
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 14 June 2022
Sec. Regulatory Science
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.893400
 Jorn Mulder1*
Jorn Mulder1* Odile C. van Stuijvenberg1
Odile C. van Stuijvenberg1 Paula B. van Hennik1
Paula B. van Hennik1 Emile E. Voest2,3
Emile E. Voest2,3 Anna M. G. Pasmooij1
Anna M. G. Pasmooij1 Violeta Stoyanova-Beninska1Anthonius de Boer1,4
Violeta Stoyanova-Beninska1Anthonius de Boer1,4There are currently four anti-cancer medicinal products approved for a tissue-agnostic indication. This is an indication based on a common biological characteristic rather than the tissue of origin. To date, the regulatory experience with tissue-agnostic approvals is limited. Therefore, we compared decision-making aspects of the first tissue-agnostic approvals between the Food and Drug Administration (FDA), European Medicines Agency (EMA) and Pharmaceuticals and Medical Devices Agency (PMDA). Post-marketing measures (PMMs) related to the tissue-agnostic indication were of specific interest. The main data source was the publicly available review documents. The following data were collected: submission date, approval date, clinical trials and datasets, and PMMs. At the time of data collection, the FDA and PMDA approved pembrolizumab, larotrectinib, and entrectinib for a tissue-agnostic indication, while the EMA approved larotrectinib and entrectinib for a tissue-agnostic indication. There were differences in analysis sets (integrated vs. non-integrated), submission dates and requests for data updates between agencies. All agencies had outstanding issues that needed to be addressed in the post-market setting. For pembrolizumab, larotrectinib and entrectinib, the number of imposed PMMs varied between one and eight, with the FDA requesting the most PMMs compared to the other two agencies. All agencies requested at least one PMM per approval to address the remaining uncertainties related to the tissue-agnostic indication. The FDA and EMA requested data from ongoing and proposed trials, while the PMDA requested data from use-result surveys. Confirmation of benefit in the post-marketing setting is an important aspect of tissue-agnostic approvals, regardless of agency. Nonetheless, each approach to confirm benefit has its inherent limitations. Post-marketing data will be essential for the regulatory and clinical decisions-making of medicinal products with a tissue-agnostic indication.
On May 23, 2017, the Food and Drug Administration (FDA) approved for the first time a medicinal product for a tissue-agnostic indication, i.e., an indication based on a common biological characteristic rather than the tissue of origin (1). This approval concerned pembrolizumab for the treatment of patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors. During the following years, the FDA approved three other medicinal products for a tissue-agnostic indication and pembrolizumab for an additional tissue-agnostic indication (2–5). The European Medicines Agency (EMA) and Pharmaceuticals and Medical Devices Agency (PMDA), as well as other agencies, also recommended approval for medicinal products for a tissue-agnostic indication. These recommendations resulted in approvals granted by the European Commission (EC) and the Japanese Ministry of Health, Labor and Welfare (MHLW), respectively.
Pembrolizumab for the treatment of MSI-H/dMMR solid tumors was granted ‘accelerated approval’ by the FDA (1). Accelerated approval is based on an effect on a surrogate endpoint that is likely to predict clinical benefit (6). For this type of approval, the drug developer is obligated to provide additional data in the post-marketing setting to confirm clinical benefit. Hence, confirmatory data will be expected for pembrolizumab for the abovementioned indication. To accommodate similar cases, the EMA also established – years ago – an expeditated approval pathway comparable to that of the FDA called “conditional marketing authorization.” Conditional marketing authorization is based on less comprehensive data than normally required (7). Importantly, both expedited programs are developed to address an unmet medical need (6, 7).
As there are currently few tissue-agnostic approvals, regulatory experience is limited. The EMA and PMDA have issued guidance documents that include information on master protocol designs and the registration of medicinal products for tissue-agnostic indications (8, 9). Another relevant document is the FDA guidance for industry on developing targeted therapies in low-frequency molecular subsets of a disease (10). However, detailed information on data requirements for tissue-agnostic approvals or how to confirm the tissue-agnostic indication in the post-marketing setting is currently not available. Therefore, we believe that evaluation of the first tissue-agnostic approvals will allow to better define the requirements for this type of indication.
The main purpose of our study was to compare decision-making aspects of the first tissue-agnostic approvals between regulatory agencies. We were specifically interested in the post-marketing measures (PMMs) imposed to resolve outstanding issues related to the tissue-angostic indication, and whether these PMMs were different between regulatory agencies. Other aspects of interest were the datasets supporting each approval and the submission and approval dates. We limited our research to the founding regulatory members of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, i.e., the FDA, EMA and PMDA.
The primary data source was the publicly available review documents. These included the Multi-Discipline Reviews from the FDA, the European Public Assessment Reports (EPARs) from the EMA, and the Review Reports from the PMDA. Other documents used in this study were the Approval Letters from the FDA, Summary of Product Characteristics (SmPCs) from the EMA, and the List of Approved Products from the PMDA. Review documents are available at the website of each respective agency. Review documents are available at: https://www.ema.europa.eu/en/medicines, https://www.accessdata.fda.gov/scripts/cder/daf/, and https://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0001.html.
The following data were collected from the review documents: submission date, approval date, (pivotal) clinical trials, clinical datasets, and PMMs. For procedures reviewed by the EMA and the PMDA, the approval date was the date the EC and MHLW granted approval, respectively. The clinical datasets were those initially included in the submission, i.e., without any updates. For the post-marketing activities the overarching term “post-marketing measures” was used. This includes the “postmarked requirements,” “post-authorization measures” and “post-marketing investigations” as defined by the FDA, EMA and PMDA, respectively. The final date for data collections was 14 May 2021 and last check for tissue-agnostic approvals per agency was 1 October 2021.
The submission and approval dates, clinical data packages, and the PMMs were descriptively compared between agencies. No statistics were used in this study.
Both the FDA and MHLW approved pembrolizumab for the treatment of patients with MSI-H or dMMR solid tumors (Figure 1). The application was first submitted to the FDA, with a difference of 571 days between submissions. The FDA granted “accelerated approval” and the MHLW granted “partial change approval.” Of note, on July, 2021 an extension of indication was applied for at the EMA for pembrolizumab as monotherapy in the treatment of unresectable or metastatic MSI-H or dMMR colorectal, endometrial, gastric, small intestine, biliary, or pancreatic cancer in adults who have received prior therapy (11). The review was ongoing at the final check for data availability (October 1, 2021). The FDA also approved pembrolizumab for the treatment of patients with Tumor Mutational Burden (TMB)-High solid tumors, i.e., a second tissue-agnostic indication for this medicinal product (12). However, no review documents were available for this approval at the final check for data availability.
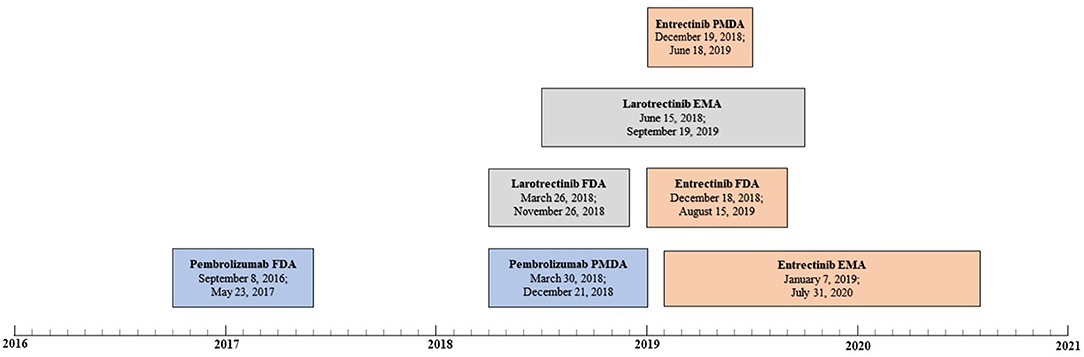
Figure 1. Review timelines for medicinal products for tissue-agnostic indications per agency (pembrolizumab in blue, larotrectinib in gray, and entrectinib in orange). For each review, the submission date and approval data are displayed, respectively.
The FDA and EC approved larotrectinib for the treatment of solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion (Figure 1). The application was first submitted to the FDA, and the difference between submissions was 82 days. The FDA granted an “accelerated approval,” and the EC granted a “conditional marketing authorization.” On March 23, 2021, the MHLW approved larotrectinib, but no review documents were available at the final check for data availability (13).
The FDA, EC and MHLW approved entrectinib for the treatment of solid tumors that have a NTRK gene fusion (Figure 1). The application was first submitted to the FDA but the difference between the submission dates was small – 21 days or less. The FDA granted an “accelerated approval,” the EC granted a “conditional marketing authorization” and the MHLW granted a “new approval.”
Finally, on August 17, 2021, the FDA granted an “accelerated approval” for dostarlimab for the treatment of patients with dMMR recurrent or advanced solid tumors. Since only the FDA approved dostarlimab for a tissue-agnostic indication no comparison could be made between the three agencies. Therefore, dostarlimab was not included in our analysis.
The FDA and PMDA approved pembrolizumab for MSI-H/dMMR solid tumors based on five and two clinical trials, respectively (Table 1). Data from KEYNOTE158 and KEYNOTE164 were submitted to both agencies, but different data cut-off dates were used. The most frequent tumor types included in the efficacy datasets were colorectal cancer and endometrial cancer (Supplementary Table 1). The application submitted to the FDA included pooled efficacy data from five clinical trials and pooled safety data from two trials, i.e., KEYNOTE016A and KEYNOTE164. The pooled efficacy population consisted of 149 patients. The primary endpoint was overall response rate (ORR). ORR was 35.6% (95% CI: 27.9, 43.8). The pooled safety population consisted of 89 patients. During the review, the FDA received updated efficacy data from KEYNOTE158 and KEYNOTE164. The data cut-off dates for the updated efficacy data were August 7, 2016 and November 23, 2016 for KEYNOTE158 and August 3, 2016 and November 23, 2016 for KEYNOTE164. Updated safety data was also provided, but the safety review was based on the initial dataset. The application submitted to the PMDA included separately presented efficacy and safety data from KEYNOTE158 and KEYNOTE164. ORR was the primary endpoint in both studies. Response rates were 34.9% (95% CI: 24.8, 46.2) and 27.9% (95% CI: 17.1, 40.8), respectively.
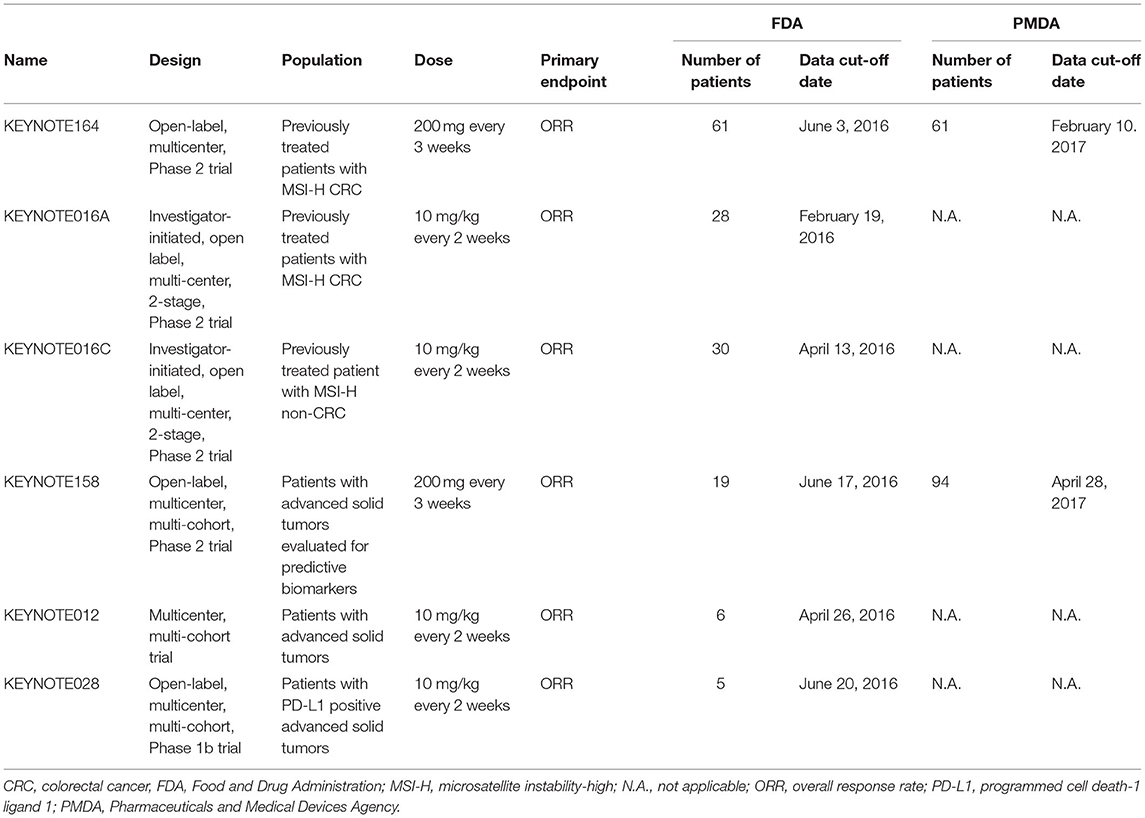
Table 1. Clinical trials supporting the approval of pembrolizumab for the treatment of MSI-H/dMMR tumors.
The FDA and EMA approved larotrectinib for NTRK fusion-positive solid tumors on the basis of three clinical trials (Table 2). The most frequent tumor types included in the efficacy datasets were sarcoma and salivary gland (Supplementary Table 1). Both applications included pooled efficacy and safety data from the three clinical trials, but there were small differences in data cut-off dates and population sizes. The primary endpoint of the integrated analysis was ORR. The agencies focused their review on different analysis sets, which resulted in differently sized efficacy populations. The FDA focused on the primary analysis set (PAS), which consisted of the first 55 patients with NTRK fusion-positive solid tumors enrolled across the three studies. ORR was 75% (95% CI: 61, 85). The pooled safety population consisted of 144 patients from three studies. During the review, the FDA received updated efficacy and safety data. The updated clinical data cut-off date was February 19, 2018. The EMA initially focused their review on the PAS – same as the FDA – but considered the second extended PAS (ePAS2) to be the main analysis set. This analysis set consisted of 93 patients from the same clinical trials but was based on updated efficacy data. ORR in the ePAS2 was 72% (95% CI: 62, 81). The pooled safety dataset consisted of 176 patients from three studies During the review, the EMA also received updated safety data. The updated clinical cut-off date was July 30, 2018.
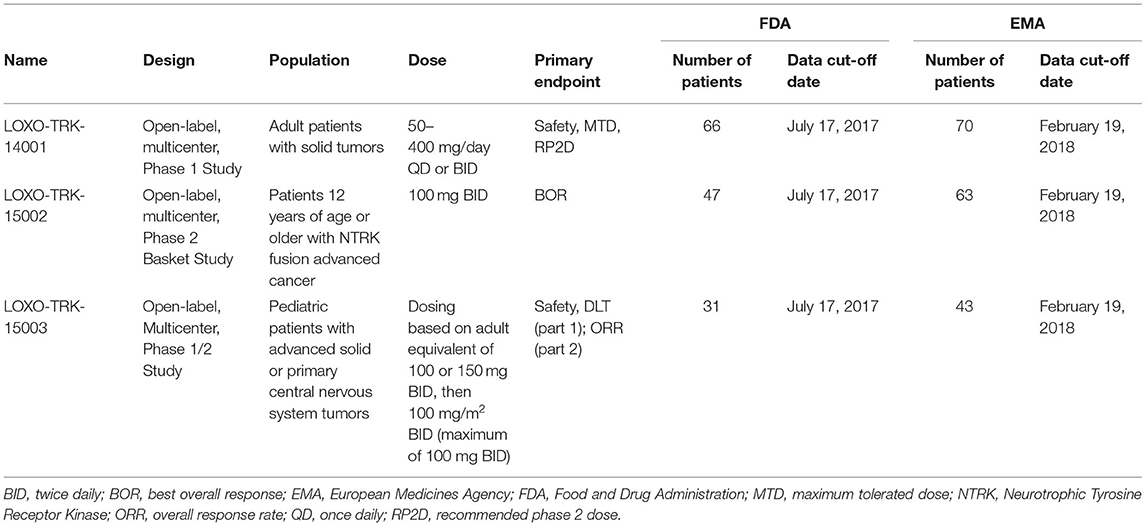
Table 2. Clinical trials supporting the approval of larotrectinib for the treatment of NTRK tumors.
The FDA and EMA approved entrectinib for NTRK fusion-positive solid tumors on the basis of four clinical trials, while the PMDA approved entrectinib for NTRK fusion-positive solid tumors on the basis of three clinical trials (Table 3). The most frequent tumor types included in the efficacy datasets were sarcoma and salivary gland (Supplementary Table 1). The applications submitted to the FDA and EMA included pooled efficacy and safety data from these clinical trials. The primary endpoint for the integrated efficacy analysis was ORR. The FDA and EMA focused on different analysis sets, which resulted in differently sized efficacy populations. The FDA focused on the PAS, which consisted of the first 54 patients enrolled in ALKA-372-001, RXDX-101-01, and RXDX-101-02. ORR was 57.4% (95% CI: 43.2, 70.8). The pooled safety population consisted of 355 patients from four studies. Updated efficacy and safety data was submitted. The updated clinical data cut-off date was October 31, 2018. The EMA initially focused on the PAS, but considered the extended PAS to be the main analysis set. This analysis set consisted of 74 patients from ALKA-372-001, RXDX-101-01, and RXDX-101-02, but was based on updated efficacy data. ORR in the extended PAS was 63.5% (95% CI: 51.5, 74.4). The pooled safety population consisted of 355 patients from four studies. During the review, the EMA also received updated safety data. The updated clinical data cut-off date was Oct 31, 2018. The application submitted to the PMDA included efficacy data from RXDX-101-02 and separately presented safety data from ALKA-372-001, RXDX-101-01, and RXDX-101-02.The PMDA focused on the PAS that consisted of 51 patients from study RXDX-101-02. The primary endpoint was ORR. Response rate was 56.9% (95% CI: 42.3, 70.7).
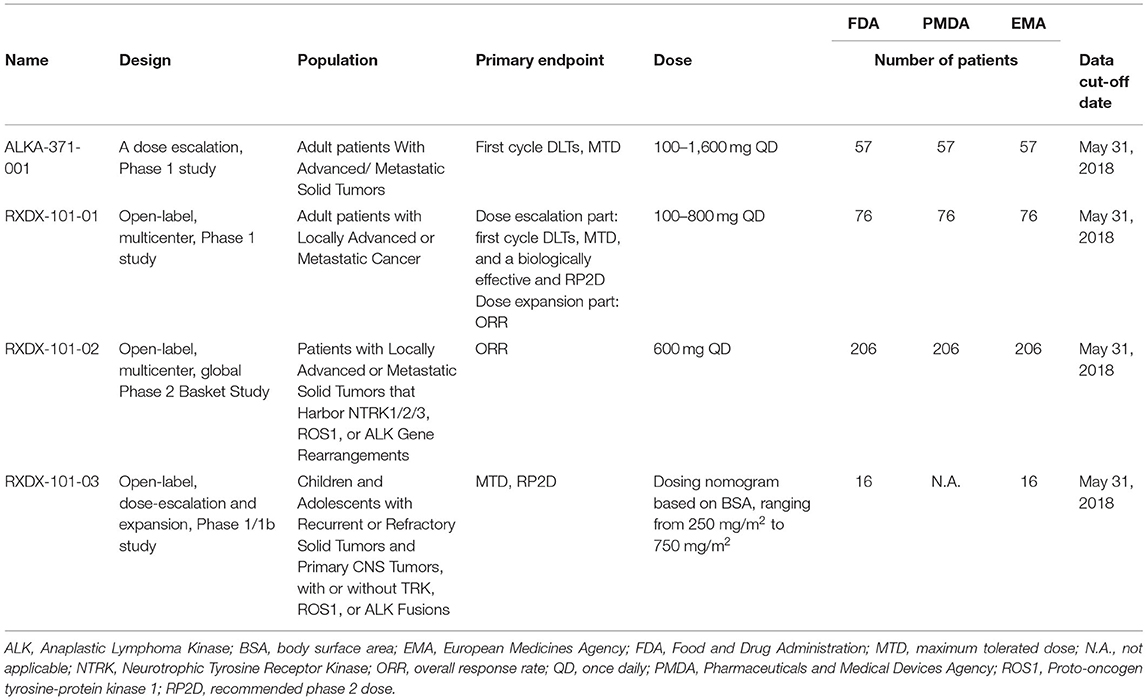
Table 3. Clinical trials supporting the approval of entrectinib for the treatment of NTRK tumors.
The FDA imposed four PMMs and the PMDA imposed one PMM for pembrolizumab (Table 4 and Supplementary Table 2). Both agencies imposed a PMM to address the small sample size of patients with MSI-H/dMMR solid tumors other than colorectal cancer. To resolve this issue, the FDA requested the final study reports from KEYNOTE 158 and KEYNOTE 164. In contrast, the PMDA requested an use-result survey. The FDA imposed three other PMMs, which are related to safety in pediatric patients and use of companion diagnostics. The latter was not relevant to the PMDA review, as a companion diagnostic assay was approved in Japan on 10 September 2018.
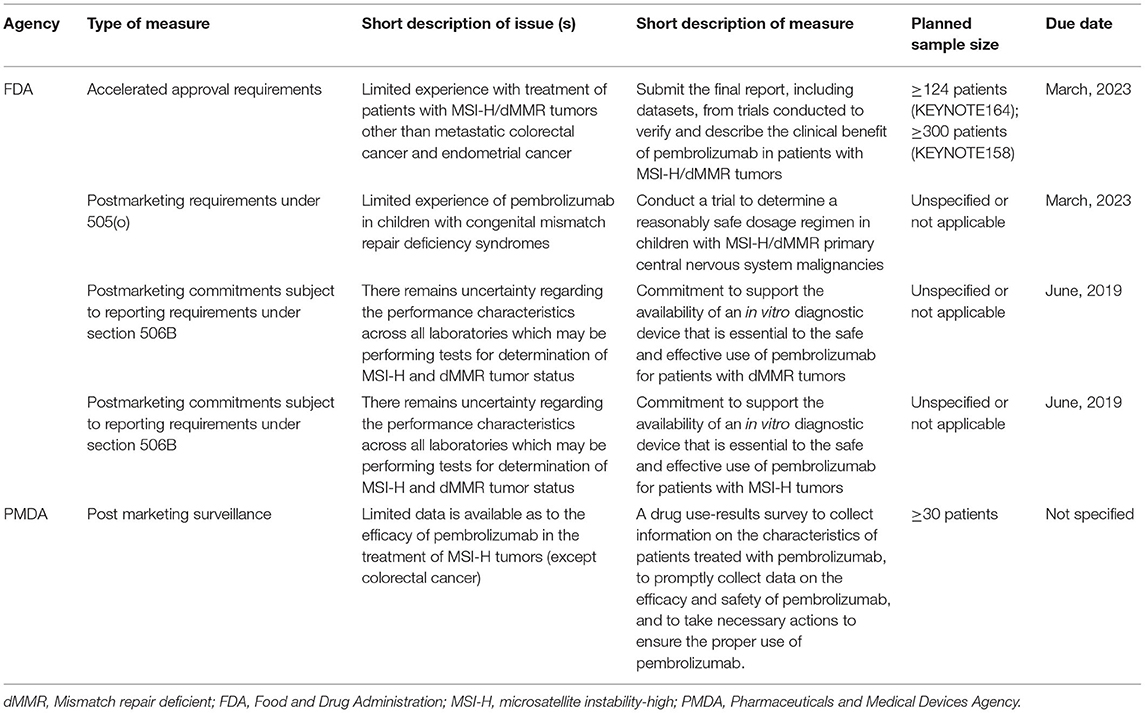
Table 4. Post-marketing measures for pembrolizumab for the treatment of MSI-H/dMMR tumors.
The FDA imposed seven PMMs and the EMA imposed three PMMs for larotrectinib. Both agencies imposed a PMM to address the small sample size of patients with NTRK fusion-positive tumors in relation to the complexity of the tissue-agnostic indication (Table 5 and Supplementary Table 2). Both agencies requested a larger dataset from (ongoing) clinical trials to resolve this issue. Unique to the PMM imposed by the EMA is that it will address two additional issues, i.e., lack of prospectively studied cohorts and secondary NTRK mutations that cause resistance to larotrectinib. In addition, both agencies imposed a PMM to address the limited safety data in pediatric patients, which resulted in comparable measures. The other PMMs were imposed by only one of the two agencies, and addressed issues related to: (1) duration of response (FDA only); (2) the dose in pediatric patients (EMA only); (3) the third dosage modification (FDA only); (4) moderate CYP inhibitors/inducers (FDA only); and (5) companion diagnostics (FDA only). The underlying issues were not identified by the other agency or were not relevant for the review. The approval of a companion diagnostic was not required by the EMA, but it is indicated in the SmPC that the presence of a NTRK gene fusion in a tumor specimen should be confirmed with a validated test. The risks related to co-administration with CYP inhibitors/inducers were addressed in the SmPC of Vitrakvi. The following statement was included: “No clinical data is available on the effect of a moderate inducer, but a decrease in larotrectinib exposure is expected.”
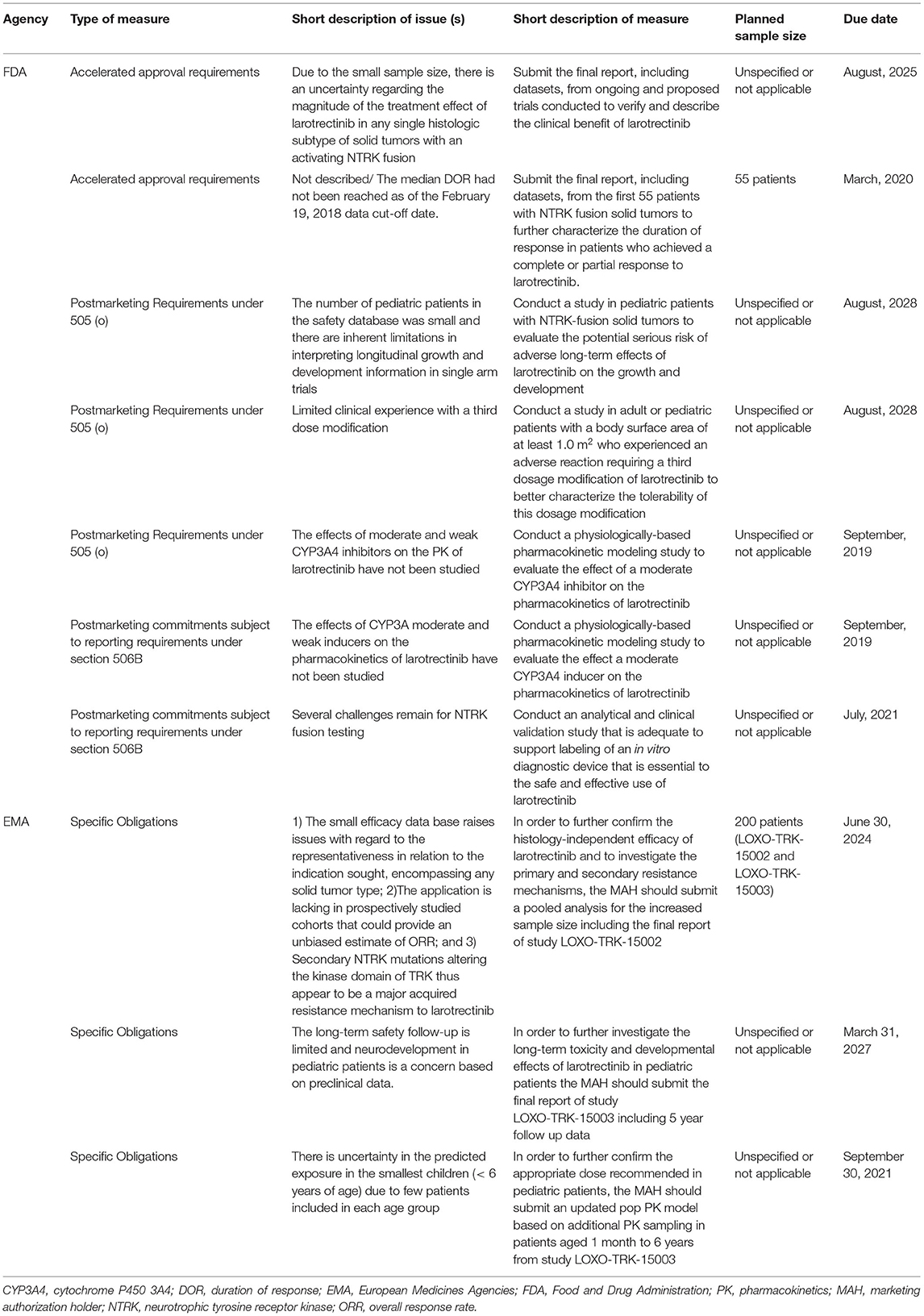
Table 5. Post-marketing measures for larotrectinib for the treatment of NTRK tumors.
The FDA imposed eight measures and the PMDA and EMA imposed two measures each for entrectinib (Table 6 and Supplementary Table 2). All three agencies imposed a PMM to address the small sample size of patients with NTRK fusion-positive tumors in relation to the complexity of a tissue-agnostic indication. The FDA and EMA both requested an expanded dataset from ongoing clinical trials. The PMDA requested an use-result survey. The agencies also imposed, sometimes integrated, PMMs to address to the risk of fractures (FDA, EMA), growth and development issues (FDA, PMDA, and EMA), and congestive heart failure (FDA, PMDA, EMA). The other PMMs were imposed by only one of the three agencies, and addressed issues related to: (1) the duration of response (FDA only); (2) off-target activity (FDA only); (3) dose in patients with hepatic impairment (FDA only); (4) companion diagnostics (FDA only); and (5) concomitant genetic mutations (EMA only). The underlying issues were not identified by the other agencies or were not relevant for the review. A companion diagnostic was already approved in Japan. The approval of a companion diagnostic was not a requirement by the EMA, but it is indicated in the SmPC that a validated assay is required for the selection of patients with NTRK gene fusion-positive solid tumors.
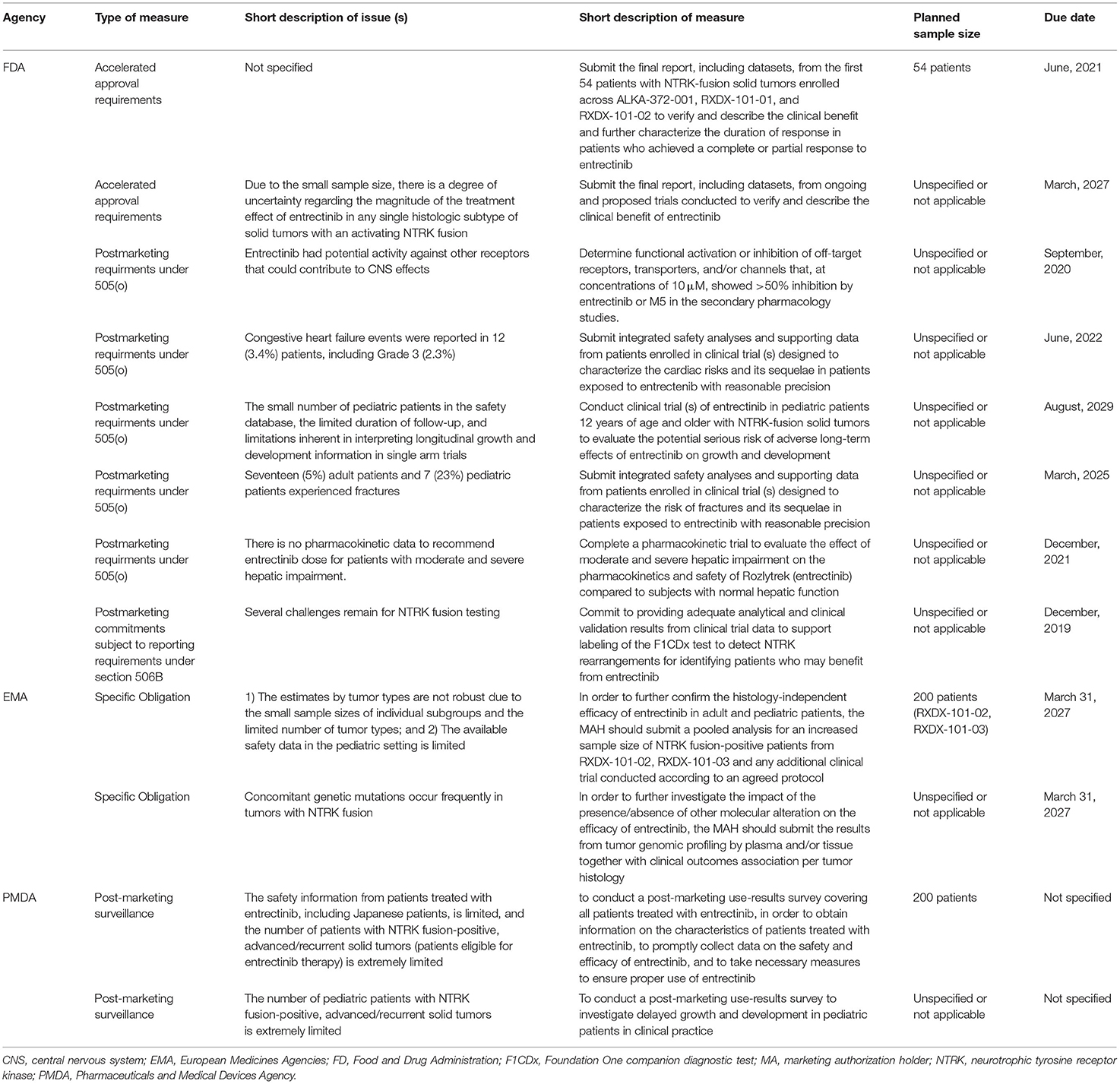
Table 6. Post-marketing measures for entrectinib for the treatment of NTRK tumors.
This study was conducted to compare decision-making aspects of tissue-agnostic approvals between regulatory agencies. Specifically, we were interested in the PMMs imposed by the regulatory agencies to resolve outstanding issues related to the tissue-agnostic indication. At the time of our analysis, pembrolizumab, larotrectinib and entrectinib received regulatory approval for a tissue-agnostic indication(s) by most, or all three, regulatory agencies. Datasets supporting the approvals were generally small, especially for the two TRK inhibitors. Numerous PMMs were imposed by the agencies, and these were mostly imposed to address issues related to efficacy, safety, and pharmacokinetics.
There remain uncertainties concerning the treatment effect across tumor types for medicinal products for tissue-agnostic indications. Our data shows that all three agencies will receive additional data to further confirm the tissue-agnostic indication in the post-marketing setting. Larger datasets will be collected from (ongoing) clinical trials or real-world studies. These studies shall include patients with tumor types that were underrepresented in the initial datasets. However, both approaches to collect data in the post-marketing setting, i.e., clinical trials vs. real-world studies, have their inherent challenges and/or limitations. For example, clinical trials may have stringent eligibility criteria, which result in limited generalizability of the study results (14). While generalization may be improved with real-world studies, these studies are not without challenges either, which can be of operational, technical and/or methodological nature (15). For instance, the quality and completeness of information is critical to describe the patients characteristics and treatment effects (16). There is currently limited guidance available on the data requirements to confirm a tissue-agnostic indication in the post-marketing setting. In the guideline of the PMDA it is recommended that high quality data, including data from cancer types not evaluated in the clinical trials, is required after marketing authorization (9). For future applications, a global strategy toward the generation of data in the post-marketing setting may be of interest, dependent on emerging insights from the current approaches.
As the FDA and EC granted expedited approval, i.e., “accelerated approval” and “conditional marketing authorization,” respectively, the drug developers are obligated to provide confirmatory data in the post-marketing setting. Importantly, regulatory action can be taken if new data alters the benefit-risk ratio, which might also impact the tissue-agnostic indication. Randomized-controlled trials remain the gold standard to determine the efficacy and safety of a medicinal product and are often preferred as confirmatory trials. However, as addressed by the FDA reviewers, it will be challenging to conduct a randomized-controlled trial in the tissue-agnostic setting (17). Some biomarker-tumor combinations are extremely rare (18). The low prevalence of a biological characteristic may lead to recruitment challenges, even for basket trials (19). In addition, shared among medicinal products approved for a tissue-agnostic indication is the “strong scientific rationale” (20). Such a scientific/biological rationale and the demonstration of large treatment effects may bring clinical equipoise into question. This explains why confirmatory data for the tissue-agnostic approvals will be obtained from ongoing single-arm (basket) trials or real-worlds studies. Recently, Seligson el al. reflected on tissue-agnostic approvals by the FDA and suggested that real-world data can be used as an alternative strategy to confirm benefit (20). Miksad et al. described that the identification of real-world patients could expand the evidence base for the NTRK population (16). Real-world data might also provide a more comprehensive understanding of long-term effects for drugs approved on the basis of earlier phase clinical trials, i.e., phase 1 or phase 2 trials (21). More general, it has been published that post-marketing studies are sometimes delayed (22). Regardless of PMM, it is important that regulators insist that the due date for these (mandatory) measures will be met, as the confirmation of benefit is an important aspect of tissue-agnostic approvals.
The first approvals set precedent for the level of evidence necessary to register medicinal products for a tissue-agnostic indication. Our results show that tissues-agnostic approvals are supported by somewhat small (efficacy) datasets – albeit sufficient for the initial assessment of the benefit-risk balance, especially considering the complexity of a tissue-agnostic indication. Furthermore, agencies have a different approach toward reviewing the submitted data, i.e., some agencies accept integrated datasets, focus on particular analysis sets and/or request updated data. Findings from literature confirm that the size of the efficacy populations supporting the tissue-agnostic approvals can be considered smaller than generally observed for registration. For example, Tenhunen et al. reported that the median number of patients in the target population was 175 for EC approvals based on results from single-arm trials (23). This is substantially larger than the initial efficacy population supporting the approval of the TRK inhibitors. The current tissue-agnostic approvals were based on a strong biological rationale and thereby the expectation of consistency in treatment effect across tumor types (20). The importance of a strong biological rationale to support a tissue-agnostic approach is highlighted in the EMA draft guidance document. It is specified in this guidance document that the assessment of homogeneity can be conducted only if a sufficient number of patients is included in the study, which is not always feasible (8). The PMMs will allow re-assessment of homogeneity based on a larger dataset in the post-marketing setting, sometimes including data from at least 200 additional patients, based on our findings. Regarding the integrated analysis sets, Seligson et al. reported that the FDA accepted pooled data due to the consistent anti-tumor activity across trials (20). However, not every agency might accept this strategy. For instance, the PMDA considered RXDX-101-02 to be the sole pivotal study to support the application of entrectinib and focused its review on the NTRK fusion-positive cohort (24). Early dialogue between developers and regulators to discuss (clinical) development plans may prevent regulatory hurdles.
Our results show that not all developers submitted an application to all agencies at the same time. The differences in submission date for pembrolizumab are the most noticeable. For instance, only recently an extension of indication for six MSI-h tumors for pembrolizumab was submitted to the EMA (11). Remarkably, the developer did not apply for a tissue-agnostic indication in the European Union (EU) (25); a different strategy compared to the applications submitted to FDA and PMDA. Understanding why the developer did not seeking a tissue-indication is of interest, but can only be speculated on. Likewise, while the EC approved dostarlimab for the treatment of patients with dMMR/MSI-H recurrent or advanced endometrial cancer, the applicant did not apply for a tissue-agnostic indication in the EU (26). A delay in submission is one of the factors that may contribute to disparity in approved therapies between countries or continents. In the absence of regulatory approval, patients in Europe are often dependent of clinical trials or patients access programs (27). Initiatives such as the personalized reimbursement scheme incorporated in the Drug Rediscovery Protocol enable access to promising medicinal products for unregistered indications, but only on a national level (28).
To our knowledge, this is the first comparison of tissue-agnostic approvals between three independent regulatory agencies. However, our study is limited to the products that received regulatory approval for a tissue-agnostic indication(s). Additional approvals might make such a study more sensitive to determine differences in regulatory decision-making between agencies. Despite this limitation, we consider that the currently available information is sufficiently abundant to compare at least some aspects related to decision-making for tissue-agnostic approvals.
The current approvals set precedent for the level of evidence necessary to register medicinal products for tissue-agnostic indication. A strong biological rationale and consistency in treatment effect across tumor types may allow for (expedited) approval, but confirmation of benefit in the post-marketing setting remains an important aspect of the tissue-agnostic approvals. There are different approaches to further confirm the tissue-agnostic indication in the post-marketing setting, albeit these are not without inherent limitations. These approaches to collect post-marketing data could complement existing data packages, but it will be of importance that “new” data are of high quality. For future applications, a global strategy toward data generation post-marketing may be of interest, dependent on emerging insights from the current approaches. Eventually, only the post-marketing data will continue to shed light on the appropriateness of the tissue-agnostic indication.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
JM, AP, VS-B, and AB designed the study. JM and OS performed the research. JM, OS, AP, and VS-B analyzed the data. JM, OS, PH, EV, AP, VS-B, and AB wrote the manuscript.
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of, or reflecting the position of the EMA or one of its committees, working parties, or any of the national agencies.
EV initiated and led the DRUP as one of the principal investigators.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.893400/full#supplementary-material
1. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. (2019) 25:3753–8. doi: 10.1158/1078-0432.CCR-18-4070
2. Food and Drug Administration. FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online at: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed March 3, 2022).
3. Food and Drug Administration. FDA Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. Available online at: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions (accessed March 3, 2022).
4. Food and Drug Administration. FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC. Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed March 3, 2022).
5. Foord and Drug Administration. FDA Grants Accelerated Approval to Dostarlimab-gxly for dMMR Advanced Solid Tumors. Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors (accessed April 30, 2022).
6. U.S. Food and Drug administration. Expedited Programs for Serious Conditions – Drugs and Biologics. (2014). Available online at: https://www.fda.gov/files/drugs/published/Expedited-Programs-for-Serious-Conditions-Drugs-and-Biologics.pdf (accessed March 3, 2022).
7. European Medicines Agency. Guideline on the Scientific Application and the Practical Arrangements Necessary to Implement Commission Regulation (EC) No 507/2006 on the Conditional Marketing Authorisation for Medicinal Products for Human Use Falling Within the Scope of Regulation (EC) No 726/2004. (2016). Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-application-practical-arrangements-necessary-implement-commission-regulation-ec/2006-conditional-marketing-authorisation-medicinal-products-human-use-falling_en.pdf (accessed March 3, 2022).
8. European Medicine Agency. Draft Guideline on the Clinical Evaluation of Anticancer Medicinal Products. Rev 6. Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-evaluation-anticancer-medicinal-products-man-revision-6_en.pdf (accessed March 3, 2022).
9. Minami H, Kiyota N, Kimbara S, Ando Y, Shimokata T, Ohtsu A, et al. Guidelines for clinical evaluation of anti-cancer drugs. Cancer Sci. (2021) 112:2563–77. doi: 10.1111/cas.14967
10. U.S. Food and Drug Administration. Guidance for industry. Developing Targeted Therapies in Low-Frequency Molecular Subsets of a Disease. (2018). Available online at: https://www.fda.gov/media/117173/download (accessed March 3, 2022).
11. Horizonscan Geneesmiddelen. Pembrolizumab. (2021). Available online at: https://www.horizonscangeneesmiddelen.nl/geneesmiddelen/pembrolizumab-oncologie-en-hematologie-oncologie_overig%5B5%5D/versie1 (accessed March 3, 2022).
12. U.S. Food and Drug Administration. Pembrolizumab Label. (2021). Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125514s112lbl.pdf (accessed March 3, 2022).
13. Bayer. Bayer Receives Approval for Vitrakvi™ in Japan. Available online at: https://media.bayer.com/baynews/baynews.nsf/id/F082D994FC24A468C12586A1003513D1?open&ref=irrefndcd (accessed March 3, 2022).
14. Booth CM, Karim S, Mackillop WJ. Real-world data: towards achieving the achievable in cancer care. Nat Rev Clin Oncol. (2019) 16:312–25. doi: 10.1038/s41571-019-0167-7
15. Cave A, Kurz X, Arlett P. Real-world data for regulatory decision making: challenges and possible solutions for Europe. Clin Pharmacol Ther. (2019) 106:36–9. doi: 10.1002/cpt.1426
16. Miksad RA, Samant MK, Sarkar S, Abernethy AP. Small but mighty: the use of real-world evidence to inform precision medicine. Clin Pharmacol Ther. (2019) 106:87–90 doi: 10.1002/cpt.1466
17. Center for Drug Evaluation and Research. Rozlytrek Multi-Discipline Review. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212725Orig1s000,%20212726Orig1s000MultidisciplineR.pdf (accessed March 3, 2022).
18. Flaherty KT, Le DT, Lemery S. Tissue-agnostic drug development. Am Soc Clin Oncol Educ Book. (2017) 37:222–30. doi: 10.1200/EDBK_173855
19. Park JHJ, Hsu G, Siden EG, Thorlund K, Mills EJ. An overview of precision oncology basket and umbrella trials for clinicians. Ca Cancer J Clin. (2020) 70:125–37. doi: 10.3322/caac.21600
20. Seligson ND, Knepper TC, Ragg S, Walko CM. Developing drugs for tissue-agnostic indication: a paradigm shift in leveraging cancer biology for precision medicine. Clin Pharmacol Ther. (2021) 109:334–42. doi: 10.1002/cpt.1946
21. Blumenthal GM, Kleutz PG, Schneider J, Goldberg KB, McKee AE, Pazdur R. Oncology drug approvals: evaluating endpoints and evidence in an era of breakthrough therapies. Oncologist. (2017) 22:762–7. doi: 10.1634/theoncologist.2017-0152
22. Woloshin S, Schwartz LM, White B, Moore TJ. The fate of FDA postapproval studies. N Engl J Med. (2017) 277:1114–7. doi: 10.1056/NEJMp1705800
23. Tenhunen O, Lasch F, Schiel A, Turpeinen M. Single-arm clinical trials as pivotal evidence for cancer drug approval: a retrospective cohort study of centralized European marketing authorizations between 2010 and 2019. Clin Pharmacol Ther. (2020) 108:653–60. doi: 10.1002/cpt.1965
24. Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau. Rozlytrek Report on the Deliberation Result. Available online at: https://www.pmda.go.jp/files/000232794.pdf (accessed March 3, 2022).
25. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) Minutes for the Meeting on 11-14 October 2021. Available online at: https://www.ema.europa.eu/en/documents/minutes/minutes-chmp-meeting-11-14-october-2021_en.pdf (accessed March 3, 2022).
26. European Medicines Agency. Assessment Report Jemperli. Available online at: https://www.ema.europa.eu/en/documents/assessment-report/jemperli-epar-public-assessment-report_en.pdf (accessed April 28, 2022).
27. Kok M, Chalabi M, Haanen J. How I treat MSI cancers with advanced disease. ESMO Open. (2019) 4:e000511. doi: 10.1136/esmoopen-2019-000511
28. van Waalwijk van Doorn-Khosrovani SB, Pisters-van Roy A, van Saase L, van der Graaff M, Gijzen J, Sleijfer S Hoes LR et al. Personalised reimbursement: a risk-sharing model for biomarker-driven treatment of rare subgroups of cancer patients. Ann Onocl. (2019) 30:663–4. doi: 10.1093/annonc/mdz119
Keywords: FDA, EMA, PMDA, tissue-agnostic indication, post-marketing measures
Citation: Mulder J, van Stuijvenberg OC, van Hennik PB, Voest EE, Pasmooij AMG, Stoyanova-Beninska V and de Boer A (2022) A Comparison of Post-marketing Measures Imposed by Regulatory Agencies to Confirm the Tissue-Agnostic Approach. Front. Med. 9:893400. doi: 10.3389/fmed.2022.893400
Received: 20 April 2022; Accepted: 10 May 2022;
Published: 14 June 2022.
Edited by:
Sandor Kerpel-Fronius, Semmelweis University, HungaryReviewed by:
Jan Trøst Jørgensen, Dx-Rx Institute, DenmarkCopyright © 2022 Mulder, van Stuijvenberg, van Hennik, Voest, Pasmooij, Stoyanova-Beninska and de Boer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorn Mulder, ai5tdWxkZXJAY2JnLW1lYi5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.