JB. Serum interleukin 17 as an early prognostic biomarker ofsevere acute pancreatitis receiving continuous blood purification. Int J Artif Organs. (2015) 38:192–8. doi: 10.5301/ijao.5000406
56. Honda K, Wada H, Nakamura M, Nakamoto K, Inui T, Sada M, et al. IL-17A synergistically stimulates TNF-α-induced IL-8 production in human airway epi-thelial cells: a potential role in amplifying airway inflammation. Exp Lung Res. (2016) 42:205–16. doi: 10.1080/01902148.2016.1190796
57. Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH oxidase mediates synergistic effects of IL-17 and TNF-α on CXCL1 expression by epithelial cells after lung ischemia-reperfusion. Am J Physiol Lung Cell Mol Physiol. (2011) 306:L69–79. doi: 10.1152/ajplung.00205.2013
58. Shieh JM, Tseng HY, Jung F, Yang SH, Lin JC. Elevation of IL-6 and IL-33 levels in serum associated with lung fibrosis and skeletal muscle wasting in a bleomycin-induced lung injury mouse model. Mediators Inflamm. (2019) 2019:7947596. doi: 10.1155/2019/7947596
59. Ning F, Zheng H, Tian H, Wang T, Hao D, Han S, et al. Research on effect of adiponectin on sepsis-induced lung injury in rats through IL-6/STAT3 signaling pathway. Panminerva Medica. (2019) 62:184–6. doi: 10.23736/S0031-0808.19.03650-4
60. Wu XY, Tian F, Su MH, Wu M, Huang Y, Hu LH, et al. BF211, a derivative of bufalin, enhances the cytocidal effects in multiple myeloma cells by inhibiting the IL-6/JAK2/STT3 pathway. Int Immunopharmacol. (2019) 64:24–32. doi: 10.1016/j.intimp.2018.08.016
61. Anaka T, Narazaki T, Kishimoto M. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
62. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. (2009) 27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627
63. Yao L, Yago T, Shao B, Liu Z, Silasi-Mansat R, Setiadi H, et al. Elevated CXCL1 expression in gp130-deficient endothelial cells impairs neutrophil migration in mice. Blood. (2013) 122:3832–42. doi: 10.1182/blood-2012-12-473835
64. Chavez-Sanchez L, Chavez-Rueda K, Legorreta-Haquet M, Zenteno E, Ledesma-Soto Y, Montoya-Díaz E, et al. The activation of CD14, TLR4, and TLR2 by mmLDL induces IL-1β, IL-6, and IL-10 secretion in human monocytes and macrophages. Lipids Health Dis. (2010) 9:117. doi: 10.1186/1476-511X-9-117
65. Jin F, Liu D, Yu H, Qi J, You Y, Xu X, et al. Sialic acid-functionalized PEG-PLGA microspheres loading mitochondrial-targeting-modified curcumin for acute lung injury therapy. Mol Pharm. (2018) 16:71–85. doi: 10.1021/acs.molpharmaceut.8b00861
66. Cao JP, He XY, Xu HT, Zou Z, Shi XY. Autolo-gous transplantation of peripheral blood-derived circulating endothelial progenitor cells attenuates endotoxin-induced acute lung injury in rabbits by direct endothelial repair and indirect immunomodulation. Anesthesiology. (2010) 116:1278–87. doi: 10.1097/ALN.0b013e3182567f84
67. Ding N, Wang F, Xiao H, Xu L, She S. Mechanical ventilation enhances HMGB1 expression in an LPS-induced lung injury model. PLoS One. (2013) 8:e74633. doi: 10.1371/journal.pone.0074633
68. Qu L, Chen C, Chen YY, Li Y, Tang F, Huang H, et al. High-mobility group box 1 (HMGB1) and autophagy in acute lung injury (ALI): a review. Med Sci Monitor. (2019) 25:1828–37. doi: 10.12659/MSM.912867
69. Levy BD, Hickey L, Morris AJ, Larvie M, Keledjian R, Petasis NA, et al. Novel polyisoprenyl phosphates block phospholipase D and human neutrophil activation in vitro and murine peritoneal inflammation in vivo. Br J Pharmacol. (2005) 146:344–51. doi: 10.1038/sj.bjp.0706338
70. Pereda JL, Sabater L, Aparisi L, Escobar J, Sandoval J, Viña J, et al. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr Med Chem. (2006) 13:2775–87.
71. Que RS, Cao LP, Ding GP, Hu JA, Mao KJ, Wang GF. Correlation of nitric oxide and other free radicals with the severity of acute pancreatitis and complicated systemic inflammatory response syndrome. Pancreas. (2010) 39:536–40. doi: 10.1097/MPA.0b013e3181c0e199
72. Copple IM, Goldring CE, Kitteringham NR. The keap1-nrf2 cellular defense pathway: mechanisms of regulation and role in protection against drug-induced toxicity. Handb Exp Pharmacol. (2010) 196:233–66. doi: 10.1007/978-3-642-00663-0_9
73. Al-Harbi NO, Nadeem A, Ahmad SF, AlThagfan SS, Alqinyah M, Alqahtani F, et al. Sulforaphane treatment reverses corticosteroid resistance in a mixed granulocytic mouse model of asthma by upregulation of antioxidants and attenuation of Th17 immune responses in the airways. Eur J Pharmacol. (2019) 855:276–84. doi: 10.1016/j.ejphar.2019.05.026
74. Wang Y, Zhang Y, Cheng J, Ni Q, Li PW, Han W, et al. Protective effects of BML-111 on cerulein-induced acute pancreatitis-associated lung injury via activation of Nrf2/ARE signaling pathway. Inflammation. (2014) 37:1120–33. doi: 10.1007/s10753-014-9836-y
75. Chan KH, Ng MK, Stocker R. Haem oxygenase-1 and cardiovascular disease: mechanisms and therapeutic potential. Clin Sci (Lond). (2011) 120:493–504. doi: 10.1042/CS20100508
76. Kuebler WM. Inflammatory pathways and microvascular responses in the lung. Pharmacol Rep. (2005) 57(Suppl.):196–205.
77. Zhang F, Hu L, Wu YX, Fan L, Liu WT, Wang J, et al. Doxycycline alleviates paraquat-induced acute lung injury by inhibiting neutrophil-derived matrix metalloproteinase 9. Int Immunopharmacol. (2019) 72:243–51. doi: 10.1016/j.intimp.2019.04.015
78. Zinter MS, Delucchi KL, Kong MY, Orwoll BE, Spicer AS, Lim MJ, et al. Early plasma matrix metalloproteinase profiles: a novel pathway in pediatric acute respiratory distress syndrome. Am J Respir Crit Care Med. (2019) 199:181–9. doi: 10.1164/rccm.201804-0678OC
79. Zhang X, Sun CY, Zhang YB, Guo HZ, Feng XX, Peng SZ, et al. Kegan Liyan oral liquid ameliorates lipopolysaccharide-induced acute lung injury through inhibition of TLR4-mediated NF-κB signaling pathway and MMP-9 expression. J Ethnopharmacol. (2016) 186:91–102. doi: 10.1016/j.jep.2016.03.057
80. Chakrabarti S, Patel KD. Matrix metalloproteinase-2 (MMP-2) and MMP-9 in pulmonary pathology. Exp Lung Res. (2005) 31:599–621. doi: 10.1080/019021490944232
81. Fan YX, Liu HJ, Li YX, Zhang Y, Liu W, Zhang G. Ouercetin ameliorates postoperative pain by suppressing matrix metalloproteinase in microglia. J China Pharm Univ. (2017) 48:272–81. doi: 10.1002/ejp.1116
82. Xu XL, Ji H, Gu SY, Huang Q, Chen Y. Effects of astragaloside IV on experimental ventricular remodeling in mice and its mechanism from matrixmetalloproteinase. Aspect. J China Pharm Univ. (2010) 41:70–5.
83. Chen LJ, Ding YB, Ma PL. The protective effect of lidocaine on lipopolysaccharide-induced acute lung injury in rats through NF-κB and p38 MAPK signaling pathway and excessive inflammatory responses. Eur Rev Med Pharmacol Sci. (2018) 22:2099–108. doi: 10.26355/eurrev-201804-14743
84. Ricou B, Nicod L, Lacraz S, Welgus HG, Suter PM, Dayer JM. Matrix metalloproteinases and TIMP in acute respiratory distress syndrome. Am J Respir Crit Care Med. (1996) 154:346–52. doi: 10.1164/ajrccm.154.2.8756805
85. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. (2008) 132:27–42. doi: 10.1016/j.cell.2007.12.018
86. Gukovskaya AS, Gukovsky IH, Algul A, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology. (2017) 153:1212–26. doi: 10.1053/j.gastro.2017.08.071
87. Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. (2013) 144:1199–209. doi: 10.1053/j.gastro.2013.02.007
88. Mareninova OA, Hermann K, French SW, O’Konski MS, Pandol SJ, Webster P, et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. Clin Invest. (2009) 119:3340–55. doi: 10.1172/JCI38674
89. Vrolyk V, Singh B. Animal models to study the role of pulmonary intravascular macrophages in spontaneous and induced acute pancreatitis. Cell Tissue Res. (2020) 380:207–22. doi: 10.1007/s00441-020-03211-y
90. Menger MD, Plusczyk T, Vollmar B. Microcirculatory derangements in acute pancreatitis. J Hepatobiliary Pancreat Surg. (2001) 8:187–94. doi: 10.1007/s005340170015
91. Frossard JL, Saluja A, Bhagat L, Lee HS, Bhatia M, Hofbauer B, et al. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology. (1999) 116:694–701. doi: 10.1016/s0016-5085(99)70192-7
92. Zhao X, Dib M, Wang X, Widegren B, Andersson R. Influence of mast cells on the expression of adhesion molecules on circulating and migrating leukocytes in acute pancreatitis-associated lung injury. Lung. (2005) 183:253–64. doi: 10.1007/s00408-004-2538-8
93. Gao ZM, Sui JD, Fan R, Qu W, Dong X, Sun D. Emodin protects against acute pancreatitis associated lung injury by Inhibiting NLPR3 inflammasome activation via Nrf2/HO-1 signaling. Drug Des Devel Ther. (2020) 14:1971–82. doi: 10.2147/DDDT.S247103
94. Han X, Wang Y, Chen H, Zhang J, Xu C, Li J, et al. Enhancement of ICAM-1 via the JAK2/STAT3 signaling pathway in a rat model of severe acute pancreatitis-associated lung injury. Exp Ther Med. (2016) 11:788–96. doi: 10.3892/etm.2016.2988
95. Sochor M, Richter S, Schmidt A, Hempel S, Hopt UT, Keck T. Inhibition of matrix metalloproteinase-9 with doxycycline reduces pancreatitis-associated lung injury. Digestion. (2009) 80:65–73. doi: 10.1159/000212080
96. Yang S, Song Y, Wang Q, Liu Y, Wu Z, Duan X, et al. Daphnetin ameliorates acute lung injury in mice with severe acute pancreatitis by inhibiting the JAK2–STAT3 pathway. Sci Rep. (2021) 11:1–11. doi: 10.1038/s41598-021-91008-6
97. Bumbasirevic V, Radenkovic D, Jankovic Z. Severe acute pancreatitis: overall and early versus late mortality in intensive care units. Pancreas. (2009) 38:122–5. doi: 10.1097/MPA.0b013e31818a392f
98. Zou XP, Chen M, Wei W, Cao J, Chen L, Tian M. Effects of enteral immunonutrition on the maintenance of gut barrier function and immune function in pigs with severe acute pancreatitis. JPEN J Parenter Enteral Nutr. (2010) 34:554–66. doi: 10.1177/0148607110362691
99. Kylanpaa ML, Repo H, Puolakkainen PA. Inflammation and immunosuppression in severe acute pancreatitis. World J Gastroenterol. (2010) 16:2867–72. doi: 10.3748/wjg.v16.i23.2867
100. Huang L, Jiang Y, Sun Z, Gao Z, Wang J, Zhang D. Autophagy strengthens in testinal mucosal barrier by attenuating oxidative stress in severe acute pancreatitis. Dig Dis Sci. (2018) 63:910–9. doi: 10.1007/s10620-018-4962-2
101. Schietroma M, Pessia B, Carlei F, Mariani P, Amicucci G. Intestinalperme ability and systemic endotoxemia in patients with acute pancreatitis. Ann Ital Chir. (2016) 87:138–44.
102. Tian R, Tan JT, Wang RL, Xie H, Qian YB, Yu KL. The role of intestinal mucosa oxidative stress in gut barrier dysfunction of severe acute pancreatitis. Eur Rev Med Pharmacol Sci. (2013) 17:349–55.
103. Tiszlavicz Z, Szabolcs A, Takacs T, Farkas G, Kovacs-Nagy R, Szantai E, et al. Polymorphisms of beta defensins are associated with the risk of severe acute pancreatitis. Pancreatology. (2010) 10:483–90. doi: 10.1159/000276987
104. Arike L, Hansson GC. The densely O-Glycosylated MUC2 mucin protects the intestine and provides food for the commensal bacteria. J Mol Biol. (2016) 428:3221–9. doi: 10.1016/j.jmb.2016.02.010
105. Feng DX, Peng W, Chen YF, Chen T, Tian JY, Si HR, et al. Down-regulation of aquaporin 1 in rats with experimental acute necrotizing pancreatitis. Pancreas. (2012) 41:1092–8. doi: 10.1097/MPA.0b013e318249938e
106. Esrefoglu M. Experimental and clinical evidence of antioxidant therapy in acute pancreatitis. World J Gastroenterol. (2012) 18:5533–41.
107. Yang F, Xie J, Wang W, Xie Y, Sun H, Jin Y, et al. Regional arterial infusion with lipoxin A4 attenuates experimental severe acute pancreatitis. PLoS One. (2014) 9:e108525. doi: 10.1371/journal.pone.0108525
108. Dong Z, Shang H, Chen YQ, Xu L, She S. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2-mediated oxidative stress and NLRP3 inflammatory pathway. Oxid Med Cell Longev. (2016) 2016:7864150. doi: 10.1155/2016/7864150
109. Yu QH, Zhang PX, Liu Y, Liu W, Yin N. Hyperbaric oxygen preconditioning protects the lung against acute pancreatitis induced injury via attenuating inflammation and oxidative stress in a nitric oxide dependent manner. Biochem Biophys Res Commun. (2016) 478:93–100. doi: 10.1016/j.bbrc.2016.07.087
110. Alfieri A, Ong AC, Kammerer RA, Solanky T, Bate S, Tasab M, et al. Angiopoietin-1 regulates microvascular reactivity and protects the microcirculation during acute endothelial dysfunction: role of eNOS and VE-cadherin. Pharmacol Res. (2014) 80:43–51. doi: 10.1016/j.phrs.2013.12.008
111. Kim S, Kwon J. COMP-Ang1 inhibits apoptosis as well as improves the attenuated osteogenic differentiation of mesenchymal stem cells induced by advanced glycation end products. Biochim Biophys Acta. (2013) 1830:4928–34. doi: 10.1016/j.bbagen.2013.06.035
112. Anand SK, Singh JA, Tikoo SK. Effect of bovine adenovirus 3 on mitochondria. Vet Res. (2014) 45:45. doi: 10.1186/1297-9716-45-45
113. Chan DC. Dynamic organelles in disease, aging, and development. Cell. (2006) 125:1241–52. doi: 10.1016/j.cell.2006.06.010
114. Niklison MV, Chirou F, Dupuy L, Gallego SM, Barreiro-Arcos ML, Avila C, et al. Microcin J25 triggers cytochrome crelease through irreversible damage of mitochondrial proteins and lipids. Int Biochem Cell Biol. (2010) 42:273–81. doi: 10.1016/j.biocel.2009.11.002
115. Mittal A, Hickey AJ, Chai CC, Loveday BP, Thompson N, Dare A, et al. Early organ-specific mitochondrial dysfunction of jejunum and lung found in rats with experimental acute pancreatitis. HPB (Oxford). (2011) 13:332–41. doi: 10.1111/j.1477-2574.2010.00290.x
116. Sanna MI, Jyrki MK. Effect of acute pancreatitis on porcine intestine: a morphological study. Uitrastruct Pathol. (2013) 37:127–38. doi: 10.3109/019131232012.745638
117. Blenkiron C, Askelund KJ, Shanbhag ST, Chakraborty M, Petrov MS, Delahunt B, et al. MicroRNAs in mesenteric lymph and plasma during acute pancreatitis. Ann Surg. (2014) 260:341–7. doi: 10.1097/SLA.0000000000000447
118. Zheng XL, Yang Y, Wang BJ, Tang HQ, Zhou XY. To discuss the exterior and interior relationship between lung and large intestine from the perspective of changes of the pulmonary andintestinal function under pathological state. China J Tradit Chin Med Pharm. (2014) 29:120–3.
119. Zhu JY, Li Y, Zhang M, Li J, Li JY, Wu FS, et al. Pharmacokinetics and pharmacodynamics of Shengjiang decoction in rats with acute pancreatitis for protecting against multiple organ injury. World J Gastroenterol. (2017) 23:8169–81. doi: 10.3748/wjg.v23.i46.8169
120. Liu H, Li W, Wang X, Li J, Yu W. Early gut mucosal dysfunction in patients with acute pancreatitis. Pancreas. (2008) 36: 192–6.
121. Zhang XP, Zhang J, Song QL, Chen HQ. Mechanism of acute pancreatitis complicated with injury of intestinal mucosa barrier. J Zhejiang Univ Sci B. (2007) 8:888–95. doi: 10.1631/jzus.2007.B0888
122. Xu GF, Guo MZ, Tian Q, Wu GZ, Zou XP, Zhang WJ. Increased of serum high-mobility group box chromosomal protein 1 correlated with intestinal mucosal barrier injury in patients with severe acute pancreatitis. World J Emerg Surg. (2014) 9:61. doi: 10.1186/1749-7922-9-61
123. Chen X, Zhao HX, Bai C, Zhou XY. Blockade of high-mobility group box 1 attenuates intestinal mucosal barrier dysfunction in experimental acute pancreatitis. Sci Rep. (2017) 7:1–10. doi: 10.1038/s41598-017-07094-y
124. Kang R, Lotze MT, Zeh HJ, Billiar TR, Tang D. Cell death and DAMPs in acute pancreatitis. Mol Med. (2014) 20:466–77. doi: 10.2119/molmed.2014.00117
125. Lin Y, Lin LJ, Jin Y, Cao Y, Zhang Y, Zheng CQ, et al. Correlation between serum levels of high mobility group box-1 protein and pancreatitis: a meta-analysis. Biomed Res Int. (2015) 2015:430185. doi: 10.1155/2015/430185
126. Capurso G, Zerboni G, Signoretti M, Valente R, Stigliano S, Piciucchi M, et al. Role of the gut barrier in acute pancreatitis. World J Gastroenterol. (2012) 26:2187–93. doi: 10.3748/wjg.v26.i18.2187
127. Guo ZZ, Wang P, Yi ZH, Huang ZY, Tang CW. The crosstalk between gut inflammation and gastrointestinal disorders during acute pancreatitis. Curr Pharm Des. (2014) 20:1051–62. doi: 10.2174/13816128113199990414
128. Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. (2002) 45:173–202. doi: 10.1053/pcad.2002.130041
129. Chen GS, Huang KF, Huang CC, Wang JY. Thaliporphine derivative improves acute lung injury after traumatic brain injury. Biomed Res Int. (2015) 2015:729831. doi: 10.1155/2015/729831
130. Sakai H, Sagara A, Matsumoto K, Hasegawa S, Sato K, Nishizaki M, et al. 5-fluorouracil induces diarrhea with changes in the expression of inflammatory cytokines and aquaporins in mouse intestines. PLoS One. (2013) 8:e54788. doi: 10.1371/journal.pone.0054788
131. Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. (2002) 82:245–89. doi: 10.1152/physrev.00026.2001
132. Penalva JC, Martinez J, Laveda R, Esteban A, Muñoz C, Sáez J, et al. A study of intestinal permeability in relation to the inflammatory response and plasma endocab IgM levels in patients with acute pancreatitis. J Clin Gastroenterol. (2004) 38:512–7. doi: 10.1097/01.mcg.0000129060.46654.e0
133. Guo YY, Li HX, Zhang Y, He WH. Hypertriglyceridemia-induced acute pancreatitis: progress on disease mechanisms and treatment modalities. Discov Med. (2019) 27:101–9.
134. Yu J, Lan N, Zhang XY, Zhang J, Abdel-Razek O, Wang G. Surfactant prorein D dampens lung injury by suppressing NLRP3 inflammasome activation NF-κB signaling in acute pancreatitis. Shock. (2019) 51:557–68. doi: 10.1097/SHK.0000000000001244
135. Awla D, Abdulla A, Regn S. TLR4 but not TLR2 regulates inflammation and tissue damage in acute pancreatitis induced by retrograde infusion of taurocholate. Inflamm Res. (2011) 60:1093–8. doi: 10.1007/s00011-011-0370-1
136. Chen WW, Yuan CC, Lu YY. Tanshinone IIA protects against acute pancreatitis in mice by inhibiting oxidative stress via the Nrf2/ROS pathway. Oxid Med Cell Longev. (2020) 2020:5390482. doi: 10.1155/2020/5390482
Keywords: acute pancreatitis, lung injury, intestinal injury, inflammation response, oxidative stress, endocrine disorders
Citation: Liu D, Wen L, Wang Z, Hai Y, Yang D, Zhang Y, Bai M, Song B and Wang Y (2022) The Mechanism of Lung and Intestinal Injury in Acute Pancreatitis: A Review. Front. Med. 9:904078. doi: 10.3389/fmed.2022.904078
Received: 25 March 2022; Accepted: 10 May 2022;
Published: 07 July 2022.
Edited by:
Luigi M. Terracciano, University of Basel, SwitzerlandReviewed by:
Vera Luiza Capelozzi, University of São Paulo, BrazilZhigang Chang, Peking University, China
Copyright © 2022 Liu, Wen, Wang, Hai, Yang, Zhang, Bai, Song and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongfeng Wang, d3lmQGdzenkuZWR1LmNu
†These authors have contributed equally to this work