 Zhifen Wu1
Zhifen Wu1 Jurong Yang
Jurong Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 17 May 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.883028
This article is part of the Research Topic Immunological Landscape and Management of Kidney Diseases View all 7 articles
Sepsis-associated acute kidney injury (SA-AKI) is common in patients with severe sepsis, and has a high incidence rate and high mortality rate in ICU patients. Most patients progress to AKI before drug treatment is initiated. Early studies suggest that the main mechanism of SA-AKI is that sepsis leads to vasodilation, hypotension and shock, resulting in insufficient renal blood perfusion, finally leading to renal tubular cell ischemia and necrosis. Research results in recent years have shown that programmed cell death such as apoptosis, necroptosis, pyroptosis and autophagy play important roles. In the early stage of sepsis-related AKI, autophagy bodies form and inhibit various types of programmed cell death. With the progress of disease, programmed cell death begins. Apoptosis promoter represents caspase-8-induced apoptosis and apoptosis effector represents caspase-3-induced apoptosis, however, caspase-11 and caspase-1 regulate gasdermin D-mediated pyroptosis. Caspase-8 and receptor interacting kinase 1 bodies mediate necroptosis. This review focuses on the pathophysiological mechanisms of various programmed cell death in sepsis-related AKI.
Sepsis is a syndrome characterized by physiological, pathological and biochemical abnormalities caused by infection (1). It is characterized by severe systemic inflammation and multiple organ dysfunction (such as of the blood vessels, lungs, liver, kidney, heart and brain) due to the host's unregulated response to infection. Sepsis is an acute onset, severe condition with high mortality, and can involve multiple organs of the whole body. Multidrug resistant bacteria, drug side effects, and adverse events complicate the treatment of sepsis (2–5). Sepsis-associated acute kidney injury (SA-AKI) is the most common complication of sepsis, which significantly increases sepsis mortality (6). Sepsis complicated with AKI is multifactorial, and its specific mechanism has not been fully clarified. However, it has been proven that an excessive inflammatory response, intrarenal hemodynamic changes, coagulation dysfunction, microvascular endothelial dysfunction and renal tubular epithelial cell (RTECs) injury are all related to this process. A large number of studies have confirmed that the main injury mode of RTECs in acute renal injury caused by renal ischemia-reperfusion (I/R) is renal cell necrosis (7). For a long time, it was agreed that in SA-AKI, pathological changes in the kidney were characterized by ischemic necrosis of RTECs (8). With further study of sepsis-related acute renal injury, it was found that the reduction of renal blood flow was rarely confirmed in patients with sepsis (9–11), and there was no extensive tubular cell necrosis in autopsy analysis in the kidneys of patients with severe SA-AKI (12). In large animal models of septic shock, AKI can occur even if renal blood flow increases or remains unchanged (13, 14). In addition, pathological staining and protein level detection of renal tissue shows that the main pathological manifestation of SA-AKI is renal tubular cell apoptosis (11, 15–17). Necrosis, pyroptosis and autophagy-dependent cell death also play important roles. Necroptosis and ferroptosis are recently discovered modes of programmed cell death. At present, there are few reports on SA-AKI, and the roles of necroptosis and ferroptosis in SA-AKI need to be confirmed in further research.
There are many types of cell death, that can be divided into programmed cell death (PCD) and non-programmed cell death (Figure 1). Non-programmed cell death is a biologically uncontrolled process, typically represented by cell necrosis. PCD involves tightly structured signal cascade reactions and molecularly defined effect mechanisms, including apoptosis, pyroptosis, necroptosis, ferroptosis, autophagy-dependent cell death, entotic cell death, netotic cell death, parthanatos, lysosome-dependent cell death, alkaliptosis and oxeiptosis (18). To date, studies have found that cell death modes such as necrosis, apoptosis, pyroptosis, necroptosis, ferroptosis, and autophagy-dependent cell death play an important role in the pathophysiological process of SA-AKI. In this review, we will introduce the definition, epidemiology and pathophysiological mechanisms of various types of programmed cell death in SA-AKI.
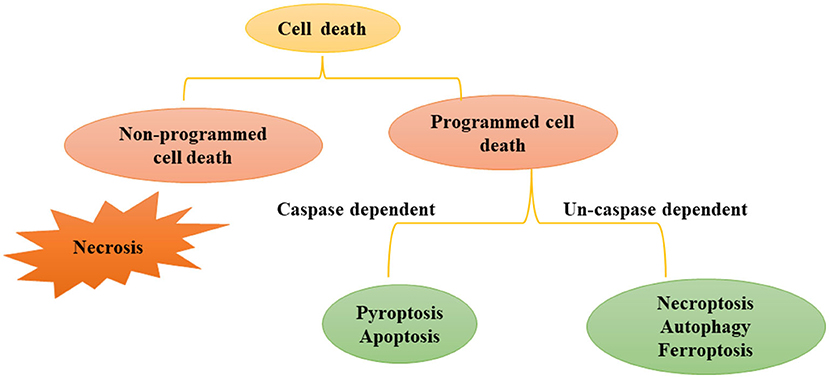
Figure 1. Modes of Cell death: programmed cell death and non-programmed cell death. Cell death includes programmed cell death and non-programmed cell death. Non programmed cell death represents cell necrosis. Programmed cell death can be divided into caspase dependent cell death (such as apoptosis and pyroptosis) and non-caspase dependent cell death (For instance necroptosis, ferrotosis, autophagy, etc.).
Sepsis is a global public health challenge (19). According to statistics, the survival rate of sepsis is only 30%. According to statistics of the World Health Organization, at least 6 million patients die of sepsis infection every year. Among the complications of sepsis, AKI is the most common and serious, and its mortality is the highest (6, 20, 21). Renal failure occurs in 30–50% of patients with sepsis, and the mortality of patients with AKI is 45–70%. Those who survive have a significantly increased risk of developing chronic kidney disease (20, 22–24). According to statistics, more than 60% of sepsis cases are related to AKI, and about 50% of AKI cases are related to sepsis (25, 26). Despite timely and active intervention, the mortality rate is still 45–70% (27). However, the exact cause of sepsis leading to this injury is unclear. The pathogenesis of SA-AKI is extremely complex, including inflammation, hemodynamics, microvascular dysfunction and renal tubular injury. Although some progress has been made in this field, there is still no standardized and satisfactory treatment strategy for SA-AKI, and specific and effective treatment methods are lacking, apart from blood purification and supportive treatment (28–30). Therefore, it is urgent to further explore the exact mechanism of SA-AKI, which can provide new insights for clinical treatment (31).
At present, the forms of cell death related to AKI can be roughly divided into apoptosis, pyroptosis, ferroptosis, necroptosis, and autophagy-dependent cell death, necrosis (32–36). The occurrence of SA-AKI involves a variety of cell death pathways. Although the molecular mechanisms of these pathways differ, they are interrelated. Therefore, in the following chapters, we will briefly describe various cell death pathways and their relationships in SA-AKI.
Apoptosis refers to a series of programmed cell death processes that activate caspase cascade apoptosis under the regulation of apoptotic genes. This process is often accompanied by chromatin condensation, DNA breakage, phosphatidylserine valgus, and the emergence of apoptotic bodies (18, 37). At present, there are three known pathways of apoptosis (Figure 2) an exogenous pathway mediated by death receptors, an endogenous pathway mediated by mitochondria, and an endoplasmic reticulum pathway.
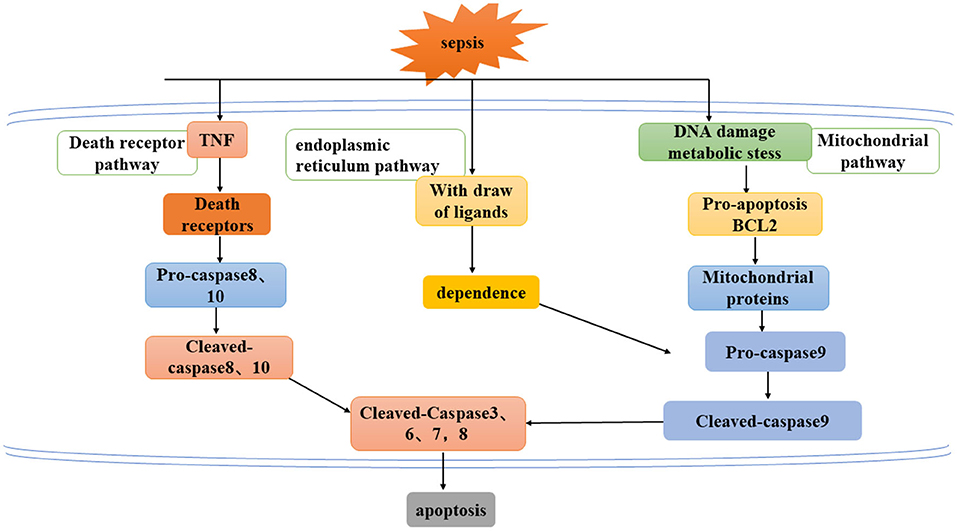
Figure 2. Three ways to mediate apoptosis: exogenous pathway mediated by death receptor, endogenous pathway mediated by mitochondria, and endoplasmic reticulum pathway.
Sepsis-related acute renal injury was initially recognized as renal tubular cell ischemia and necrosis caused by insufficient renal blood perfusion owing to sepsis. Therefore, for a long time, treatment was focused on increasing perfusion pressure and renal blood flow. However, a series of animal experiments and human data showed that most patients with sepsis-related acute renal injury do not have decreased renal blood flow perfusion. Interestingly, normal or increased renal blood flow was found in animal models of sepsis and in patients with sepsis (11, 15, 37–39). Lerolle et al. found that apoptotic cells were observed in the proximal and distal tubules of all patients with septic shock and anuria AKI through renal biopsy after death, which was compared with patients who died of non-infectious shock without renal injury in the ICU and patients with trauma without renal injury, Renal tubular cell apoptosis and capillary leukocyte infiltration were increased significantly, which strongly proved that apoptosis plays an important role in the pathogenesis of septic shock AKI (11). Similar results were obtained by Aslan et al. (17). However, in animal models of sepsis, pathological manifestations of the kidney are different from those of human patients, as shown in Table 1. The reasons for this may be: (1) in the animal models, especially mouse model, the body is small and the whole body blood circulation is rapid, resulting in inconsistent pathological damage between human patients and animals; (2) human specimens are collected after the patients' death, and blood circulation stops after the patient's death, leading to rapid organ death (42, 43).

Table 1. Modes of sepsis-associated acute kidney injury(SA-AKI).
Mariano et al. treated renal tubular cells and podocytes with plasma from patients with severe burn and SA-AKI, and apoptosis was significantly increased compared with patients without AKI. This indicates that circulatory system factors lead to renal apoptosis (16, 40). Thus, the pathogenesis of sepsis-related AKI is complex, involving multiple factors, and apoptosis is dominant (11, 15–17). However, the specific mechanism of initiating apoptosis is not clear.
Lipopolysaccharide (LPS), a well-known circulating endotoxin and a component of the bacterial cell wall, is the main cause of septic shock and may lead to acute renal failure by directly inducing apoptosis of renal tubular cells through systemic cytokine release (40, 41). Guo et al. emphasized the role of LPS in AKI-induced apoptosis of renal tubular cells in mouse experiments (44). Intervention with caspase-3 inhibitor, could protect renal tubular cells from acute renal failure caused by LPS. Interestingly, in that model, caspase-3 inhibitors could not only prevent apoptosis, but could also reduce renal inflammation, confirming that caspase plays an important role in renal tubular cell apoptosis (45). In addition to epithelial cell injury, macrophages and endothelial cells are also a target in sepsis. Endothelial cell injury destroys microvascular blood flow, resulting in renal hypoperfusion, hypoxia and epithelial cell ischemia. In sepsis, macrophages can cause spontaneous death while scavenging microorganisms, but excessive apoptosis of macrophages may inhibit immune function and aggravate renal damage (46). The activation of glomerular endothelial cells increases vascular permeability. In gram-negative sepsis, LPS and tumor necrosis factor (TNF-α) both induced apoptosis of glomerular endothelial cells, in renal endothelial cells, TNF-α Receptor 1 induces apoptosis via a caspase-8-dependent pathway (47), and may promote sepsis-induced AKI in vivo (44, 48). In addition, Bannerman et al. proposed that LPS can also directly aggregate Fas-related death domain (FADD) and TNF receptor related death domain (TRADD) through the death effector domain (DED), thereby inducing the cleavage of Caspase-3, 6,7, and 8, apoptosis occurs finally (49). The activation of Caspase-3 signaling leads to the inhibition of protein hydrolysis and irreversible cell death. The translocation of cleaved caspase-3 into the nucleus leads to substrate cleavage, DNA degradation, and protein modification, and finally leads to the emergence of apoptotic bodies. Endotoxin stimulation and/or oxygen restriction leads to the production of reactive oxygen species (ROS), which can initiate apoptosis. Excessive ROS can stimulate p53 and induce apoptosis. B-cell lymphoma-2-associated X protein (Bax) is activated by ROS and/or p53 and is transferred to the outer membrane of mitochondria, resulting in the leakage of cytochrome c (Cytc), which is a strong inducer of apoptosis (50).
It can be seen that apoptosis plays a vital role in SA-AKI. Blocking apoptosis is expected to reduce mortality patients with SA-AKI and block the progression to chronic kidney disease (CKD). Recent study reported that apoptosis belongs to immune silent death, and no damage related molecular patterns (DAMPs) was released by apoptotic cells (51, 52). Studies have confirmed that necroptosis and ferroptosis play a very important role in the pathophysiological progress of SA-AKI (53).
Necroptosis is a recently discovered programmed cell death mode that is morphologically different from apoptosis. Studies have confirmed that necroptosis can cause the death of renal cells, which is closely related to inflammation (53). It is characterized by plasma membrane rupture, cell content leakage and organelle swelling (54–56). Necroptosis is not dependent on caspase, but is involved in regulation through multiple cellular signal transduction proteins, including receptor interacting protein kinase 1 (RIPK1), receptor interacting protein kinase 3 (RIPK3) and its downstream target mixed lineage kinase domain like (MLKL) (57) (Figure 3A). Studies have shown that many cell mediators can activate the necroptosis pathway, such as death receptors, interferons, Toll like receptor (TLRs), and RNA or DNA sensors (58).
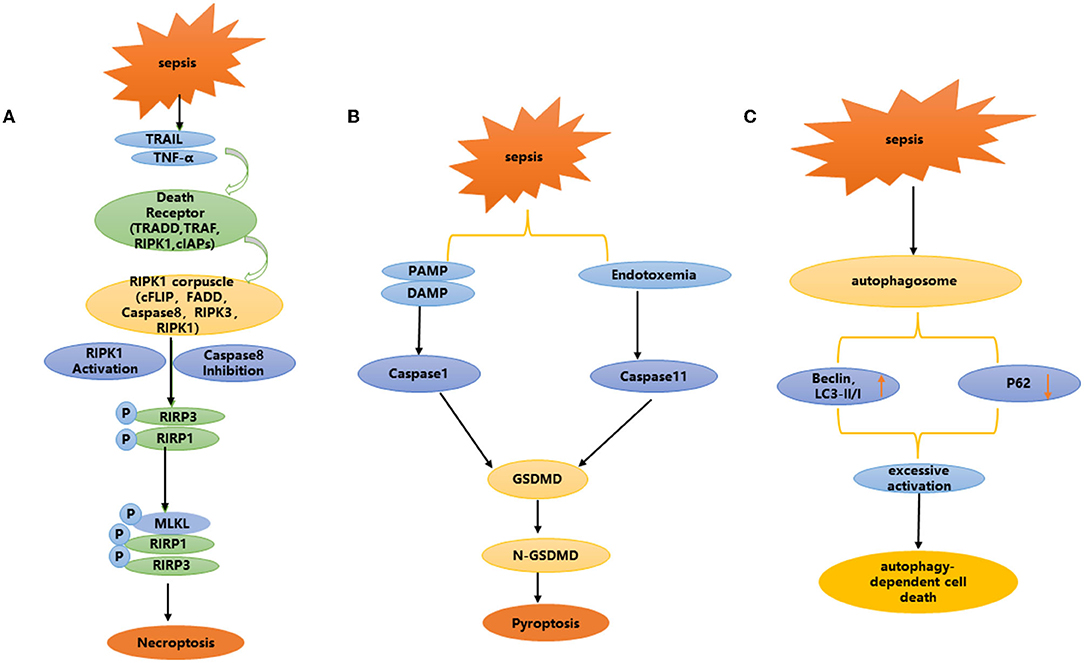
Figure 3. (A–C) Non-apoptotic programmed cell death: necroptosis, pyroptosis, and autophagy dependent cell death.
The activation of RIPK1 is widely considered to be the main mediator of necrotic apoptosis mainly through its binding to TNF receptor (TNFR) (59). RIPK1 further activates RIPK3 through the homologous interaction motif (RHIM) and then activates MLKL (60–62). After activation, oligomers of phosphorylated mixed lineage kinase-like (MLKL) protein are formed and transferred to the plasma membrane. This structure leads to cell swelling, rupture and the release of damage-associated molecular patterns (DAMPs) (62, 63). The pathophysiology of septic AKI is different from that of other AKI models. After systemic injection of lethal doses of lipopolysaccharide (LPS), no significant RTECs necrosis is observed in septic kidneys (64). Therefore, the role of necroptosis in septic AKI has attracted interest in current research. In vitro experiment, Sureshbabu et al. (65) showed that the expression levels of phosphorylated RIPK3 (p-RIPK3), RIPK3 and p-MLKL increased in a time-dependent manner under LPS stimulation in human renal proximal tubular cells (HK-2 cells). In mice undergoing CLP surgery, the expression levels of p-RIPK3, RIPK3, and MLKL in renal tissue were higher 6 h after treatment. These results suggest that RTECs necroptosis is also involved in the occurrence and development of SA-AKI. However, there are few studies on the necroptosis of endothelial cells and RTECs during sepsis. Here, we propose to emphasizing the necroptosis regulation of renal vascular endothelial cells and RTECs.
Endothelial cells and macrophages also play an important role in SA-AKI necrotic apoptosis. In addition, programmed cell necrosis was also observed in cisplatin, I/R and contrast medium induced nephrotoxicity. At present, there are few reports on necroptosis in sepsis, especially sepsis related AKI. Further research is needed to deepen our understanding and provide a new direction for clinical treatment. In addition, necroptosis is involved in various other pathological conditions, such as I/R injury, nonalcoholic steatohepatitis and atherosclerosis (66).
Pyroptosis is a type of pro-inflammatory programmed cell death. It is considerably different from apoptosis and autophagy in cell morphology and function. It is activated by members of the inflammation related caspase family, which cleave gasdermin family proteins to expose their N-terminus and transfer to the cell membrane, resulting in cell membrane damage and 1.1–2.4 nm perforation. By changing the osmotic pressure inside and outside the cell, the cell becomes swollen and ruptured, and membrane integrity is lost, which eventually leads to the release of inflammatory factors [including interleukin-1 (IL-1) and IL-18] mediated by cell collapse and pyroptosis (67, 68). However, during this process, the structure and function of mitochondria remain intact (69).
Pyroptosis is involved in the occurrence and development of a variety of diseases, especially related to the occurrence and development of kidney diseases. Focal death is mainly involved in renal disease in two ways: via the caspase-1-mediated classical focal death pathway and the caspase-11 mediated non-classical focal death pathway (Figure 3B). Caspase-1 mediated classical focal death is a regulated form of cell death, which depends on the activation of caspase 1.When the host is infected and generates a danger signal, such as damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), nod like receptors (NLRs) recognize these signals and trigger focal death (70, 71). NLRs, usually involve NLRP3 or NLRP4, and NLRP3 is one of the most important intracellular receptors, that can be expressed by NF-κ B transcription induction. Activation of the NLRP3 inflammasome plays a key role in focal death (72–75). After recognizing the ligand, the adaptor protein apoptosis-associated speck-like protein (ASC) is captured by NLRP3, while activating caspase-1, resulting in inflammatory factors such as IL-18 and IL-1β (76–78). Cleaved-caspase-1 cleaves the N-terminal sequence of gasdermin D(GSDMD), binds to the membrane, and forms membrane pores, resulting in cell swelling, membrane rupture and cell death. In addition, the production of reactive oxygen species (ROS), is a potential trigger for NLRP3 inflammasome assembly (79). Due to infection, inflammation or mitochondrial dysfunction, the level of mitochondrial reactive oxygen species (mtROS) increases, and mitochondrial DNA is released into the cytoplasm in the form of oxidation. This occurs through direct interaction between oxidized mitochondrial DNA (ox-tmDNA) and NLRP3, triggering assembly and activation of the NLRP3 inflammasome (80–82). In a rat model of sepsis, Yang et al. found that the caspase-1 inhibitor AC-YVAD-CMK significantly reduced the expression of GSDMD in renal tissue, and protected against acute renal injury caused by sepsis by blocking the expression of NLRP1 inflammatory body, reducing the scorch death of RTECs induced by the inflammatory body, increasing antioxidant enzyme activity, and weakening oxidation products (83).
In the non-classical pyroptosis pathway, caspase-4, 5, and 11 can be directly activated after interacting with bacterial LPS, resulting in the cleavage of GSDMD, independent of the activation of NLRP3 and caspase-1 (84–86). Caspase-11 is an intracellular cysteine protease that mediates the response of host cells to gram-negative bacterial pathogens and sepsis. Host cells sense LPS in gram-negative bacteria through a non-standard inflammasome pathway, which eventually leads to caspase-11 activation and pyroptosis (76, 78, 87–89). A series of studies have shown that caspase-11 plays a key role in pyroptosis (76, 78, 87–92). Ye et al. used LPS induced sepsis to produce soluble death of RTECs, accompanied by increased expression of focal death-related proteins caspase-11 and GSDMD. Through caspase-11 gene knockout, those authors further confirmed that caspase-11 mediated focal death plays an important role in SA-AKI. LPS induced focal death-related protein expression increases and weakens the focal death of RTECs induced by caspase-11 (93).
In SA-AKI, pyroptosis is also observed in macrophages and endothelial cells in addition to renal tubular cells (94, 95). Compared with in vitro experiments, the in vivo experiments show more severe focal death in renal tubular cells. Current evidence suggests that inhibition of pyroptosis signaling can improve SA-AKI in mice.
Ferroptosis is a unique programmed cell death mode discovered by Stockwell et al. (96) (Figure 3C). It is characterized by excessive accumulation of iron, enhanced lipid peroxidation and ineffective clearance of lipid peroxide (97), mainly characterized by smaller mitochondria and increased membrane density (98), without nuclear condensation, DNA fragmentation and caspase activation. Glutathione peroxidase 4 (GPX4) and prostaglandin endoperoxide synthase 2 (PTGS2) are well-known markers of ferroptosis. SystemXc- (SXC), glutathione (GSH) metabolism regulating GPX4 activity and ROS production, and participate in the regulation of ferroptosis (96–98).
Recent studies have shown that ferroptosis is involved in the occurrence and development of a variety of diseases, including degenerative brain disease, and injury to the heart, liver, intestines, and other organs (99). Xiao et al. reported that after cecal ligation and puncture (CLP), the expression of GPX4 decreased significantly and the expression of PTGS2 increased. This was the first confirmation that ferroptosis was involved in SA-AKI (100), but the mechanism of ferroptosis in SA-AKI needs to be further studied.
Although there are few reports on ferroptosis in SA-AKI, it is worth noting that ferroptosis was first found in the kidney, and ferroptosis is closely related to inflammation. It is necessary to further study ferroptosis in SA-AKI.
Autophagy, as one of the main models of cell death, has been considered key to maintaining intracellular homeostasis and the stress response. Autophagy plays an important role in maintaining and promoting cell survival and metabolism (101–103). Autophagy is also a degradation system via which intracellular pathogens, damaged or long-lived proteins, and dysfunctional organelles are encapsulated within autophagosomes and are degraded in lysosomes (104).
Autophagy plays an important role in maintaining intracellular homeostasis in SA-AKI (105). LPS, a mediator of gram-negative bacterial sepsis, induces autophagy in RTECs in vivo and in vitro (106, 107). Wu et al. found that the expression levels of autophagy related proteins Beclin1 and LC3 II/I increased significantly and the expression level of autophagy substrate p62 decreased in a mouse model of polymicrobial sepsis induced by CLP (108). Compared with mice in CLP group, there was less renal injury in CLP + 3-MA (autophagy inhibitor) group, and the postoperative survival rate was significantly higher than that in the CLP group, The autophagy inhibitor chloroquine increases the sensitivity of mice to the lethal effect of CLP, indicating that sepsis can activate autophagy in vivo (108, 109). After determining endotoxemia, the administration of ticrolimus inhibitor can prevent the occurrence of LPS induced AKI by inducing autophagy. These results suggest that autophagy is activated and provides protection in sepsis associated acute renal injury (106, 107). Other studies have found that in mice receiving CLP, renal autophagy was enhanced in the early stage (6 h), indicating that the accumulation of LC3-II and autophagic flow increased, and then decreased at a later time point (24 h). Further induction of autophagy by rapamycin can improve renal function and apoptosis of RTECs in CLP mice (110). In a study using a rat model of CLP induced sepsis, autophagy initially increased at 4 h and then began to decrease after 9 h. The decrease of autophagy led to the apoptosis of proximal renal tubular cells during sepsis (111). This shows that autophagy can protect the body from sepsis induced AKI (108).
Leventhal et al. (106) and Mei et al. (107) used renal proximal tubular epithelial cell specific Atg7 gene knockout mice and found that PTAtg7 knockout mice had more severe renal dysfunction and parenchymal injury than wild-type mice, and there was evidence of increased IL-6 and STAT activation in renal tissue caused by LPS (106). In vitro experiments showed that autophagy function was impaired and IL-6 production was enhanced in Atg7 knockout RTECs, which was related to LPS (106). These studies showed that Atg7 gene knockout enhanced TNF- α induced cell death, whereas the autophagy activator rapamycin inhibited the death of renal tubular cells in vitro (111). These studies suggest that enhanced autophagy provides a general renal protective mechanism in septic AKI by reducing apoptosis, inflammation and oxidative stress (54).
Autophagy dependent cell death is a regulated form of cell death, which depends on the autophagy mechanism, and may occur in a specific environment, contributing to the pathogenesis of disease (68, 112).
In addition to participating in regulated cell death, autophagy can affect other basic processes associated with aging or disease, including the regulation of inflammation, innate immunity and host defense (113, 114). Studies have shown that the autophagy mechanism critical for the degradation of cell death components and regulatory factors. Autophagy also plays a role as a scaffold of necrosis. Inhibiting lysosomal fusion can aggravate cell death. Importantly, these two functions can lead to competitive effects, that is, inhibiting or increasing the possibility of cell death.
In the early study of SA-AKI, it is agreed that SA-AKI is the change of renal hemodynamics caused by septic blood, which leads to renal ischemia and hypoxia and acute tubular necrosis (ATN) (64). The Pathological results showed that ATN was discontinuous distributed in some renal biopsy tissues of some septic patients, and the morphology of most patients was stage 2 ATN, vacuolization, renal tubular edema, epithelial flattening and some apoptotic renal tubular cells (17). However, a series of subsequent studies confirmed that Multiple programmed cell death play a more important role in SA-AKI. This paper mainly expounds the role of programmed cell death in SA-AKI, and does not make too much statement for necrosis.
Apoptosis, pyroptosis, necroptosis, and autophagy are involved in the pathophysiological progress of SA-AKI, each with unique morphological, cell biological and biochemical characteristics. These modes exist simultaneously in SA-AKI (Table 2). The latest research reports that ferroptosis also plays an important role in SA-AKI. For example, apoptosis and necrosis are found in renal tissue in SA-AKI (11). Guo et al. (115) and Liu et al. (116) found that apoptosis and pyroptosis coexist in CLP mice. Li and Liu confirmed the same results (117, 118). Yu et al. (108) found that autophagy existed in a CLP mouse model, in the early stage, autophagy was stronger, and gradually weakened over time. In addition, Chen et al. reported that RIPK3 mediated necroptosis and GSDMD mediated pyroptosis play a synergistic role in sepsis, aggravating septic inflammatory response and tissue damage (119). These evidences suggests that all types of programmed cell death may occur in the kidney at the same time.
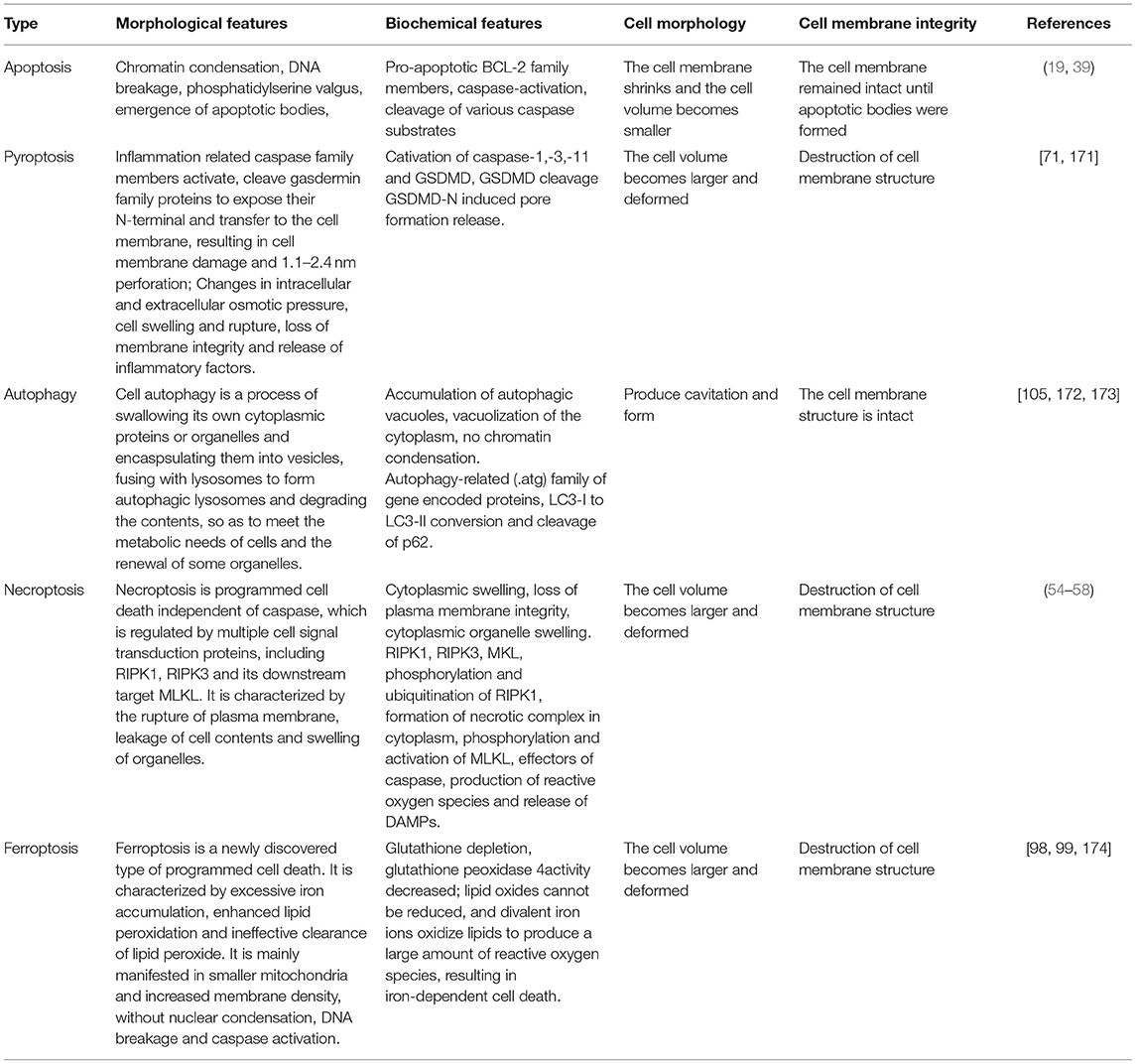
Table 2. Modes cell death.
Apoptosis, autophagy, pyroptosis, and necroptosis also interact. With many interrelated events among them. The most typical is caspase-8, which is involved in different cell death pathways. Caspase-8 is thought to interact with ASC to activate apoptosis (108, 120) and strengthen the apoptotic cascade leading to the inhibition of necroptosis (121). Fritsch et al. (122) found that the expression of inactive caspase-8 led to the death of mouse embryos, which may be due to the enhancement of necroptosis and focal death. MLKL deficiency could rescue the cardiovascular phenotype, but unexpectedly led to perinatal death in caspase8 +/+ mice, suggesting that focal death occurs when apoptosis and necrotic apoptosis are inhibited simultaneously (122). In addition, in caspase-8+/+ MLKL–/– ASC–/– or caspase-8+/+MLKL–/– caspase-1–/–mice, early death and embryo lethality were completely prevented. This suggests that caspase-8 represents a molecular switch that controls apoptosis, necroptosis and focal death (122).
Apoptosis is a type of cell death, that occurs by activating caspases. Autophagy and apoptosis are mutually regulated. Hou et al. (123) found that autophagy is involved in the regulation of cysteine enzyme activity. In Bax–/– HCT116 cells, treated together with cell death stimulator (for example: TNF-related apoptosis-inducing ligand: TRAIL), knockdown Beclin-1 or vps34 at the same time, and inhibits the occurrence of autophagy. Study results showed that the expression of cleaved caspase-8 increased significantly in the cytoplasm. A small amount of activated caspase-8 can be eliminated by lysosomal degradation and can inhibit the occurrence of apoptosis. Further studies showed that the transition from autophagy to apoptosis mainly depends on caspase-8, because knockdown of other caspases in these cells did not prevent autophagy from inhibiting the observed apoptotic transition. In addition, Fortunato et al. found that inhibition of autophagy can induce caspase-8 activity and promote apoptosis, and impaired fusion of autophagosomes and lysosomes can also lead to apoptosis (124). CD95/Fas mediated apoptosis is enhanced by autophagic degradation of protein tyrosine phosphatase non-receptor type 13 (PTPN13). In the same cells, autophagy inhibits apoptosis through TRAIL death receptors (125).
The relationship between autophagy and necroptosis is complex. Various studies have shown that autophagy flux or lysosomal dysfunction is key to the spread of necroptosis. Necroptosis usually occurs simultaneously with autophagy activation (54, 126). Knockout of Atg5, Atg7, or Beclin 1 can prevent necroptosis (127, 128), whereas depletion of Atg16L1 or Atg7, which reduced autophagy flow, can promote necroptosis (129, 130). Recently, autophagy has been shown to be involved in the conversion of complexes containing TRIF, RIPK1, RIPK3, and Z-DNA-binding protein 1 (ZBP1) (131). Deletion of autophagy receptor optineurin (OPTN) can lead to RIPK3-dependent necroptosis (112, 132). Rapamycin treatment and increased autophagy flux reduce RIPK1 expression and protect photoreceptor cells from Zvad-induced necroptosis (133). The negative regulator of necroptosis is caspase-8 (134). Active caspase-8 can cut RIPK1 and RIPK3, and then block necroptosis (135–137).
Ferroptosis is also regulated by autophagy. The increase in ferritin phages allows for the accumulation of free iron and promotes iron sagging (138). Knocking down regulatory factors of autophagy, such as Atg3, Atg5, Atg7, or Atg13, can inhibit ferroptosis (138, 139). A study by Zhang et al. showed that the kinase inhibitor sorafenib promoted the increase of autophagy flux and ferritin deposition (140). A study by Gao et al. showed that inhibiting autophagy could prevent ferritin deposition (139). Bafilomycin A1 treatment inhibited autophagy while significantly reducing erastin induced ferroptosis (141, 142). GPX4 can also be selectively degraded by chaperone mediated autophagy to promote ferroptosis (143). Although autophagy can prevent cell death by scavenging ROS levels, the role of autophagy in ferroptosis usually involves helping to initiate cell death.
Various studies have also reported the interdependence between autophagy and pyroptosis. In particular, autophagy can reduce endotoxin mediated proinflammatory cell kinase IL-1 β and IL-18 secretion. After Atg16L1 or Atg7 knockout, caspase 1 activity and cytokine secretion were enhanced by LPS (82, 144). A unique feature of autophagy regulating pyroptosis is through the degradation of NLRP3 inflammatory bodies and bacterial pathogens. The NLRP3 inflammasome acts as a sensor of mitochondrial dysfunction, demonstrating the link between mitochondrial damage, autophagy / mitochondrial phagocytosis and inflammation (145). The NLRP3 inflammasome can also be selectively degraded by autophagy, limiting cytokine secretion and reducing pyroptosis (79, 146). On the contrary, NRLP3 can inhibit autophagy (147). Some reports have shown that activation of autophagy can prevent t pyroptosis in a variety of situations (148–151), or inhibition of autophagy can enhance pyroptosis (152, 153). It is worth noting that in order to prevent pyroptosis, autophagosomes often degrade large protein complexes or organelles, rather than just a single signal molecule. Therefore, autophagy produces very different features of pyroptosis regulation compared with ferroptosis and necroptosis.
When sepsis associated acute renal injury occurs, all modes of programmed cell death are involved and interrelated. Inflammatory stimulation and increased expression of body death irritants such as TNF-α, the body to produce death receptors (RIPK1 and TRADD, TRAF and cIAPs), and RIPK1 ubiquitinates to form RIPK1 bodies (FADD, caspase-8, cFLIP, RIPK1, and RIPK3) (134, 154–158). The latter cleaves the apoptosis promoter caspase-8 and then activates downstream apoptosis effectors, such as caspase-3, to promote apoptosis (69, 159, 160). Caspase 8 can also degrade cell death stimuli such as TRAIL by autophagy to prevent apoptosis (123). When caspase-8 activity is inhibited or RIPK1 is activated, RIPK1 and RIPK3 are phosphorylated, resulting in necroptosis (123, 161). In addition, during the early stage of inflammation, the body initiates autophagy to protect itself from inflammatory stimulation. Autophagy bodies form to mark the occurrence of autophagy. Autophagy bodies block caspase-8 cleavage by inhibiting RIPK1 bodies, so as to prevent apoptosis. Studies have confirmed that autophagy and necroptosis occur simultaneously, and reducing autophagy flow can enhance necroptosis (129, 138, 162). In addition, autophagy can degrade damaged mitochondria and inhibitors of apoptosis proteins (IAPs) inhibit the occurrence of necroptosis pathways. Autophagy is also involved in the conversion of complexes containing TRIF, RIPK1, RIPK3, and ZBP1, thus promoting necroptosis (131). In addition, autophagy can also interfere with NLRP3 inflammatory bodies, induce the degradation of NLRP3 inflammatory bodies and damaged mitochondria, limit cytokine secretion, inhibit caspase-1 activity, and thereby inhibit the occurrence of pyroptosis (79, 130, 146, 163). In contrast, the NLRP3 inflammasome can also inhibit autophagy to promote pyroptosis (147). Autophagy can also inhibit ferroptosis by inhibiting GPX4 and selectively degrading damaged mitochondria or ferritin. Autophagy can also degrade toxic oxidized lipids that cause damage to cellular DNA and proteins to inhibit ferroptosis (131). When the body continues to be stimulated by infection and inflammation, autophagy decreases, renal tubular cells are damaged, and apoptosis, pyroptosis, necroptosis and inflammatory factors of the circulatory system act together, resulting in acute renal injury.
Sepsis is a multisystem acute infectious disease. The pathological mechanism of acute renal injury is complex and involves many aspects, including renal injury caused by inflammatory factors in the circulatory system. Apoptosis plays a major role in renal injury. However, the study found that apoptosis can not fully explain the serious clinical manifestations of the kidney. With the further study, People have a better understanding of sepsis related acute renal injury. More research confirmed that apoptosis plays a core role in SA-AKI, and non-apoptotic programmed cell death such as pyroptosis necroptosis plays a very important role in SA-AKI. Autophagy dependent cell death also plays an important role in the pathophysiological progression of SA-AKI. Recent studies have reported that ferroptosis also plays an indispensable role in SA-AKI. However, the mechanism of non-apoptotic programmed cell death, especially ferroptosis in SA-AKI is unclear, it needs further research and exploration. Information on the regulatory process between several programmed cell death is limited. This makes the prevention and treatment of SA-AKI more difficult. However, there is limited information on the regulatory process between these types of programmed cell death. This makes the prevention and treatment of SA-AKI more difficult. According to various signaling pathways, the mutual regulation of various types of cell death may occur at different times and developmental stages in the progression of sepsis, as well as in different types of cells. Understanding the interaction between different cell death pathways and clarifying the exact mechanism behind cell death is important to developing potential therapies for the treatment of SA-AKI. This review focuses on the mechanism of programmed cell death in SA-AKI, to provide new insights for the diagnosis and treatment of SA-AKI.
At present, studies have confirmed that there are many kinds of PCD in SA-AKI, but whether these kinds of PCD occur at the same time remains to be further explored. It has been reported that there are three kinds of programmed cell death including apoptosis, pyroptosis and necroptosis in infectious diseases, they have a common upstream regulatory target ZBP1, which is named ZBP1 dependent PANoptosis (164). However, whether this new PCD death model exists in renal disease has not been reported. Whether there is an upstream target that can regulate more PCD modes at the same time needs to be further explored.
ZW wrote the paper, collected literature, and produced the chart. JY modified the paper. All authors participated in this review and approved the final version of the manuscript.
National Natural Science Foundation of China (No. 81770682), Basic and Frontier Research Program of Chongqing(cstc2017jcyjBX0014), the Chongqing Talent Program Project (cstc2021ycjh-bgzxm0090), Scientific Research Incubation Project of the Third Affiliated Hospital of Chongqing Medical University (KY20078), and Scientific Research Incubation Project of the Third Affiliated Hospital of Chongqing Medical University (KY08031).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the students and teachers in the laboratory.
SA-AKI, Sepsis-associated acute kidney injury. RTECs
1. Zheng Y, Shi X, Hou J, Gao S, Chao Y, Ding J, et al. Integrating metabolomics and network pharmacology to explore Rhizoma Coptidis extracts against sepsis-associated acute kidney injury. J Chromatogr B Analyt Technol Biomed Life Sci. (2021) 1164:122525. doi: 10.1016/j.jchromb.2021.122525
2. Mayr FB, Prescott HC. Identifying sepsis survivors at risk for adverse cardiovascular outcomes. Am J Respir Crit Care Med. (2021) 204:500–1. doi: 10.1164/rccm.202105-1322ED
3. Liu Q, Song H, Andersson ML, Magnusson P, Fang F. Psychiatric disorders are associated with increased risk of sepsis following a cancer diagnosis. Cancer Res. (2020) 80:canres.0502.2020. doi: 10.1158/0008-5472.CAN-20-0502
4. Eisen D, Leder K, Woods R, Lockery J, McGuinness S, Wolfe R, et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): a randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir Med. (2021) 9:186–95. doi: 10.1016/S2213-2600(20)30411-2
5. Lee S, Hsu T, Lee M, Chao C, Lee W, Lai C, et al. Nationwide trend of sepsis: a comparison among octogenarians, elderly, and young adults. Crit Care Med. (2018) 46:926–34. doi: 10.1097/CCM.0000000000003085
6. Wijayaratne D, Wijewickrama E. Acute kidney injury in sepsis. Sri Lanka J Surg. (2017) 35:15. doi: 10.4038/sljs.v35i1.8348
7. Hultstrm M. Neurohormonal interactions on the renal oxygen delivery and consumption in haemorrhagic shock-induced acute kidney injury. Acta Physiologica. (2013) 209:11–25. doi: 10.1111/apha.12147
8. Haddy FJ. Acute renal failure and sepsis. New Engl J Med. (2004) 351:2347–9. doi: 10.1056/NEJM200411253512224
9. Prowle JR, Molan MP, Hornsey E, Bellomo R. Measurement of renal blood flow by phase-contrast magnetic resonance imaging during septic acute kidney injury: a pilot investigation. Crit Care Med. (2012) 40:1768–76. doi: 10.1097/CCM.0b013e318246bd85
10. Brenner M, Schaer GL, Mallory DL, Su Ff Redini AF, Parrillo JE. Detection of renal blood flow abnormalities in septic and critically ill patients using a newly designed indwelling thermodilution renal vein catheter. Chest. (1990) 98:170–9. doi: 10.1378/chest.98.1.170
11. Lerolle N, Nochy D, Guérot E, Bruneval P, Fagon JY, Diehl JL, et al. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med. (2010) 36:471–8. doi: 10.1007/s00134-009-1723-x
12. Legrand M, Bezemer R, Kandil A, Demirci C, Ince C. The role of renal hypoperfusion in development of renal microcirculatory dysfunction in endotoxemic rats. Intensive Care Med. (2011) 37:1534–42. doi: 10.1007/s00134-011-2267-4
13. Frithiof R, Soehnlein O, Eriksson S, Fenhammar J, Hjelmqvist H, Lindbom L, et al. The effects of isoflurane anesthesia and mechanical ventilation on renal function during endotoxemia. Acta Anaesthesiol Scand. (2014) 55:401–10. doi: 10.1111/j.1399-6576.2011.02406.x
14. Lipcsey M, Bellomo R. Septic acute kidney injury: hemodynamic syndrome, inflammatory disorder, or both? Critical Care. (2011) 15:1008. doi: 10.1186/cc10525
15. Klenzak J, Himmelfarb J. Sepsis and the Kidney. Crit Care Clin. (2005) 21:211–22. doi: 10.1016/j.ccc.2005.01.002
16. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. (1999) 27:1230–51. doi: 10.1097/00003246-199907000-00002
17. Aslan A, van den Heuvel M, Stegeman C, Popa E, Leliveld A, Molema G, et al. Kidney histopathology in lethal human sepsis. Critical care (London, England). (2018) 22:359. doi: 10.1186/s13054-018-2287-3
18. Tang D, Kang R, Berghe T, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
19. Kadri S, Lai Y, Warner S, Strich J, Babiker A, Ricotta E, et al. Inappropriate empirical antibiotic therapy for bloodstream infections based on discordant in-vitro susceptibilities: a retrospective cohort analysis of prevalence, predictors, and mortality risk in US hospitals. Lancet Infect Dis. (2021) 21:241–51. doi: 10.1016/S1473-3099(20)30477-1
20. Zhang J, Wang C, Kang K, Liu H, Yu K. Loganin attenuates septic acute renal injury with the participation of AKT and Nrf2/HO-1 signaling pathways. drug design, development and therapy. Volume. (2021) 15:501–13. doi: 10.2147/DDDT.S294266
21. Zhou P, Ma B, Xu S, Zhang S, Tang H, Zhu S, et al. Knockdown of Burton's tyrosine kinase confers potent protection against sepsis-induced acute lung injury. Cell Biochem Biophys. (2014) 70:1265. doi: 10.1007/s12013-014-0050-1
22. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. (2015) 41:1411–23. doi: 10.1007/s00134-015-3934-7
23. Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol. 7:209–17. doi: 10.1038/nrneph.2011.13
24. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. (2019) 96:1083–99. doi: 10.1016/j.kint.2019.05.026
25. Bagshaw SM, Lapinsky S, Dial S, Arabi Y, Dodek P, Wood G, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. (2009) 35:871–81. doi: 10.1007/s00134-008-1367-2
26. Uchino S, Kellum JA, Bellomo R, Doig GS, Ronco C. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. (2005) 294:813–8. doi: 10.1001/jama.294.7.813
27. Bagshaw SM, Laupland KB, Doig CJ, Mortis G. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Critical Care. (2005) 9:R700. doi: 10.1186/cc3879
28. Sevransky, Investigators E, Brokowski C. Effect of vitamin C, thiamine, and hydrocortisone on ventilator- and vasopressor-free days in patients with sepsis: the VICTAS randomized clinical trial. JAMA. (2021) 325:742–50. doi: 10.1001/jama.2020.24505
29. Panigrahi P, Parida S, Nanda N, Satpathy R, Pradhan L, Chandel D, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature. (2017) 548:407–12. doi: 10.1038/nature23480
30. Geiger S, Traba J, Richoz N, Farley T, Brooks S, Petermann F, et al. Feeding-induced resistance to acute lethal sepsis is dependent on hepatic BMAL1 and FXR signalling. Nat Commun. (2021) 12:2745. doi: 10.1038/s41467-021-22961-z
31. Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim KE, Attia SM. Bruton's tyrosine kinase inhibition attenuates oxidative stress in systemic immune cells and renal compartment during sepsis-induced acute kidney injury in mice. Int Immunopharmacol. (2020) 90:107123. doi: 10.1016/j.intimp.2020.107123
32. Liu D, Shu G, Jin F, Qi J, Du Y. ROS-responsive chitosan-SS31 prodrug for AKI therapy via rapid distribution in the kidney and long-term retention in the renal tubule. Sci Adv. (2020) 6:eabb7422. doi: 10.1126/sciadv.abb7422
33. Xia W, Li Y, Wu M, Jin Q, Jia Z. Gasdermin E deficiency attenuates acute kidney injury by inhibiting pyroptosis and inflammation. Cell Death Dis. (2021) 12:139. doi: 10.1038/s41419-021-03431-2
34. Yang Q, Ren GL, Wei B, Jin J, Huang XR, Shao W, et al. Conditional knockout of TGF-βRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics. (2019) 9:8277–93. doi: 10.7150/thno.35686
35. Mulay SR, Honarpisheh MM, Foresto-Neto O, Shi C, Anders HJ. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol. (2019) 30:1857–69. doi: 10.1681/ASN.2018121218
36. Lin Q, Li S, Jiang N, Jin H, Ni Z. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. (2020) 1–16. doi: 10.1080/15548627.2020.1848971
37. Wan L, Ba Gshaw SM, Langenberg C, Saotome T, May C, Bellomo R. Pathophysiology of septic acute kidney injury: what do we really know? Crit Care Med. (2008) 36:198–203. doi: 10.1097/CCM.0b013e318168ccd5
38. Langenberg C, Bellomo R, May CN, Egi M, Wan L, Morgera S. Renal vascular resistance in sepsis. Nephron Physiol. (2006) 104:p1–11. doi: 10.1159/000093275
39. Dear JW, Yasuda H, Hu X, Hieny S, Yuen P, Hewitt SM, et al. Sepsis-induced organ failure is mediated by different pathways in the kidney and liver: acute renal failure is dependent on MyD88 but not renal cell apoptosis. Kidney Int. (2006) 69:832–6. doi: 10.1038/sj.ki.5000165
40. Mariano F, Cantaluppi V, Stella M. Circulating plasma factors induce tubular and glomerular alterations in septic burns patients. Critical Care. (2008) 12:R42. doi: 10.1186/cc6848
41. KCNQ1 K Channels are Involved in Lipopolysaccharide-induced Apoptosis of Distal Kidney Cells. Cell Physiol Biochem. (2010) 25:367–78. doi: 10.1159/000303041
42. Xia S, Lin H, Liu H, Lu Z, Wang H, Fan S, et al. Honokiol attenuates sepsis-associated acute kidney injury via the inhibition of oxidative stress and inflammation. Inflammation. (2019) 42:826–34. doi: 10.1007/s10753-018-0937-x
43. Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. (2009) 119:2868–78. doi: 10.1172/JCI39421
44. Guo R. Acute renal failure in endotoxemia is dependent on caspase activation. J Am Soc Nephrol. (2004) 15:3093–102. doi: 10.1097/01.ASN.0000145530.73247.F5
45. Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. (2004) 172:2629–35. doi: 10.4049/jimmunol.172.4.2629
46. Otto G, Sossdorf M, Claus R, Rödel J, Menge K, Reinhart K, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Critical Care (London, England). (2011) 15:R183. doi: 10.1186/cc10332
47. Wu X, Guo R, Chen P, Quan W, Cunningham PN. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am J Physiol Renal Physiol. (2009) 297:316–26. doi: 10.1152/ajprenal.00089.2009
48. Messmer UK, Briner VA, Pfeilschifter J. Tumor necrosis factor-alpha and lipopolysaccharide induce apoptotic cell death in bovine glomerular endothelial cells. Kidney Int. (1999) 55:2322. doi: 10.1046/j.1523-1755.1999.00473.x
49. Meßmer UK, Winkel G, Briner VA, Pfeilschifter J. Glucocorticoids potently block tumour necrosis factor-α- and lipopolysaccharide-induced apoptotic cell death in bovine glomerular endothelial cells upstream of caspase 3 activation. Br J Pharmacol. (1999) 127:1633–40. doi: 10.1038/sj.bjp.0702726
50. Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. (2003) 284:L899. doi: 10.1152/ajplung.00338.2002
51. Nagata S, Segawa K. Sensing and clearance of apoptotic cells. Curr Opin Immunol. (2021) 68:1–8. doi: 10.1016/j.coi.2020.07.007
52. Perry J, Morioka S, Medina CB, Etchegaray JI, Ravichandran KS. Interpreting an apoptotic corpse as anti-inflammatory involves a chloride sensing pathway. Nat Cell Biol. (2019) 21:1532–43. doi: 10.1038/s41556-019-0431-1
53. Maremonti F, Meyer C, Linkermann A. Mechanisms and models of kidney tubular necrosis and nephron loss. J Am Soc Nephrol. (2022) 33:472–86. doi: 10.1681/ASN.2021101293
54. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
55. Galluzzi L, Kepp O, Chan F, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. (2017) 12:103–30. doi: 10.1146/annurev-pathol-052016-100247
56. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. (2014) 35:24–32. doi: 10.1016/j.semcdb.2014.02.006
57. Kaiser W, Sridharan H, Huang C, Mandal P, Upton J, Gough P, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
58. Upton J, Kaiser W, Mocarski E. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. (2010) 7:302–13. doi: 10.1016/j.chom.2010.03.006
59. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren R, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. (2020) 580:391–5. doi: 10.1038/s41586-020-2129-8
60. Wang J, Liu M, Wang F, Wei B, Yang Q, Cai Y, et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci (London, England: 1979). (2019) 133:1609–27. doi: 10.1042/CS20190599
61. Wu W, Wang J, Li Z, Wei B, Jin J, Gao L, et al. 7-Hydroxycoumarin protects against cisplatin-induced acute kidney injury by inhibiting necroptosis and promoting Sox9-mediated tubular epithelial cell proliferation. Phytomedicine. (2020) 69:153202. doi: 10.1016/j.phymed.2020.153202
62. Conos SA, Chen KW, Nardo DD, Hara H, Vince JE. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Nat Acad Sci. (2017) 114:201613305. doi: 10.1073/pnas.1613305114
63. Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III Acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. (2017) 169:286–300. doi: 10.1016/j.cell.2017.03.020
64. Takasu O, Gaut J, Watanabe E, To K, Fagley R, Sato B, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. (2013) 187:509–17. doi: 10.1164/rccm.201211-1983OC
65. Sureshbabu A, Patino E, Ma K, Laursen K, Finkelsztein E, Akchurin O, et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI insight. (2018) 3:e98411. doi: 10.1172/jci.insight.98411
66. Hadifar S, Behrouzi A, Fateh A, Khatami S, Rahimi Jamnani F, Siadat S, et al. Comparative study of interruption of signaling pathways in lung epithelial cell by two different Mycobacterium tuberculosis lineages. J Cell Physiol. (2019) 234:4739–53. doi: 10.1002/jcp.27271
67. Nei L, Krbi R, Amidi L, Gajanin R, Jaevi V. Protective effects of simvastatin on endotoxin-induced acute kidney injury through activation of tubular epithelial cells' survival and hindering cytochrome C-mediated apoptosis. Int J Mol Sci. 21:7236. doi: 10.3390/ijms21197236
68. Galluzzi L, Vitale I, Aaronson S, Abrams J, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
69. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2010) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
70. Cervantes J, Nagata T, Uchijima M, Shibata K, Koide Y. Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol. (2010) 10:41–52.
71. Wang Y, Li Y, Xu Y. Pyroptosis in kidney disease. J Mol Biol. (2022) 434:167290. doi: 10.1016/j.jmb.2021.167290
72. Kufer TA, Sansonetti PJ. Sensing of bacteria: NOD a lonely job. Curr Opin Microbiol. (2007) 10:62–9. doi: 10.1016/j.mib.2006.11.003
73. Goncalves AC, Ferreira LS, Manente FA, Jellmayer JA, de Faria CM, Polesi MC, et al. NLRP3 inflammasome contribution to host protection during infection by sporothrix schenckii. Cytokine. (2016) 87:84–84.
74. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. (2017) 17:151–64. doi: 10.1038/nri.2016.147
75. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
76. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. (2013) 341:1250–3. doi: 10.1126/science.1240988
77. Eldridge M, Sanchez-Garrido J, Hoben G, Goddard P, Shenoy A. The atypical ubiquitin E2 conjugase UBE2L3 is an indirect caspase-1 target and controls IL-1β secretion by inflammasomes. Cell Rep. (2017) 18:1285–97. doi: 10.1016/j.celrep.2017.01.015
78. Kayagaki N, Wong M, Stowe I, Ramani S, Gonzalez L, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (New York, NY). (2013) 341:1246–9. doi: 10.1126/science.1240248
79. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469:221–5. doi: 10.1038/nature09663
80. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
81. Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
82. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. (2008) 456:264–8. doi: 10.1038/nature07383
83. Degterev A, Hitomi J, Germscheid M., Ch En IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. (2008) 4:313–21. doi: 10.1038/nchembio.83
84. Khweek AA, Amer AO. Pyroptotic and non pyroptotic effector functions of caspase. Immunol Rev. (2020) 297:39–52. doi: 10.1111/imr.12910
85. Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. (2017) 7:45126. doi: 10.1038/srep45126
86. Dolunay A, Senol S, Temiz-Resitoglu M, Guden D, Sari A, Sahan-Firat S, et al. Inhibition of NLRP3 inflammasome prevents LPS-induced inflammatory hyperalgesia in mice: contribution of NF-κB, caspase-1/11, ASC, NOX, and NOS isoforms. Inflammation. (2017) 40:366–86. doi: 10.1007/s10753-016-0483-3
87. Yuan J, Najafov A, Py BF. Roles of caspases in necrotic cell death. Cell. (2016) 167:1693. doi: 10.1016/j.cell.2016.11.047
88. Kayagaki N, Stowe I, Lee B, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
89. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
90. Broz P. Immunology: Caspase target drives pyroptosis. Nature. (2015) 526:642–3. doi: 10.1038/nature15632
91. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. (2013) 339:975–8. doi: 10.1126/science.1230751
92. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
93. Ye Z, Zhang L, Li R, Dong W, Liang X. Caspase-11 mediates pyroptosis of tubular epithelial cells and septic acute kidney injury. Kidney Blood Press Res. (2019) 44:1–14. doi: 10.1159/000499685
94. Fu Y, Wang D, Wang S, Zhang Q, Liu H, Yang S, et al. Blockade of Macrophage-Associated Programmed Death 1 Inhibits the Pyroptosis Signalling Pathway in Sepsis (2021) 70:993–1004. doi: 10.1007/s00011-021-01493-8
95. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (New York, NY). (2016) 352:1232–6. doi: 10.1126/science.aaf3036
96. Dixon S, Lemberg K, Lamprecht M, Skouta R, Zaitsev E, Gleason C, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
97. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
98. Stockwell B, Friedmann Angeli J, Bayir H, Bush A, Conrad M, Dixon S, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
99. Stockwell B, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. (2020) 30:478–90. doi: 10.1016/j.tcb.2020.02.009
100. Xiao J, Yang Q, Zhang Y, Xu H, Ye Y, Li L, et al. Maresin conjugates in tissue regeneration-1 suppresses ferroptosis in septic acute kidney injury. Cell Biosci. (2021) 11:221. doi: 10.1186/s13578-021-00734-x
101. Oehme I, Linke JP, Böck B, Milde T, Lodrini M, Hartenstein B, et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci USA. (2013) 110:E2592–2601. doi: 10.1073/pnas.1300113110
102. Zhang Y, Wang L, Meng L, Cao G, Wu Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell cycle (Georgetown, Tex). (2019:1–12. doi: 10.1080/15384101.2019.1568746
103. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. (2015) 22:367–76. doi: 10.1038/cdd.2014.143
104. Yang T, Feng X, Zhao Y, Zhang H, Cui H, Wei M, et al. via dexmedetomidine enhances autophagy α2-AR/AMPK/mTOR pathway to inhibit the activation of NLRP3 inflammasome and subsequently alleviates lipopolysaccharide-induced acute kidney injury. Front Pharmacol. (2020) 11:790. doi: 10.3389/fphar.2020.00790
105. Bhatia D, Choi M. Autophagy in kidney disease: Advances and therapeutic potential. Prog Mol Biol Transl Sci. (2020) 172:107–33. doi: 10.1016/bs.pmbts.2020.01.008
106. Leventhal JS Ni J, Osmond M, Lee K, Ross MJ. Autophagy limits endotoxemic acute kidney injury and alters renal tubular epithelial cell cytokine expression. PLoS ONE. (2016) 11:e0150001. doi: 10.1371/journal.pone.0150001
107. Mei S, Livingston M, Hao J, Li L, Mei C, Dong Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep. (2016) 6:22171. doi: 10.1038/srep22171
108. Wu Y, Wang L, Meng L, Cao G, Zhao Y, Zhang Y. Biological effects of autophagy in mice with sepsis-induced acute kidney injury. Exp Ther Med. (2019) 17:316–22.
109. Takahashi W, Watanabe E, Fujimura L, Watanabe-Takano H, Hatano M. Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit Care. (2013) 17:1–13. doi: 10.1186/cc12839
110. Sunahara S, Watanabe E, Hatano M, Swanson PE, Oami T, Fujimura L, et al. Influence of autophagy on acute kidney injury in a murine cecal ligation and puncture sepsis model. Sci Rep. (2018) 8:1050. doi: 10.1038/s41598-018-19350-w
111. Hsiao H, Tsai K, Wang L, Chen Y, Chiang P, Chuang S, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock (Augusta, Ga). (2012) 37:289–96. doi: 10.1097/SHK.0b013e318240b52a
112. Galluzzi L, Baehrecke E, Ballabio A, Boya P, Bravo-San Pedro J, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
113. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
114. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nature Reviews Immunology. (2013) 13:722–37. doi: 10.1038/nri3532
115. Guo LP, Liu SX, Yang Q, Liu HY, Zhang XQ. Erratum to “Effect of Thymoquinone on Acute Kidney Injury Induced by Sepsis in BALB/c Mice”. BioMed Res Int. (2020). (2020) 2020:1–2. doi: 10.1155/2020/3182919
116. Liu R, Wang SC, Li M, Ma XH, Yu KJ. Erratum to “An Inhibitor of DRP1 (Mdivi-1) Alleviates LPS-Induced Septic AKI by Inhibiting NLRP3 Inflammasome Activation”. BioMed Res Int. (2020) 2020:1–3. doi: 10.1155/2020/8493938
117. Zhang Y, Zhang Y, Yang A, Xia F. Downregulation of IRF2 alleviates sepsis-related acute kidney injury in vitro and in vivo. Drug Des Devel Ther. (2021) 15:5123–32. doi: 10.2147/DDDT.S334518
118. Guo J, Wang R, Liu D. viaBone marrow-derived mesenchymal stem cells ameliorate sepsis-induced acute kidney injury by promoting mitophagy of renal tubular epithelial cells the SIRT1/Parkin Axis. Front Endocrinol. (2021) 12:639165. doi: 10.3389/fendo.2021.639165
119. Chen H, Li Y, Wu J, Li G, Tao X, Lai K, et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. (2020) 27:2568–85. doi: 10.1038/s41418-020-0524-1
120. Vajjhala PR, Lu A, Brown DL, Pang SW, Stacey KJ. The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J Biol Chem. (2015) 290:29217–30. doi: 10.1074/jbc.M115.687731
121. Priante G, Gianesello L, Ceol M, Prete DD, Anglani F. Cell death in the kidney. Int J Mol Sci. (2019) 20:3598. doi: 10.3390/ijms20143598
122. Fritsch M, Günther S, Schwarzer R, Albert M, Schorn F, Werthenbach J, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
123. Hou W, Han J, Lu C, Goldstein L, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. (2010) 6:891–900. doi: 10.4161/auto.6.7.13038
124. Fortunato F, Bürgers H, Bergmann F, Rieger P, Büchler M, Kroemer G, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. (2009) 137:350–60, 360.e351–5. doi: 10.1053/j.gastro.2009.04.003
125. Gump J, Staskiewicz L, Morgan M, Bamberg A, Riches D, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. (2014) 16:47–54. doi: 10.1038/ncb2886
126. Ye Y, Yu L, Wang H, Tashiro S, Onodera S, Ikejima T. TNFα-induced necroptosis and autophagy via supression of the p38-NF-κB survival pathway in L929 cells. J Pharmacol Sci. (2011) 117:160–9. doi: 10.1254/jphs.11105FP
127. Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 6:1221–8. doi: 10.1038/ncb1192
128. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science (New York, NY). (2004) 304:1500–2. doi: 10.1126/science.1096645
129. Matsuzawa-Ishimoto Y, Shono Y, Gomez L, Hubbard-Lucey V, Cammer M, Neil J, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. (2017) 214:3687–705. doi: 10.1084/jem.20170558
130. Zhou X, Xie L, Xia L, Bergmann F, Büchler M, Kroemer G, et al. RIP3 attenuates the pancreatic damage induced by deletion of ATG7. Cell Death Dis. (2017) 8:e2918. doi: 10.1038/cddis.2017.313
131. Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife. (2019) 8:e44452. doi: 10.7554/eLife.44452.036
132. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (New York, NY). (2016) 353:603–8. doi: 10.1126/science.aaf6803
133. Ding J, Yang N, Yan Y, Wang Y, Wang X, Lu L, et al. Rapamycin inhibited photoreceptor necroptosis and protected the retina by activation of autophagy in experimental retinal detachment. Curr Eye Res. (2019) 44:739–45. doi: 10.1080/02713683.2019.1588331
134. Oberst A, Dillon C, Weinlich R, McCormick L, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. (2011) 471:363–7. doi: 10.1038/nature09852
135. O'Donnell M, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. (2011) 13:1437–42. doi: 10.1038/ncb2362
136. Feng S, Yang Y, Mei Y, Ma L, Zhu D, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. (2007) 19:2056–67. doi: 10.1016/j.cellsig.2007.05.016
137. Rébé C, Cathelin S, Launay S, Filomenko R, Prévotat L., L'Ollivier C, et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood. (2007) 109:1442–50. doi: 10.1182/blood-2006-03-011585
138. Frank D, Vaux D, Murphy J, Vince J, Lindqvist L. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J Cell Sci. (2019) 132:jcs220996. doi: 10.1242/jcs.220996
139. Hou W, Xie Y, Song X, Sun X, Lotze M, Zeh H, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
140. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
141. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. (2018) 14:2083–103. doi: 10.1080/15548627.2018.1503146
142. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. (2018) 503:1550–6. doi: 10.1016/j.bbrc.2018.07.078
143. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. (2018) 9:753. doi: 10.1038/s41419-018-0794-4
144. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
145. Pu Q, Gan C, Li R, Li Y, Tan S, Li X, et al. PseudomonasAtg7 deficiency intensifies inflammasome activation and pyroptosis in sepsis. J Immunol (Baltimore, Md: 1950). (2017) 198:3205–13. doi: 10.4049/jimmunol.1601196
146. Shi C, Shenderov K, Huang N, Kabat J, Abu-Asab M, Fitzgerald K, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. (2012) 13:255–63. doi: 10.1038/ni.2215
147. Mehto S, Jena KK, Nath P, Chauhan S, Kolapalli SP, Das SK, et al. The Crohn's disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell. (2019) 73:1–17. doi: 10.1016/j.molcel.2018.11.018
148. Wei Q, Zhu R, Zhu J, Zhao R, Li M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol Res. (2019) 27:827–34. doi: 10.3727/096504018X15462920753012
149. Wang Z, Li Y, Yang X, Zhang L, Shen H, Xu W, et al. Protective effects of rapamycin induced autophagy on CLP septic mice. Comp Immunol Microbiol Infect Dis. (2019) 64:47–52. doi: 10.1016/j.cimid.2019.01.009
150. Li M, Zhu X, Zhao B, Shi L, Wang W, Hu W, et al. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis. (2019) 10:489. doi: 10.1038/s41419-019-1728-5
151. Tu Y, Guo C, Song F, Huo Y, Geng Y, Guo M, et al. Mild hypothermia alleviates diabetes aggravated cerebral ischemic injury via activating autophagy and inhibiting pyroptosis. Brain Res Bull. (2019) 150:1–12. doi: 10.1016/j.brainresbull.2019.05.003
152. Zi Y, Yi-An Y, Bing J, Yan L, Jing T, Chun-Yu G, et al. Sirt6-induced autophagy restricted TREM-1-mediated pyroptosis in ox-LDL-treated endothelial cells: relevance to prognostication of patients with acute myocardial infarction. Cell Death Discov. (2019) 5:88. doi: 10.1038/s41420-019-0168-4
153. Yu P, Wang H, Tian M, Li A, Chen X, Wang X, et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. (2019) 40:1237–44. doi: 10.1038/s41401-019-0222-z
154. Qiu T, Pei P, Yao X, Jiang L, Wei S, Wang Z, et al. Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death Dis. (2018) 9:946. doi: 10.1038/s41419-018-1004-0
155. McComb S, Cheung H, Korneluk R, Wang S, Krishnan L, Sad S. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. (2012) 19:1791–801. doi: 10.1038/cdd.2012.59
156. Moulin M, Anderton H, Voss A, Thomas T, Wong W, Bankovacki A, et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. (2012) 31:1679–91. doi: 10.1038/emboj.2012.18
157. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. (2011) 18:656–65. doi: 10.1038/cdd.2010.138
158. Yabal M, Müller N, Adler H, Knies N, Groß C, Damgaard R, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. (2014) 7:1796–808. doi: 10.1016/j.celrep.2014.05.008
159. Cheng K, Xiong S, Ye Z, Hong Z, Di A, Tsang K, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. (2017) 127:4124–35. doi: 10.1172/JCI94495
160. Fink S, Bergsbaken T, Cookson B. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. (2008) 105:4312–7. doi: 10.1073/pnas.0707370105
161. Liu S, Li Y, Choi H, Sarkar C, Koh E, Wu J, et al. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. (2018) 9:476. doi: 10.1038/s41419-018-0469-1
162. Bray K, Mathew R, Lau A, Kamphorst J, Fan J, Chen J, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS ONE. (2012) 7:e41831. doi: 10.1371/journal.pone.0041831
163. Chuang S, Yang C, Chou C, Chiang Y, Chuang T, Hsu L. TLR-induced PAI-2 expression suppresses IL-1β processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci USA. (2013) 110:16079–84. doi: 10.1073/pnas.1306556110
Keywords: sepsis, acute kidney injury, apoptosis, pyroptosis, necroptosis, autophagy, sepsis-associated acute kidney injury
Citation: Wu Z, Deng J, Zhou H, Tan W, Lin L and Yang J (2022) Programmed Cell Death in Sepsis Associated Acute Kidney Injury. Front. Med. 9:883028. doi: 10.3389/fmed.2022.883028
Received: 24 February 2022; Accepted: 21 March 2022;
Published: 17 May 2022.
Edited by:
Qinggang Li, Chinese PLA General Hospital, ChinaReviewed by:
Yanfang Xu, First Affiliated Hospital of Fujian Medical University, ChinaCopyright © 2022 Wu, Deng, Zhou, Tan, Lin and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jurong Yang, eWpyOTIzQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.