 Lifeng Jiang1
Lifeng Jiang1 Dmitry A. Goncharov1
Dmitry A. Goncharov1 Yuanjun Shen1
Yuanjun Shen1 Derek Lin1Baojun Chang2Andressa Pena2
Derek Lin1Baojun Chang2Andressa Pena2 Horace DeLisser3
Horace DeLisser3 Elena A. Goncharova1†
Elena A. Goncharova1† Tatiana V. Kudryashova1*†
Tatiana V. Kudryashova1*†- 1Lung Center, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Internal Medicine, School of Medicine, University of California, Davis, Davis, CA, United States
- 2Pittsburgh Heart, Lung, and Blood Vascular Medicine Institute, University of Pittsburgh, Pittsburgh, PA, United States
- 3Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Hyper-proliferation of pulmonary arterial vascular smooth muscle cells (PAVSMC) is an important pathological component of pulmonary vascular remodeling in pulmonary arterial hypertension (PAH). Lipogenesis is linked to numerous proliferative diseases, but its role in PAVSMC proliferation in PAH remains to be elucidated. We found that early-passage human PAH PAVSMC had significant up-regulation of key fatty acids synthesis enzymes ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), and increased unstimulated proliferation compared to control human PAVSMC. Treatment with an allosteric ACC inhibitor 5-tetradecyloxy-2-furoic acid (TOFA) significantly decreased proliferation and induced apoptosis of human PAH PAVSMC. Intracellular lipid content and proliferation of PAH PAVSMC were not reduced by incubation in lipid-depleted media but suppressed by a non-metabolizable analog of glucose 2-Deoxy-D-glucose (2-DG) and partially restored by addition of pyruvate. Protein kinase Akt was upregulated in human PAH PAVSMC in a sirtuin 7 (SIRT7)- and c-Jun N-terminal kinase (JNK)-dependent manner. Pharmacological inhibition of Akt down-regulated ACLY and ACC, significantly reduced intracellular lipid content, inhibited proliferation and induced apoptosis of human PAH PAVSMC. Taken together, these data demonstrate that human PAH PAVSMC have up-regulated lipogenesis, which is supported in an Akt- and glycolysis-dependent manner and is required for increased proliferation and survival. Our data suggest that there is a mechanistic link between glycolysis, lipogenesis, and the proliferation of human PAH PAVSMC and call for further studies to determine the potential attractiveness of a SIRT7/JNK-Akt-lipogenesis axis as a target pathway to inhibit PAVSMC hyper-proliferation in PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a devastating progressive disease leading to a deteriorating quality of life and high morbidity and mortality rates (1–3). Continuous vasoconstriction and excessive remodeling of pulmonary arteries (PA) cause an increase in pulmonary arterial pressure and pulmonary vascular resistance, leading to elevated right ventricular (RV) afterload and ultimately right heart failure and death (4, 5). At present, available vasodilatory therapies do not stop disease progression, and currently there are no vascular remodeling-focused therapies available for PAH patients (4, 6). All three layers of small pulmonary arteries—intima, media and adventitia—contribute to pulmonary vascular remodeling (7, 8). One of the important features of pulmonary vascular remodeling is the increased proliferation and survival of pulmonary arterial vascular smooth muscle cells (PAVSMC) (9, 10) in the medial layer of small muscular PAs (11). Over the last decades significant progress was achieved in dissecting the signaling molecules and pathways supporting the pathological pro-proliferative/pro-survival nature of PAVSMC in PAH. However, the underlying mechanisms are still not completely understood.
We and others previously demonstrated that PAH PAVSMC undergo a complex metabolic reprogramming required to maintain energy consuming pro-proliferative phenotype (9, 10, 12, 13). In contrast to non-diseased cells, PAH PAVSMC demonstrate increased ATP generation, proliferation and survival which depend predominantly on glycolytic metabolism (10) and undergo a metabolic shift from mitochondrial oxidative phosphorylation to glycolysis, similar to the “Warburg effect” in cancer cells (9, 10, 14). Multiple crucial metabolic regulators and enzymes, including AMP-activated protein kinase (AMPK), mechanistic target of rapamycin (mTOR) complex 2 (mTORC2) (10), hypoxia-induced factor (HIF) 1α (9), nuclear factor of activated T-cells (NFAT) (15), peroxisome proliferator-activated receptor (PPAR)γ (16), pyruvate dehydrogenase (PDH) (17), PDH kinase (PDK) (18), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) (19), pyruvate carboxylase (PC) (20), and enolase (21), support this glycolytic shift in PAH PAVSMC (9, 10, 22). A stable isotope metabolomics-based study confirmed that PAH PAVSMC have increased glucose uptake and utilization by glycolysis and the pentose shunt, but intriguingly no changes in fatty acid or glutamine uptake or utilization were detected (23).
Besides increased ATP levels, hyper-proliferative cells require increased amounts of intracellular essential “building blocks” such as lipids, proteins, and nucleic acids (24). The fact that highly proliferative PAH PAVSMC do not demonstrate increased fatty acid uptake (23) indicates that lipid metabolism in these cells might be re-organized to produce and accumulate the required amount of lipids internally. Indeed, several lines of evidence indicate that lipid metabolism is deregulated in PAH PAVSMC (25). RNAseq-based analysis identified up-regulation of fatty acid biosynthesis and metabolism pathways in isolated human PAH PASMC (26). Up-regulation of a key enzyme in fatty acid synthesis, fatty acid synthase (FASN) (27), and a key rate-limiting enzyme of mitochondrial fatty acid β-oxidation, carnitine palmitoyltransferase (CPT) (28), was reported in PAVSMC in rats with monocrotaline (MCT)-induced PH. Interestingly, a similar metabolic adaptation to satisfy high lipids demand is observed in most cancer cells, which have elevated endogenous fatty acid synthesis supported by the increased glycolysis and an increased ability to synthesize lipids (29, 30). De novo fatty acid synthesis makes a major contribution to the intracellular fatty acid pool in tumor cells (31) suggesting that PAVSMC in PAH might use similar strategies to support their highly proliferative phenotype. However, in contrast to the role of lipid metabolism in right ventricle (RV) function in PAH (25), the potential role of the lipogenic process and underlying mechanisms in PAH PAVSMC require further investigation.
In this study, we aimed to evaluate the status and role of lipogenesis in PAH PAVSMC proliferation and survival. Our data demonstrate that the pro-proliferative/pro-survival phenotype of PAVSMC in PAH is supported by glycolysis-dependent de novo lipid synthesis and suggest the potential role of Sirtuin 7 (SIRT7)- c-Jun N-terminal kinase (JNK)-Akt axis as a regulator of lipogenesis and a potential molecular target for anti-remodeling therapy.
Materials and Methods
Cell Culture
Early-passage (3–8 passage) human PAVSMC isolated from small (≤1 mm outer diameter) PAs of patients with PAH and non-diseased subjects were provided by UC Davis Lung Center Pulmonary Vascular Disease Program human specimens biobank, University of Pittsburgh Vascular Medicine Institute Cell Processing Core, and the Pulmonary Hypertension Breakthrough Initiative (PHBI) under approved protocols in accordance with Institutional Review Board (IRB) and Committee for Oversight of Research and Clinical Training Involving Decedents (CORID) policies. Cells isolation, characterization and maintenance were performed under PHBI-approved protocols as described previously (10, 32). Cells were maintained in complete PromoCell Smooth Muscle Cell Growth Medium 2 with SupplementPack and Antibiotic-Antimycotic. Before experiments, cells were incubated for 24–48 h in basal media supplemented with 0.1% bovine serum albumin (BSA) if not stated otherwise. For functional experiments (Ki-67, cell counts, TUNEL) a minimum of three technical replicas was performed within one experiment, minimum of 100 cells/subject was analyzed.
Exogenous Lipids Removal
To achieve the lipid-deprived cell culture condition, cell culture grade fetal bovine serum (FBS) and BSA were delipidated using Cleanascite™ Lipid Removal Reagent (Biotech Support Group; Monmouth Junction, NJ, United States) according to the manufacturer’s protocol with modification. Briefly, Cleanascite™ was added to the FBS or BSA (1:4 volume ratio). The mixture was incubated for four hours at 4°C with gentle shaking and centrifuged at 16,000 g for 10 min. Supernatant was collected, then a second dose of Cleanascite™ Lipid Removal Reagent was added (1:4 volume ratio), and incubation and centrifugation steps were repeated as described above. The supernatants, consisting of lipid depleted FBS or BSA, were then used as cell culture media supplements to prepare lipid-depleted media.
Intracellular Lipid Detection
Intracellular lipid detection was performed using a fluorescent probe for lipid droplets BODIPY 493/503 as previously described in Qiu et al. (33) with modifications. Briefly, cells were washed twice with PBS and then incubated with 2 μM BODIPY 493/503 staining solution (#D3922 Thermo Fisher Scientific, Chicago, IL, United States) for 15 min at 37°C. Then cells were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS), followed by DAPI staining to detect nuclei. Immunostaining was visualized and images were taken using a Keyence BZ-X800 (Keyence Corporation of America, Itasca, IL, United States) and Zeiss LSM700 confocal microscope (White Plains, NY, United States). A minimum of 100 cells per condition was analyzed.
Cell Proliferation Assay
Cell proliferation was assessed using Ki-67 immunostaining as described previously (10, 32, 34). Briefly, cells were washed with PBS, fixed in 4% paraformaldehyde/PBS, and permeabilized using Triton X-100/PBS solution. Then cells were incubated in blocking solution (2% BSA/PBS), followed by overnight incubation with primary anti-Ki-67 antibody (#9129, Cell Signaling, Danvers, MA, United States) in blocking solution. Next day, the slides were washed with PBS, followed by incubation with secondary chicken anti-rabbit IgG (H + L) Alexa Fluor 594 antibody (#A-21442, Invitrogen, Waltham, MA, United States) and 4′,6-diamidino-2-phenylindole (DAPI) to detect nuclei. Images were taken using a Keyence BZ-X800 microscope.
Cell Growth Assay
Cell growth analysis was performed as described previously (10, 32, 34). Briefly, 300,000 cells per well were plated in a six well plate in cell culture media supplemented with 5% FBS. Forty-eight hours later, when cells attached and spread (day 0), the media was changed to 0.1% BSA with or without lipids and incubated for six more days (media was renewed every 48 h). Then, cell counting was performed using the Countess II FL Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, United States) according to manufacturer’s protocol.
Apoptosis Analysis
Apoptosis analysis was performed using the In situ Cell Death Detection Kit (Roche, Nutley, NJ, United States) based on terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) technology following the manufacturer’s protocol.
Immunoblot Analysis
Immunoblot analysis was performed as described previously (10, 32, 34). Antibodies for ACLY (#4332), P-S79-ACC (#11818), ACC (#3676), FASN (#3180), P-S473-Akt (#4060), P-T450-Akt (#12178), Akt (#9272), PThr183/Tyr185-JNK (#4668), JNK (#9252), α/β-Tubulin (#2148), CPT1A (#97361), hexokinase II (HKII) (#2867), phosphofructokinase (PFKP) (#8164) were purchased from Cell Signaling (Danvers, MA, United States). The antibody for P-S455-ACLY (#PA5-97395) was purchased from Thermo Fisher Scientific (Chicago, IL, United States). The antibody for malonyl CoA decarboxylase (MLYCD) (#15265-1-AP) were purchased from Proteintech (Rosemont, IL, United States). Secondary HRP conjugated anti-mouse antibody (ab205719) was purchased from Abcam (Boston, MA, United States), secondary HRP-conjugated anti-rabbit antibody (#7074) was purchased from Cell Signaling (Danvers, MA, United States).
Inhibitors and Activators
2-Deoxy-D-glucose (2-DG, D6134), IL-6 (I1395), PDGF-BB (GF149), and Akt inhibitor VIII (SIAL-124018) were purchased from Millipore Sigma (St. Louis, MO, United States), 5-tetradecyloxy-2-furoic acid (TOFA, sc-200653) was purchased from Santa Cruz Biotechnology (Dallas, TX, United States), JNK inhibitor (bentamapimod, HY-14761) was purchased from MedChemExpress (Monmouth Junction, NJ, United States).
Lactate Assay
To measure intracellular lactate amount, the lactate assay kit (#MAK064, Millipore Sigma) was used according to the manufacturer protocol with modifications. Briefly, 2 × 106 cell were homogenized in 200 μL of lactate assay buffer and centrifuged at 21,000 g for 5 min to remove cell debris. The supernatant was deproteinized with a 10 kDa MWCO spin filter (#UFC5003, Millipore Sigma) to remove lactate dehydrogenase, and the lactate assay was performed. Protein concentrations were determined in the supernatants before deproteinization using the BCA protein assay kit (#23227, Thermo Fisher Scientific) and lactate content was normalized to the amount of total protein used for the assay.
Data Analysis
Immunoblots, BODIPY 493/503, Ki-67 and apoptosis assays were analyzed using ImageJ (NIH, Bethesda, MD, United States), StatView (SAS Institute, Cary, NC, United States) and GraphPad Prizm 9.02 (GraphPad Software, San Diego, CA, United States). Immunocytochemical analyses were detected and captured using the Keyence BZ-X800 system and software (Keyence Corporation of America, Itasca, IL, United States) and Zeiss LSM700 confocal microscope and software (ZEN, 2009; White Plains, NY, United States). Statistical comparisons between the two groups were performed by Mann–Whitney U test. Statistical comparisons among three or more groups were performed by the Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test. Statistical significance was defined as p ≤ 0.05.
Results
Increased Lipid Synthesis Is Required for Hyper-Proliferation and Survival of Human PAH PAVSMC
In order to evaluate the status of lipogenesis in PAH PAVSMC, we first tested the expression of key lipogenic enzymes driving the biosynthesis of fatty acids. Immunoblot analysis demonstrated significant increase in phosphorylated ACLY, and elevated protein levels of ACC and FASN in early-passage distal human PAH PAVSMC compared to cells from non-diseased subjects (CTRL) (Figures 1A–D and Supplementary Figure 1A). Interestingly, we detected no significant differences between non-diseased and PAH PAVSMC in protein levels of CPT1A (Figures 1A,E) and MLYCD (Supplementary Figure 1B), the regulatory enzymes in fatty acid β-oxidation and synthesis, in spite of previously reported up-regulation of CPT1A in smooth muscle cells in rats with monocrotaline-induced PH and protective effect of MLYCD deletion against development of hypoxia-induced PH in mice (28, 35). Observed up-regulation of key fatty acid synthesis enzymes suggested that de novo lipid synthesis is altered in PAH PAVSMC. To confirm our observations, we performed immunocytochemical detection of lipid droplets (a major storage depot for neutral lipids, primarily triglycerides) using a fluorescent probe (BODIPY 493/503) to visualize neutral lipid accumulation in PAH PAVSMC. We found that lipid accumulation detected in PAH PAVSMC was preserved in media deprived from exogenous lipids (Figures 1F,G), indicating that PAH PAVSMC have an ability to generate lipids de novo. Importantly, PAH PAVSMC, in contrast to control cells, demonstrated increased growth not only in serum-deprived media, but in lipid-deprived serum-deprived media (Figure 1H). Moreover, 5-tetradecyloxy-2-furoic acid (TOFA), an allosteric inhibitor of ACC, a key enzyme regulating fatty acid synthesis, significantly decreased proliferation and induced apoptosis in PAH PAVSMC (Figures 1I,J), demonstrating that lipogenesis is required for PAH PAVSMC hyper-proliferation and survival. Taken together, these data show that de novo lipid synthesis is up-regulated and required for maintaining the pro-proliferative/pro-survival phenotype of human PAH PAVSMC.
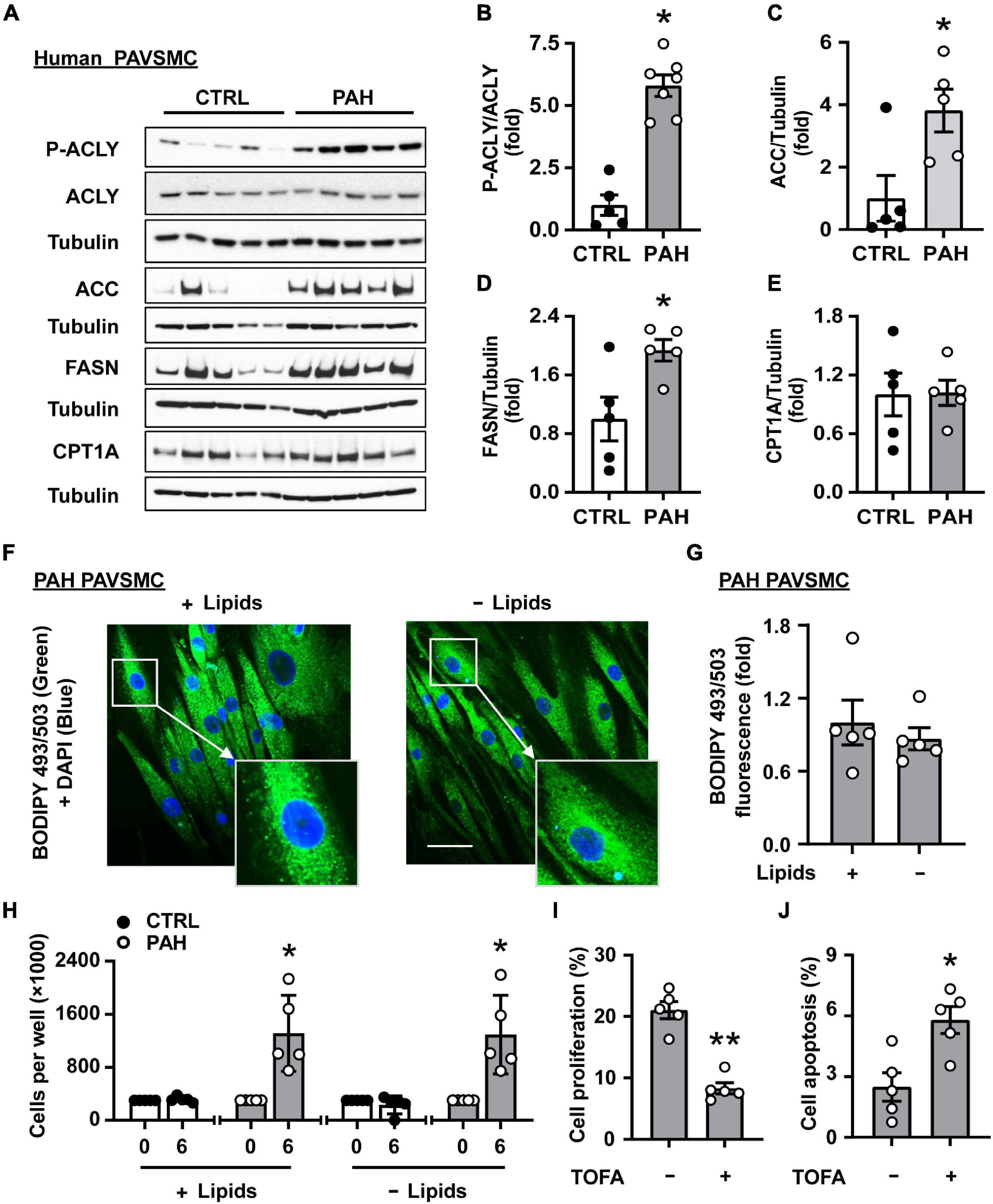
Figure 1. Increased lipid synthesis is required for hyper-proliferation and survival of human PAH PAVSMC. (A–E) Early passage distal primary human PAVSMC from non-diseased (CTRL) and PAH subjects were serum-deprived for 48 hours and subjected to immunoblot analysis to detect indicated proteins. n = 5 (CTRL), n = 7 (PAH for P-ACLY, ACLY, see Supplementary Figure 1A for additional immunoblots used for statistical analysis) or n = 5 (PAH for ACC, FASN, CPT1A). Data are means ± SE, fold to control. (F,G) Human PAH PAVSMC were incubated for 48 h in serum deprived media supplemented with regular (+ Lipids) or lipid-deprived (-Lipids) 0.1% BSA and subjected to fluorescent BODIPY 493/503 staining (green) to detect intracellular neutral lipids followed by DAPI (blue) staining to detect nuclei. Representative images with enlarged area (F) and statistical analysis (G) are shown. Bar equals 50 μm. Data are means ± SE from n = 5 subjects/group, fold to cells incubated in regular 0.1% BSA (+ Lipids) group. (H) Equal amount of human PAVSMC from five PAH and five control subjects were seeded to 6 well plates. 48 h later (day 0), media was changed to the serum-free media supplemented with regular (+ Lipids) or lipid-deprived (-Lipids) 0.1% BSA; six days later, cell count assay was performed. Data are means ± SE, n = 5 subjects/group. (I) PAH PAVSMC were treated with ACC inhibitor TOFA (20 μM) for 48 h followed by proliferation analysis (Ki-67). Data are means ± SE, percentage of Ki-67 positive cells/total number of cells, n = 5 subjects/group. (J) PAH PAVSMC were treated with ACC inhibitor TOFA (20 μM) for 48 h followed by apoptosis analysis. Data are means ± SE, percentage of TUNEL-positive cells/total number of cells, n = 5 subjects/group. *p < 0.05, **p < 0.01 by Mann–Whitney U test.
Increased Lipid Synthesis in Human PAH PAVSMC Is Glucose-Dependent
Since fatty acid uptake is not altered in PAH PAVSMC (23) and we found that PAH PAVSMC growth was maintained in lipid-deprived media, we hypothesized that glucose could be a potential source for de novo lipid synthesis in PAH. We found that intracellular lipid content in PAH PAVSMC in lipid-deprived conditions was significantly depleted by the treatment with the non-metabolizable analog of glucose 2-DG. Importantly, co-treatment with glucose metabolite pyruvate significantly attenuated 2-DG-dependent inhibition of intracellular lipid accumulation in human PAH PAVSMC (Figures 2A,B). Furthermore, 2-DG-induced inhibition of PAH PAVSMC proliferation in lipid-deprived conditions was also partially reversed by pyruvate (Figure 2C). These data demonstrate that glucose, metabolized through the glycolysis, serves as the main source for de novo lipid synthesis and supports lipid accumulation and increased proliferation of PAH PAVSMC. Confirming up-regulation of glycolysis, intracellular lactate levels were significantly higher in PAH PAVSMC compared to non-diseased cells (Figure 2D). Moreover, protein levels of key glycolytic enzymes PFKP and HKII were significantly higher in human PAH PAVSMC compared to controls (Figures 2E–G), and HIF1α over accumulation was detected in six out of seven PAVSMC from PAH patients compared to one out of five non-diseased (control) subjects (Supplementary Figure 2). This is in good agreement with previously published data demonstrating the importance of glycolysis for PH (19, 36, 37). Together, our data demonstrate that glycolysis-metabolized glucose serves as a main source for increased de novo lipid synthesis in PAH PAVSMC, supporting a pro-proliferative cell phenotype.
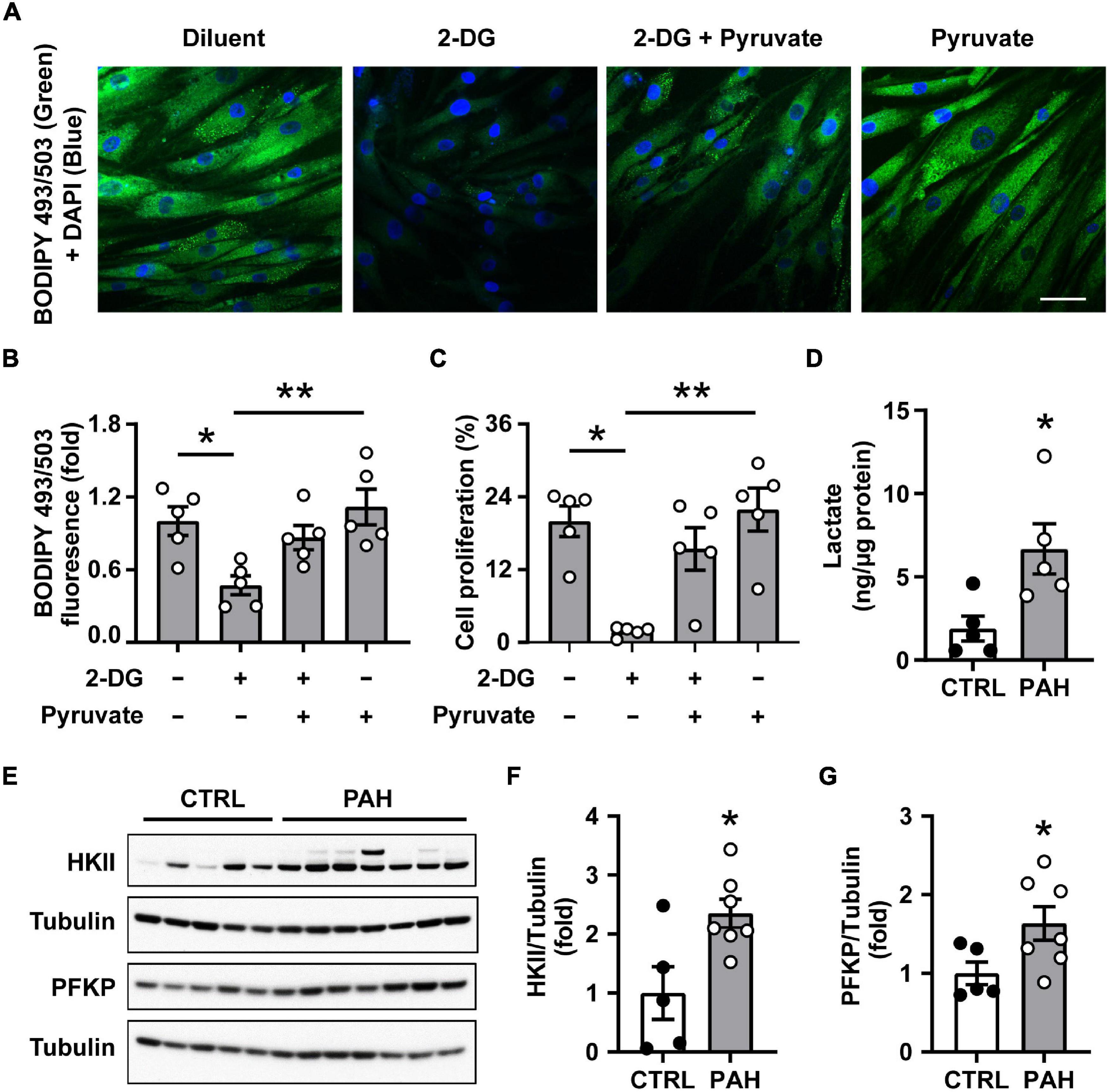
Figure 2. Increased lipid synthesis in human PAH PAVSMC is glucose dependent. (A–C) Human PAH PAVSMC were incubated in serum deprived media supplemented with 0.1% lipid-deprived BSA in presence of 2-Deoxy-D-glucose (2-DG, 100 mM) and/or pyruvate (10 mM) or vehicle (−). Forty-eight hours later neutral lipid accumulation (A,B) was evaluated by fluorescent BODIPY 493/503 staining (green) followed by DAPI co-staining (blue) to detect nuclei and cell proliferation (Ki-67) (C) was analyzed. Representative images (A) and statistical analysis (B,C) are shown. Bar equals 50 μm. Data are means ± SE, n = 5 subjects/group, fold to vehicle-treated group (B), percentage of Ki-67 positive cells/total (C), *p < 0.05, **p < 0.01 by Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test. (D) Intracellular lactate levels were measured in PAVSMC from five non-diseased and five PAH subjects. (E–G) Early passage distal primary human PAVSMC from five non-diseased (CTRL) and seven PAH subjects were serum deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. (D,F,G) Data are means ± SE, fold to control (F,G), *p < 0.05 by Mann–Whitney U test.
Akt Supports ACLY and ACC Up-Regulation in Human PAH PAVSMC
One of the key regulators of glucose and lipid metabolism is a serine/threonine kinase Akt (38–42), which stimulates glucose uptake and glycolysis (43) and promotes de novo lipid synthesis through sterol regulatory binding protein (SREBP)-dependent expression of lipogenic enzymes (43, 44). Accumulating evidence from multiple groups, including ours, demonstrate pathological role of Akt as a regulator of pulmonary vascular cell metabolism, proliferation, pulmonary vascular remodeling, and overall PH (10, 45, 46). This allowed us to hypothesize that Akt might coordinate glucose metabolism and de novo lipid synthesis supporting PAVSMC hyper-proliferation in PAH. In agreement with published studies reporting Akt activation in PAH (10, 32), we found that S473-Akt phosphorylation was significantly increased in human PAH PAVSMC (Figures 3A,B). Importantly, Akt inhibitor VIII suppressed activation of both key lipogenesis enzymes, ACLY and ACC; ACLY deactivation was detected by decrease in S455-ACLY phosphorylation (47), and ACC deactivation was detected by an increase in inhibitory S79-ACC (48) phosphorylation (Figures 3C–F and Supplementary Figures 3A,B), suggesting that Akt supports increased lipogenesis in human PAH PAVSMC. Moreover, Akt inhibitor suppressed phosphorylation of ribosomal protein S6 (Figures 3C,G), the main downstream effector of mTOR complex 1 (mTORC1), a known activator of cell growth, proliferation, and lipid biogenesis (49). In summary, these data demonstrate that de novo lipid synthesis in PAH PAVSMC is regulated by Akt.
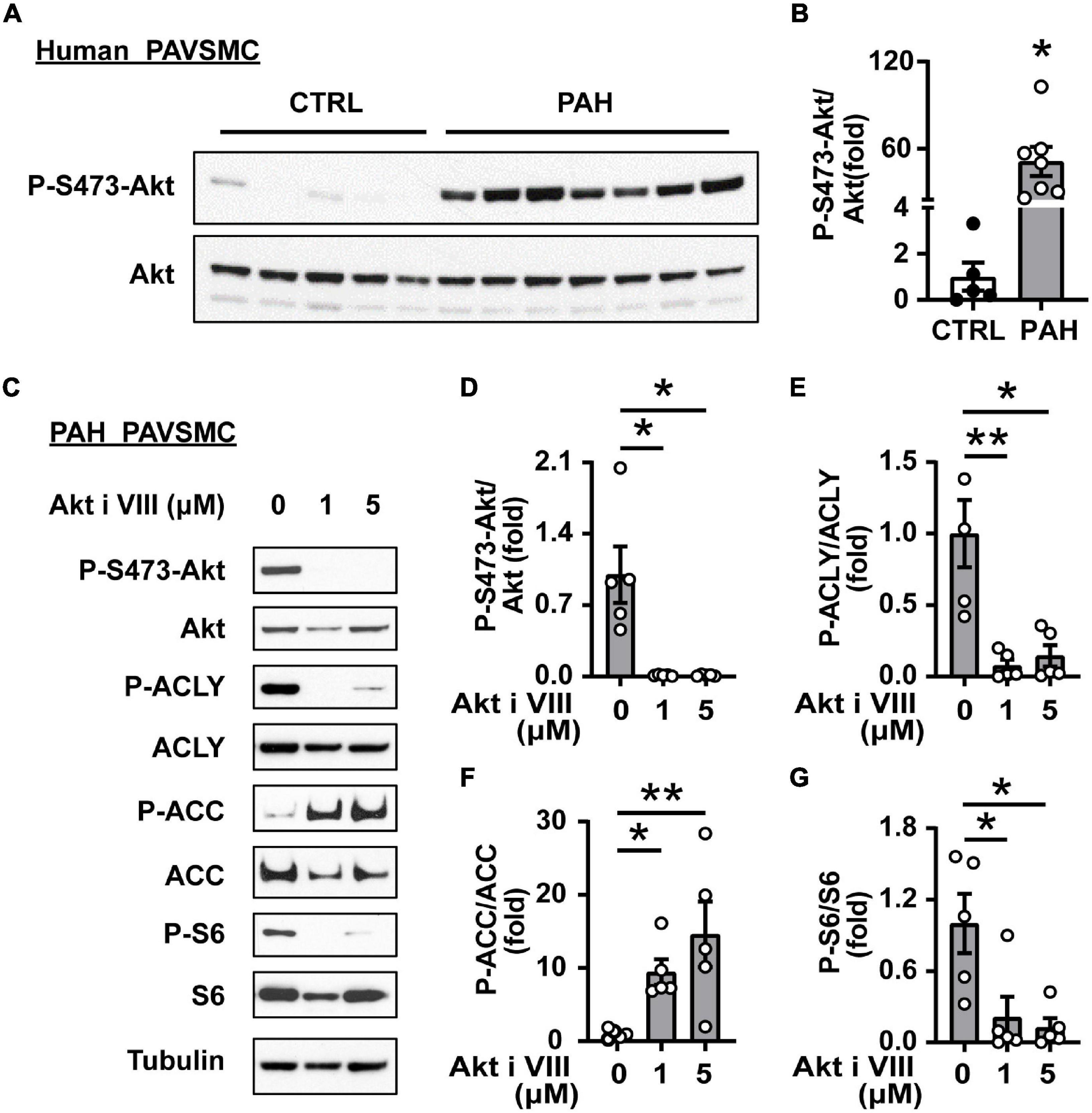
Figure 3. Akt supports ACLY and ACC up-regulation in human PAH PAVSMC. (A,B) Early passage distal human PAVSMC from five non-diseased (CTRL) and seven PAH subjects were serum deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. Immunoblots (A) and statistical analysis (B) are shown. Data are means ± SE, fold to control, *p < 0.05 by Mann–Whitney U test. (C–G) Early passage human PAH PAVSMC were treated with Akt inhibitor VIII (1, 5 μM) or vehicle (0) for 24 h followed by immunoblot analysis. Data are fold to vehicle-treated group, n = 5 subjects/group, *p < 0.05, **p < 0.01 by Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test.
SIRT7 Up-Regulates Akt and Lipogenic Enzymes in Human PAH PAVSMC
Next, we aimed to identify the factor(s) regulating de novo Akt-dependent lipid synthesis in PAH. Several studies demonstrate that lysine deacetylase SIRT7 coordinates cellular metabolic balance by regulating glucose sensing/homeostasis, glycolysis, mitochondria and ribosomal biogenesis, DNA damage response, fatty acid synthesis and overall lipid metabolism (50–56). Moreover, it was recently shown that SIRT7 modulates aortic vascular smooth muscle cell proliferation in wire injury model of the femoral artery (57) and promotes cancer progression by activating Akt (56, 58) and mTORC1 effector S6K1 (59). We found that SIRT7 protein levels were significantly higher in human PAH PAVSMC compared to non-diseased PAVSMC (Figures 4A,B). Importantly, shRNA-induced depletion of SIRT7 in PAH PAVSMC significantly reduced S473 Akt phosphorylation (Figures 4C,D) and activatory phosphorylation of ACLY, and significantly increased inhibitory phosphorylation of ACC in human PAH PAVSMC (Figures 4C,E,F), suggesting that Akt-mediated lipogenesis in human PAH PAVSMC is regulated by SIRT7.
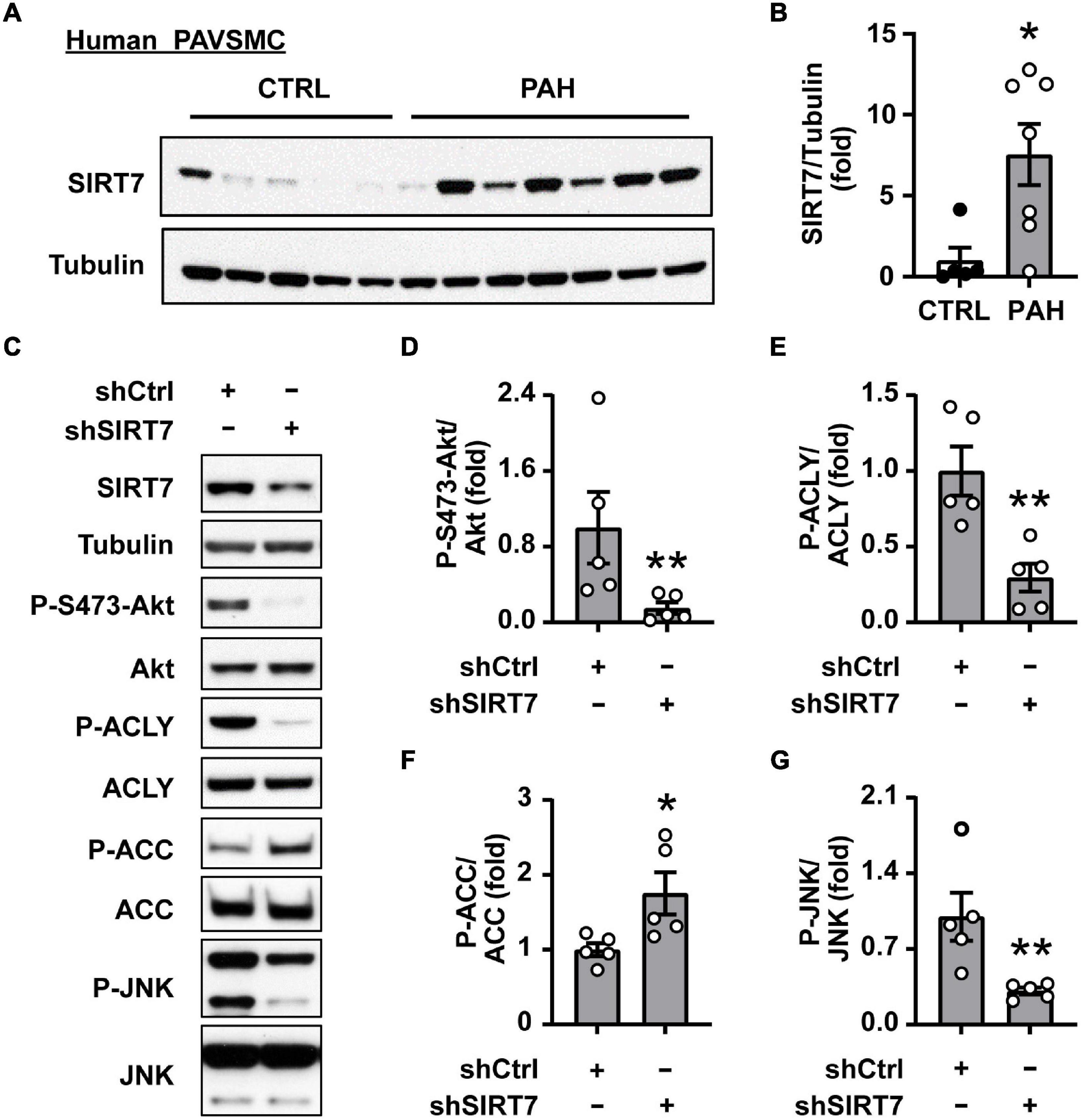
Figure 4. SIRT7 regulates Akt status and lipogenic enzymes activation in human PAH PAVSMC. (A,B) Early passage distal human PAVSMC from five non-diseased (CTRL) and seven PAH subjects were serum deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. Immunoblots (A) and statistical analysis (B) are shown. Data are means ± SE, fold to control. (C–G) Human PAH PAVSMC were transfected with shRNA SIRT7 (shSIRT7), or control scramble shRNA (shCtrl) for 72 h followed by immunoblot analysis. Data represent fold to shCtrl. Data are means ± SE, n = 5 subjects/group. *p < 0.05, **p < 0.01 by Mann–Whitney U test.
SIRT7-Dependent JNK Activation Is Required for Akt and Lipogenic Enzymes Up-Regulation in Human PAH PAVSMC
Since SIRT7 does not possess kinase activity, its regulation of Akt phosphorylation status most likely is mediated by intermediate player(s) and one of the potential SIRT7 pro-survival targets is JNK (54). Indeed, we found that shSIRT7 significantly decreased phosphorylation of JNK in PAH PAVSMC (Figures 4C,G), also suggesting that SIRT7 regulates Akt and lipogenesis through JNK. Previous studies have shown that Akt activation via phosphorylation at S473 is achieved through a series of phosphorylation steps, and the initial priming phosphorylation of T450 can be regulated by JNK (60). We found a significant increase in T183/185 JNK phosphorylation in human PAH PAVSMC compared to control PAVSMC (Figures 5A,B), indicating that JNK is activated in PAH PAVSMC. JNK inhibitor HY-14761 significantly reduced both S473 and priming Thr450 Akt phosphorylation in PAH PAVSMC (Figures 5C–E). JNK inhibitor-mediated deactivation of Akt was associated with de-activation of lipogenic enzymes ACLY and ACC in human PAH PAVSMC (Figures 5C,F,G and Supplementary Figures 3C,D). These data indicate that Akt-mediated lipogenesis in human PAH PAVSMC is at least in part regulated by JNK.
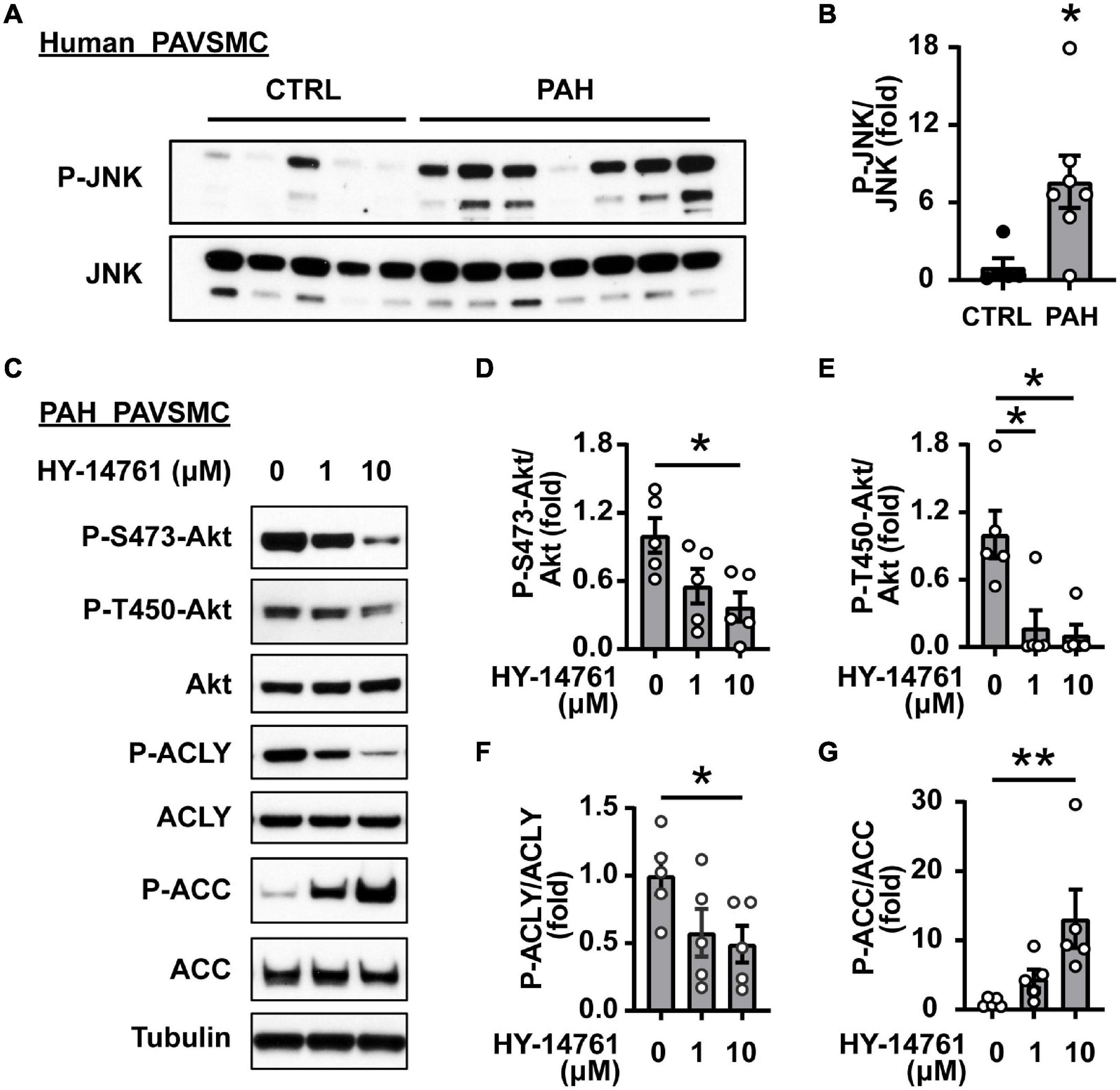
Figure 5. JNK regulates Akt and lipogenic enzymes in human PAH PAVSMC. (A,B) Early passage distal human PAVSMC from five non-diseased (CTRL) and seven PAH subjects were serum deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. Immunoblots (A) and statistical analysis (B) are shown. Data are means ± SE, fold to control, *p < 0.05 by Mann–Whitney U test. (C–G) Early passage human PAH PAVSMC were treated with JNK inhibitor HY-14761 (1, 10 μM) or vehicle (0) for 48 h followed by immunoblot analysis. Data represent fold to vehicle-treated group, n = 5 subjects/group, *p < 0.05, **p < 0.01 by Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test.
In PAH, Akt activation in resident pulmonary vascular cells could be induced by excessive amounts of growth factors and pro-inflammatory mediators. To start determining pro-PH agents modulating SIRT7/JNK axis in PAVSMC, we treated non-diseased human PAVSMC with PDGF and IL-6. Interestingly, while both PGDF and IL-6 significantly increased JNK phosphorylation, only PDGF induced significant increase of SIRT7 protein levels (Supplementary Figures 4A,B), similar to those seen in human PAH PAVSMC. Together, these data suggest that SIRT7/JNK-dependent Akt activation in PAVSMC could be induced by PDGF.
Akt Supports Lipid Accumulation, Proliferation and Survival of Human PAH PAVSMC
To further clarify the role of Akt in lipid accumulation, hyper-proliferation, and survival of human PAH PAVSMC, we treated cells with Akt inhibitor VIII. Immunocytochemical analysis demonstrated that intracellular lipid accumulation in PAH PAVSMC, maintained in lipid-deprived media, was significantly downregulated by Akt inhibitor VIII (Figures 6A,B). Moreover, treatment with Akt inhibitor VIII significantly reduced proliferation and promoted apoptosis of PAH PAVSMC (Figures 6C,D), demonstrating that Akt supports increased cell proliferation, survival, and lipogenesis in PAH PAVSMC (Figure 6E). Furthermore, inhibition of Akt did not augment 2-DG-dependent inhibition of PAH PAVSMC proliferation (Supplementary Figures 5A,B), suggesting that Akt regulates PAH PAVSMC proliferation at the level of or downstream of glycolytic enzymes. Taken together, our data demonstrate that human PAH PAVSMC have up-regulated lipogenesis supported in an Akt- and glycolysis-dependent manner to sustain increased proliferation and survival, and that Akt signaling is regulated, at least in part, by SIRT7-JNK axis (Figure 6E).
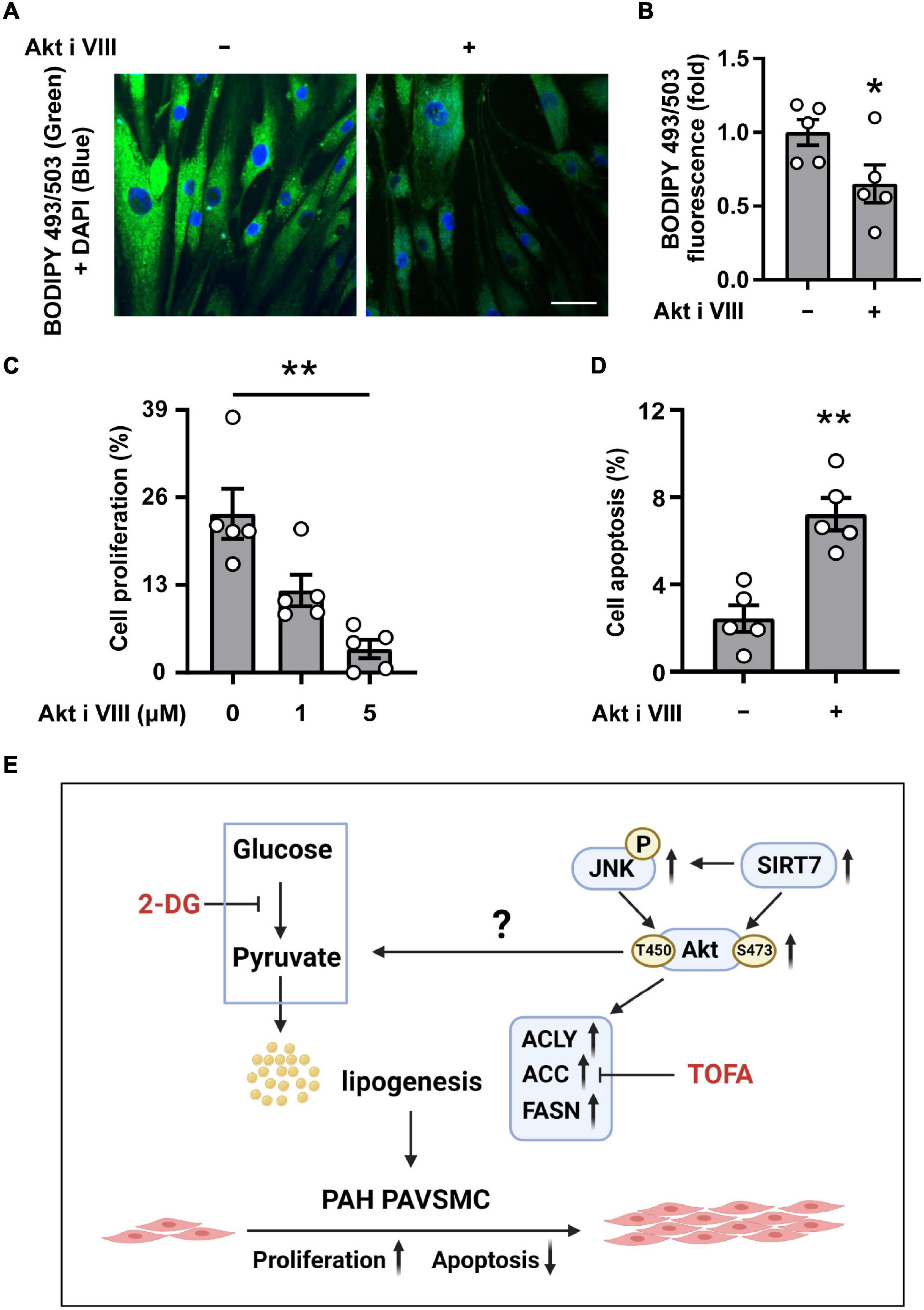
Figure 6. Akt supports intracellular lipid levels, proliferation and survival of human PAH PAVSMC. (A,B) Human PAH PAVSMC were incubated in serum deprived media supplemented with 0.1% lipid deprived BSA in presence of Akt inhibitor VIII (10 μM) or diluent. Forty-eight hours later neutral lipid accumulation was evaluated by fluorescent BODIPY 493/503 staining (green) followed by DAPI co-staining (blue) to detect nuclei. Representative images (A) and statistical analysis (B) are shown. Bar equals 50 μm. Data are means ± SE, fold to the diluent-treated cells, n = 5 subjects/group, *p < 0.05 by Mann–Whitney U test. (C) Human PAH PAVSMC were serum deprived in media, supplemented with regular 0.1% BSA, and were treated with 1, 5 μM (C) or 10 μM (D) Akt inhibitor VIII or vehicle (0/-) for 48 h followed by proliferation (Ki-67) (C) or apoptosis (D) analyses. Data are percentage of Ki-67- or TUNEL- positive cells/total number of cells, means ± SE, n = 5 subjects/group. *p < 0.05 Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test (C) or by Mann–Whitney U test (D). (E) Schematic representation of the proposed regulation of the de novo lipid synthesis, required for increased proliferation and survival of PAVSMC in PAH.
Discussion
Increased proliferation and survival of PAVSMC in small PAs are critical components of pulmonary vascular remodeling in PAH, the mechanisms of which are not completely understood. Here we report that human PAH PAVSMC have up-regulated lipogenesis, which is required to support increased cell proliferation and survival. We also show that observed de novo lipid synthesis is glucose-dependent and is regulated by Akt. We further demonstrate that SIRT7 and JNK are up-regulated in PAH PAVSMC, and support Akt activation and lipogenesis. Lastly, we show that Akt activation is required for increased lipid accumulation, cell proliferation and survival of PAH PAVSMC. Overall, although further studies are needed, our data suggest that the SIRT7/JNK-Akt-lipogenesis axis could be considered as a potential target pathway for developing novel anti-remodeling therapy for PAH.
Metabolic alterations in glucose homeostasis and glycolysis, similar to the “Warburg effect” in cancer cells, support increased PAVSMC proliferation and pulmonary vascular remodeling in PAH (9, 10, 12). Increased glucose uptake coupled with unaltered fatty acid uptake by PAH PAVSMC (23) indicates that intracellular demand in lipids in these highly proliferative cells is fulfilled by alternative internal pathways. This phenomenon is well known in cancer, since tumor cells demonstrate elevated endogenous fatty acid synthesis, supported by the increased glycolysis, to maintain hyper-proliferation (29). Glucose serves as major source supporting lipid synthesis. Glucose is converted to pyruvate through glycolysis. Pyruvate, in turn, enters mitochondria for citrate generation. Through mitochondrial carrier Slc25a1, citrate can be exported into the cytosol, wherein it is cleaved by ACLY to produce acetyl-CoA, which is processed for de novo lipogenesis by ACC and FASN (61). We demonstrate that all the key enzymes of fatty acid synthesis, ACLY, ACC, and FASN are up-regulated in human PAH PAVSMC. Moreover, we found that PAH PAVSMC maintain hyper-proliferation and glucose-dependent accumulation of intracellular lipids even in the absence of an extracellular lipid source but require activation of intracellular de novo lipid synthesis machinery. This is an important observation, confirming that lipid synthesis plays a crucial role not only in RV disfunction in PAH (62, 63) but also in smooth muscle proliferation and pulmonary vascular remodeling. It also supports previous studies suggesting that targeting lipid metabolism in PAH VSM needs to be considered for developing on anti-remodeling therapeutic options for PAH treatment (27, 28).
Besides glucose, glutamine can also contribute carbon to lipogenic acetyl-coenzyme A (acetyl-CoA) through glutamine-derived α-ketoglutarate (α-KG). Generated α-KG could be, in turn, converted into citrate via an isocitrate dehydrogenase-1 (IDH1)-dependent mitochondrial or cytosolic pathway (64, 65). In our present study, we have shown that PAH PAVSMC de novo lipogenesis depends on glucose, but whether α-KG contributes to this process remains unstudied.
Akt signaling is tightly related to the regulation of glycolysis and lipogenesis (66–68). Akt stimulates aerobic glycolysis in cancer cells, supporting continued growth and survival, mediates bioenergetic stability in epithelial cells (68), and stimulates hepatic SREBP1c and lipogenesis through mTORC1-dependent and independent pathways (69), suggesting that Akt might be involved in regulation of lipid synthesis and accumulation in PAH PAVSMC. Our data demonstrate that Akt not only supports glycolysis-driven lipogenesis in PAH PAVSMC, but also regulates proliferation and survival of PAH PAVSMC. Since Akt is a major player, coordinating multiple processes directly involved in cell survival, growth, metabolism, proliferation, migration and differentiation (70), pharmacological targeting of Akt is a highly attractive therapeutic approach for proliferative diseases such as cancer. Multiple Akt inhibitors are now in various stages of clinical development (71, 72). However, since Akt activation occurs through various mechanisms, clinical efficacies of Akt inhibitors are limited (73), and Akt targeting still remains a challenge. Thus, inhibiting Akt via modulating its upstream regulators might represent an alternative, potentially attractive strategy for therapeutic intervention.
The members of the sirtuin family of lysine deacylases are important regulators of metabolic pathways and energy homeostasis (50, 74) which are also known regulators of Akt (75). Sirtuins have been implicated in multiple metabolic diseases, including aging, cancers, cardiovascular diseases, obesity, and diabetes mellitus (50) and are considered potential targets for the therapeutic interventions (76). Unlike other sirtuins, SIRT7 demonstrates a relatively weak deacetylase activity, but is involved in regulating cellular energy metabolism homeostasis, lipid metabolism, cell migration, and was recently shown to control VSMC proliferation (50, 57, 77, 78). SIRT7 is also considered an attractive target for anti-cancer therapy (79). Despite being the least well-characterized member of the sirtuin family, accumulating evidence shows that SIRT7 acts as an Akt upstream regulator in other cell types (58, 59, 80). Here we demonstrate that SIRT7 acts as an upstream positive regulator of Akt in PAH PAVSMC and supports Akt phosphorylation in a JNK-dependent manner. We found that pharmacological inhibition of JNK significantly reduced both S473 and T450 Akt phosphorylation in PAH PAVSMC. This finding is in agreement with previous studies, demonstrating that JNK regulates Akt reactivation through T450 phosphorylation in cardiomyocyte survival after hypoxia (60).
Our study, however, has limitations. Although use of cells from PAH and non-diseased human subjects strongly suggests the translational significance of our findings, we only verified the role of SIRT7-JNK-Akt-de novo lipid synthesis in vitro, and further studies are needed to determine the role of this axis in vivo. Also, further pre-clinical studies are needed to evaluate whether the link between SIRT7-JNK and Akt-dependent de novo lipid synthesis, required for increased proliferation and survival of PAH PAVSMC, could be a potential target pathway for therapeutic intervention.
In summary, we found that human PAH PAVSMC have up-regulated lipogenesis supported in an Akt- and glycolysis-dependent manner to sustain increased cell proliferation. We also show that inhibition of the Akt-lipogenesis axis reduces proliferation and induces apoptosis of human PAH PAVSMC. In aggregate, our data provide a link between glycolysis, lipogenesis and proliferation of human PAH PAVSMC and call for further studies to determine the potential attractiveness of the SIRT7/JNK-Akt-lipogenesis axis as a target pathway for therapeutic intervention.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
EG and TK: conception and design. LJ, TK, HD, EG, DG, AP, BC, YS, and DL: experimental work, analysis, and interpretation. LJ, EG, and TK: drafting the manuscript and intellectual content. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH/NHLBI R01HL113178 (EG), R01HL130261 (EG), and R01HL150638 (EG). The Pulmonary Hypertension Breakthrough Initiative is supported by NIH/NHLBI R24HL123767.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.886868/full#supplementary-material
Supplementary Figure 1 | (A,B) Early passage distal primary human PAVSMC from non-diseased (CTRL) and PAH subjects were serum-deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. (A) n = 5 (CTRL), n = 7 (PAH) (see Figure 1B for statistical analysis). (B) Data are means ± SE, fold to control, n = 5 subjects/group.
Supplementary Figure 2 | Early passage PAVSMC from five non-diseased (CTRL) and seven PAH subjects were serum deprived for 48 h and subjected to immunoblot analysis to detect indicated proteins. Immunoblots (A) and statistical analysis (B) are shown. (B) Data are means ± SE, fold to control.
Supplementary Figure 3 | Early passage human PAH PAVSMC were treated with Akt inhibitor VIII, JNK inhibitor HY-14761, or diluent (0) and subjected to immunoblot analysis to detect indicated proteins. Data are fold to diluent-treated group. *p < 0.05 by Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test.
Supplementary Figure 4 | Early passage human control (non-diseased) PAVSMC were treated with (A) PDGF-BB (60 ng/ml) or (B) IL-6 (20 ng/ml) and appropriate vehicles for 48 h followed by immunoblot analyses of indicated proteins. Representative immunoblots and statistical analyses are shown. Data are means ± SE, n = 3 subjects/group, *p < 0.05, by Mann–Whitney U test.
Supplementary Figure 5 | Early passage human PAH PAVSMC were incubated in serum deprived media in presence of 2-Deoxy-D-glucose (2-DG) 10 mM (A) or 1 mM (B) and/or Akt inhibitor VIII (1 μM) or vehicle (−). Forty-eight hours later cell proliferation (Ki-67) was analyzed. Data are means ± SE, percentage of Ki-67-positive cells/total. *p < 0.05, **p < 0.01 by Kruskal–Wallis rank test with Dunn pairwise comparison post hoc test.
References
1. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. (2014) 115:115–30. doi: 10.1161/circresaha.115.301146
2. Besinque GM, Lickert CA, Pruett JA. The myth of the stable pulmonary arterial hypertension patient. Am J Manag Care. (2019) 25:S47–52.
3. Sarzynska K, Swiatoniowska-Lonc N, Dudek K, Jonas K, Kopec G, Gajek J, et al. Quality of life of patients with pulmonary arterial hypertension: a meta-analysis. Eur Rev Med Pharmacol Sci. (2021) 25:4983–98. doi: 10.26355/eurrev_202108_26455
4. Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. (2019) 53:1801887. doi: 10.1183/13993003.01887-2018
5. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. (2018) 360:j5492. doi: 10.1136/bmj.j5492
6. Sommer N, Ghofrani HA, Pak O, Bonnet S, Provencher S, Sitbon O, et al. Current and future treatments of pulmonary arterial hypertension. Br J Pharmacol. (2021) 178:6–30. doi: 10.1111/bph.15016
7. Hassoun PM. Pulmonary arterial hypertension. N Engl J Med. (2021) 385:2361–76. doi: 10.1056/NEJMra2000348
8. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. (2017) 367:643–9. doi: 10.1007/s00441-016-2539-y
9. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. (2008) 294:H570–8. doi: 10.1152/ajpheart.01324.2007
10. Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, et al. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation. (2014) 129:864–74. doi: 10.1161/circulationaha.113.004581
11. Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Mol Med. (2014) 20:191–201. doi: 10.2119/molmed.2013.00165
12. Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med. (2012) 185:260–6. doi: 10.1164/rccm.201108-1536PP
13. Kudryashova TV, Goncharov DA, Pena A, Ihida-Stansbury K, DeLisser H, Kawut SM, et al. Profiling the role of mammalian target of rapamycin in the vascular smooth muscle metabolome in pulmonary arterial hypertension. Pulm Circ. (2015) 5:667–80. doi: 10.1086/683810
14. Xu W, Janocha AJ, Erzurum SC. Metabolism in pulmonary hypertension. Annu Rev Physiol. (2021) 83:551–76. doi: 10.1146/annurev-physiol-031620-123956
15. Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. (2007) 104:11418–23. doi: 10.1073/pnas.0610467104
16. Hansmann G, Calvier L, Risbano MG, Chan SY. Activation of the metabolic master regulator PPARgamma: a potential PIOneering therapy for pulmonary arterial hypertension. Am J Respir Cell Mol Biol. (2020) 62:143–56. doi: 10.1165/rcmb.2019-0226PS
17. Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl). (2011) 89:771–83. doi: 10.1007/s00109-011-0762-2
18. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. (2017) 9:eaao4583. doi: 10.1126/scitranslmed.aao4583
19. Kovacs L, Cao Y, Han W, Meadows L, Kovacs-Kasa A, Kondrikov D, et al. PFKFB3 in smooth muscle promotes vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med. (2019) 200:617–27. doi: 10.1164/rccm.201812-2290OC
20. Valuparampil Varghese M, James J, Eccles CA, Niihori M, Rafikova O, Rafikov R. Inhibition of anaplerosis attenuated vascular proliferation in pulmonary arterial hypertension. J Clin Med. (2020) 9:443. doi: 10.3390/jcm9020443
21. Dai J, Zhou Q, Chen J, Rexius-Hall ML, Rehman J, Zhou G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat Commun. (2018) 9:3850. doi: 10.1038/s41467-018-06376-x
22. Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med. (2017) 195:425–37. doi: 10.1164/rccm.201606-1226PP
23. Hernandez-Saavedra D, Sanders L, Freeman S, Reisz JA, Lee MH, Mickael C, et al. Stable isotope metabolomics of pulmonary artery smooth muscle and endothelial cells in pulmonary hypertension and with TGF-beta treatment. Sci Rep. (2020) 10:413. doi: 10.1038/s41598-019-57200-5
24. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. (2019) 20:436–50. doi: 10.1038/s41580-019-0123-5
25. Culley MK, Chan SY. Mitochondrial metabolism in pulmonary hypertension: beyond mountains there are mountains. J Clin Invest. (2018) 128:3704–15. doi: 10.1172/JCI120847
26. Mumby S, Perros F, Hui C, Xu BL, Xu W, Elyasigomari V, et al. Extracellular matrix degradation pathways and fatty acid metabolism regulate distinct pulmonary vascular cell types in pulmonary arterial hypertension. Pulm Circ. (2021) 11:2045894021996190. doi: 10.1177/2045894021996190
27. Singh N, Manhas A, Kaur G, Jagavelu K, Hanif K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br J Pharmacol. (2016) 173:2030–45. doi: 10.1111/bph.13495
28. Zhuang W, Lian G, Huang B, Du A, Gong J, Xiao G, et al. CPT1 regulates the proliferation of pulmonary artery smooth muscle cells through the AMPK-p53-p21 pathway in pulmonary arterial hypertension. Mol Cell Biochem. (2019) 455:169–83. doi: 10.1007/s11010-018-3480-z
29. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
30. Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? J Cell Biochem. (2004) 91:47–53. doi: 10.1002/jcb.10708
31. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Progr Lipid Res. (2013) 52:585–9. doi: 10.1016/j.plipres.2013.08.005
32. Kudryashova TV, Goncharov DA, Pena A, Kelly N, Vanderpool R, Baust J, et al. HIPPO-Integrin-linked Kinase cross-talk controls self-sustaining proliferation and survival in pulmonary hypertension. Am J Respir Crit Care Med. (2016) 194:866–77. doi: 10.1164/rccm.201510-2003OC
33. Qiu B, Simon MC. BODIPY 493/503 staining of neutral lipid droplets for microscopy and quantification by flow cytometry. Bio Protoc. (2016) 6:e1912. doi: 10.21769/BioProtoc.1912
34. Kudryashova TV, Dabral S, Nayakanti S, Ray A, Goncharov DA, Avolio T, et al. Noncanonical HIPPO/MST signaling via BUB3 and FOXO drives pulmonary vascular cell growth and survival. Circ Res. (2022) 130:760–78. doi: 10.1161/CIRCRESAHA.121.319100
35. Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. (2010) 2:44ra58. doi: 10.1126/scitranslmed.3001327
36. Zhang YL, Zhang R, Shen YF, Huang KY, He YY, Zhao JH, et al. 3-Bromopyruvate attenuates experimental pulmonary hypertension via inhibition of glycolysis. Am J Hypertens. (2019) 32:426–32. doi: 10.1093/ajh/hpy191
37. He S, Zhu T, Fang Z. The role and regulation of pulmonary artery smooth muscle cells in pulmonary hypertension. Int J Hypertens. (2020) 2020:1478291. doi: 10.1155/2020/1478291
38. Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract. (2007) 77(Suppl. 1):S49–57. doi: 10.1016/j.diabres.2007.01.033
39. Tappy L, Dussoix P, Iynedjian P, Henry S, Schneiter P, Zahnd G, et al. Abnormal regulation of hepatic glucose output in maturity-onset diabetes of the young caused by a specific mutation of the glucokinase gene. Diabetes. (1997) 46:204–8. doi: 10.2337/diab.46.2.204
40. Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. (2011) 14:9–19. doi: 10.1016/j.cmet.2011.06.003
41. Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. (2010) 21:268–76. doi: 10.1016/j.tem.2010.01.001
42. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. (2008) 8:224–36. doi: 10.1016/j.cmet.2008.07.007
43. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
44. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
45. Garat CV, Crossno JT Jr., Sullivan TM, Reusch JE, Klemm DJ. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J Cardiovasc Pharmacol. (2013) 62:539–48. doi: 10.1097/FJC.0000000000000014
46. Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, et al. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol. (2013) 48:568–77. doi: 10.1165/rcmb.2012-0429OC
47. Baardman J, Verberk SGS, van der Velden S, Gijbels MJJ, van Roomen C, Sluimer JC, et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat Commun. (2020) 11:6296. doi: 10.1038/s41467-020-20141-z
48. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. (2002) 415:339–43. doi: 10.1038/415339a
49. Goncharova EA. mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J. (2013) 27:1796–807. doi: 10.1096/fj.12-222224
50. Yan WW, Liang YL, Zhang QX, Wang D, Lei MZ, Qu J, et al. Arginine methylation of SIRT7 couples glucose sensing with mitochondria biogenesis. EMBO Rep. (2018) 19:e46377. doi: 10.15252/embr.201846377
51. Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. (2013) 5:654–65. doi: 10.1016/j.celrep.2013.10.007
52. Yoshizawa T, Karim MF, Sato Y, Senokuchi T, Miyata K, Fukuda T, et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metab. (2014) 19:712–21. doi: 10.1016/j.cmet.2014.03.006
53. Tang M, Tang H, Tu B, Zhu WG. SIRT7: a sentinel of genome stability. Open Biol. (2021) 11:210047. doi: 10.1098/rsob.210047
54. Kiran S, Oddi V, Ramakrishna G. Sirtuin 7 promotes cellular survival following genomic stress by attenuation of DNA damage, SAPK activation and p53 response. Exp Cell Res. (2015) 331:123–41. doi: 10.1016/j.yexcr.2014.11.001
55. Wu D, Li Y, Zhu KS, Wang H, Zhu WG. Advances in cellular characterization of the sirtuin isoform, SIRT7. Front Endocrinol (Lausanne). (2018) 9:652. doi: 10.3389/fendo.2018.00652
56. Tang X, Li G, Shi L, Su F, Qian M, Liu Z, et al. Combined intermittent fasting and ERK inhibition enhance the anti-tumor effects of chemotherapy via the GSK3beta-SIRT7 axis. Nat Commun. (2021) 12:5058. doi: 10.1038/s41467-021-25274-3
57. Kimura Y, Izumiya Y, Araki S, Yamamura S, Hanatani S, Onoue Y, et al. Sirt7 deficiency attenuates neointimal formation following vascular injury by modulating vascular smooth muscle cell proliferation. Circ J. (2021) 85:2232–40. doi: 10.1253/circj.CJ-20-0936
58. Zhao Y, Ye X, Chen R, Gao Q, Zhao D, Ling C, et al. Sirtuin 7 promotes non-small cell lung cancer progression by facilitating G1/S phase and epithelial-mesenchymal transition and activating AKT and ERK1/2 signaling. Oncol Rep. (2020) 44:959–72. doi: 10.3892/or.2020.7672
59. Li H, Tian Z, Qu Y, Yang Q, Guan H, Shi B, et al. SIRT7 promotes thyroid tumorigenesis through phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene. (2019) 38:345–59. doi: 10.1038/s41388-018-0434-6
60. Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ Res. (2006) 98:111–8. doi: 10.1161/01.RES.0000197781.20524.b9
61. Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. (2009) 100:1369–72. doi: 10.1038/sj.bjc.6605007
62. Agrawal V, Lahm T, Hansmann G, Hemnes AR. Molecular mechanisms of right ventricular dysfunction in pulmonary arterial hypertension: focus on the coronary vasculature, sex hormones, and glucose/lipid metabolism. Cardiovasc Diagn Ther. (2020) 10:1522–40. doi: 10.21037/cdt-20-404
63. Koop AC, Bossers GPL, Ploegstra MJ, Hagdorn QAJ, Berger RMF, Sillje HHW, et al. Metabolic remodeling in the pressure-loaded right ventricle: shifts in glucose and fatty acid Metabolism-A systematic review and meta-analysis. J Am Heart Assoc. (2019) 8:e012086. doi: 10.1161/JAHA.119.012086
64. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. (2011) 481:380–4. doi: 10.1038/nature10602
65. Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. (2013) 6:1353–63. doi: 10.1242/dmm.011338
66. Lamming DW, Sabatini DM. A central role for mTOR in lipid homeostasis. Cell Metab. (2013) 18:465–9. doi: 10.1016/j.cmet.2013.08.002
67. Martinez Calejman C, Trefely S, Entwisle SW, Luciano A, Jung SM, Hsiao W, et al. mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat Commun. (2020) 11:575. doi: 10.1038/s41467-020-14430-w
68. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. (2004) 64:3892–9. doi: 10.1158/0008-5472.Can-03-2904
69. Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. (2011) 14:21–32. doi: 10.1016/j.cmet.2011.06.002
70. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
71. Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT inhibitors: new weapons in the fight against breast cancer? Front Pharmacol. (2021) 12:662232. doi: 10.3389/fphar.2021.662232
72. Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther. (2017) 172:101–15. doi: 10.1016/j.pharmthera.2016.12.001
73. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. (2021) 6:425. doi: 10.1038/s41392-021-00828-5
74. Mei Z, Zhang X, Yi J, Huang J, He J, Tao Y. Sirtuins in metabolism, DNA repair and cancer. J Exp Clin Cancer Res. (2016) 35:182. doi: 10.1186/s13046-016-0461-5
75. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. (2014) 114:368–78. doi: 10.1161/CIRCRESAHA.113.300536
76. Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, et al. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics. (2016) 8:61. doi: 10.1186/s13148-016-0224-3
77. Blank MF, Grummt I. The seven faces of SIRT7. Transcription. (2017) 8:67–74. doi: 10.1080/21541264.2016.1276658
78. Chen KL, Li L, Yang FX, Li CM, Wang YR, Wang GL. SIRT7 depletion inhibits cell proliferation, migration, and increases drug sensitivity by activating p38MAPK in breast cancer cells. J Cell Physiol. (2018) 233:6767–78. doi: 10.1002/jcp.26398
79. Kim JH, Kim D, Cho SJ, Jung KY, Kim JH, Lee JM, et al. Identification of a novel SIRT7 inhibitor as anticancer drug candidate. Biochem Biophys Res Commun. (2019) 508:451–7. doi: 10.1016/j.bbrc.2018.11.120
Keywords: pulmonary arterial hypertension, lipogenesis, vascular smooth muscle, proliferation, apoptosis, JNK, SIRT7, Akt
Citation: Jiang L, Goncharov DA, Shen Y, Lin D, Chang B, Pena A, DeLisser H, Goncharova EA and Kudryashova TV (2022) Akt-Dependent Glycolysis-Driven Lipogenesis Supports Proliferation and Survival of Human Pulmonary Arterial Smooth Muscle Cells in Pulmonary Hypertension. Front. Med. 9:886868. doi: 10.3389/fmed.2022.886868
Received: 01 March 2022; Accepted: 31 May 2022;
Published: 28 June 2022.
Edited by:
Soban Umar, University of California, Los Angeles, United StatesReviewed by:
Olivier Boucherat, Laval University, CanadaWerner Seeger, University of Giessen, Germany
Copyright © 2022 Jiang, Goncharov, Shen, Lin, Chang, Pena, DeLisser, Goncharova and Kudryashova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatiana V. Kudryashova, dGt1ZEB1Y2RhdmlzLmVkdQ==
†These authors share senior authorship