
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 09 June 2022
Sec. Dermatology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.875492
This article is part of the Research Topic Insights in Dermatology: 2021 View all 15 articles
 Hideyuki Ujiie1
Hideyuki Ujiie1 David Rosmarin2
David Rosmarin2 Michael P. Schön3,4
Michael P. Schön3,4 Sonja Ständer5
Sonja Ständer5 Katharina Boch6
Katharina Boch6 Martin Metz7,8
Martin Metz7,8 Marcus Maurer7,8
Marcus Maurer7,8 Diamant Thaci9
Diamant Thaci9 Enno Schmidt6,10
Enno Schmidt6,10 Connor Cole11,12
Connor Cole11,12 Kyle T. Amber11,12Dario Didona13
Kyle T. Amber11,12Dario Didona13 Michael Hertl13
Michael Hertl13 Andreas Recke6
Andreas Recke6 Hanna Graßhoff14Alexander Hackel14Anja Schumann14
Hanna Graßhoff14Alexander Hackel14Anja Schumann14 Gabriela Riemekasten14
Gabriela Riemekasten14 Katja Bieber10
Katja Bieber10 Gant Sprow15,16
Gant Sprow15,16 Joshua Dan15,16
Joshua Dan15,16 Detlef Zillikens6
Detlef Zillikens6 Tanya Sezin17Angela M. Christiano17
Tanya Sezin17Angela M. Christiano17 Kerstin Wolk18,19
Kerstin Wolk18,19 Robert Sabat18,19
Robert Sabat18,19 Khalaf Kridin10,20
Khalaf Kridin10,20 Victoria P. Werth15,16
Victoria P. Werth15,16 Ralf J. Ludwig6,10*
Ralf J. Ludwig6,10*An estimated 20–25% of the population is affected by chronic, non-communicable inflammatory skin diseases. Chronic skin inflammation has many causes. Among the most frequent chronic inflammatory skin diseases are atopic dermatitis, psoriasis, urticaria, lichen planus, and hidradenitis suppurativa, driven by a complex interplay of genetics and environmental factors. Autoimmunity is another important cause of chronic skin inflammation. The autoimmune response may be mainly T cell driven, such as in alopecia areata or vitiligo, or B cell driven in chronic spontaneous urticaria, pemphigus and pemphigoid diseases. Rare causes of chronic skin inflammation are autoinflammatory diseases, or rheumatic diseases, such as cutaneous lupus erythematosus or dermatomyositis. Whilst we have seen a significant improvement in diagnosis and treatment, several challenges remain. Especially for rarer causes of chronic skin inflammation, early diagnosis is often missed because of low awareness and lack of diagnostics. Systemic immunosuppression is the treatment of choice for almost all of these diseases. Adverse events due to immunosuppression, insufficient therapeutic responses and relapses remain a challenge. For atopic dermatitis and psoriasis, a broad spectrum of innovative treatments has been developed. However, treatment responses cannot be predicted so far. Hence, development of (bio)markers allowing selection of specific medications for individual patients is needed. Given the encouraging developments during the past years, we envision that many of these challenges in the diagnosis and treatment of chronic inflammatory skin diseases will be thoroughly addressed in the future.
Chronic, non-communicable skin inflammation can be caused by many different diseases. Herein, we categorized these into (i) chronic inflammatory diseases (atopic dermatitis, psoriasis, lichen planus, chronic prurigo, and hidradenitis suppurativa), (ii) autoimmune diseases (alopecia areata, vitiligo, chronic spontaneous urticaria, pemphigus, bullous pemphigoid, mucous membrane pemphigoid, and epidermolysis bullosa acquisita), (iii) autoinflammatory diseases (cryopyrin-associated periodic syndrome and Schnitzler's syndrome), and (iv) rheumatic diseases (cutaneous lupus erythematosus, dermatomyositis, and systemic sclerosis). This categorization is based on the main driving pathomechanism(s) of each disease. However, a clear classification of the pathologic driver is challenging as in lichen planus and psoriasis autoreactive T- and B- cells potentially contribute to disease pathogenesis (1, 2). This classification is also expected to change over time, as it will need to adopt and consider new data on disease pathogenesis. Alternatively, to the here used classification, chronic inflammatory skin diseases may be categorized based on key driving molecules. For example, Janus kinases (JAK) in atopic dermatitis, alopecia areata, vitiligo, and cutaneous lupus erythematosus. Furthermore, as detailed below, for many chronic skin inflammatory diseases the clinical presentation varies greatly even within the same disease, as with psoriasis or bullous pemphigoid (3, 4). With the emerge of multidimensional datasets, it has been proposed to classify inflammatory skin diseases based on molecular patterns (5). The increasing understanding of (molecular) disease pathogenesis and availability of appropriate biomarkers for their identification, we expect a more complex, but more tailored categorization of molecular disease pathogenesis is leading to the emergence of potential biomarkers, and a more categorization of chronic, non-communicable skin inflammatory diseases. These diseases are a major medical burden because of their high and, in many cases, increasing prevalence (Table 1), diagnostic challenges, lack of curative treatments, co-morbidity, as well as significant economic impact. We here selected 17 chronic, non-communicable skin inflammatory diseases that collectively affect 15–20% of the population (Table 1). For each disease, the current diagnostic and therapeutic challenges are outlined. Furthermore, a perspective is given on how these challenges may be met in the future.
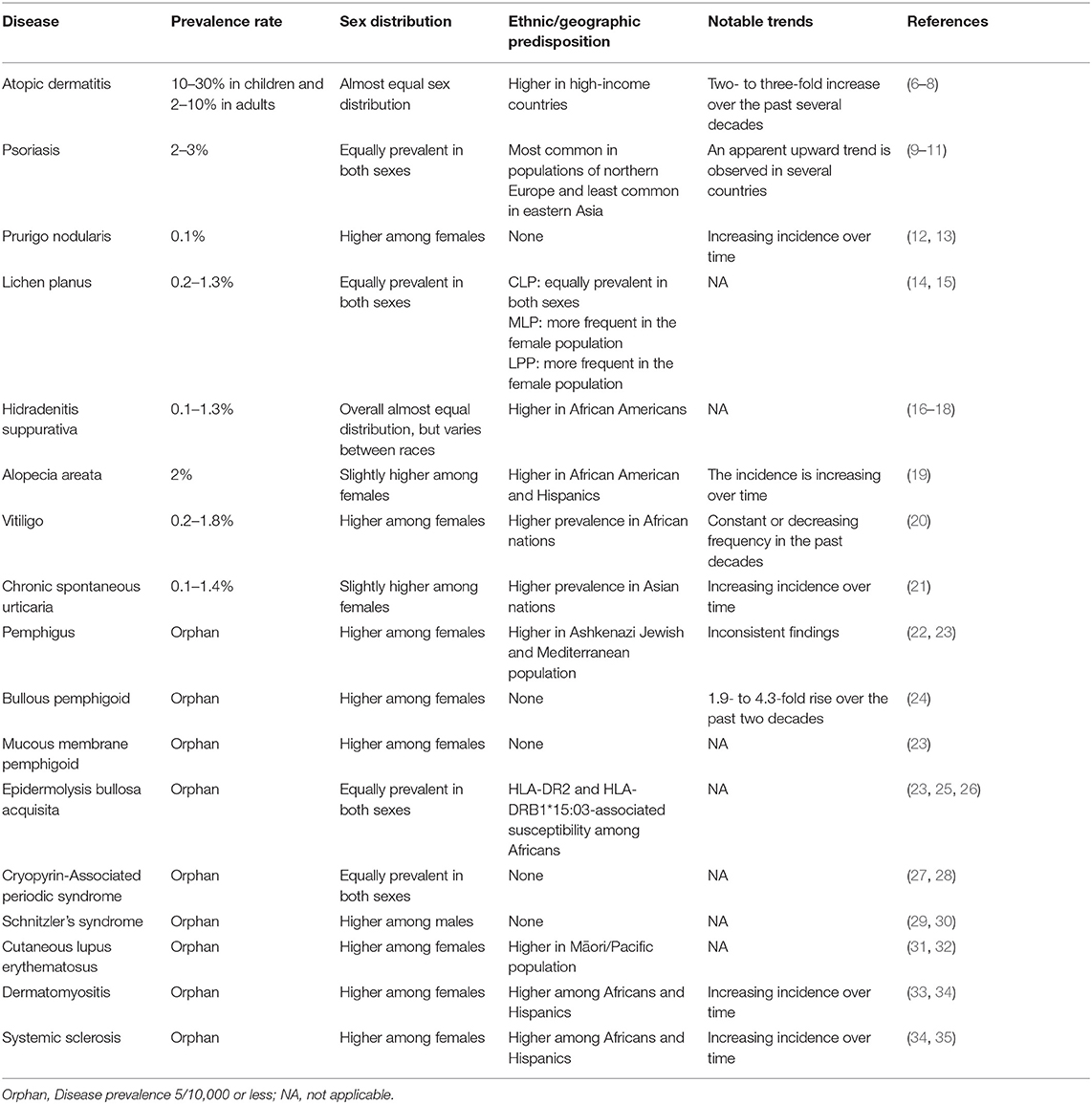
Table 1. Epidemiology of selected chronic inflammatory skin diseases.
Atopic Dermatitis (AD) or atopic eczema is a common, chronic, relapsing inflammatory disease, affecting up to 30% of the pediatric population and 2–10% of adults (36). While most commonly symptoms start in the first 5 years of life, it is now recognized that onset can occur at any age. There can be a significant effect on patient's quality of life and sleep due to itch and pain (37). There are also significant effects on patients' mental health with higher incidence of depression and suicide (38). The high burden of disease can interfere with work productivity, not only from the baseline disease but particularly from flares (39). Patient also have many out-of-pocket costs, including cleaning products, clothing, moisturizers, and other expenses (40). In AD, there is an interplay between barrier dysfunction, immune dysregulation, and the microbiome (41). Both genetics and environmental factors play a role in the pathogenesis (42). In AD, the stratum corneum, composed of the terminally differentiated enucleated keratinocytes called corneocytes, is often compromised. Among European Caucasians, filaggrin mutations are associated with early-onset and severe AD (43). Filaggrin is broken down into compounds that constitute natural moisturizing factor which is important for appropriate hydration, desquamation, plasticity, acidity, and the commensal microbiome (44, 45). Patients with AD have a higher burden of Staphylococcus aureus which contributes to the inflammation (46). As allergens penetrate the defective skin barrier in AD, pro-inflammatory cytokines are released. While a type 2 immune response with elevated levels of IL-4 and IL-13 predominate in the acute phase, chronically, a mixed response of Th1, Th17, and Th22 immune cells is observed (47). IL-31 is particularly implicated in pruritus (48).
AD is usually diagnosed based on clinical experience (Figure 1A). There are diagnostic criteria, but no simple test for definitive diagnosis (49). When patients manifest in atypical locations, develop lesions later in life, have uncommon morphologies or other overlying skin diseases the diagnosis can be challenging. AD is heterogenous and can show racial variation (50). Asians may manifest with well-demarcated lesions and skin of color patients may have increased xerosis, follicular eczema, and post-inflammatory pigmentation changes (51). Allergic contact dermatitis may overly AD, so patch testing should be considered in those with recalcitrant atopic dermatitis. AD is notably associated with other atopic disorders such as asthma, allergic rhinitis, and food allergies. There also has been an association with obesity, malignancy, and cardiovascular disease (52–54). For a precision medicine approach, validated and reliable biomarkers are needed to individually tailor treatment (55).
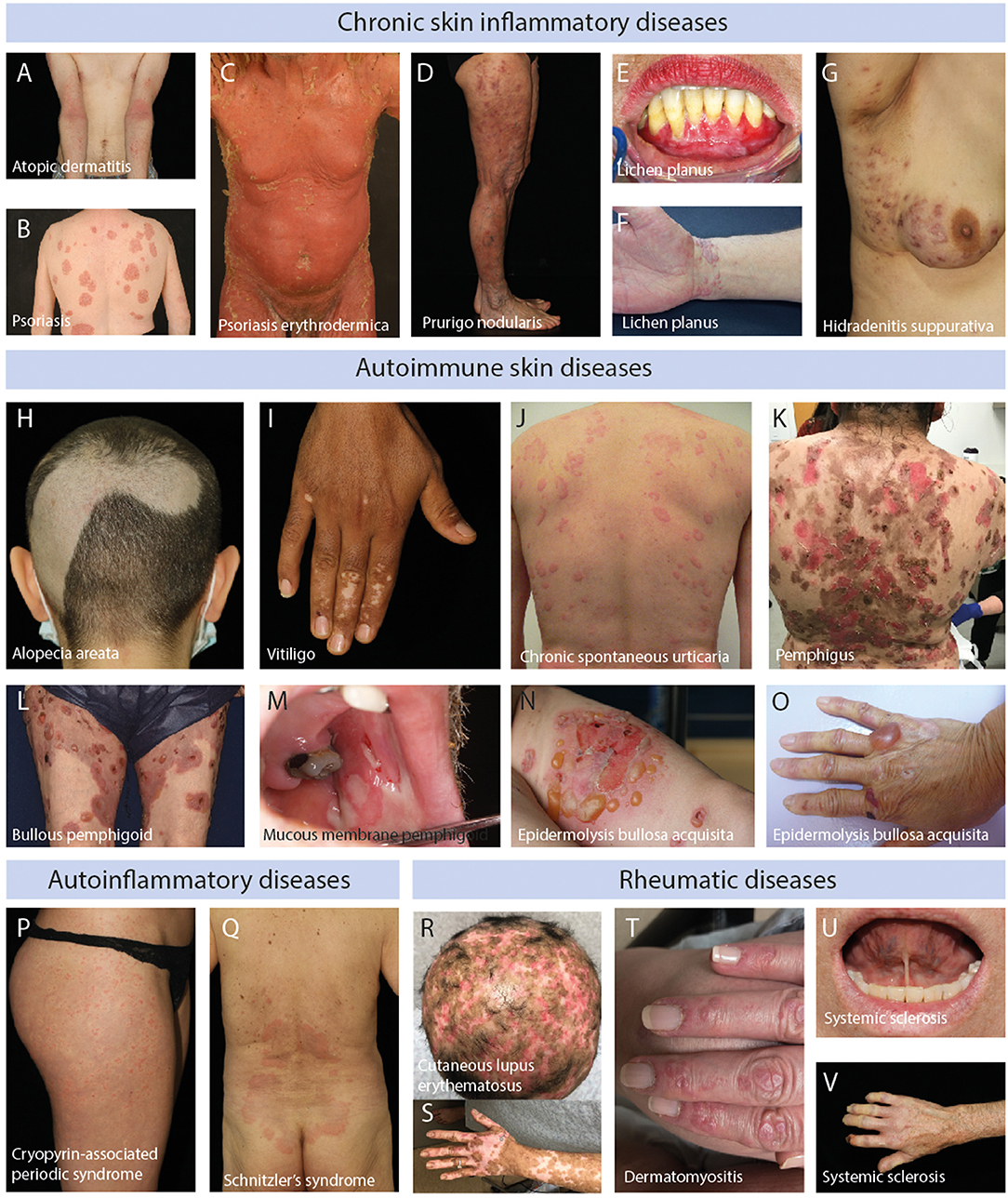
Figure 1. Clinical images of patients with chronic skin inflammatory diseases. (A) Blurry erythema and lichenification at the inside the bend of the elbows and arms of a patient with atopic dermatitis. (B) Sharply demarked, scaling, erythematous plaques at the back of a patient with psoriasis vulgaris. (C) Generalized erythema and scaling in a patient with psoriasis. (D) Erythematous nodules, partially excoriated, in a patient with prurigo nodularis. (E) Erosions of the lower gums in a patient with mucosal lichen planus. (F) Polygonal, scaling, reddish-violet plaques at the wrist of a patient with cutaneous lichen planus. (G) Scaring, nodules and pustules located at the sub-axillary region of a patient with hidradenitis suppurativa. (H) Sharply demarked hair loss at the back of the head in a patient with alopecia areata. (I) Sharpley demarked white maculae at the hands of a patient with vitiligo. (J) Wheals at the back of a patient with chronic spontaneous urticaria. (K) Brown macules and erosions at the back of a patient with muco-cutaneous pemphigus vulgaris. (L) Tense blisters on erythematous skin on the legs of a patient with bullous pemphigoid. (M) Oral erosions in a patient with mucous membrane pemphigoid. (N) Tense blisters on erythema on the arm of a patient with inflammatory/non-mechano-bullous epidermolysis bullosa acquisita. (O) Tense blister and scaring on the hand of a patient with predominant mechano-bullous epidermolysis bullosa acquisita. (P) Wheals at the leg of a patient with cryopyrin-associated periodic syndrome. (Q) Urticarial exanthema at the lower back of a patient with Schnitzler's syndrome. (R) Alopecia and erythema at the head of a patient with cutaneous lupus erythematosus. (S) Erythema and depigmentation at the arm of a patient with cutaneous lupus erythematosus. (T) Gottron papules in a patient with dermatomyositis. (U) Shortening of the sublingual frenulum in a patient with systemic sclerosis. (V) Raynaud's phenomenon (anemic color of the fingers) and necrosis of the index finger in a patient with systemic sclerosis.
Treatment is mainly aimed at restoring the skin barrier and modulating the abnormal immune response. Education on skin hygiene strategies is important for all patients, ideally with written action plans. There is uncertainty as to ideal bathing recommendations as it may improve skin hydration and provide some symptom relief while use of detergents may have a dehydrating effect. Emollients are a cornerstone of treatment and can lead to a decrease in the amount of prescription topical agents needed to treat AD (56). However, it is not known the optimal amount or frequency of emollient application. Additionally, there are some moisturizers that may irritate the skin of individual patients. Besides emollients, topical agents including corticosteroids are first-line therapy. Non-steroidal options such as topical calcineurin inhibitors (TCIs) are useful for areas of sensitive skin such as face, neck, and genitals. Calcineurin inhibitors can also be used as maintenance twice a week to reduce the frequency and severity of flares (57). While there was initial concern regarding the use of TCIs and the risk of malignancy, post-marketing research has been reassuring as to the safety of these treatments (58). In patients who fail topical treatment, phototherapy, oral immunosuppressants, and targeted biologics are indicated. In particular, the anti-IL4 receptor alpha inhibitor dupilumab has changed the way we treat AD in both pediatric and adult patient. While sedating antihistamines for short-term use can assist with sleep disturbance caused by pruritus, there is a lack of evidence to support the use of non-sedating and sedating antihistamines for generalized, extended use. Due to cumulative side effects, oral corticosteroids should be avoided in the long-term and in children. There are many exciting new mechanisms of action in development (or very recently approved) to treat atopic dermatitis including aryl hydrocarbon receptor agonists, commensal bacteria, JAK inhibitors (JAKi), and new biologics that target IL-13, IL-31, IL-33, and OX-40 (59, 60).
Atopic dermatitis is one of the most common inflammatory skin disorders and there are still multiple unmet needs and educational gaps. Instructing patients and caregivers regarding skin hygiene with liberal use of emollients is essential for all. Additionally reassuring fears of corticosteroids is an important task of providers. There is no generally accepted goal of treatment, so currently plans are individualized for patients with a need for biomarkers and research into personalized medicine. Adherence to therapy remains a long-term challenge as care of atopic dermatitis can be quite time consuming and costly. There are an increasing number of therapeutic options that are being developed due to our improved understanding about the pathogenesis of AD and with it, improved hope at helping more patients who suffer from atopic dermatitis.
Over the past three decades, psoriasis has become a model disease for the study of chronic inflammatory diseases. Several new drugs have been and are being developed first for psoriasis and then extended to other indications (61). Central to our current understanding of the pathogenesis of psoriasis is a close interaction between components of the innate and adaptive immune systems (62, 63). For example, the former branch is represented by macrophages, neutrophilic granulocytes, and (plasmacytoid) dendritic cells; the latter by T lymphocytes, primarily Th17 cells. Communication between these immune cells is mediated by various cytokines including TNFα, IL-17, and IL-23 which have become targets of multiple biologic therapies (64, 65).
Psoriasis is diagnosed based on history and clinical presentation—only rarely a biopsy is needed to confirm the diagnosis. Comorbidity, especially psoriatic arthritis should be excluded at diagnosis and during follow-up (62). However, psoriatic disease is not a uniform disease entity (Figures 1B,C). Although current drugs were developed and approved for the so-called chronic plaque psoriasis, we encounter psoriasis in the clinic as a spectrum ranging from acute exanthematous to chronic stable, from classically scaly and sharply demarcated plaques to highly inflammatory, pustular or erythrodermic forms, or from forms restricted to a few predilection sites to generalized or inverse forms. Specific underlying genetic patterns have now been identified for some of these manifestations; e.g., in IL36RN (3, 66). The involvement of other organ systems (comorbidity) and provoking factors in psoriasis as a systemic disease also influence the disease process. To account for the increased inflammation throughout the organism with (possible) systemic impairment of several other organ systems, we now tend to refer to it as psoriatic disease.
There are well-defined guidelines for the treatment of psoriasis (67, 68). The following drugs are FDA or EMA approved for psoriasis and/or psoriatic arthritis: TNFα inhibitors etanercept, infliximab, adalimumab, certolizumab pegol, and golimumab; IL-17a inhibitors secukinumab and ixekizumab; IL-17A and IL-17F dual inhibitor bimekizumab; IL-17 receptor A/C inhibitor brodalumab; IL-12 and IL-23 inhibitor, ustekinumab; and IL-23 inhibitors guselkumab, tildrakizumab, and risankizumab (69–72). Very recent developments also include mirikizumab and netakimab (73). The development of specific therapeutics against essential cytokines in the IL-23/IL-17 axis is a good illustration of how basic and translational immunological research has led to the development of highly potent drugs that can effectively and safely treat most patients with at least moderate psoriasis. These therapies have been and continue to be included in current guidelines (67, 68, 74, 75). In addition, small-molecule drugs have been and are being developed, such as apremilast against phosphodiesterase 4 (PDE4), deucravacitinib against tyrosine kinase 2 (Tyk2) and piclidenoson, a Gi protein-associated A3 adenosine receptor agonist (73). Compared to the era before biologics, the impact has been so great that it is fair to label these treatments revolutionary. Thus, we are now in a fairly comfortable position with regard to the treatment psoriatic disease: there are now numerous effective and well-tolerated preparations available and due to competitive pressure and the increasing availability of biosimilars, the price will (hopefully) come down in such a way that more and more patients can be treated with these systemic therapeutics. Still, there are still unmet medical needs for psoriasis that related to diagnostics and treatment. Specifically, in terms of personalized and precision medicine, however, there is definitely still a need for development here, since by no means do all of our patients meet the “standard” of chronic plaque psoriasis, which is usually considered in registration studies. On this basis, later extensions of indications are also conceivable for diseases that have similar pathogenetic features and for which, due to their relative rarity, large prospective clinical trials are usually not conducted (76). The categorization and characterization of inflammatory and autoimmunity patterns, which is already under development, may help in this regard (77). There is also not yet enough data on combination therapies for psoriasis (78). In particular, combinations of modern and conventional therapies could in some cases increase effectiveness and reduce costs. Similar considerations apply to individualized dosing regimens and terminations of therapies.
It is thus clear that the pathogenesis of psoriasis is complex. Increasingly, it is becoming clear that the overall pattern of inflammatory mediators and cells, which may well shift over the course of the “disease career,” is ultimately responsible for the individual form of manifestation. Therefore, it is reasonable to strive for a more detailed understanding of these inflammatory patterns and the factors regulating them on a “holistic” level. Hence, the establishment and clinical validation of biomarkers and molecular genetic patterns could enable predictions of response or loss of efficacy of specific therapies in individual patients (79). If successful, patients could quickly and sustainably receive the most appropriate therapy for them and we would avoid unnecessary delays, side effects and costs. This is only beginning to happen and should improve over time (80). Regarding therapeutics, development is proceeding in two major directions: On the one hand, further mediators are being inhibited in a targeted manner and with more refined methods and reagents. The most recently approved example is an antibody that blocks the effects of both IL-17A and IL-17F (bimekizumab) (81). The clinical effectiveness of this approach is convincingly good. The development was prompted by scientific findings that although IL-17A, which was initially considered, has a much higher affinity at the receptor, that the homologous IL-17F isoform is present in much greater amounts in psoriatic skin. In addition, IL-17A and IL-17 F may also act as heterodimers at the receptor (82). Other interesting developments also include mediators that have been primarily attributed to more innate immune mechanisms that may factor more strongly in therapeutic considerations and developments. A good example of this is the development of IL-36 antagonists or blockade of other members of the IL-1 family (83), which are proving to be promising in pustular and highly inflammatory forms of psoriasis (84). Besides biologics, small-molecules which are orally available and able to penetrate cells are being pursued. They inhibit central signaling pathways in pathogenetically relevant cell types. Due to their small molecular size, these substances are in principle also suitable for topical application. In contrast to the older preparations such as methotrexate, retinoids, or fumaric acid esters, whose effects are quite broad and where mechanisms have not yet been completely clarified, the newer preparations have a more selective effect. Despite supposed selectivity, however, the effect is sometimes more pleiotropic and there are more off-target effects than with biologics. The first compound approved for the treatment of psoriasis in this group is the PDE4 inhibitor apremilast. In late stages of clinical development is the TYK2 inhibitor deucravacitinib, which in clinical trials offers quite convincing clinical effects with a good safety profile. The background for the latter development is the recognition that many mediators, for example type I interferons or IL-23, mediate their inflammatory signals via Janus kinases (JAK, to which TYK2 also belongs) (85, 86). Thus, by blocking these signaling molecules, an (indirect) inhibition of inflammatory mechanisms can be achieved.
To address most of these challenges, it is likely that proteogenomic approaches will have to be expanded, implemented and/or developed in conjunction with sophisticated immunological-functional studies. These will need to be supported by comprehensive “real world” data, for example from registry studies, to capture the full actual spectrum of psoriatic disease. Only in this way will it be possible to stratify the heterogeneous population of patients with psoriatic disease even more precisely in cross-section and to characterize it down to the level of the individual. It will also enable a more accurate longitudinal characterization of the disease over its lifelong course. The ultimate goal must be to find and apply the most effective, and at the same time best tolerated therapy for each individual patient at any point in their “disease career,” sometimes in a quite variable manner (87).
Prurigo nodularis (PN) belongs to the spectrum of chronic prurigo and it is the dominant phenotype for 70% of patients with chronic prurigo (13, 88, 89). Chronic prurigo is a persistent and burdensome neuroinflammatory dermatosis associated with severe itching, permanent scratching behavior and diverse comorbidities. Among the comorbidities, for example atopic predisposition, atopic dermatitis, bullous pemphigoid, lichen planus, chronic kidney disease, hepatobiliary diseases, diabetes mellitus, chronic iron deficiency, HIV, and solid tumors have been reported to be causally related to PN (90). However, the role of these comorbidities as etiological factor in PN is still debated and needs additional studies. PN affects both sexes, all races and all ages with a preference for females above age of 60 years (90). Children may be affected, but this is very rare. Epidemiological studies are infrequent and report different prevalences depending on the method and population included (13, 91, 92). For example, Poland reported an estimated prevalence of 6.5/100,000, USA of 36.7 to 43.9/100,000 and Germany up to 100/100,000. However, all studies seem to argue for the fact that PN is not really a rare disease. Currently, neuroimmune mechanisms are considered the dominant mechanism underlying PN (93). In PN skin, different helper T cell phenotypes have been identified including Th1, Th2, Th17, and Th22 cells. Especially Th2 cytokines such as interleukin (IL)-4 and IL-31 are abundantly present in PN skin. Cutaneous sensory C- and Ad- nerve fibers express corresponding IL receptors. This enables a close neuroimmune communication with continuous stimulation of nerve fibers, their release of neuropeptides and induction of itch. Interestingly, IL31 has a prominent role not only enhancing the inflammation, but also leading to epidermal hyperplasia and fibrosis formation of the dermal collagen tissue. In addition to IL-31, also upregulated periostin might promote fibrosis formation by releasing IL31 from various immune cells (94, 95) from various immune cells. Fibrosis is a prominent feature in PN and distinguishes the disease histologically from atopic dermatitis. In addition, IL-4 plays a major role in fostering neuroimmune communication and neuronal hypersensitivity via the IL-4Rα/JAK pathway. Recent studies suggested that nerve fiber dysfunction and structural neuroanatomical changes are present in PN skin which are induced by scratching and maintained by inflammation (96, 97).
The diagnosis of PN is made clinically (98). PN is characterized by symmetrically distributed nodules; papules, and plaques may occur (Figure 1D). The severity of PN can range from some single nodules to several hundreds. All lesions are itchy and subject to scratching, formation of excoriations and bleeding. Lesions are found mainly on the extensor surfaces of the extremities and trunk with a typical butterfly sign at the back (lack of lesions at the central back which can be explained by the patient's inability to scratch these skin areas). Palms, soles and the face are rarely affected. PN can be documented by a PN-specific, validated investigator global assessment (99). The validated investigator questionnaire Prurigo Activity and Severity Score (PAS) assesses several parameters of the disease such as type, number, and distribution of pruriginous lesions, proportion of PN lesions with excoriations and proportion of healed lesions (100). Itch intensity is monitored best with validated instruments such as the numerical rating scale (NRS) for example as it is also done in other types of pruritus (101). The health-related quality of life can be documented either by Dermatology Life Quality Index (DLQI) or the itch-specific ItchyQol (102).
The first international guideline on diagnostic and therapy of PN recommends a laddered approach to treat PN (98). The first two steps comprise topical and intralesional corticosteroids, topical calcineurin inhibitors, capsaicin, systemic antihistamines and UV-phototherapy. In the third step, either neuronal therapies or immunosuppressants are advised such as gabapentinoids, antidepressants as well as cyclosporine and azathioprine. The clinical findings (inflamed vs. non-inflamed nodules) and quality of itch (itch with pain, burning and stinging) may guide to the right therapy.
Novel, effective, safe and approved therapies are urgently needed as patients are in general dissatisfied with the currently medical care (103). Currently, opioid modulators and investigational substances (dupilumab, nemolizumab) are recommended as off-label treatments in refractory cases (104, 105). For some of these substances, clinical trials are currently being conducted. For example, opioid modulators as well as IL4 and IL31 receptor antibodies are in current pharmaceutical development. IL31 signals through a heterodimeric receptor complex consisting of IL-31 receptor α (IL-31RA) and oncostatin M receptor β (OSMRβ). Novel substances target each of the two receptor components and are in phase II and phase III stage of development.
Lichen planus (LP) is a chronic immune-mediated disease which affects skin, mucosa and skin appendages. LP is the prototype of a lichenoid dermatosis which is characterized by a dense dermal T cellular and macrophage-rich infiltrate. LP is a common disease with an incidence in the general population is up to 1.27%. While LP is most common in the third and sixth decade, it may occur at any age. Mucosal LP (MLP) shows a prevalence of 0.89% and it is more commonly diagnosed in the female population. Involvement of the scalp is also more often reported in female patients, with a sex ratio close to 5:1. Clinically, we recognize three major subtypes of LP: cutaneous LP (CLP), MLP, and LP of the scalp, classically called lichen planopilaris (LPP) (14, 106, 107). CLP is classically characterized by violaceous, polygonal, slightly scaling and extremely pruriginous flat papules, which affect mostly the extremities (Figures 1E,F). Typically, CLP lesions show Wickham striae, whitish net-like lines that represent the clinical expression of the histologically seen epidermal hypergranulosis. Furthermore, several variants of CLP have been reported in the literature, including annular LP, atrophic LP, and LP verrucosus. A rare entity is represented by LP pemphigoides, which is clinically characterized by papules and blisters and serologically by the detection of IgG autoantibodies against BP180 and BP230 (108–110). MLP affects most frequently the oral mucosa. It has been more often described in female patients in the fourth decade (107). Clinically, MLP of the oral cavity is characterized by Wickham striae, erythematous macules, and, in some aggressive cases, by ulcerations (Figure 1E). A concomitant genital involvement has been reported in every second female patient affected by oral MLP (106). LP can involve other mucosal sites, including ocular, laryngeal, and esophageal mucosa. In the last case, the presence of dysphagia or odynophagia has been frequently reported, in 80 and 30% of cases, respectively (111). LPP is clinically characterized by red papules or plaques and perifollicular erythema. The chronic inflammation leads to destruction of hair follicles and to development of scarring alopecia. Patients affected by LPP may experience itching, burning of the scalp, and hair fragility. LPP requires an intensive and long-lasting therapy because of the characteristic refractory course of the disease. A variety of drugs may trigger LP, including antibiotics (e.g., dapsone and tetracycline), antifungal, and antimalarial drugs. Therefore, a detailed pharmacologic history is mandatory. The typical histological feature of LP is a band-like lymphohistiocytic infiltrate at the dermal-epidermal junction and in the upper dermis. Furthermore, hypergranulosis, irregular hyperplasia of the rete ridges with a classical saw-toothed pattern, and basilar vacuolar degeneration have been typically reported. Apoptosis of epidermal keratinocytes leads to the development of Civatte bodies, described as rounded, homogenous, eosinophilic cellular deposits in the upper dermis that can be identified by PAS staining and direct immunofluorescence microscopy (112).
The diagnosis should be performed according to the clinical and histopathological features. In addition, several differential diagnoses should be excluded, such as lichenoid drug eruptions, lichen planus pemphigoides, graft-vs. host disease, granuloma annulare, oral candidiasis, and oral leukoplakia (113). However, diagnosis is often delayed because of the highly variable clinical appearance and inconsistent histopathological findings in LP (114).
At times, LP may pose a therapeutic challenge. Indeed, some clinical variants are characterized by a refractory course, especially LPP, ulcerative oral LP and genital LP (14, 109). In CLP topical steroid treatments usually in combination with UVB or PUVA phototherapy are recommended. In recalcitrant cases, oral prednisone or oral retinoids may be useful (115). Oral LP can be initially treated with topical potent corticosteroids (e.g., clobetasol propionate 0.05%). Intralesional injection of corticosteroids can be useful in ulcerative oral LP. In addition, an off-label therapy with topical application of pimecrolimus or tacrolimus can be used. In case of severe involvement of the oral mucosa, several systemic therapies have been tried, including systemic corticosteroids, azathioprine, methotrexate, and retinoids (115, 116). In LPP an early and rapid control of inflammation is of pivotal importance to prevent the development of scaring alopecia. Topically, potent corticosteroids can be used in moderate cases (115). Alternatively, treatment with topical calcineurin inhibitors or with topical JAKi (e.g., tofacitinib) have been shown to be effective (109, 117). In more aggressive cases, a concomitant treatment with systemic corticosteroids is recommended. Alternatively, hydroxychloroquine or methotrexate can be used as second-line treatment. In recalcitrant cases, mycophenolate mofetil or cyclosporine A can be used as off-label treatment (118).
Recently, the use of anti-IL-17, anti-IL-12/IL-23, and anti-IL-23 monoclonal antibodies was reported to lead to an improvement of oral ulcerations in extremely refractory cases (109). This off-label use of these therapeutics was based on the observation of a Th1/Th17-dominated cell response in the peripheral blood of LP patients (1). To address, if this pathway is amendable to pharmacological interventions, a total of five patients with lichen planus were treated in a compassionate use trial. Of these, three received secukinumab, one patient ustekinumab and one guselkumab. In all cases, marked improvement was documented within the 12-week observation period. Of note, the clinical improvement was accompanied by a strong reduction of the Th1 and Th17/Tc17 cellular mucosal infiltrate, suggesting that IL-17-producing T cells are central to disease pathogenesis (109) At this regard, an open label, parallel, randomized, multi- center, phase II trial to evaluate the efficacy, ssafety, and tolerability of guselkumab in patients with oral LP is now ongoing (EudraCT Number: 2021-000271-36). In addition, a phase II study to evaluate the efficacy, safety, and tolerability of secukinumab 300 mg over 32 weeks in adult patients with biopsy-proven clinical variants of LP is ongoing (EudraCT number 2019-003588-24). Furthermore, JAKi have emerged as promising therapeutic agents in LP (14, 117).
Patients with hidradenitis suppurativa (HS) suffer from chronic painful inflammatory skin lesions in intertriginous sites (119) (Figure 1G). The manifestation of the disease mostly occurs around the age of 25. The average prevalence rate of HS is 0.2–0.4%, with highest rates in the Caucasian (0.75%) and the African American populations (1.3%) (17). Both sexes are affected with similar frequencies (16, 18, 120). Besides skin alterations, HS patients commonly suffer from numerous systemic comorbidities such as metabolic syndrome, spondyloarthritis or spondyloarthropathy (SpA), mental depression, and inflammatory bowel disease (121–125). HS leads to profound impairment in the quality of life of affected people, which is much more pronounced than the impairment caused by other dermatoses (16, 126). Furthermore, HS is associated with patients‘ body image impairment and increased suicidal behaviors (127–129). Ischemic heart diseases as well as accidents and violence (incl. suicides) contribute to the massively shortened (~15 years) life expectancy of patients with HS (130).
The diagnosis of HS is based solely on the physical examination and medical history (119). HS is confirmed when the following criteria are met: (i) typical skin alterations such as inflammatory nodules, abscesses, inflamed and draining tunnels (sinus tracts or fistulas), and rope-like scarring, (ii) in typical localizations such as in axillary (armpits), inguinal (groin), gluteal, perianal, and submammary (women) areas of the body, and (iii) typical occurrence, i.e., persistent (at least 6 months) or recurrent (>2 skin lesions occurring or recurring within 6 months) (119). Surprisingly, diagnosis is frequently delayed, although the diagnostic criteria are very clear (120). In Germany, the average duration between the manifestation of first symptoms and the HS diagnosis is 10 years (120). Importantly, the longer the delay of diagnosis, the more misdiagnoses, the greater the disease severity at diagnosis, and the higher the number of concomitant diseases (120). Various clinical scores are used to assess the severity of HS skin alterations. The International Hidradenitis Suppurativa Severity Score System (IHS4) is becoming increasingly important. It is a dynamic score based on the number of nodules, abscesses, and draining tunnels and allows dividing the severity of the disease into mild, moderate, severe (131). Patient-reported outcome measures, like DLQI, are also often used to assess the disease impact (132–134). Several blood biomarkers reflecting the activity of the immune system have been suggested, but none of them is currently used in everyday practice (135–137).
Treatment options for HS lesions include pharmacological therapies (local and systemic) and surgical treatments (119). The choice of therapy depends on the type and severity of the skin lesions as well as the patient's expectations. Individual inflamed nodules can be treated with topical antiseptic or antibiotic ointments or creams. If there are several inflamed nodules or abscesses, local therapy is supplemented by systemic pharmacological treatment. Irreversible skin alterations such as tunnels and scars can be effectively treated by surgery. Therefore, it is highly important to prevent such alterations from occurring through timely and effective pharmacological therapy. A combination of clindamycin and rifampicin is commonly used for systemic treatment. However, surgical and conventional pharmacologic therapies of HS are not associated with long-lasting improvement of patients' quality of life (132). Furthermore, the anti-TNF-a antibody adalimumab is the only approved systemic treatment for HS so far (119). Thus, one of significant challenges in HS care is the lack of further systemic treatment options. This limitation is basically due to our limited understanding of the molecular and immunological processes underling the formation and persistence of skin alterations in HS (138). TNF-a, IL-1, 5-lipoxygenase and G-CSF are thought to have a role in HS pathogenesis, but the pathophysiology is not well-understood (139–142).
Due to the long delay in diagnosis, the enormous impairment of the quality of life caused by HS and the limited range of evidence-based therapies, patients with HS have an enormous unmet medical need. To change this situation, first and foremost the time interval between first symptoms and diagnosis must be significantly shortened. This is extremely important because of the progressive nature of the disease that over time leads to irreversible skin destruction. To this end, the patients must be pharmacologically treated as soon as the first symptoms appear. Training of doctors such as general physicians, dermatologists, surgeons, and gynecologists, as well as programs to raise/create the awareness of the disease within large parts of the society are needed. It is gratifying that many clinical trials with a focus on new systemic pharmacological treatment are currently being carried out. However, these are often without a well-founded scientific rationale and a better understanding of disease mechanisms is needed. Thus, we need extensive, well-founded translational research into the pathogenesis of the HS as the basis for the development of targeted systemic therapies for HS. A further aspect is a holistic view of the patient to include awareness of systemic inflammation evaluation, treatment of systemic comorbidities and pain, and psychological care for the patient. The last aspect is to motivate and support the patient in changing lifestyle factors that can contribute to the persistence of HS, such as smoking and obesity. Structured patient counseling that provides information about these associations, including referral to smoking cessation programs and weight loss might be helpful.
Alopecia areata (AA) is an autoimmune skin disease that affects ~2% of the worldwide population (19). In AA T cells attack the hair follicles causing an inflammatory, non-scarring, hair loss that is typically manifested in patches as a single or multiple well-demarcated areas. Patients with the patchy form of alopecia areata (AAP) commonly exhibit hair loss on the scalp but may also present hair loss in other hair-bearing areas of the body (Figure 1H). The disease course varies greatly between AA-affected individuals in terms of disease severity, duration, and prognosis. Hence, AA patients may present patchy, diffuse, confluent, or mosaic patterns of hair loss during a single episode, or recurrent disease episodes. Additionally, while, up to 75% of the AAP patients exhibit a spontaneous regrowth of hair within a few months (143), in up to 25% of all AAP patients, the disease progresses to its more severe form, and the hair loss extends to the entire scalp (alopecia totalis; AT) or body (alopecia universalis; AU) (144). In addition to hair loss, nail abnormalities, most commonly pitting and trachyonychia, are observed in patients with AA and are more prevalent in patients with AT and AU (145).
The diagnosis of AA is typically made based on the patient's medical history and a clinical examination that determines the location and the extent of hair loss, and differentiates AA from other potential causes of hair loss, thereby providing a more accurate prognosis and identifying a favorable line of treatment (146). The clinical examination is often supported by a positive hair pull test at the periphery of the lesion, especially in patients with active disease. Additionally, the clinical diagnosis is frequently accompanied by trichoscopy examination that is used to examine the hair follicle, hair shaft, and the surrounding skin, and establish the phase of the disease (147). Dermatoscopic findings in AA may vary depending on the specific disease phase (146). In the acute phase of AA, exclamation point hairs that are located at the border of the plaque and broken hairs that are thicker proximal to the scalp are typically observed, while in the chronic stage of AA, dystrophic hairs, uniform black dots, and yellow dots, are predominantly present. In cases with an unclear clinical presentation, clinical diagnosis is supported by a histological examination of a horizontally sectioned 4 mm scalp biopsy taken from an area of active hair loss (148). In the acute phase, a peribulbar infiltrate is observed that consists predominantly of CD4+/CD8+ T cells and Langerhans cells as well as eosinophils, mast cells, and plasma cells, in a typical “swarm of bees” pattern. In addition, pigment incontinence may also be present, due to the destruction of melanocytes in the apex of the dermal papilla. Other histological signs of AA characteristic of acute and the subacute phases include hair follicle miniaturization, a decreased anagen-to-telogen ratio, and a decreased terminal-to-vellus hair ratio (148).
The first lines of therapy in most AA patients include corticosteroids and/or immunotherapy that is aimed at containing the inflammation and promote the recovery of dystrophic hair follicles. The type of treatment assigned is determined based on the age of the patient, and the extent and the severity of hair loss. The first line of therapy in AAP patients with active disease include intralesional (triamcinolone acetonide, triamcinolone hexacetonide, and hydrocortisone acetate) and topical corticosteroids (desoximetasone, betamethasone valerate, and clobetasol propionate), which show low solubility and promote maximum local anti-inflammatory actions with minimal systemic side effects (149). The adverse effects of topical and intralesional corticosteroids include folliculitis, reversible skin atrophy, telangiectasia, and hypopigmentation. In more severe cases, to contain rapidly progressing hair loss in AAP patients, systemic high-dose pulsed oral, or intravenous glucocorticoids (prednisolone) are recommended (150–152). However, one major drawback of this line of therapy includes recurrence of hair loss after therapy is discontinued (153). In AT, AU, and AAP patients with a chronic disease or in AAP patients with an active disease who fail to respond to topical or intralesional corticosteroids, topical application of contact allergens is recommended. In this line of therapy, potent contact allergens such as 1-chloro, 2, 4, dinitrobenzene (DNCB), diphenylcyclopropenone (DPCP), or squaric acid dibutyl ester (SADBE) are applied weekly onto the lesion to induce mild contact dermatitis, which via yet incompletely understood molecular mechanism, results in regrowth of hair (154, 155). Several side effects associated with this treatment include severe contact dermatitis, occipital or cervical lymphadenopathy, urticaria, dyschromia, and vitiligo (156, 157). Lastly, systemic glucocorticoids and systemic immunosuppressives (methotrexate, sulfasalazine, and azathioprine) can be used in patients with active AT and AU. Recently, a new class of small molecules known as JAKi were shown to be effective in AA. JAKi are especially effective in AA since they target a family of tyrosine kinases JAK1/2 and JAK1/3 that transduce cytokine-mediated signaling in T cells, which were shown to play a critical role in AA (158). Blockade of JAK1/3 and JAK 1/2 by the oral selective inhibitors, tofacitinib, and ruxolitinib, respectively, was shown to be effective in inducing regrowth of hair in AAP and AT/AU patients with active and chronic disease (159, 160). Although, no adverse side effects of these drugs were reported in AA patients, increased risk of infections and neoplasia were observed in rheumatoid arthritis patients treated with tofacitinib (161). Thus, future investigations into the potential side effects of prolonged treatment with JAKi, as well as examining the efficacy of topical JAKi formulations in AA, are required.
The heterogeneous clinical presentation, variability in the rate of spontaneous remission, and differences in disease prognosis still pose significant difficulties in assessing the efficacy of therapy in AA, making it challenging to generalize a certain line of treatment for different AA patients. Future, well-powered randomized placebo-controlled trials are required to systematically assess the efficacy of existing lines of therapy and facilitate the development of FDA-approved treatment options in AA. Large randomized placebo-controlled trials are underway for at least 3 JAKi in AA (Baracitinib, deuterated Ruxolitinib, and Ritlecitinib) with several others already approved for other inflammatory diseases (162, 163). Despite the significant improvements in our understanding of the pathophysiology of AA, future research is warranted to understand the contribution of environmental triggers to AA pathogenesis, since only a 55% concordance rate was observed in monozygotic twins, suggesting other factors contribute to disease onset (164).
Vitiligo is an autoimmune depigmenting disorder of the skin. The depigmentation results from the loss of epidermal melanocytes. Clinically presenting with well-demarcated white patches on the body, vitiligo can be cosmetically very disabling and create a psychological burden (165, 166). There has been a great advance in understanding the pathological basis due to current research. JAK kinase signaling pathways and the cytokines involved in the Th1 pathway are the focus of the upcoming vitiligo treatments, followed by antioxidant and repigmenting agents (167).
Vitiligo is usually diagnosed clinically (168). Occasionally skin biopsy may be recommended (169). A characteristic histological hallmark is the absence of melanocytes and epidermal pigment (Figure 1I). Screening to assess potential autoimmune diseases is recommended.
Therapy of vitiligo is currently unsatisfactory. Topical treatments include corticosteroid and calcineurin inhibitors (170). Phototherapy, ranging from broadband, or narrowband UVB to psoralen plus UVA, may be another option (171). In severe or treatment-refractory cases systemic treatments include mini-pulses of oral steroids, methotrexate, cyclosporin or mycophenolate mofetil. Currently, there are several drugs available, alone or combination, aiming to arrest progression and induce repigmentation of the skin. The degrees of repigmentation vary (172). Of note, there is no approved treatment for vitiligo repigmentation and current off-label therapies have limited efficacy. This emphasizes the need for better treatment options.
It is essential to increase awareness of the comorbidities associated with the disorder. The most common comorbid conditions of vitiligo are thyroid disease, diabetes mellitus, Addison's disease, pernicious anemia, rheumatoid arthritis, inflammatory bowel disease, ocular and audiological abnormalities, alopecia areata, systemic lupus erythematosus, Sjögren's syndrome, dermatomyositis, scleroderma, psoriasis, and atopic dermatitis (173). Among emerging treatments that may meet the need for safe and effective vitiligo treatments, JAK inhibitors (topical and oral) are the most promising new class of drugs currently available and act best in conjunction with phototherapy (174–177). The result from the phase III TRuE-V clinical trial program (NCT04052425 and NCT04057573), evaluating the topical JAKi ruxolitinib (Opzelura™ cream) showed a substantial repigmentation of vitiligo lesions. Hence, approval in the U.S. and Europe is expected in the upcoming months. Further treatment potential options like phosphodiesterase inhibitors (PDE4) or abatacept, a fully human fusion protein of CTLA-4 and the Fc portion of human IgG1 are sometimes used off-label. Considering the role of PD-1 ligand (PD-L1, a PD-1 agonist) and CTLA-4 in maintaining immune balance, targeting this pathway could be a therapeutic option. Furthermore, it was shown, that IL-15 acts via JAK STAT signaling pathways and has been recently implicated in oxidative stress mediated destruction of melanocyte. Thus, the future of vitiligo treatment may rely on the development of more specific drugs (167).
Chronic spontaneous urticaria (CSU) is defined by the occurrence of itchy wheals, angioedema, or both for longer than 6 weeks (178). In most patients, CSU lasts for several years and then shows spontaneous remission. Because of the severe pruritus and the unpredictability of the occurrence of the signs and symptoms, most patients who are not adequately treated are severely affected in their quality of life (179). CSU is a mast cell-driven disease, and its signs and symptoms occur in response to the activation of skin mast cells and their subsequent release of histamine and other mediators. The exact underlying pathomechanisms of skin mast cell activation in CSU are not fully understood. Based on recent evidence, three subtypes of CSU have been described, type I autoimmunity (or “autoallergy”), type IIb autoimmunity (“classical autoimmunity”), and CSU due to unknown cause (180). In addition, other factors such as acute infections, certain drugs or stress modulate mast cell activation and drive exacerbations or worsening of CSU.
In most patients, the diagnosis of CSU is straightforward, with spontaneously recurring itchy wheals, angioedema, or both, for longer than 6 weeks (Figure 1J). The current guideline on the definition, classification, diagnosis, and management of urticaria recommends a detailed patient history, physical examination (including pictures from patients) and a basic diagnostic workup consisting of a complete blood count with differential, CRP, IgG anti-TPO and total IgE (178). The questions and investigations are mainly aimed at ruling out rare differential diagnoses, for example urticaria vasculitis, autoinflammatory syndromes or bradykinin-mediated angioedema, assessing patients for underlying causes and modifying conditions, and identifying comorbid diseases and consequences of having CSU (180). Based on the answers to the respective questions, additional investigations such as histological examination of a skin biopsy or further laboratory analyses may be necessary. An important aspect of the diagnosis is the assessment of CSU activity, impact, and control. For this purpose, the urticaria activity score (UAS), the chronic urticaria quality of life questionnaire (CU-Q2oL) and the urticaria control test (UCT) should be used (178). In CSU patients with angioedema, the angioedema activity score (AAS), the angioedema quality of life questionnaire (AE-QoL), and the angioedema control test (AECT) should also be used (178).
The goal of any treatment in CSU is the absence of signs and symptoms, complete disease control and a normal quality of life. To achieve this, an effective prophylactic treatment is required for all patients. The use of a 2nd generation H1-antihistamine is the recommended first-line treatment for CSU, first at standard dose and then, if needed, at up to 4-fold the standard dose (178). While 2nd generation antihistamines have proven to be a very safe long-term treatment, also at higher than standard doses (181), many patients with CSU do not achieve complete response. For those patients, the second step in the treatment algorithm is the addition of the monoclonal anti-IgE antibody omalizumab, which has been shown to be effective and safe in many H1-antihistamine refractory CSU patients (182). A significant proportion of CSU patients do not achieve complete control with omalizumab. Recent data indicate that patients with markers of type IIb autoimmune CSU, e.g., low total IgE and elevated levels of IgG anti-TPO, show slow and poor response to omalizumab treatment (180, 183). In patients who do not respond to omalizumab within 6 months of treatment (or earlier, if symptoms are unbearable), cyclosporin up to 5 mg/kg body weight is recommended in addition to antihistamines. Due to the poor safety profile, this is not possible in all patients and potential side effects should be rigorously monitored.
Better treatments are needed for CSU and several are currently under investigation (184), most of them mast cell-targeted (185, 186). These treatments aim to inhibit mast cell mediators, prevent mast cell activation (187), silence mast cells via inhibitory receptors, or deplete mast cells. One of the biggest challenges in treating CSU patients in the future will be to figure out which patients benefit best from which treatment. For example, fenebrutinib, an oral Bruton's tyrosine kinase inhibitor, has been shown to be most effective in type IIb autoimmune CSU (180). The identification of reliable and easy to analyze biomarker for response to treatment will thus be an important task for future research.
Pemphigus refers to a group of rare autoimmune blistering diseases characterized by autoantibodies targeting desmosomal cadherins: most commonly desmoglein-1 (Dsg1) and desmoglein-3 (Dsg3). It presents with localized or widespread flaccid bullae which can rupture and progress to post-bullous erosions and crusts (Figure 1K). There are two major types: Pemphigus vulgaris (PV) and pemphigus foliaceus (PF). These subtypes are differentiated by oral and/or mucous membrane involvement in PV, which is absent in PF. The histological hallmark of pemphigus is acantholysis, caused by loss of adhesion between effected keratinocytes (188). Overall, pemphigus is associated with significant morbidity and mortality (22, 189).
The diagnosis can be made by direct immunofluorescence (IF) microscopy of a perilesional skin biopsy, revealing deposition of IgG autoantibodies and/or C3 on the cell surface of keratinocytes (190). Detection of antibodies against Dsg1 or Dsg3 using ELISA, or use of indirect immunofluorescence microscopy against monkey esophagus allows serologic characterization (188). Significant delays in diagnosis are unfortunately common (191). Barriers to obtaining direct immunofluorescence microscopy serve as a roadblock in the diagnosis of pemphigus, particularly in the developing world (192). Immunohistochemical approaches, and even desmoglein ELISA have significant sensitivity limitations furthering diagnostic delays when direct immunofluorescent microscopy is not feasible (193). In the so far largest multicenter prospective study, anti-Dsg1/ Dsg3 serum antibodies were, however, detected in 329 (98.5%) of 333 pemphigus sera diagnosed by the clinical picture and direct IF microscopy using widely available assays.
The first-line treatment for pemphigus is systemic corticosteroids, often used in conjunction with other immunosuppressive agents (194). More recently, evidence suggests the use of the anti-CD20 monoclonal antibody rituximab as an alternative first-line agent used alongside corticosteroids (195, 196). Additional therapies such as intravenous immunoglobulin (IVIg) and immunoadsorption can be used as adjuvant treatments, either in combination with first-line medications or when contraindications are present (194). However, despite the significant advances in the treatment of pemphigus in recent years, there are still numerous limitations in current therapies. To achieve clinical response during the acute phase of disease, high dose corticosteroids are generally required (197). While novel treatments such as rituximab may reduce cumulative steroid dosages, they do not work quickly. Furthermore, relapses are also frequently encountered (198).
Thus, there is a need for short-term agents that can minimize the need for high dose steroids. Once achieving complete remission, relapses remain common, though this can be decreased with more aggressive protocols utilizing additional rituximab infusions (199–201). An alternative approach, for example targeting autoantibody-induced tissue pathology have emerged (202, 203). In addition, attempts to incorporate precision medicine into the treatment of pemphigus are on the horizon (19). However, optimism must be tempered by the contributory role of non-desmoglein autoantibodies in pemphigus and aberrant cell signaling, which contribute toward the pathogenesis (204–206).
Bullous pemphigoid (BP) is one of the most common autoimmune blistering skin diseases, and it is characterized clinically by tense blisters with itchy urticarial erythema on the trunk and extremities (Figure 1L) (207). Mucosal surfaces can also be affected. It is most prevalent in the elderly (late 70s), but can appear in younger people (208). The molecules targeted by BP autoantibodies are the two hemidesmosomal proteins type XVII collagen (COL17, also called BP180) and BP230, and the former molecule has been recognized to be the major autoantigen. Triggering factors for BP include ultraviolet rays and other radiation, burns, trauma, and regulatory T-cell dysfunction (209–211). Dipeptidyl peptidase-4 (DPP-4) inhibitors have recently gained attention as a cause of BP (212–214).
BP is diagnosed based on the clinical, histological, and immunological findings (215, 216). In addition to the clinical features of tense blisters and urticarial erythema and the histological feature of subepidermal blistering, the detection of tissue binding and/or circulating autoantibodies against the dermal-epidermal junction (DEJ) is essential. Direct IF microscopy of perilesional skin is the most sensitive method for detecting autoantibodies in BP, with a linear IgG and/or C3 deposition at the DEJ. To detect circulating autoantibodies, indirect IF microscopy using cryosections of normal human skin or 1M NaCl-split human skin is useful. To confirm the target antigen of autoantibodies, an ELISA using recombinant BP180 NC16A is widely used. A full-length BP180 ELISA (217) and a BP230 ELISA are also useful. Diagnostic challenges are infrequently encountered in patients presenting with “classical” BP lesions, i.e., tense blisters on erythematous skin. By contrast, atypical clinical presentations, which occur in least 20% of all BP patients, diagnosis is often delayed by several months, if not years (4, 218, 219).
In clinically localized or mild cases, superpotent topical corticosteroids (clobetasol propionate) are applied to lesions only or to the whole body except the face as a first choice (215, 216, 220). Low-dose oral corticosteroids, tetracycline (and nicotinamide) and dapsone are also used. In generalized or moderate/severe cases, oral corticosteroids (0.5–1.0 mg/kg/day) or superpotent topical corticosteroids are the mainstay treatment. If sufficient efficacy cannot be achieved, immunosuppressants (e.g., azathioprine, mizoribine, cyclophosphamide, cyclosporin, mycophenolate mofetil, methotrexate), steroid pulse therapy, plasma exchange/immunonoadsorption, or intravenous immunoglobulins should be added as appropriate (215, 216, 221, 222). A randomized controlled trial demonstrated the efficacy of doxycycline (200 mg/day) as an initial treatment for BP. Non-inferiority was shown in comparison with oral prednisolone (0.5 mg/kg/day), and the safety was significantly higher (223). Whilst all these treatments, especially those using topical or systemic corticosteroids, induce remission in over 90% of the patients within 4 weeks, relapses during tapering corticosteroids or after stopping treatment are frequent (220, 222, 224). This necessitates prolonged treatment with corticosteroids. In turn, this long-term use of oral corticosteroids frequently causes severe side effects, particularly in the elderly. In addition, although most BP cases are well-controlled by standard therapies, intractable and recurrent cases still exist. Therefore, new treatments that can suppress the disease activity and reduce or replace (oral) corticosteroids are much anticipated.
Based on the clinical and immunological characteristics, some molecules are considered as promising targets for BP therapies. As in pemphigus, the anti-CD20 antibody rituximab has been reported as effective against BP (225, 226). The pathogenicity of IgE autoantibodies has been described in many studies (227–229), and the efficacy of the anti-IgE antibody omalizumab against BP has been reported (227, 230, 231). Furthermore, the anti-IL-4 receptor alpha dupilumab has been reported as an alternative to prednisolone (232, 233). Several clinical trials targeting these molecules are under way, which may provide new treatment options for BP in the near future (234). Regarding the early diagnosis, continued education of healthcare providers, especially outside dermatology, is important to raise awareness for (atypical) BP (219), as well as forms of drug-induced BP (24). One important pillar in raising the awareness for BP and other rare skin blistering autoimmune diseases is the International Pemphigus & Pemphigoid Foundation (IPPF), the largest patient organization for those affected by pemphigus or pemphigoid.
Mucous membrane pemphigoid (MMP) is a subepithelial/ subepidermal blistering autoimmune disease with predominant involvement of orifice-close mucosal surfaces and autoantibodies against proteins of the dermal-epidermal junction (Figure 1M) (235). The main target antigens are BP180 (type XVII collagen) and laminin 332 recognized in about 80 and 10–20% of patients, respectively. In <5% of MMP patients, type VII collagen is targeted and individual patients with reactivity against a6b4 integrin have been described (207, 236). The incidence of MMP has been estimated to 1.3 and 2.0/ million/year in France and Germany (237–239) and its prevalence was calculated to be 24.6 patients/million, i.e., about 2,000 patients in Germany in 2014 (23) MMP mainly occurs between the age of 60–80 years and is extremely rare in children and adolescents (240, 241). The oral cavity and conjunctivae are the most frequently affected mucosal surfaces followed by nasopharynx and genitalia, and more rarely, larynx, esophagus, and trachea. In about 30% of patients, additional skin lesions may occur (241). MMP is associated with a considerable morbidity including pain, difficulties in food intake and breathing as well as visual impairments that can lead to blindness (241). Further studies are needed to assemble more data about the incidence and prevalence of MMP in different geographical regions. So far, epidemiological studies have been mostly limited to central Europe. In the Schleswig-Holstein registry of autoimmune blistering diseases including all newly diagnosed patients in the most northern German province (www.sh-register-pemphigoid-pemphigus.de) we are prospectively mining the annual incidences of MMP since 2016.
For the management of MMP the recent S3 guidelines of the European Academy of Dermatology and Venereology will be instrumental (241, 242). Diagnosis of MMP is based on the presence of predominant mucosal lesions and the detection of tissue-bound and/or circulating autoantibodies (242). Direct IF microscopy of a biopsy taken form perilesional tissue or unaffected oral mucosa is the diagnostic gold standard with a sensitivity of 60–90% (241–243). Like in all pemphigoid disorders, in MMP, it reveals linear deposits of IgG, IgA, and/or C3 at the subepithelial basement membrane zone (BMZ). Repeated biopsies for direct IF can increase the sensitivity from 70 to 95% (243, 244). Indirect IF microscopy on human salt-split skin is a convenient and sensitive screening assay for circulating autoantibodies against the subepithelial BMZ and allows the differentiation between IgG/IgA that binds to the roof of the artificial split, i.e., antibodies against BP180, BP230, and a6b4 integrin and IgG/IgA that labels the blister floor as seen with reactivity against laminin 332 and type VII collagen (245–247). Widely available antigen-specific test systems include ELISA and/or indirect IF applying the recombinant NC16A domain of BP180 NC16A, the NC1-domain of type VII collagen, a C-terminal stretch of BP230, and the laminin 332 heterotrimer (242, 248–252). In particular, detection of anti-laminin 332 antibodies is essential since-anti-laminin 332 MMP is associated with a malignancy in 25–30% of patients. After the initial observation of solid malignancies in 2 of 5 MMP patients with serum autoantibodies against laminin 332 by Leverkus et al. (253), Egan et al. reported malignancies in 10 of 35 patients (29%) (254). This important clinical association has then been corroborated by several other studies (249, 253–259). In contrast, using an in-house ELISA Bernard et al., did not recognize the association of anti-laminin 332 IgG and malignancies (260). Recently, in a large multicenter study, a 6.8-fold higher risk of malignancy has been calculated in anti-laminin 332 MMP patients compared to the general population (237). However, serological diagnosis is limited by relatively low autoantibody levels. In addition, no standardized assay is widely available for serum IgG against the BP180 ectodomain outside the NC16A domain, an immunodominant stretch in anti-BP180 MMP. Since IgA reactivity is frequently seen in MMP, the lack of widely available test systems for IgA reactivity against BP180, BP230, and type VII collagen is further limiting the diagnostic power.
The European S3 guidelines recommend the first-line use of topical corticosteroids with or without dapsone, methotrexate or tetracyclines for mild and moderate MMP and for severe MMP, dapsone in combination with systemic cyclophosphamide with or without systemic corticosteroids (242). However, apart from two small phase IIa trials comparing dapsone with cyclophosphamide and prednisone with cyclophosphamide, respectively, in MMP patients with ocular disease, no randomized control trials have been performed in MMP (261). Hence, well-designed clinical trials are urgently needed to identify the best current available treatment options for MMP patients.
The highly standardized indirect IF test based on the expression of recombinant laminin 332 in a human cell line has become widely available (249). This assay will be instrumental for the in-depth analysis of the occurrence of malignancies in patients with anti-laminin 332 MMP, an association that has not yet been widely recognized in the community. In this sense, the recommendation of the S3 guidelines to assay for anti-laminin 332 reactivity in all patients with negative or dermal binding by indirect IF microscopy on salt-split skin will propel our knowledge. For the management of anti-BP180 MMP only the anti-BP180 NC16A IgG ELISA is widely available. Since in MMP the NC16A domain is not an immunodominant region and IgA reactivity is frequently found, assays for the detection of serum IgA and IgG against other parts of the BP180 ectodomain are urgently needed. Considerable progress is being awaited on our understanding of the disease mechanisms in MMP using a recently established mouse model of anti-laminin 332 MMP (262). In contrast to the previously reported model by Lazarova et al. this model depends on Fc receptor-mediated inflammatory pathways and C5aR1 (262, 263). The future use of the novel model to preclinically evaluate future therapeutic strategies has recently been supported by the observation that dapsone, first-line treatment in MMP, resulted in a significant reduction of oral and cutaneous lesions compared to vehicle-treated mice (264). Nonetheless, until a mouse model for anti-BP180 MMP, that represents the large majority of MMP cases, has been developed, it will remain unclear whether the anti-laminin 332 MMP model fully represents experimental MMP. In any case, the present anti-laminin 332 MMP mouse model opens the possibility to pre-clinically test anti-inflammatory agents and as such pave the way for randomized controlled trials in MMP.
Epidermolysis bullosa acquisita (EBA) is caused by autoantibodies targeting type VII collagen (COL7) which is a major component of anchoring fibrils (265, 266). Despite this singular key pathogenic principle, the clinical presentation of EBA is broad (Figures 1N,O). The disease may present as fragile skin with subsequent scaring, or as a widespread inflammatory disease with blistering and erosions. In addition to the skin and mucous membranes, internal organs may be affected (267). For example, strictures of the esophagus are relatively common (268). Thus, EBA imposes a high burden on the patients affected by this rare disease.
EBA is confirmed if linear deposits of immunoglobulins and/or C3 are detected by direct immunofluorescence (IF) microscopy or perilesional skin biopsy and if an u-serrated pattern is seen in direct IF microscopy, or circulating COL7 autoantibodies are detected (247, 269). Due to the heterogeneous clinical presentation, EBA is often not considered as a differential. Thus, the challenge is to raise awareness for this rare disease because once considered as a differential, diagnosis can readily be obtained using direct IF microscopy and serology.
There are no controlled clinical trials for EBA treatment which is thus based on expert recommendation. Unspecific immunosuppression is the mainstay of EBA treatment. Most commonly, systemic corticosteroids are used. In many cases additional immunosuppressants are added to systemic corticosteroids, most commonly azathioprine or cyclosporine are used (270). Overall, management of EBA is notoriously challenging—median time to remission is 9 months. In the same study, complete remissions were achieved in 45% of patients, with another 45% in partial remission and 10% with ongoing active disease−6 years after the initial diagnosis was made (271). Thus, treatments that induce remissions more reliably and faster are urgently needed to relieve the burden imposed by EBA.
In a metanalysis of over 1,000 EBA cases, use of the CD20 antibody rituximab or high dose intravenous immunoglobulin G (IVIG) were, compared to all other treatments, more often associated with the induction of remissions (270). These observations are a basis to establish protocols for clinical trials in EBA, evaluating the impact of either rituximab or IVIG. In addition, this also indicates that drugs targeting the B cells, such as the BTK inhibitor PRN1008, or compounds modulating the half-live of IgG, such as FcRn inhibitors, could be also effective in EBA (234). In addition, use of pre-clinical model systems has identified and validated a number of novel therapeutic targets in EBA (272–276). Based on these findings in pre-clinical EBA models, controlled clinical trials are currently performed—albeit in bullous pemphigoid patients (234).
Cryopyrin-associated periodic syndrome (CAPS) comprises a group of rare diseases that, despite certain clinical similarities, were previously considered separate disorders. These include Familial Cold Urticaria Syndrome (FCAS) which was first described in 1940 (277), Muckle-Wells Syndrome (MWS), and Chronic Infantile Neurologic Cutaneous and Articular or Neonatal Onset Multisystem Inflammatory Disease (CINCA/NOMID). Its prevalence is about 1–2 per million inhabitants in Europe and the USA.These diseases are characterized by an attack-like course with fever episodes lasting up to a few days and an enormous increase of inflammatory laboratory parameters, usually an accompanying urticaria-like exanthema, conjunctivitis, joint and muscle pain as well as hepatomegaly and splenomegaly (Figure 1P). In severe cases, cartilage growth leads to joint dysfunction. In MWS and NOMID/CINCA, central nervous involvement with mental retardation, epilepsy and hearing loss is found. The expression of the disease pattern varies from individual to individual. A long-term complication is the development of AA amyloidosis, which affects multiple organs, most prominently the heart and the kidney. As the cause for these diseases, heterozygous monogenetic deficiency in the NLRP3 (278), NLRP12 (279), PLCG2 (280), and NLRC4 (281) genes have been identified. However, a considerable number of cases are caused by mosaicism in the respective genes, such as in NLRP3 (282).
The diagnosis is based on a careful history and observation of the clinical course, especially of the inflammatory parameters, as well as genetic testing, preferably requiring NGS panel diagnostics. It may be necessary to assess whether the disease can be influenced by corticosteroids, NSAIDs, and ultimately IL-1b inhibitors in an individual patient. The additional use of clinical scores, such as the EUROFEVER/PRINTO score (283), is helpful, although, given the rarity of the disease, a systematic approach is needed to differentiate it from other diseases. A special difficulty are cases where the disease is caused by somatic mosaicism or by a not yet identified unknown genetic defect. In such cases the diagnosis may not be made satisfyingly, leading to delay in efficient, but often expensive, treatment options. In addition to diagnosis, monitoring of disease activity is also of high importance. While ESR and CRP are routine diagnostics, the measurement of calprotectin levels or amyloid A in the serum, for example, are helpful but much less available. Another problem is the early diagnosis of AA amyloidosis and determination of its extent. Here, nuclear medicine methods such as a PET-CT scan with 18F-florbetaben have been described (284) but are also not yet routine.
The IL-1β inhibitors anakinra and canakinumab are approved and available for the treatment of CAPS. However, other cytokines such as IL-18 have been described to be important in autoinflammatory diseases (285, 286). Hence, breakthrough attacks carried by IL-18 are not inhibited by current treatments. On the long run, secondary AA amyloidosis poses a challenge. Although it can be indirectly alleviated by inhibition of IL-1β signaling, targeted resolution of amyloid deposits is not possible to date (287).
Over the past 20 years, genetic defects have been identified for a variety of autoinflammatory diseases. Especially challenging are cases in which no classical germline mutation is present. In such cases, classical genetic methods reach their limits. The development of third generation sequencing methods such as nanopore sequencing with the simultaneous development of bioinformatics and advances in IT infrastructures could provide the solution for these cases as well (288). In addition to inhibition of secondary proinflammatory messengers such as IL-1β, inhibition of NLRP3 by small-molecule agents may also show promise (289, 290). To inhibit the action of IL-18, which is important in addition to IL-1β and plays a role in macrophage activation in particular, a promising drug might be available in the form of IL-18 binding protein (tadekinig alfa) (291).
Schnitzler's syndrome is a late-onset autoinflammatory diseases that has been described first in 1972 by Schnitzler (292). The disease is characterized by the combination of urticaria-like exanthema (neutrophilic urticarial exanthema) and gammopathy, associated with fever, joint, muscle or bone pain, elevated inflammation markers, morphologic bone changes, hepato-splenomegaly, and palpable lymph nodes (Figure 1Q) (29). It is considered to be a rare entity—with only about 100 patients described in the literature—although a retrospective database search for urticarial exanthema associated with dysproteinemia led to the identification of 16 patients at Mayo Clinic (293), pointing toward a much higher incidence. The etiology and pathogenesis of the disease is unknown. A somatic mutation is assumed (293), which is comparable to the pathogenesis of mastocytosis. In the case of mastocytosis, even low frequencies of mutant cKit that are barely detectable by means of digital PCR can lead to pronounced symptoms (294). The possible causal relationship between gammopathy and Schnitzler syndrome is also unclear.
The diagnosis of Schnitzler's syndrome should be considered in patients with gammopathy and urticarial exanthema, especially those without itching, increased inflammation markers and fever (295). On the other hand, chronic spontaneous urticaria (CSU) is a much more common diagnosis, which renders the differentiation very difficult. Especially, pressure urticaria may present with systemic symptoms such as fever and myalgia (296). On the other hand, CSU is such a much more common disease than Schnitzler syndrome that not every gammopathy in combination with urticarial exanthema should be misdiagnosed as Schnitzler syndrome. Unfortunately, the diagnosis of chronic urticaria is not established by specific biomarkers that would allow differentiation from other entities including allergic forms. Nevertheless, a lack of response to antihistamines, biologics such as omalizumab, or even corticosteroids may serve as further evidence of an autoinflammatory syndrome. The Strasbourg criteria are helpful in establishing the diagnosis, although they are still considered provisional and specificity and sensitivity have not been adequately determined (297). The dermatohistopathological differentiation between neutrophil-rich infiltrates in Schnitzler syndrome (298), urticarial vasculitis, and urticaria is not straightforwardly possible in practical settings, although a morphologic criterion, neutrophilic epidermotropism, might be specific for autoinflammatory diseases such a Schnitzler's syndrome. Moreover, the gammopathy strictly required by the Strasbourg criteria may be not that absolutely necessary, as cases that appear to be clearly late-onset autoinflammatory diseases may present without it (299, 300). Gammopathy may also develop later in the course of the disease. Hence, establishment of validated diagnostic criteria for Schnitzler's syndrome is needed.
IL-1β inhibiting treatment with anakinra and also canakinumab is highly efficient and can lead to resolution of symptoms within a few hours (301). Other treatment modalities such as colchicine, hydroxychloroquine, pefloxacin (29), and IL-6 inhibition with tocilizumab (302) are described. Like CAPS, Schnitzler's syndrome may lead to AA amyloidosis (303). AL amyloidosis due to gammopathy occurs, although rarely (304). Both types are difficult to treat, and no specific amyloid resolving treatment is known.
At the time being, only 9 controlled studies for Schnitzler's syndrome are listed in ClinicalTrials.gov, 5 of which that are using established IL-1β and IL-6 inhibitors have the status of being completed. A novel IL-1β inhibitor with affinity to IL-1α and IL-1Ra is recruiting, and a study that tests the histone deacetylase inhibitor ITF2357 (305) has an unknown status. Drugs that target the inflammasome may be able to prevent the activation of the autoinflammatory cascade (290). A pilot study using the NLPR3 inhibitor dapansutrile is listed as recruiting. Interestingly, a clinical observation of a resolution of Schnitzler's syndrome after haematopoietic stem cell transplantation may hint at the pathogenesis, e.g., a somatic mutation in bone marrow cells (306). This observation may also hint at a similar pathogenic pattern as in systemic mastocytosis, where myeloablative conditioning followed by (allogenic) stem-cell transplantation is used for treatment (307).
Lupus erythematosus (LE) is a multisystem autoimmune condition that ranges from skin to multiorgan involvement. While systemic lupus erythematosus (SLE) involves many systems, cutaneous lupus erythematosus (CLE) affects the skin and/or mucosal surfaces (Figures 1R,S). CLE can present with a variety of cutaneous manifestations and is accordingly subdivided into three major categories: acute CLE (ACLE), subacute CLE (SCLE), and chronic CLE (CCLE). Discoid lupus (DLE), a subset of CCLE, and SCLE are the most common forms of cutaneous lupus. Skin lesions are often a cause of significant disability and may be associated with underlying multisystem involvement secondary to SLE activity (308, 309). There are several pathways involved in the pathomechanism of CLE. Excess production of type I interferons (IFNs) has been implicated in the pathogenesis of SLE (29224681). Both plasmacytoid dendritic cells (pDCs) and cytotoxic CD8+T cells are known modulators of type 1 IFNs and seem to be critical in disease progression (310). Additionally, type I IFNs induce JAK/STAT signaling which are commonly upregulated in lesional skin (311).
There are no standardized diagnostic criteria for CLE, though preliminary criteria have been developed for DLE (312). The diagnosis of CLE is largely based on clinical presentation, laboratory serologies, and histopathological findings (312). Hallmark cutaneous manifestations include malar erythema for ACLE, psoriasiform or annular lesions with central clearing for SCLE, and erythematous, scarring lesions for CCLE. Other clinical symptoms seen especially with DLE include scarring or non-scarring alopecia, scarring, and dyspigmentation. While these findings are suggestive, CLE is often misdiagnosed (313), especially as other autoimmune connective tissue diseases such as dermatomyositis (DM) (314). Diagnosis is supported with serologies demonstrating positive antinuclear antibody (ANA) or antibodies to double-stranded DNA (dsDNA), and anti-Smith (anti-Sm), but these are frequently absent. ANA is largely ubiquitous among rheumatologic conditions; only one-third of positive ANA serologies correspond with a diagnosis of LE (315). While a positive ANA is regarded as highly sensitive for SLE, there are numerous cases of ANA negative CLE with systemic findings that in the past would have been classified as SLE (316). Autoantibodies against dsDNA and Sm are more specific for SLE, though their median prevalence ranges from 30 to 70% (317, 318). Histopathological findings are used to aid in the diagnosis of CLE but are similarly not specific for CLE. Patterns such as interface dermatitis, dermal mucin deposition, and periadnexal lymphocytic infiltrates are present in both dermatomyositis and CLE. Even characteristic CLE findings on direct immunofluorescence (DIF), including granular immunoglobulin and complement deposition, are found in DM (314). Misdiagnosing CLE not only delays treatment resulting in more skin damage but prevents screening for potentially serious organ involvement.
Since the approval of hydroxychloroquine in 1955, the Food and Drug Administration (FDA) has included three additional therapies for SLE: belimumab, a B-lymphocyte stimulator inhibitor, Anifrolumab, an anti-IFNAR receptor antibody, and voclosporin, a calcineurin inhibitor (319, 320). Belimumab and voclosporin are specifically approved for lupus nephritis. Currently, hydroxychloroquine is the only FDA-approved for CLE (313). Despite this, antimalarials [hydroxychloroquine (HCQ), chloroquine, and quinacrine] and topical corticosteroids remain first-line for the treatment of CLE. Topical calcineurin inhibitors may be used as an alternative to corticosteroids for sensitive areas of the skin and long-term use (313). About 65% of patients with CLE respond to some variation of these therapies (321). In CLE refractory to antimalarials, methotrexate (MTX), and mycophenolate mofetil (MMF) are the most effective immunosuppressives, but they may not be tolerated (322, 323). There are several reports of Azathioprine treating CLE, though MTX and MMF are typically more effective (324). Dapsone may be considered in recalcitrant CLE as there is some evidence of its success (325). Retinoids have demonstrated success in CLE as well, though long-term use is required, which increases the risk of adverse effects (326). Lenalidomide, a thalidomide analog, has recently been used for patients with refractory CLE (327). It shares similar efficacy to thalidomide with an improved safety profile (328). Though these therapies effectively reduce disease burden in patients, off-label use makes them difficult to obtain. For example, patients must pay out of pocket for quinacrine, a drug that has shown efficacy in patients that do not respond to HCQ alone (313). As there are no curative therapies for CLE, the medications listed above are intended only to mitigate disease burden. Even when properly managed, damage that developed due to previous disease activity is notoriously difficult to resolve.
Although only approved for SLE, Anifrolumab demonstrated improvements in cutaneous disease and may benefit those who meet criteria SLE with cutaneous involvement. There are several clinical trials measuring improvement of cutaneous disease in CLE as a primary outcome. As with Anifrolumab, these novel therapies frequently target the type I interferon pathway, identified as a leading driver of cutaneous lesions. One such monoclonal antibody, BIIB059, causes internalization of the blood dendritic cell antigen 2 receptor on plasma dendritic cells (PDCs), subsequently inhibiting type I interferons and other pro-inflammatory modulators. A phase 2 trial testing this therapy met its primary outcome, which measured improvement of cutaneous LE compared to placebo (329) and a phase 3 trial will begin shortly. VIB7734, another monoclonal antibody that targets PDCs, showed efficacy in CLE in a phase 1 trial (330) and is of interest for future trials in CLE, although an ongoing phase 2 trial is in SLE. Janus Kinase (JAK) inhibitors have demonstrated improvement in a range of dermatological conditions and are actively being investigated for SLE (331, 332). Further studies that include patients with moderate to severe skin disease are necessary to elucidate their potential benefit in CLE. Iberdomide, a potent thalidomide analog, recently demonstrated an impressive reduction in cutaneous activity in patients with SLE. Improvement in SCLE and a trend to improvement with DLE activity was seen, although those with ACLE did not show improvement (333). The variable response based on CLE subtype may highlight the need for subgroup analysis in future trials. It is important to note that CLE is difficult to classify, especially early in disease, and 20% of CLE patients have more than one subtype of CLE. As clinical trials continue to focus on cutaneous disease as a primary endpoint, new, well-supported therapies may be identified for use in CLE.
Dermatomyositis (DM) is thought to result from environmental triggers such as UV exposure, medications, infections, or malignancies in genetically predisposed individuals. Regarding genetic predisposition, mostly associations with HLA have been reported (334). Several characteristic cutaneous findings may be seen, including but not limited to symmetric macular erythema of the elbows, knees, or dorsal hands (Gottron's sign), papules of the dorsal metacarpophalangeal or interphalangeal joints (Gottron's papules), periorbital violaceous erythema (heliotrope rash), periungual telangiectasias, and macular erythema of the upper back (Shawl sign) and V-area of the upper chest (V-sign) (Figure 1T) (335). DM can also affect several other organ systems, potentially involving the skeletal muscle, lungs, heart, and esophagus. Especially interstitial lung disease is prevalent in almost 60% of DM patients (336). Thus, DM can have a large impact on patients' quality of life which tends to correlate with the amount of skin disease activity (337).
The EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies (IIM) and their major subgroups, including DM, are the only validated DM classification criteria (338). Patients are scored based on age of symptom onset, presence of muscle weakness, skin manifestations, other clinical manifestations, laboratory testing, and muscle biopsy features and can be further subcategorized into a form of IIM, like DM, if their score meets the cut-off probability of 55% (338). Despite validated criteria and characteristic cutaneous features, many clinicians do not accurately diagnose DM patients as evidenced by a retrospective study that showed that 56% of DM patients referred to an academic medical center were incorrectly diagnosed, the majority of whom were labeled as lupus or undifferentiated connective tissue disease (339). Patients with no muscle involvement and at least 2 of 3 possible skin-related items (heliotrope rash, Gottron's papules, and Gottron's sign) can be classified by the EULAR/ACR criteria as having amyopathic DM (ADM), and a skin biopsy is encouraged in such patients (339). However, these criteria have limitations as at least one retrospective study showed that 26% of patients with confirmed ADM would not meet the EULAR/ACR classification criteria due to the specific cutaneous findings required (340). It is suggested that cancer-screening investigations should be undertaken once a diagnosis of DM is established due to the associated increased risk of malignancy (341). However, no evidence-based malignancy screening protocol for DM patients currently exists. Thorough characterization of the autoantibody response in DM patients is also important in this regard, as certain autoantibodies are associated with a high risk of cancer (342). In addition, a detailed characterization of the autoantibody response in DM also allows to differentiate organ involvement and prognosis (343). In summary, missing awareness, lack of definite diagnostic criteria and no evidence-based recommendation for cancer screening are diagnostic challenges in DM.
Systemic corticosteroids, with or without immunosuppressives, are the mainstay of DM treatment when muscle disease is confirmed and often allows patients to improve their muscle symptoms (344). Many patients unfortunately have persistent cutaneous disease despite aggressive topical and systemic therapy. A common therapeutic ladder for the treatment of cutaneous DM involves antimalarials like hydroxychloroquine or chloroquine with or without quinacrine followed by methotrexate or mycophenolate mofetil and then IVIG, currently the only FDA-approved therapeutic for DM (345). However, even these more commonly used options have several limitations as DM patients have increased risk of cutaneous reactions to hydroxychloroquine than lupus patients, while steroid-sparing agents can have many side effects which are intolerable to patients (346). Very few randomized controlled trials have been performed in DM. However, other therapies with some evidence to support their use include topical corticosteroids, topical tacrolimus (347), topical pimecrolimus (348), tofacitinib (349), dapsone (350), and thalidomide (351), among others. One challenge facing the development of new therapies for DM is that clinical trials often use primary outcome measures which heavily weigh muscle involvement, like the TIS score. In the recently completed phase 3 study of lenabasum, the primary outcome was not met despite improvement in skin disease activity, highlighting the need for careful consideration of outcomes (352).
Much work is currently ongoing to overcome the diagnostic and therapeutic challenges facing DM. A recent international project that developed skin-focused classification criteria for DM that is more inclusive than the EULAR/ACR criteria while still excluding disease mimickers like lupus is undergoing prospective validation (353). While no consensus guidelines exist for cancer screening in DM patients, The International Myositis Assessment and Clinical Studies Group has an ongoing effort to create evidence-based malignancy screening guidelines for IIM patients (354). There is hope for improved therapeutic options for DM patients based on ongoing clinical trials as well as promising proof-of-concept studies. Additional therapeutics with trials that are ongoing or have reported data include JAK inhibitors, anti-interferon beta, subcutaneous immunoglobulin, and KZR-616 (349, 355).
Systemic sclerosis (SSc) is an autoimmune disease belonging to the connective tissue/rheumatic diseases, characterized by a triad of vasculopathy, inflammation, and fibrosis. SSc is a rare disease with a prevalence of 40–200:1,000,000 inhabitants (35). Clinically, SSc is characterized by a wide heterogeneity, ranging from skin findings to severe organ damage including gastrointestinal dysfunction, interstitial lung disease, pulmonary arterial hypertension, cardiac inflammation, arrhythmias, neurological deficits, or end-stage renal failure. Skin findings can include oedema, scleroderma as well as acral ulcers, necrosis, or gangrene (Figures 1U,V). In recent years, the understanding of pathomechanisms in SSc and concomitantly, the therapeutic options for the treatment of affected patients have improved, especially regarding pulmonary artery hypertension and interstitial lung diseases. This has led to a reduction in disease-related mortality (356). Nevertheless, a multinational study examining mortality in patients with SSc between 2005 and 2014 continued to demonstrate early patient death (357). In addition, health-related quality of life is significantly lower in patients with SSc compared to healthy controls (358) and compared to other autoimmune diseases (359). The involvement of the gastrointestinal tract, pulmonary arterial hypertension, Raynaud's phenomenon and digital ulcers represent disease manifestations that affect the quality of life of patients (360). In addition, symptoms such as pain, dyspnea or impaired hand function are frequently reported by the patients as determinants for the quality of life. In addition, symptoms such as erectile dysfunction, pruritus, psychological problems such as anxiety received insufficient consideration in diagnostics and the development of treatment strategies for patients. Therefore, despite significant improvements in the understanding of the disease and expansion of therapeutic options, there is still a high unmet medical need in SSc.
Years before the development of disease-defining symptoms in SSc, a risk for disease development in the presence of Raynaud's phenomenon or puffy fingers can be predicted by determination of biomarkers such as antinuclear antibodies and changes in capillary microscopy (361). These changes are summarized in the concept of “early SSc.” Currently, patients at risk receive close clinical follow-ups. However, it is unclear whether and which early treatment would attenuate the course of the disease (362). Due to the heterogeneity of disease progression in SSc, the identification of biomarkers is central to predict the development and severity of organ manifestations, disease progression, and response to treatment. Although numerous biomarkers have been investigated in studies, only a few of these biomarkers have found their way into routine clinical practice (e.g., AT1R autoantibodies, ETAR autoantibodies) in specialized centers (363, 364). The identification of biomarkers for individual prediction of organ manifestations and severity of disease progression represents the basis for establishing the concept of individualized medicine in SSc.
To date, drug treatment of patients follows a manifestation-based approach according to the EULAR recommendations (365). However, sufficient, evidence-based treatment strategies are lacking for several disease manifestations of SSc. One obstacle in the development of appropriate treatment options is that the pathophysiological mechanisms leading to specific disease manifestations are poorly understood to date, leaving only symptomatic treatment options available. These include exemplarily treatment of contractures, calcinosis cutis, acral necrosis, gastrointestinal involvement, fatigue, arthritis, or enthesitis. The highest agreement on treatment recommendations for patients with SSc is to consider immunosuppressive therapies particularly for the early inflammatory phase of the disease including autologeous stem cell transplantation. The use of angiotensin-converting enzyme inhibitors is recommended for scleroderma renal crisis. Prostacyclins, endothelin receptor blockers, phosphodiesterase-V inhibitors, and stimulators of soluble guanylate cyclase (sGC) were shown to be effective in the therapy of the obliterative vasculopathy. Here, combination therapies are increasingly applied particularly for the therapy of pulmonary arterial hypertension (PAH). Recently, nintedanib, a small molecule tyrosine-kinase inhibitor was approved for the therapy of SSc-associated lung fibrosis. Of note, the anti-CD20 antibody rituximab has recently been demonstrated to have beneficial effects on skin and lung fibrosis and seems to be effective also in PAH. It is approved in Japan for the treatment of SSc (366, 367). Since these disease manifestations affect most patients, studies are urgently needed to decipher pathophysiology and develop causal therapeutic approaches. Furthermore, medication adherence is poorly investigated. Only a few clinical studies address compliance of SSc patients. Improving drug adherence could increase remission rates and prevent secondary disease complications. Moreover, since there are options for the therapy of cardiac arrhythmias, such as pacemakers or defibrillators, the establishment of a structured assessment for corresponding diagnosing is required. In addition to the insufficiently investigated treatment options for organ-related disease manifestations, the global disease activity cannot yet be adequately controlled with the approved therapeutic agents in all patients. Due to the described heterogeneity of organ manifestations, an interprofessional and interdisciplinary team is necessary to achieve optimal management of individual disease manifestations. A survey of patients with SSc conducted in the USA with regard to their individual unmet medical needs revealed deficits with regard to the psychological care of patients (368). In many places, there is a lack of structures that ensure interdisciplinary treatment of patients, which also includes psychological co-care of patients. Besides psychological co-care patients require physiotherapy and physical therapy to attenuate contracture development. Therefore, a prioritized objective for the next few years should be to create awareness of the need for interdisciplinary treatment and to establish appropriate structures on this basis. While diagnostics and therapy often focus on organ manifestations leading to the high disease-associated mortality, affected patients often evaluate pain, fatigue as well as alleviation of Raynaud's phenomenon and gastrointestinal symptoms as a treatment priority (360, 369). Therefore, practitioners need to define the individual treatment goal with the patient, considering not only global health but also health-related quality of life.
Despite the progress made in deciphering the pathogenesis of SSc in recent years, the triggers of the disease and the mechanisms that lead to the heterogeneous disease manifestations and disease severity remain poorly understood. However, since an understanding of these mechanisms is the basis for the identification of key molecules in pathogenesis and thus new therapeutic options, deciphering the disease-driving mechanisms of SSc is urgently needed. A key requirement for this is the enrollment of patients in international registries as well as a close collaboration between patients, clinicians, and scientists.
In conclusion, there are a multitude of challenges for the diagnosis and treatment of chronic skin inflammation. These are, however, different for each disease: At a generalized glance, for the common inflammatory skin diseases, especially psoriasis, atopic dermatitis and lichen planus, disease heterogenicity and the identification of biomarkers that allow to predict treatment responses are at the forefront of the medical needs. We assume that with the advent of more and more detailed molecular data from these patients, a stratification allowing personalized treatment options are on the horizon (63).
For the rare and orphan chronic skin inflammatory diseases, medical practitioners from all specialties need to be made aware of these diagnoses, for example pemphigus or dermatomyositis. Patient organizations, such as the International Pemphigus & Pemphigoid Foundation (IPPF), that educate patients and medical practitioners alike are key for this. This will then also lead to an earlier and more validated diagnosis, which are both essential to start the appropriate treatments. Regarding these, there is a high need to develop more selective, and potentially causal, treatments for chronic skin inflammation. The use of the chimeric antigen receptor (CAR)-T cell technology to selectively deplete autoreactive B cells in pre-clinical models of pemphigus is a milestone in reaching this goal (370).
These differences in unmet medical needs across the here discussed chronic, non-communicable inflammatory skin diseases, do, however, not allow to determine which of these diseases has the “most” or the “highest” unmet medical need. In a generalized manner, one could approach this open issue by a systematic and longitudinal assessment of patient reported outcomes across a wide range of chronic inflammatory skin diseases. This would allow to determine the burden of individual diseases at diagnosis, as well as at a time point where treatment should have had a positive impact on both objective and subjective disease symptoms.
Another challenge that is observed across almost all chronic (skin) inflammatory diseases is comorbidity. At the forefront of these are metabolic syndrome, (cardio)vascular and mental health diseases (348, 371–374). One hypothesis is that chronic skin inflammation drives the associated comorbidity (279, 375). By contrast, others provided evidence that the environment is a key driver for the observed comorbidity in chronic (skin) inflammation (376).
Thus, in perspective, we believe that we will observe significant changes how chronic skin inflammation is diagnosed and treated during the next years. Overall, this will improve the quality of life of patients. We also envision the emerge of curative treatments for those autoimmune skin diseases, where culprit cells can specifically be targeted, i.e., autoreactive B cells.
RL: conceptualization. KBi (lead), HU, DR, MS, SS, MMe, MMa, DT, ES, CC, KA, DD, MH, AR, HG, AH, AS, GR, GS, JD, DZ, TS, AC, KW, RS, KK, VW, and RL: visualization. All authors: writing–original draft and review and editing. All authors contributed to the article and approved the submitted version.
This work has been financially supported by the Cluster of Excellence Precision Medicine in Chronic Inflammation (EXC 2167) and the Collaborative Research Center Pathomechanisms of Antibody-mediated Autoimmunity (SFB 1526), the Research Units PruSearch (FOR 2690) and PEGASUS (FOR 2497), the Individual Research Grant RI/1056-11, all from the Deutsche Forschungsgemeinschaft; the Schleswig-Holstein Excellence-Chair Program from the State of Schleswig Holstein, an EADV research fellowship grant 2019, the Department of Veterans Affairs Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases) R01AR071653 (VPW) and R01AR076766 (VPW), and Sinergia Unravel principles of self-organization in injured tissue (CRSII5_202301/1) from the Swiss National Science Foundation.
HU has received research grants from JB, Otsuka, Taiho, Boehringer Ingelheim, Kyowa Kirin, Kaken, Sun Pharma, Shionogi, Teijin, Mitsubishi Tanabe, Nihon Zoki, Eisai, Torii and Tokiwa, consultant fees from Ono, Nihon-Pharmaceutical, Sun Pharma, argenx and Ishin Pharma, and speaker's fees from Nihon-Pharmaceutical, Maruho, Eli Lilly, Abbie, Eisai, Sanofi, Janssen, Kyowa Kirin, Ono, UCB, Novartis, Sun Pharma, Torii, Taiho, Mitsubishi Tanabe, and Boehringer Ingelheim during the last 3 years, DR has received honoraria or research support from AbbVie, Abcuro, AltruBio, Amgen, Boehringer-Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant, Dermira, Galdrema, Incyte, Janssen, Kyowa Kirin, Lilly, Merck, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharmaceuticals, UCB, and VielaBio during the last 3 years, MS has received consulting/speakers' fees, or grant support from AbbVie, Almirall, Amgen, Biogen, BMS, Janssen, Novartis, and UCB during the last 3 years, SS has received funding and personal fees from Celldex, Clexio, Dermasence, Galderma, GSK, Kiniksa, Menlo, Trevi, Novartis, Sanofi (investigator all), Abbvie, Almirall, Beiersdorf, Bellus Health, Benevolent, Bionorica, Cara, Celgene, CelloHealth, Clexio, DS Biopharma, Eli Lilly, Escient, Galderma, Grünenthal, Kiniksa, Klinge Pharma, Menlo, Sanofi, Sienna, Trevi, P.G. Unna Academy, Perrigo, Pfizer, Vanda, Vifor, WebMD (Consultancy/Advisory board) and Almirall, Eli Lilly, Sanofi, Galderma, Menlo, Omnicuris, Beiersdorf, Leo Pharma, Novartis, P. G. Unna Academy, Pfizer, Pierre Fabre (speaker) during the last 3 years, MMe has received honoraria as a speaker and/or consultant for Amgen, Aralez, argenx, Bayer, Beiersdorf, Celgene, Escient, Galderma, GSK, Menlo, Moxie, Novartis, Pharvaris, Pfizer, Roche, Sanofi, Siennabio, and Uriach, MMa was a speaker and/or advisor for and/or has received research funding from Allakos, Amgen, Aralez, ArgenX, AstraZeneca, Celldex, Centogene, CSL Behring, FAES, Genentech, GIInnovation, Innate Pharma, Kyowa Kirin, Leo Pharma, Lilly, Menarini, Moxie, Novartis, Roche, Sanofi/Regeneron, Third HarmonicBio, UCB, and Uriach during the last 3 years, is a consultant, advisory board member, and/or investigator for AbbVie, Almirall, Amgen, Beiersdorf, BMS, Boehringer Ingelheim, Eli Lilly, Galapagos, Janssen-Cilag, LEO Pharma, MorphoSys, Novartis, Pfizer, Regeneron Pharmaceuticals, Inc., Samsung, Sandoz, Sanofi, Sun Pharma, and UCB, ES has received research grants from UCB, Incyte, Biotest, ArgenX, Dompe, Fresenius Medical Care, Bayer, AstraZenca, and Euroimmun and honoraria from Biotest, Thermo Fisher, ArgenX, Fresenius Medical care, Topas, Leo, Chugai, AstraZenca, and Almirall during the last 3 years, MH has received honoraria from Novartis, Sanofi, Celgene, and unrestricted grants from Biotest, Janssen Cilag and Topas durimg the last 3 years, KBi has received research funds from ArgenX during the last 3 years, DZ has received support for research and development work, lecturing and consulting from Euroimmun AG, UCB Pharma, ArgenX, Biotest, Abbvie, Janssen, Sanofi in the last 3 years, KW has received research grants, travel grants, consulting honoraria or lecturer's honoraria from Amgen, Bristol Myers Squibb, Celgene, Charité Research Organization, Flexopharm, Janssen-Cilag, Novartis Pharma, Sanofi-Aventis, and Trial Form Support during the last 3 years, RS has received research grants or honoraria for participation in advisory boards, clinical trials, or as speaker for one or more of the following: AbbVie, Amgen, Bayer, Boehringer Ingelheim Pharma, Bristol Myers Squibb, Celgene, Charité Research Organization, CSL Behring, Dr. Willmar Schwabe, Flexopharm, Incyte, Janssen-Cilag, La Roche-Posay Laboratoire Dermatologique, Novartis Pharma, Parexel International, Sanofi–Aventis, TFS, and UCB Biopharma during the last 3 years, VW has received grants from Celgene, Amgen, Janssen, Biogen, Gilead, Viela; Horizon therapeutics, Pfizer, Corbus, CSL Behring, consulted Astra-Zeneca, Pfizer, Biogen, Celgene, Resolve, Janssen, Gilead, Lilly, BMS, Nektar, Abbvie, Viela, GSK, EMD Serona, Sanofi, Anaptysbio, Amgen, Merck, Pfizer, Janssen, Neovacs, Idera, Octapharma, CSL Behring, Corbus, Novartis, Romefor during the last 3 years, RL has received honoraria for speaking or consulting or has obtained research grants from Novartis, Lilly, Bayer, Dompe, Synthon, Argen-X, and Incyte during the last 3 years.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schmidt T, Solimani F, Pollmann R, Stein R, Schmidt A, Stulberg I, et al. TH1/TH17 cell recognition of desmoglein 3 and bullous pemphigoid antigen 180 in patients with lichen planus. J Allergy Clin Immunol. (2018) 142:669–72.e7. doi: 10.1016/j.jaci.2018.02.044
2. Arakawa A, Siewert K, Stöhr J, Besgen P, Kim SM, Rühl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. (2015) 212:2203–12. doi: 10.1084/jem.20151093
3. Christophers E, van de Kerkhof PCM. Severity, heterogeneity and systemic inflammation in psoriasis. J Eur Acad Dermatol Venereol. (2019) 33:643–7. doi: 10.1111/jdv.15339
4. Meijer JM, Lamberts A, Luijendijk HJ, Diercks GFH, Pas HH, Zuidema SU, et al. Prevalence of pemphigoid as a potentially unrecognized cause of pruritus in nursing home residents. JAMA Dermatol. (2019) 155:1423–4. doi: 10.1001/jamadermatol.2019.3308
5. Eyerich K, Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. (2018) 32:692–703. doi: 10.1111/jdv.14673
6. Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab. (2015) 66 (Suppl. 1):8–16. doi: 10.1159/000370220
7. Bylund S, Kobyletzki LB, Svalstedt M, Svensson Å. Prevalence and incidence of atopic dermatitis: a systematic review. Acta Derm Venereol. (2020) 100:adv00160. doi: 10.2340/00015555-3510
8. Deckers IA, McLean S, Linssen S, Mommers M, van Schayck CP, Sheikh A. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS ONE. (2012) 7:e39803. doi: 10.1371/journal.pone.0039803
9. Armstrong AW, Mehta MD, Schupp CW, Gondo GC, Bell SJ, Griffiths CEM. Psoriasis prevalence in adults in the United States. JAMA Dermatol. (2021) 157:940–6. doi: 10.1001/jamadermatol.2021.2007
10. Iskandar IYK, Parisi R, Griffiths CEM, Ashcroft DM, Global PA. Systematic review examining changes over time and variation in the incidence and prevalence of psoriasis by age and gender. Br J Dermatol. (2021) 184:243–58. doi: 10.1111/bjd.19169
11. Damiani G, Bragazzi NL, Aksut CK, Wu D, Alicandro G, McGonagle D, et al. The global, regional, and national burden of psoriasis: results and insights from the global burden of disease 2019 study. Front Med. (2021) 8:743180. doi: 10.3389/fmed.2021.743180
12. Ständer S, Ketz M, Kossack N, Akumo D, Pignot M, Gabriel S, et al. Epidemiology of prurigo nodularis compared with psoriasis in germany: a claims database analysis. Acta Derm Venereol. (2020) 100:adv00309. doi: 10.2340/00015555-3655
13. Ständer S, Augustin M, Berger T, Elmariah S, Korman NJ, Weisshaar E, et al. Prevalence of prurigo nodularis in the United States of America: a retrospective database analysis. JAAD Int. (2021) 2:28–30. doi: 10.1016/j.jdin.2020.10.009
14. Boch K, Langan EA, Kridin K, Zillikens D, Ludwig RJ, Bieber K. Lichen planus. Front Med. (2021) 8:737813. doi: 10.3389/fmed.2021.737813
15. Le Cleach L, Chosidow O. Clinical practice. Lichen planus. N Engl J Med. (2012) 366:723–32. doi: 10.1056/NEJMcp1103641
16. Schneider-Burrus S, Lux G, van der Linde K, Barbus S, Huss-Marp J, Tsaousi A, et al. Hidradenitis suppurativa - prevalence analyses of German statutory health insurance data. J Eur Acad Dermatol Venereol. (2021) 35:e32–5. doi: 10.1111/jdv.16783
17. Sachdeva M, Shah M, Alavi A. Race-Specific prevalence of hidradenitis suppurativa. J Cutan Med Surg. (2021) 25:177–7. doi: 10.1177/1203475420972348
18. Liang YT, Yeh CJ, Huang JY, Wei JC. Epidemiology of hidradenitis suppurativa in Taiwan: a 14-year nationwide population-based study. J Dermatol. (2021) 48:613–9. doi: 10.1111/1346-8138.15811
19. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/JCI138416
20. Zhang Y, Cai Y, Shi M, Jiang S, Cui S, Wu Y, et al. The prevalence of vitiligo: a meta-analysis. PLoS ONE. (2016) 11:e0163806. doi: 10.1371/journal.pone.0163806
21. Fricke J, Ávila G, Keller T, Weller K, Lau S, Maurer M, et al. Prevalence of chronic urticaria in children and adults across the globe: systematic review with meta-analysis. Allergy. (2020) 75:423–32. doi: 10.1111/all.14037
22. Kridin K, Schmidt E. Epidemiology of pemphigus. JID Innov. (2021) 1:100004. doi: 10.1016/j.xjidi.2021.100004
23. Hübner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–8. doi: 10.1016/j.jid.2016.07.013
24. Kridin K, Ludwig RJ. The growing incidence of bullous pemphigoid: overview and potential explanations. Front Med. (2018) 5:220. doi: 10.3389/fmed.2018.00220
25. Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. (2017) 66:6–17. doi: 10.1007/s12026-017-8975-2
26. Zumelzu C, Le Roux-Villet C, Loiseau P, Busson M, Heller M, Aucouturier F, et al. Black patients of African descent and HLA-DRB1*15:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol. (2011) 131:2386–93. doi: 10.1038/jid.2011.231
27. Cuisset L, Jeru I, Dumont B, Fabre A, Cochet E, Le Bozec J, et al. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: epidemiological study and lessons from eight years of genetic analysis in France. Ann Rheum Dis. (2011) 70:495–9. doi: 10.1136/ard.2010.138420
28. Mehr S, Allen R, Boros C, Adib N, Kakakios A, Turner PJ, et al. Cryopyrin-associated periodic syndrome in Australian children and adults: epidemiological, clinical and treatment characteristics. J Paediatr Child Health. (2016) 52:889–95. doi: 10.1111/jpc.13270
29. Simon A, Asli B, Braun-Falco M, De Koning H, Fermand JP, Grattan C, et al. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy. (2013) 68:562–8. doi: 10.1111/all.12129
30. van Leersum FS, Potjewijd J, van Geel M, Steijlen PM, Vreeburg M. Schnitzler's syndrome - a novel hypothesis of a shared pathophysiologic mechanism with Waldenström's disease. Orphanet J Rare Dis. (2019) 14:151. doi: 10.1186/s13023-019-1117-2
31. Jarrett P, Werth VP. A review of cutaneous lupus erythematosus: improving outcomes with a multidisciplinary approach. J Multidiscip Healthc. (2019) 12:419–28. doi: 10.2147/JMDH.S179623
32. Jarrett P, Thornley S, Scragg R. Ethnic differences in the epidemiology of cutaneous lupus erythematosus in New Zealand. Lupus. (2016) 25:1497–502. doi: 10.1177/0961203316651745
33. Kronzer VL, Kimbrough BA, Crowson CS, Davis JM, Holmqvist M, Ernste FC. Incidence, prevalence, and mortality of dermatomyositis: a population-based cohort study. Arthritis Care Res. (2021). doi: 10.1002/acr.24786. [Epub ahead of print].
34. Goonesekera S, Bansal A, Tadwalkar S, Isherwood A. The Prevalence of Systemic Sclerosis, Dermatomyositis/Polymyositis, Giant Cell Arteritis in the United States by Race Ethnicity: An Analysis Using Electronic Health Records. (2020). Available online at: https://acrabstracts.org/abstract/the-prevalence-of-systemic-sclerosis-dermatomyositis-polymyositis-and-giant-cell-arteritis-in-the-united-states-by-race-and-ethnicity-an-analysis-using-electronic-health-records/
35. Bairkdar M, Rossides M, Westerlind H, Hesselstrand R, Arkema EV, Holmqvist M. Incidence and prevalence of systemic sclerosis globally: a comprehensive systematic review and meta-analysis. Rheumatology. (2021) 60:3121–33. doi: 10.1093/rheumatology/keab190
36. Vakharia PP, Silverberg JI. Adult-Onset atopic dermatitis: characteristics and management. Am J Clin Dermatol. (2019) 20:771–9. doi: 10.1007/s40257-019-00453-7
37. Langenbruch A, Radtke M, Franzke N, Ring J, Foelster-Holst R, Augustin M. Quality of health care of atopic eczema in Germany: results of the national health care study atopichealth. J Eur Acad Dermatol Venereol. (2014) 28:719–26. doi: 10.1111/jdv.12154
38. Halvorsen JA, Lien L, Dalgard F, Bjertness E, Stern RS. Suicidal ideation, mental health problems, and social function in adolescents with eczema: a population-based study. J Invest Dermatol. (2014) 134:1847–54. doi: 10.1038/jid.2014.70
39. Wei W, Ghorayeb E, Andria M, Walker V, Schnitzer J, Kennedy M, et al. A real-world study evaluating adeQUacy of existing systemic treatments for patients with moderate-to-severe atopic dermatitis (QUEST-AD): baseline treatment patterns and unmet needs assessment. Ann Allergy Asthma Immunol. (2019) 123:381–8.e2. doi: 10.1016/j.anai.2019.07.008
40. Zink AGS, Arents B, Fink-Wagner A, Seitz IA, Mensing U, Wettemann N, et al. Out-of-pocket costs for individuals with atopic eczema: a cross-sectional study in nine European countries. Acta Derm Venereol. (2019) 99:263–7. doi: 10.2340/00015555-3102
41. David Boothe W, Tarbox JA, Tarbox MB. Atopic dermatitis: pathophysiology. Adv Exp Med Biol. (2017) 1027:21–37. doi: 10.1007/978-3-319-64804-0_3
42. Khan SJ, Dharmage SC, Matheson MC, Gurrin LC. Is the atopic march related to confounding by genetics and early-life environment? A systematic review of sibship and twin data. Allergy. (2018) 73:17–28. doi: 10.1111/all.13228
43. Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38:441–6. doi: 10.1038/ng1767
44. Kezic S, Kemperman PM, Koster ES, de Jongh CM, Thio HB, Campbell LE, et al. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J Invest Dermatol. (2008) 128:2117–9. doi: 10.1038/jid.2008.29
45. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. (2017) 9: eaah4680. doi: 10.1126/scitranslmed.aah4680
46. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. (2012) 22:850–9. doi: 10.1101/gr.131029.111
47. Cork MJ, Danby SG, Ogg GS. Atopic dermatitis epidemiology and unmet need in the United Kingdom. J Dermatolog Treat. (2020) 31:801–9. doi: 10.1080/09546634.2019.1655137
48. Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. (2006) 117:411–7. doi: 10.1016/j.jaci.2005.10.033
49. De D, Kanwar AJ, Handa S. Comparative efficacy of Hanifin and Rajka's criteria and the UK working party's diagnostic criteria in diagnosis of atopic dermatitis in a hospital setting in North India. J Eur Acad Dermatol Venereol. (2006) 20:853–9. doi: 10.1111/j.1468-3083.2006.01664.x
50. Garmhausen D, Hagemann T, Bieber T, Dimitriou I, Fimmers R, Diepgen T, et al. Characterization of different courses of atopic dermatitis in adolescent and adult patients. Allergy. (2013) 68:498–506. doi: 10.1111/all.12112
51. Chan TC, Sanyal RD, Pavel AB, Glickman J, Zheng X, Xu H, et al. Atopic dermatitis in Chinese patients shows TH2/TH17 skewing with psoriasiform features. J Allergy Clin Immunol. (2018) 142:1013–7. doi: 10.1016/j.jaci.2018.06.016
52. Brunner PM, Suárez-Fariñas M, He H, Malik K, Wen HC, Gonzalez J, et al. The atopic dermatitis blood signature is characterized by increases in inflammatory and cardiovascular risk proteins. Sci Rep. (2017) 7:8707. doi: 10.1038/s41598-017-09207-z
53. Silverberg JI. Association between adult atopic dermatitis, cardiovascular disease, and increased heart attacks in three population-based studies. Allergy. (2015) 70:1300–8. doi: 10.1111/all.12685
54. Arana A, Wentworth CE, Fernández-Vidaurre C, Schlienger RG, Conde E, Arellano FM. Incidence of cancer in the general population and in patients with or without atopic dermatitis in the U.K. Br J Dermatol. (2010) 163:1036–43. doi: 10.1111/j.1365-2133.2010.09887.x
55. Bieber T, D'Erme AM, Akdis CA, Traidl-Hoffmann C, Lauener R, Schäppi G, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: where are we, and where should we go. J Allergy Clin Immunol. (2017) 139:S58–64. doi: 10.1016/j.jaci.2017.01.008
56. Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. (2014) 71:116–32. doi: 10.1016/j.jaad.2014.03.023
57. Wollenberg A, Reitamo S, Girolomoni G, Lahfa M, Ruzicka T, Healy E, et al. Proactive treatment of atopic dermatitis in adults with 0.1% tacrolimus ointment. Allergy. (2008) 63:742–50. doi: 10.1111/j.1398-9995.2008.01683.x
58. Asgari MM, Tsai AL, Avalos L, Sokil M, Quesenberry CP. Association between topical calcineurin inhibitor use and keratinocyte carcinoma risk among adults with atopic dermatitis. JAMA Dermatol. (2020) 156:1066–73. doi: 10.1001/jamadermatol.2020.2240
59. Newsom M, Bashyam AM, Balogh EA, Feldman SR, Strowd LC. New and emerging systemic treatments for atopic dermatitis. Drugs. (2020) 80:1041–52. doi: 10.1007/s40265-020-01335-7
60. Puar N, Chovatiya R, Paller AS. New treatments in atopic dermatitis. Ann Allergy Asthma Immunol. (2021) 126:21–31. doi: 10.1016/j.anai.2020.08.016
61. Tseng JC, Chang YC, Huang CM, Hsu LC, Chuang TH. Therapeutic development based on the immunopathogenic mechanisms of psoriasis. Pharmaceutics. (2021) 13:1064. doi: 10.3390/pharmaceutics13071064
63. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker JNWN. Psoriasis. Lancet. (2021) 397:1301–15. doi: 10.1016/S0140-6736(20)32549-6
64. Ghoreschi K, Balato A, Enerbäck C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet. (2021) 397:754–66. doi: 10.1016/S0140-6736(21)00184-7
65. Schön MP, Erpenbeck L. The interleukin-23/Interleukin-17 axis links adaptive and innate immunity in psoriasis. Front Immunol. (2018) 9:1323. doi: 10.3389/fimmu.2018.01323
66. Schön MP. Adaptive and innate immunity in psoriasis and other inflammatory disorders. Front Immunol. (2019) 10:1764. doi: 10.3389/fimmu.2019.01764
67. Nast A, Altenburg A, Augustin M, Boehncke WH, Härle P, Klaus J, et al. German S3-guideline on the treatment of psoriasis vulgaris, adapted from EuroGuiDerm - part 1: treatment goals and treatment recommendations. J Dtsch Dermatol Ges. (2021) 19:934–150. doi: 10.1111/ddg.14508
68. Nast A, Altenburg A, Augustin M, Boehncke WH, Härle P, Klaus J, et al. German S3-Guideline on the treatment of Psoriasis vulgaris, adapted from EuroGuiDerm—part 2: treatment monitoring and specific clinical or comorbid situations. J Dtsch Dermatol Ges. (2021) 19:1092–115. doi: 10.1111/ddg.14507
69. Armstrong AW, Soliman AM, Betts KA, Wang Y, Gao Y, Puig L, et al. Comparative Efficacy and relative ranking of biologics and oral therapies for moderate-to-severe plaque psoriasis: a network meta-analysis. Dermatol Ther. (2021) 11:885–905. doi: 10.1007/s13555-021-00511-1
70. Ivanic MG, Ahn GS, Herndon P, Wu JJ. Update on biologics for psoriasis in clinical practice. Cutis. (2021) 108:15–8. doi: 10.12788/cutis.0317
71. Landeck L, Sabat R, Ghoreschi K, Man XY, Fuhrmeister K, Gonzalez-Martinez E, et al. Immunotherapy in psoriasis. Immunotherapy. (2021) 13:605–19. doi: 10.2217/imt-2020-0292
72. Sbidian E, Chaimani A, Garcia-Doval I, Doney L, Dressler C, Hua C, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. (2021) 4:CD011535. doi: 10.1002/14651858.CD011535.pub4
73. Bellinato F, Gisondi P, Girolomoni G. Latest advances for the treatment of chronic plaque psoriasis with biologics and oral small molecules. Biologics. (2021) 15:247–53. doi: 10.2147/BTT.S290309
74. Eisert L, Augustin M, Bach S, Dittmann M, Eiler R, Fölster-Holst R, et al. S2k guidelines for the treatment of psoriasis in children and adolescents - short version part 1. J Dtsch Dermatol Ges. (2019) 17:856–70. doi: 10.1111/ddg.13907
75. Eisert L, Augustin M, Bach S, Dittmann M, Eiler R, Fölster-Holst R, et al. S2k guidelines for the treatment of psoriasis in children and adolescents - short version part 2. J Dtsch Dermatol Ges. (2019) 17:959–73. doi: 10.1111/ddg.13936
76. Aghamajidi A, Raoufi E, Parsamanesh G, Jalili A, Salehi-Shadkami M, Mehrali M, et al. The attentive focus on T cell-mediated autoimmune pathogenesis of psoriasis, lichen planus and vitiligo. Scand J Immunol. (2021) 93:e13000. doi: 10.1111/sji.13000
77. Gudjonsson JE, Kabashima K, Eyerich K. Mechanisms of skin autoimmunity: cellular and soluble immune components of the skin. J Allergy Clin Immunol. (2020) 146:8–16. doi: 10.1016/j.jaci.2020.05.009
78. Arora S, Das P, Arora G. Systematic review and recommendations to combine newer therapies with conventional therapy in psoriatic disease. Front Med. (2021) 8:696597. doi: 10.3389/fmed.2021.696597
79. Honma M, Nozaki H. Molecular pathogenesis of psoriasis and biomarkers reflecting disease activity. J Clin Med. (2021) 10:3199. doi: 10.3390/jcm10153199
80. Magee C, Jethwa H, FitzGerald OM, Jadon DR. Biomarkers predictive of treatment response in psoriasis and psoriatic arthritis: a systematic review. Ther Adv Musculoskelet Dis. (2021) 13:1759720X211014010. doi: 10.1177/1759720X211014010
81. Freitas E, Blauvelt A, Torres T. Bimekizumab for the treatment of psoriasis. Drugs. (2021) 81:1751–1762. doi: 10.1007/s40265-021-01612-z
82. Lauffer F, Eyerich K, Boehncke WH, Asadullah K, Beissert S, Ghoreschi K, et al. Cytokines of the IL-17 family in psoriasis. J Dtsch Dermatol Ges. (2020) 18:675–81. doi: 10.1111/ddg.14124
83. Conti P, Pregliasco FE, Bellomo RG, Gallenga CE, Caraffa A, Kritas SK, et al. Mast cell cytokines IL-1, IL-33, and IL-36 mediate skin inflammation in psoriasis: a novel therapeutic approach with the anti-inflammatory cytokines IL-37, IL-38, and IL-1Ra. Int J Mol Sci. (2021) 22:8076. doi: 10.3390/ijms22158076
84. Iznardo H, Puig L. Exploring the role of IL-36 cytokines as a new target in psoriatic disease. Int J Mol Sci. (2021) 22:4344. doi: 10.3390/ijms22094344
85. Fragoulis GE, Brock J, Basu N, McInnes IB, Siebert S. The role for JAK inhibitors in the treatment of immune-mediated rheumatic and related conditions. J Allergy Clin Immunol. (2021) 148:941–952. doi: 10.1016/j.jaci.2021.08.010
86. Słuczanowska-Głabowska S, Ziegler-Krawczyk A, Szumilas K, Pawlik A. Role of janus kinase inhibitors in therapy of psoriasis. J Clin Med. (2021) 10:4307. doi: 10.3390/jcm10194307
87. Das D, Akhtar S, Kurra S, Gupta S, Sharma A. Emerging role of immune cell network in autoimmune skin disorders: an update on pemphigus, vitiligo and psoriasis. Cytokine Growth Factor Rev. (2019) 45:35–44. doi: 10.1016/j.cytogfr.2019.01.001
88. Zeidler C, Pereira MP, Ständer S. Chronic prurigo: similar clinical profile and burden across clinical phenotypes. Front Med. (2021) 8:649332. doi: 10.3389/fmed.2021.649332
89. Pereira MP, Steinke S, Zeidler C, Forner C, Riepe C, Augustin M, et al. European academy of dermatology and venereology European prurigo project: expert consensus on the definition, classification and terminology of chronic prurigo. J Eur Acad Dermatol Venereol. (2018) 32:1059–65. doi: 10.1111/jdv.14570
90. Boozalis E, Tang O, Patel S, Semenov YR, Pereira MP, Stander S, et al. Ethnic differences and comorbidities of 909 prurigo nodularis patients. J Am Acad Dermatol. (2018) 79:714–9.e3. doi: 10.1016/j.jaad.2018.04.047
91. Ryczek A, Reich A. Prevalence of prurigo nodularis in Poland. Acta Derm Venereol. (2020) 100:adv00155. doi: 10.2340/00015555-3518
92. Augustin M, Garbe C, Hagenström K, Petersen J, Pereira MP, Ständer S. Prevalence, incidence and presence of comorbidities in patients with prurigo and pruritus in Germany: a population-based claims data analysis. J Eur Acad Dermatol Venereol. (2021) 35:2270–6. doi: 10.1111/jdv.17485
93. Williams KA, Roh YS, Brown I, Sutaria N, Bakhshi P, Choi J, et al. Pathophysiology, diagnosis, and pharmacological treatment of prurigo nodularis. Expert Rev Clin Pharmacol. (2021) 14:67–77. doi: 10.1080/17512433.2021.1852080
94. Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. (2014) 133:448–60. doi: 10.1016/j.jaci.2013.10.048
95. Mishra SK, Wheeler JJ, Pitake S, Ding H, Jiang C, Fukuyama T, et al. Periostin activation of integrin receptors on sensory neurons induces allergic itch. Cell Rep. (2020) 31:107472. doi: 10.1016/j.celrep.2020.03.036
96. Pogatzki-Zahn EM, Pereira MP, Cremer A, Zeidler C, Dreyer T, Riepe C, et al. Peripheral sensitization and loss of descending inhibition is a hallmark of chronic pruritus. J Invest Dermatol. (2020) 140:203–211.e4. doi: 10.1016/j.jid.2019.05.029
97. Pereira MP, Pogatzki-Zahn E, Snels C, Vu TH, Üçeyler N, Loser K, et al. There is no functional small-fibre neuropathy in prurigo nodularis despite neuroanatomical alterations. Exp Dermatol. (2017) 26:969–71. doi: 10.1111/exd.13343
98. Ständer S, Pereira MP, Berger T, Zeidler C, Augustin M, Bobko S, et al. IFSI-guideline on chronic prurigo including prurigo nodularis. Itch. (2020) 5:e42. doi: 10.1097/itx.0000000000000042
99. Zeidler C, Pereira MP, Augustin M, Spellman M, Ständer S. Investigator's global assessment of chronic prurigo: a new instrument for use in clinical trials. Acta Derm Venereol. (2021) 101:adv00401. doi: 10.2340/00015555-3701
100. Pölking J, Zeidler C, Schedel F, Osada N, Augustin M, Metze D, et al. Prurigo activity score (PAS): validity and reliability of a new instrument to monitor chronic prurigo. J Eur Acad Dermatol Venereol. (2018) 32:1754–60. doi: 10.1111/jdv.15040
101. Kimel M, Zeidler C, Kwon P, Revicki D, Ständer S. Validation of psychometric properties of the itch numeric rating scale for pruritus associated with prurigo nodularis: a secondary analysis of a randomized clinical trial. JAMA Dermatol. (2020) 156:1354–8. doi: 10.1001/jamadermatol.2020.3071
102. Zeidler C, Steinke S, Riepe C, Bruland P, Soto-Rey I, Storck M, et al. Cross-European validation of the ItchyQoL in pruritic dermatoses. J Eur Acad Dermatol Venereol. (2019) 33:391–7. doi: 10.1111/jdv.15225
103. Pereira MP, Zeidler C, Wallengren J, Halvorsen JA, Weisshaar E, Garcovich S, et al. Chronic nodular prurigo: a european cross-sectional study of patient perspectives on therapeutic goals and satisfaction. Acta Derm Venereol. (2021) 101:adv00403. doi: 10.2340/00015555-3726
104. Müller S, Bieber T, Ständer S. Therapeutic potential of biologics in prurigo nodularis. Expert Opin Biol Ther. (2021) 22:47–58. doi: 10.1080/14712598.2021.1958777
105. Kovács B, Rose E, Kuznik N, Shimanovich I, Zillikens D, Ludwig RJ, et al. Dupilumab for treatment-refractory prurigo nodularis. J Dtsch Dermatol Ges. (2020) 18:618–24. doi: 10.1111/ddg.14107
106. Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. (1991) 25:593–619. doi: 10.1016/0190-9622(91)70241-S
107. Solimani F, Forchhammer S, Schloegl A, Ghoreschi K, Meier K. Lichen planus - a clinical guide. J Dtsch Dermatol Ges. (2021) 19:864–82. doi: 10.1111/ddg.14565
108. Hübner F, Langan EA, Recke A. Lichen planus pemphigoides: from lichenoid inflammation to autoantibody-mediated blistering. Front Immunol. (2019) 10:1389. doi: 10.3389/fimmu.2019.01389
109. Solimani F, Pollmann R, Schmidt T, Schmidt A, Zheng X, Savai R, et al. Therapeutic targeting of Th17/Tc17 cells leads to clinical improvement of lichen planus. Front Immunol. (2019) 10:1808. doi: 10.3389/fimmu.2019.01808
110. Didona D, Di Zenzo G. Humoral epitope spreading in autoimmune bullous diseases. Front Immunol. (2018) 9:779. doi: 10.3389/fimmu.2018.00779
111. Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. (2011) 65:175–83. doi: 10.1016/j.jaad.2010.03.029
112. Crowson AN, Magro CM, Mihm MC. Interface dermatitis. Arch Pathol Lab Med. (2008) 132:652–66. doi: 10.5858/2008-132-652-ID
113. Didona D, Fania L, Didona B, Eming R, Hertl M, Di Zenzo G. Paraneoplastic dermatoses: a brief general review and an extensive analysis of paraneoplastic pemphigus and paraneoplastic dermatomyositis. Int J Mol Sci. (2020) 21:E2178. doi: 10.3390/ijms21062178
114. Wu M, Lee G, Fischer G. Forming diagnostic criteria for vulvar lichen planus. Austral J Dermatol. (2020) 61:324–9. doi: 10.1111/ajd.13350
115. Ioannides D, Vakirlis E, Kemeny L, Marinovic B, Massone C, Murphy R, et al. European S1 guidelines on the management of lichen planus: a cooperation of the european dermatology forum with the european academy of dermatology and venereology. J Eur Acad Dermatol Venereol. (2020) 34:1403–14. doi: 10.1111/jdv.16464
116. Cosgarea R, Pollmann R, Sharif J, Schmidt T, Stein R, Bodea A, et al. Photodynamic therapy in oral lichen planus: a prospective case-controlled pilot study. Sci Rep. (2020) 10:1667. doi: 10.1038/s41598-020-58548-9
117. Damsky W, Wang A, Olamiju B, Peterson D, Galan A, King B. Treatment of severe lichen planus with the JAK inhibitor tofacitinib. J Allergy Clin Immunol. (2020) 145:1708–10.e2. doi: 10.1016/j.jaci.2020.01.031
118. Manousaridis I, Manousaridis K, Peitsch WK, Schneider SW. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. (2013) 11:981–91. doi: 10.1111/ddg.12141
119. Sabat R, Jemec GBE, Matusiak Ł, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primers. (2020) 6:18. doi: 10.1038/s41572-020-0149-1
120. Kokolakis G, Wolk K, Schneider-Burrus S, Kalus S, Barbus S, Gomis-Kleindienst S, et al. Delayed diagnosis of hidradenitis suppurativa and its effect on patients and healthcare system. Dermatology. (2020) 236:421–30. doi: 10.1159/000508787
121. Sabat R, Chanwangpong A, Schneider-Burrus S, Metternich D, Kokolakis G, Kurek A, et al. Increased prevalence of metabolic syndrome in patients with acne inversa. PLoS ONE. (2012) 7:e31810. doi: 10.1371/journal.pone.0031810
122. Kurek A, Johanne Peters EM, Sabat R, Sterry W, Schneider-Burrus S. Depression is a frequent co-morbidity in patients with acne inversa. J Dtsch Dermatol Ges. (2013) 11:743–9. doi: 10.1111/ddg.12067
123. Shavit E, Dreiher J, Freud T, Halevy S, Vinker S, Cohen AD. Psychiatric comorbidities in 3207 patients with hidradenitis suppurativa. J Eur Acad Dermatol Venereol. (2015) 29:371–6. doi: 10.1111/jdv.12567
124. Schneider-Burrus S, Witte-Haendel E, Christou D, Rigoni B, Sabat R, Diederichs G. High prevalence of back pain and axial spondyloarthropathy in patients with hidradenitis suppurativa. Dermatology. (2016) 232:606–12. doi: 10.1159/000448838
125. van der Zee HH, van der Woude CJ, Florencia EF, Prens EP. Hidradenitis suppurativa and inflammatory bowel disease: are they associated? Results of a pilot study. Br J Dermatol. (2010) 162:195–7. doi: 10.1111/j.1365-2133.2009.09430.x
126. Matusiak L, Bieniek A, Szepietowski JC. Psychophysical aspects of hidradenitis suppurativa. Acta Derm Venereol. (2010) 90:264–8. doi: 10.2340/00015555-0866
127. Schneider-Burrus S, Jost A, Peters EMJ, Witte-Haendel E, Sterry W, Sabat R. Association of hidradenitis suppurativa with body image. JAMA Dermatol. (2018) 154:447–51. doi: 10.1001/jamadermatol.2017.6058
128. Thorlacius L, Cohen AD, Gislason GH, Jemec GBE, Egeberg A. Increased suicide risk in patients with hidradenitis suppurativa. J Invest Dermatol. (2018) 138:52–7. doi: 10.1016/j.jid.2017.09.008
129. Gupta MA, Vujcic B, Sheridan AD, Gupta AK. Reduced risk of suicidal behaviours associated with the treatment of hidradenitis suppurativa with tumour necrosis factor alpha antagonists: results from the US FDA adverse events reporting system pharmacovigilance database. J Eur Acad Dermatol Venereol. (2020) 34:1564–8. doi: 10.1111/jdv.16224
130. Tiri H, Jokelainen J, Timonen M, Tasanen K, Huilaja L. Substantially reduced life expectancy in patients with hidradenitis suppurativa: a finnish nationwide registry study. Br J Dermatol. (2019) 180:1543–4. doi: 10.1111/bjd.17578
131. Zouboulis CC, Tzellos T, Kyrgidis A, Jemec GBE, Bechara FG, Giamarellos-Bourboulis EJ, et al. Development and validation of the international hidradenitis suppurativa severity score system (IHS4), a novel dynamic scoring system to assess HS severity. Br J Dermatol. (2017) 177:1401–9. doi: 10.1111/bjd.15748
132. Schneider-Burrus S, Tsaousi A, Barbus S, Huss-Marp J, Witte K, Wolk K, et al. Features associated with quality of life impairment in hidradenitis suppurativa patients. Front Med. (2021) 8:676241. doi: 10.3389/fmed.2021.676241
133. Kursawe Larsen C, Kjaersgaard Andersen R, Kirby JS, Tan J, Saunte DML, Jemec GBE. Convergent validity of suffering and quality of life as measured by the hidradenitis suppurativa quality of life. J Eur Acad Dermatol Venereol. (2021) 35:1577–81. doi: 10.1111/jdv.17148
134. Fabbrocini G, Ruina G, Giovanardi G, Dini V, Raone B, Venturini M, et al. Hidradenitis suppurativa in a large cohort of italian patients: evaluation of the burden of disease. Dermatology. (2021) 238:487–97. doi: 10.1159/000517412
135. Matusiak Ł, Bieniek A, Szepietowski JC. Soluble interleukin-2 receptor serum level is a useful marker of hidradenitis suppurativa clinical staging. Biomarkers. (2009) 14:432–7. doi: 10.1080/13547500903075218
136. Tsaousi A, Witte E, Witte K, Röwert-Huber HJ, Volk HD, Sterry W, et al. MMP8 is increased in lesions and blood of acne inversa patients: a potential link to skin destruction and metabolic alterations. Mediators Inflamm. (2016) 2016:4097574. doi: 10.1155/2016/4097574
137. Wolk K, Wenzel J, Tsaousi A, Witte-Händel E, Babel N, Zelenak C, et al. Lipocalin-2 is expressed by activated granulocytes and keratinocytes in affected skin and reflects disease activity in acne inversa/hidradenitis suppurativa. Br J Dermatol. (2017) 177:1385–93. doi: 10.1111/bjd.15424
138. Wolk K, Join-Lambert O, Sabat R. Aetiology and pathogenesis of hidradenitis suppurativa. Br J Dermatol. (2020) 183:999–1010. doi: 10.1111/bjd.19556
139. Wolk K, Warszawska K, Hoeflich C, Witte E, Schneider-Burrus S, Witte K, et al. Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol. (2011) 186:1228–39. doi: 10.4049/jimmunol.0903907
140. Witte-Händel E, Wolk K, Tsaousi A, Irmer ML, Mößner R, Shomroni O, et al. The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J Invest Dermatol. (2019) 139:1294–305. doi: 10.1016/j.jid.2018.11.018
141. Penno CA, Jäger P, Laguerre C, Hasler F, Hofmann A, Gass SK, et al. Lipidomics profiling of hidradenitis suppurativa skin lesions reveals lipoxygenase pathway dysregulation and accumulation of proinflammatory leukotriene B4. J Invest Dermatol. (2020) 140:2421–32.e10. doi: 10.1016/j.jid.2020.04.011
142. Wolk K, Brembach TC, Šimaite D, Bartnik E, Cucinotta S, Pokrywka A, et al. Activity and components of the granulocyte colony-stimulating factor pathway in hidradenitis suppurativa. Br J Dermatol. (2021) 185:164–76. doi: 10.1111/bjd.19795
143. Spano F, Donovan JC. Alopecia areata: part 1: pathogenesis, diagnosis, and prognosis. Can Fam Phys. (2015) 61:751–5.
144. MacDonald Hull SP, Wood ML, Hutchinson PE, Sladden M, Messenger AG, British AOD. Guidelines for the management of alopecia areata. Br J Dermatol. (2003) 149:692–9. doi: 10.1046/j.1365-2133.2003.05535.x
145. Chelidze K, Lipner SR. Nail changes in alopecia areata: an update and review. Int J Dermatol. (2018) 57:776–83. doi: 10.1111/ijd.13866
146. Rossi A, Muscianese M, Piraccini BM, Starace M, Carlesimo M, Mandel VD, et al. Italian guidelines in diagnosis and treatment of alopecia areata. G Ital Dermatol Venereol. (2019) 154:609–23. doi: 10.23736/S0392-0488.19.06458-7
148. Dy LC, Whiting DA. Histopathology of alopecia areata, acute and chronic: why is it important to the clinician. Dermatol Ther. (2011) 24:369–74. doi: 10.1111/j.1529-8019.2011.01414.x
149. Kumaresan M. Intralesional steroids for alopecia areata. Int J Trichol. (2010) 2:63–5. doi: 10.4103/0974-7753.66920
150. Kar BR, Handa S, Dogra S, Kumar B. Placebo-controlled oral pulse prednisolone therapy in alopecia areata. J Am Acad Dermatol. (2005) 52:287–90. doi: 10.1016/j.jaad.2004.10.873
151. Sharma VK. Pulsed administration of corticosteroids in the treatment of alopecia areata. Int J Dermatol. (1996) 35:133–6. doi: 10.1111/j.1365-4362.1996.tb03281.x
152. Efentaki P, Altenburg A, Haerting J, Zouboulis CC. Medium-dose prednisolone pulse therapy in alopecia areata. Dermatoendocrinology. (2009) 1:310–3. doi: 10.4161/derm.1.6.11236
153. Winter RJ, Kern F, Blizzard RM. Prednisone therapy for alopecia areata. A follow-up report. Arch Dermatol. (1976) 112:1549–52. doi: 10.1001/archderm.1976.01630350025006
154. van der Steen PH, van Baar HM, Perret CM, Happle R. Treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. (1991) 24:253–7. doi: 10.1016/0190-9622(91)70037-3
155. Rokhsar CK, Shupack JL, Vafai JJ, Washenik K. Efficacy of topical sensitizers in the treatment of alopecia areata. J Am Acad Dermatol. (1998) 39:751–61. doi: 10.1016/S0190-9622(98)70048-9
156. Tosti A, Guerra L, Bardazzi F. Contact urticaria during topical immunotherapy. Contact Dermatitis. (1989) 21:196–7. doi: 10.1111/j.1600-0536.1989.tb04737.x
157. Henderson CA, Ilchyshyn A. Vitiligo complicating diphencyprone sensitization therapy for alopecia universalis. Br J Dermatol. (1995) 133:491–7. doi: 10.1111/j.1365-2133.1995.tb02692.x
158. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
159. Jabbari A, Sansaricq F, Cerise J, Chen JC, Bitterman A, Ulerio G, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. (2018) 138:1539–45. doi: 10.1016/j.jid.2018.01.032
160. Mackay-Wiggan J, Jabbari A, Nguyen N, Cerise JE, Clark C, Ulerio G, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight. (2016) 1:e89790. doi: 10.1172/jci.insight.89790
161. Boyce EG, Vyas D, Rogan EL, Valle-Oseguera CS, O'Dell KM. Impact of tofacitinib on patient outcomes in rheumatoid arthritis - review of clinical studies. Pat Relat Outcome Meas. (2016) 7:1–12. doi: 10.2147/PROM.S62879
162. King B, Guttman-Yassky E, Peeva E, Banerjee A, Sinclair R, Pavel AB, et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J Am Acad Dermatol. (2021) 85:379–87. doi: 10.1016/j.jaad.2021.03.050
163. King B, Ko J, Forman S, Ohyama M, Mesinkovska N, Yu G, et al. Efficacy and safety of the oral janus kinase inhibitor baricitinib in the treatment of adults with alopecia areata: phase 2 results from a randomized controlled study. J Am Acad Dermatol. (2021) 85:847–53. doi: 10.1016/j.jaad.2021.05.050
164. Rodriguez TA, Fernandes KE, Dresser KL, Duvic M, National AAR. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. (2010) 62:525–7. doi: 10.1016/j.jaad.2009.02.006
165. Picardo M, Dell'Anna ML, Ezzedine K, Hamzavi I, Harris JE, Parsad D, et al. Vitiligo. Nat Rev Dis Primers. (2015) 1:15011. doi: 10.1038/nrdp.2015.11
166. Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. (2015) 386:74–84. doi: 10.1016/S0140-6736(14)60763-7
167. Karagaiah P, Valle Y, Sigova J, Zerbinati N, Vojvodic P, Parsad D, et al. Emerging drugs for the treatment of vitiligo. Expert Opin Emerg Drugs. (2020) 25:7–24. doi: 10.1080/14728214.2020.1712358
168. Eleftheriadou V. Reliability and validity of the vitiligo signs of activity score. Br J Dermatol. (2020) 183:801–2. doi: 10.1111/bjd.19164
170. Taieb A, Alomar A, Böhm M, Dell'anna ML, De Pase A, Eleftheriadou V, et al. Guidelines for the management of vitiligo: the European dermatology forum consensus. Br J Dermatol. (2013) 168:5–19. doi: 10.1111/j.1365-2133.2012.11197.x
171. Bae JM, Jung HM, Hong BY, Lee JH, Choi WJ, Lee JH, et al. Phototherapy for vitiligo: a systematic review and meta-analysis. JAMA Dermatol. (2017) 153:666–74. doi: 10.1001/jamadermatol.2017.0002
172. Kubelis-López DE, Zapata-Salazar NA, Said-Fernández SL, Sánchez-Domínguez CN, Salinas-Santander MA, Martínez-Rodríguez HG, et al. Updates and new medical treatments for vitiligo (review). Exp Ther Med. (2021) 22:797. doi: 10.3892/etm.2021.10229
173. Dahir AM, Thomsen SF. Comorbidities in vitiligo: comprehensive review. Int J Dermatol. (2018) 57:1157–64. doi: 10.1111/ijd.14055
174. Craiglow BG, King BA. Tofacitinib citrate for the treatment of vitiligo: a pathogenesis-directed therapy. JAMA Dermatol. (2015) 151:1110–2. doi: 10.1001/jamadermatol.2015.1520
175. Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the janus kinase inhibitor tofacitinib may require concomitant light exposure. J Am Acad Dermatol. (2017) 77:675–82.e1. doi: 10.1016/j.jaad.2017.05.043
176. Rothstein B, Joshipura D, Saraiya A, Abdat R, Ashkar H, Turkowski Y, et al. Treatment of vitiligo with the topical janus kinase inhibitor ruxolitinib. J Am Acad Dermatol. (2017) 76:1054–60.e1. doi: 10.1016/j.jaad.2017.02.049
177. Rosmarin D, Pandya AG, Lebwohl M, Grimes P, Hamzavi I, Gottlieb AB, et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet. (2020) 396:110–20. doi: 10.1016/S0140-6736(20)30609-7
178. Zuberbier T, Abdul Latiff AH, Abuzakouk M, Aquilina S, Asero R, Baker D, et al. The international EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy. (2021) 77:734–66. doi: 10.1111/all.15090
179. Sánchez-Borges M, Ansotegui IJ, Baiardini I, Bernstein J, Canonica GW, Ebisawa M, et al. The challenges of chronic urticaria part 1: epidemiology, immunopathogenesis, comorbidities, quality of life, and management. World Allergy Organ J. (2021) 14:100533. doi: 10.1016/j.waojou.2021.100533
180. Metz M, Altrichter S, Buttgereit T, Fluhr JW, Fok JS, Hawro T, et al. The diagnostic workup in chronic spontaneous urticaria-what to test and why. J Allergy Clin Immunol Pract. (2021) 9:2274–83. doi: 10.1016/j.jaip.2021.03.049
181. Guillén-Aguinaga S, Jáuregui Presa I, Aguinaga-Ontoso E, Guillén-Grima F, Ferrer M. Updosing nonsedating antihistamines in patients with chronic spontaneous urticaria: a systematic review and meta-analysis. Br J Dermatol. (2016) 175:1153–65. doi: 10.1111/bjd.14768
182. Maurer M, Rosen K, Hsieh HJ, Saini S, Grattan C, Gimenez-Arnau A, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N Engl J Med. (2013) 368:924–35. doi: 10.1056/NEJMoa1215372
183. Deza G, March-Rodríguez A, Sánchez S, Ribas-Llaurad, ó C., Soto D, Pujol RM, et al. Relevance of the basophil high-affinity ige receptor in chronic urticaria: clinical experience from a tertiary care institution. J Allergy Clin Immunol Pract. (2019) 7:1619–26.e1. doi: 10.1016/j.jaip.2019.01.026
184. Kolkhir P, Elieh-Ali-Komi D, Metz M, Siebenhaar F, Maurer M. Understanding human mast cells: lesson from therapies for allergic and non-allergic diseases. Nat Rev Immunol. (2022) 22:294–308. doi: 10.1038/s41577-021-00622-y
185. Metz M, Maurer M. Use of biologics in chronic spontaneous urticaria - beyond omalizumab therapy. Allergol Select. (2021) 5:89–95. doi: 10.5414/ALX02204E
186. Johal KJ, Saini SS. Current and emerging treatments for chronic spontaneous urticaria. Ann Allergy Asthma Immunol. (2020) 125:380–7. doi: 10.1016/j.anai.2019.08.465
187. Maurer M, Giménez-Arnau AM, Sussman G, Metz M, Baker DR, Bauer A, et al. Ligelizumab for chronic spontaneous urticaria. N Engl J Med. (2019) 381:1321–32. doi: 10.1056/NEJMoa1900408
188. Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. (2016) 38:57–74. doi: 10.1007/s00281-015-0541-1
189. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. (2017) 3:17026. doi: 10.1038/nrdp.2017.26
190. Schmidt E, Kasperkiewicz M, Joly J. Pemphigus. Lancet. (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
191. Daltaban Ö, Özçentik A, Akman Karakaş A, Üstün K, Hatipoglu Uzun MS. Clinical presentation and diagnostic delay in pemphigus vulgaris: a prospective study from Turkey. J Oral Pathol Med. (2020) 49:681–6. doi: 10.1111/jop.13052
192. Kasperkiewicz M, Lai O, Kim G, DeClerck B, Woodley DT, Zillikens D, et al. Immunoglobulin and complement immunohistochemistry on paraffin sections in autoimmune bullous diseases: a systematic review and meta-analysis. Am J Dermatopathol. (2021) 43:689–99. doi: 10.1097/DAD.0000000000001817
193. Petruzzi M, Lucchese A, Contaldo M, Tampoia M, Frassanito MA, Lauritano D, et al. ELISA detection of anti-desmoglein 1 and anti-desmoglein 3 and indirect immunofluorescence in oral pemphigus: a retrospective study. Oral Dis. (2021) 28:1149–56. doi: 10.1111/odi.13849
194. Amber KT, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
195. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
196. Werth VP, Joly P, Chen DM. Rituximab versus mycophenolate mofetil in pemphigus vulgaris. Reply. N Engl J Med. (2021) 385:1056. doi: 10.1056/NEJMc2111763
197. Joly P, Horwath B, Patsatsi, Uzun S, Bech R, Beissert S, et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol. (2020) 34:1900–13. doi: 10.1111/jdv.16752
198. De D, Bishnoi A, Handa S, Mahapatra T, Mahajan R. Effectiveness and safety analysis of rituximab in 146 Indian pemphigus patients: a retrospective single-center review of up to 68 months follow-up. Indian J Dermatol Venereol Leprol. (2020) 86:39–44. doi: 10.4103/ijdvl.IJDVL_848_17
199. Rashid H, Lamberts A, van Maanen D, Bolling MC, Diercks GFH, Pas HH, et al. The effectiveness of rituximab in pemphigus and the benefit of additional maintenance infusions: daily practice data from a retrospective study. J Am Acad Dermatol. (2020) 83:1503–5. doi: 10.1016/j.jaad.2020.06.024
200. Kushner CJ, Wang S, Tovanabutra N, Tsai DE, Werth VP, Payne AS. Factors associated with complete remission after rituximab therapy for pemphigus. JAMA Dermatol. (2019) 155:1404–9. doi: 10.1001/jamadermatol.2019.3236
201. Grando SA. Retrospective analysis of a single-center clinical experience toward development of curative treatment of 123 pemphigus patients with a long-term follow-up: efficacy and safety of the multidrug protocol combining intravenous immunoglobulin with the cytotoxic immunosuppressor and mitochondrion-protecting drugs. Int J Dermatol. (2019) 58:114–25. doi: 10.1111/ijd.14143
202. Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest. (2013) 123:800–11. doi: 10.1172/JCI60139
203. Burmester IAK, Flaswinkel S, Thies CS, Kasprick A, Kamaguchi M, Bumiller-Bini V, et al. Identification of novel therapeutic targets for blocking acantholysis in pemphigus. Br J Pharmacol. (2020) 177:5114–30. doi: 10.1111/bph.15233
204. Amber KT, Valdebran M, Grando SA. Non-Desmoglein antibodies in patients with pemphigus vulgaris. Front Immunol. (2018) 9:1190. doi: 10.3389/fimmu.2018.01190
205. Schmitt T, Egu DT, Walter E, Sigmund AM, Eichkorn R, Yazdi A, et al. Ca2+ signalling is critical for autoantibody-induced blistering of human epidermis in pemphigus. Br J Dermatol. (2021) 185:595–604. doi: 10.1111/bjd.20091
206. Schmitt T, Waschke J. Autoantibody-Specific signalling in pemphigus. Front Med. (2021) 8:701809. doi: 10.3389/fmed.2021.701809
207. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
208. Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris–incidence and mortality in the UK: population based cohort study. BMJ. (2008) 337:a180. doi: 10.1136/bmj.a180
209. Ständer S, Kasperkiewicz M, Thaçi D, Schmidt E, Zillikens D, Vorobyev A, et al. Prevalence and presumptive triggers of localized bullous pemphigoid. J Dermatol. (2021) 48:1257–61. doi: 10.1111/1346-8138.15912
210. Mai Y, Nishie W, Sato K, Hotta M, Izumi K, Ito K, et al. Bullous pemphigoid triggered by thermal burn under medication with a dipeptidyl peptidase-iv inhibitor: a case report and review of the literature. Front Immunol. (2018) 9:542. doi: 10.3389/fimmu.2018.00542
211. Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. (2018) 142:1818–30. doi: 10.1016/j.jaci.2018.03.014
212. Bén, é J., Moulis G, Bennani I, Auffret M, Coupe P, Babai S, et al. Bullous pemphigoid and dipeptidyl peptidase IV inhibitors: a case-noncase study in the french pharmacovigilance database. Br J Dermatol. (2016) 175:296–301. doi: 10.1111/bjd.14601
213. Arai M, Shirakawa J, Konishi H, Sagawa N, Terauchi Y. Bullous pemphigoid and dipeptidyl peptidase 4 inhibitors: a disproportionality analysis based on the japanese adverse drug event report database. Diabetes Care. (2018) 41:e130–2. doi: 10.2337/dc18-0210
214. Kridin K, Bergman R. Association of bullous pemphigoid with dipeptidyl-peptidase 4 inhibitors in patients with diabetes: estimating the risk of the new agents and characterizing the patients. JAMA Dermatol. (2018) 154:1152–8. doi: 10.1001/jamadermatol.2018.2352
215. Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the european dermatology forum consensus in collaboration with the european academy of dermatology and venereology. Br J Dermatol. (2015) 172:867–77. doi: 10.1111/bjd.13717
216. Ujiie H, Iwata H, Yamagami J, Nakama T, Aoyama Y, Ikeda S, et al. Japanese guidelines for the management of pemphigoid (including epidermolysis bullosa acquisita). J Dermatol. (2019) 46:1102–35. doi: 10.1111/1346-8138.15111
217. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody profile differentiates between inflammatory and noninflammatory bullous pemphigoid. J Invest Dermatol. (2016) 136:2201–10. doi: 10.1016/j.jid.2016.06.622
218. Lamberts A, Meijer JM, Jonkman MF. Nonbullous pemphigoid: a systematic review. J Am Acad Dermatol. (2018) 78:989–95.e2. doi: 10.1016/j.jaad.2017.10.035
219. Lamberts A, Yale M, Grando SA, Horváth B, Zillikens D, Jonkman MF. Unmet needs in pemphigoid diseases: an international survey amongst patients, clinicians and researchers. Acta Derm Venereol. (2019) 99:224–5. doi: 10.2340/00015555-3052
220. Ujiie I, Iwata H, Yoshimoto N, Izumi K, Shimizu H, Ujiie H. Clinical characteristics and outcomes of bullous pemphigoid patients with versus without oral prednisolone treatment. J Dermatol. (2021) 48:502–10. doi: 10.1111/1346-8138.15816
221. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. (2002) 346:321–7. doi: 10.1056/NEJMoa011592
222. Joly P, Roujeau JC, Benichou J, Delaporte E, D'Incan M, Dreno B, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. (2009) 129:1681–7. doi: 10.1038/jid.2008.412
223. Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet. (2017) 389:1630–8. doi: 10.1016/S0140-6736(17)30560-3
224. Fichel F, Barbe C, Joly P, Bedane C, Vabres P, Truchetet F, et al. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA Dermatol. (2014) 150:25–33. doi: 10.1001/jamadermatol.2013.5757
225. Heymann WR. Bullous pemphigoid: rituximab to the rescue [editorial]. J Am Acad Dermatol. (2020) 82:1089. doi: 10.1016/j.jaad.2020.02.058
226. Yoo DS, Lee JH, Kim SC, Kim JH. Mortality and clinical response of patients with bullous pemphigoid treated with rituximab. Br J Dermatol. (2021) 185:210–12. doi: 10.1111/bjd.19890
227. Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Invest Dermatol. (2007) 127:2605–11. doi: 10.1038/sj.jid.5700958
228. Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol. (2007) 127:1167–74. doi: 10.1038/sj.jid.5700681
229. Messingham KN, Srikantha R, Degueme AM, Fairley JA. FcR-Independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol. (2011) 187:553–60. doi: 10.4049/jimmunol.1001753
230. Balakirski G, Alkhateeb A, Merk HF, Leverkus M, Megahed M. Successful treatment of bullous pemphigoid with omalizumab as corticosteroid-sparing agent: report of two cases and review of literature. J Eur Acad Dermatol Venereol. (2016) 30:1778–82. doi: 10.1111/jdv.13758
231. Seyed Jafari SM, Gadaldi K, Feldmeyer L, Yawalkar N, Borradori L, Schlapbach C. Effects of omalizumab on FcεRI and IgE expression in lesional skin of bullous pemphigoid. Front Immunol. (2019) 10:1919. doi: 10.3389/fimmu.2019.01919
232. Kaye A, Gordon SC, Deverapalli SC, Her MJ, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. (2018) 154:1225–26. doi: 10.1001/jamadermatol.2018.2526
233. Abdat R, Waldman RA, de Bedout V, Czernik A, Mcleod M, King B, et al. Dupilumab as a novel therapy for bullous pemphigoid: a multicenter case series. J Am Acad Dermatol. (2020) 83:46–52. doi: 10.1016/j.jaad.2020.01.089
234. Izumi K, Bieber K, Ludwig RJ. Current clinical trials in pemphigus and pemphigoid. Front Immunol. (2019) 10:978. doi: 10.3389/fimmu.2019.00978
235. Du S, Patzelt S, van Beek N, Schmidt E. Mucous membrane pemphigoid. Autoimmun Rev. (2022) 21:103036. doi: 10.1016/j.autrev.2022.103036
236. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2018) 54:26–51. doi: 10.1007/s12016-017-8633-4
237. van Beek N, Kridin K, Bühler E, Kochan AS, Ständer S, Ludwig RJ, et al. Evaluation of site- and autoantigen-specific characteristics of mucous membrane pemphigoid. JAMA Dermatol. (2021) 158:84–9. doi: 10.1001/jamadermatol.2021.4773
238. Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous diseases french study group. Arch Dermatol. (1995) 131:48–52. doi: 10.1001/archderm.1995.01690130050009
239. Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in lower Franconia, Germany. J Dtsch Dermatol Ges. (2009) 7:434–40. doi: 10.1111/j.1610-0387.2008.06976.x
240. Hübner F, König IR, Holtsche MM, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases among paediatric patients in Germany. J Eur Acad Dermatol Venereol. (2020) 34:2600–5. doi: 10.1111/jdv.16467
241. Rashid H, Lamberts A, Borradori L, Alberti-Violetti S, Barry RJ, Caproni M, et al. European guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European academy of dermatology and venereology - part I. J Eur Acad Dermatol Venereol. (2021) 35:1750–64. doi: 10.1111/jdv.17397
242. Schmidt E, Rashid H, Marzano AV, Lamberts A, Di Zenzo G, Diercks GFH, et al. European guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European academy of dermatology and venereology - part II. J Eur Acad Dermatol Venereol. (2021) 35:1926–48. doi: 10.1111/jdv.17395
243. Shimanovich I, Nitz JM, Zillikens D. Multiple and repeated sampling increases the sensitivity of direct immunofluorescence testing for the diagnosis of mucous membrane pemphigoid. J Am Acad Dermatol. (2017) 77:700–5. doi: 10.1016/j.jaad.2017.05.016
244. Kamaguchi M, Iwata H, Ujiie I, Ujiie H, Kitagawa Y, Shimizu H. Direct immunofluorescence using non-lesional buccal mucosa in mucous membrane pemphigoid. Front Med. (2018) 5:20. doi: 10.3389/fmed.2018.00020
245. van Beek N, Zillikens D, Schmidt E. Bullous autoimmune dermatoses. Dtsch Arztebl Int. (2021) 118:413–20. doi: 10.3238/arztebl.m2021.0136
246. van Beek N, Zillikens D, Schmidt E. Diagnosis of autoimmune bullous diseases. J Dtsch Dermatol Ges. (2018) 16:1077–91. doi: 10.1111/ddg.13637
247. Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. (2018) 5:296. doi: 10.3389/fmed.2018.00296
248. Blocker IM, Dahnrich C, Probst C, Komorowski L, Saschenbrecker S, Schlumberger W, et al. Epitope mapping of BP230 leading to a novel enzyme-linked immunosorbent assay for autoantibodies in bullous pemphigoid. Br J Dermatol. (2012) 166:964–70. doi: 10.1111/j.1365-2133.2012.10820.x
249. Goletz S, Probst C, Komorowski L, Schlumberger W, Fechner K, van Beek N, et al. A sensitive and specific assay for the serological diagnosis of antilaminin 332 mucous membrane pemphigoid. Br J Dermatol. (2019) 180:149–56. doi: 10.1111/bjd.17202
250. Kim JH, Kim YH, Kim S, Noh EB, Kim SE, Vorobyev A, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol. (2013) 27:e224–30. doi: 10.1111/j.1468-3083.2012.04617.x
251. Komorowski L, Muller R, Vorobyev A, Probst C, Recke A, Jonkman MF, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. (2012) 68:e89–95. doi: 10.1016/j.jaad.2011.12.032
252. Sitaru C, Dahnrich C, Probst C, Komorowski L, Blocker I, Schmidt E, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. (2007) 16:770–7. doi: 10.1111/j.1600-0625.2007.00592.x
253. Leverkus M, Schmidt E, Lazarova Z, Bröcker EB, Yancey KB, Zillikens D. Antiepiligrin cicatricial pemphigoid: an underdiagnosed entity within the spectrum of scarring autoimmune subepidermal bullous diseases. Arch Dermatol. (1999) 135:1091–8. doi: 10.1001/archderm.135.9.1091
254. Egan CA, Lazarova Z, Darling TN, Yee C, Cote T, Yancey KB. Anti-epiligrin cicatricial pemphigoid and relative risk for cancer. Lancet. (2001) 357:1850–1. doi: 10.1016/S0140-6736(00)04971-0
255. Matsushima S, Horiguchi Y, Honda T, Fujii S, Okano T, Tanabe M, et al. A case of anti-epiligrin cicatricial pemphigoid associated with lung carcinoma and severe laryngeal stenosis: review of Japanese cases and evaluation of risk for internal malignancy. J Dermatol. (2004) 31:10–5. doi: 10.1111/j.1346-8138.2004.tb00497.x
256. Terra JB, Pas HH, Hertl M, Dikkers FG, Kamminga N, Jonkman MF. Immunofluorescence serration pattern analysis as a diagnostic criterion in antilaminin-332 mucous membrane pemphigoid: immunopathological findings and clinical experience in 10 dutch patients. Br J Dermatol. (2011) 165:815–22. doi: 10.1111/j.1365-2133.2011.10474.x
257. Kitagaki H, Ono N, Hayakawa K, Kitazawa T, Watanabe K, Shiohara T. Repeated elicitation of contact hypersensitivity induces a shift in cutaneous cytokine milieu from a T helper cell type 1 to a T helper cell type 2 profile. J Immunol. (1997) 159:2484–91.
258. Gasparini G, Cozzani E, Di Zenzo G, Salemme A, Dematt, é E., Vassallo C, et al. Anti-laminin 332 antibody detection using biochip immunofluorescence microscopy in a real-life cohort of Italian patients with mucous membrane pemphigoid. Eur J Dermatol. (2021). doi: 10.1684/ejd.2021.4104. [Epub ahead of print].
259. Li X, Qian H, Natsuaki Y, Koga H, Kawakami T, Tateishi C, et al. Clinical and immunological findings in 55 patients with anti-laminin 332-type mucous membrane pemphigoid. Br J Dermatol. (2021) 185:449–51. doi: 10.1111/bjd.20099
260. Bernard P, Antonicelli F, Bedane C, Joly P, Le Roux-Villet C, Duvert-Lehembre S, et al. Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid. JAMA Dermatol. (2013) 149:533–40. doi: 10.1001/jamadermatol.2013.1434
262. Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C, et al. Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and immunopathological characteristics of the human disease. J Invest Dermatol. (2017) 137:1709–18. doi: 10.1016/j.jid.2017.03.037
263. Lazarova Z, Yee C, Darling T, Briggaman RA, Yancey KB. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest. (1996) 98:1509–18. doi: 10.1172/JCI118942
264. Murthy S, Schilf P, Patzelt S, Thieme M, Becker M, Kröger L, et al. Dapsone suppresses disease in preclinical murine models of pemphigoid diseases. J Invest Dermatol. (2021) 141:2587–95.e2. doi: 10.1016/j.jid.2021.04.009
265. Kridin K, Kneiber D, Kowalski EH, Valdebran M, Amber KT. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. (2019) 18:786–95. doi: 10.1016/j.autrev.2019.06.007
266. Koga H, Prost-Squarcioni C, Iwata H, Jonkman MF, Ludwig RJ, Bieber K. Epidermolysis bullosa acquisita: The 2019 update. Front Med. (2019) 5:362. doi: 10.3389/fmed.2018.00362
267. Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. (2016) 13:157–69. doi: 10.1080/1744666X.2016.1221343
268. Lu CY, Hsieh MS, Wei KC, Ezmerli M, Kuo CH, Chen W. Gastrointestinal involvement of primary skin diseases. J Eur Acad Dermatol Venereol. (2020) 34:2766–74. doi: 10.1111/jdv.16676
269. Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol. (2018) 78:754–9.e6. doi: 10.1016/j.jaad.2017.11.029
270. Iwata H, Vorobyev A, Koga H, Recke A, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. (2018) 13:153. doi: 10.1186/s13023-018-0896-1
271. Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. (2011) 91:307–12. doi: 10.2340/00015555-1065
272. Hiroyasu S, Zeglinski MR, Zhao H, Pawluk MA, Turner CT, Kasprick A, et al. Granzyme B inhibition reduces disease severity in autoimmune blistering diseases. Nat Commun. (2021) 12:302. doi: 10.1038/s41467-020-20604-3
273. Bieber K, Ludwig RJ. Drug development in pemphigoid diseases. Acta Derm Venereol. (2020) 100: adv00055. doi: 10.2340/00015555-3400
274. Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight. (2019) 4:e128239. doi: 10.1172/jci.insight.128239
275. Stüssel P, Schulze Dieckhoff K, Künzel S, Hartmann V, Gupta Y, Kaiser G, et al. Propranolol is an effective topical and systemic treatment option for experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2020) 140:2408–20. doi: 10.1016/j.jid.2020.04.025
276. Zillikens H, Kasprick A, Osterloh C, Gross N, Radziewitz M, Hass C, et al. Topical Application of the PI3Kβ-selective small molecule inhibitor TGX-221 is an effective treatment option for experimental epidermolysis bullosa acquisita. Front Med. (2021) 8:713312. doi: 10.3389/fmed.2021.713312
277. Kile RL, Rusk HA. A case of cold urticaria with an unusual family history. J Am Med Assoc. (1940) 114:1067–8. doi: 10.1001/jama.1940.62810120003010b
278. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. (2001) 29:301–5. doi: 10.1038/ng756
279. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. (2008) 105:1614–9. doi: 10.1073/pnas.0708616105
280. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. (2012) 366:330–8. doi: 10.1056/NEJMoa1102140
281. Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med. (2014) 211:2385–96. doi: 10.1084/jem.20141091
282. Aróstegui JI, Lopez Saldaña MD, Pascal M, Clemente D, Aymerich M, Balaguer F, et al. A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. (2010) 62:1158–66. doi: 10.1002/art.27342
283. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. (2019) 78:1025–32. doi: 10.1136/annrheumdis-2019-215048
284. Papathanasiou M, Carpinteiro A, Hagenacker T, Herrmann K, Rassaf T, Rischpler C, et al. 18F-florbetaben positron emission tomography detects cardiac involvement in systemic AA amyloidosis. Eur J Nucl Med Mol Imaging. (2020) 47:3186–7. doi: 10.1007/s00259-020-04861-4
285. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:E3328. doi: 10.3390/ijms20133328
286. Poli MC. New autoinflammatory diseases. Curr Opin Pediatr. (2018) 30:837–47. doi: 10.1097/MOP.0000000000000696
287. Papa R, Lachmann HJ. Secondary, AA, amyloidosis. Rheum Dis Clin North Am. (2018) 44:585–603. doi: 10.1016/j.rdc.2018.06.004
288. Petersen LM, Martin IW, Moschetti WE, Kershaw CM, Tsongalis GJ. Third-Generation sequencing in the clinical laboratory: exploring the advantages and challenges of nanopore sequencing. J Clin Microbiol. (2019) 58:e01315–9. doi: 10.1128/JCM.01315-19
289. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
290. Aliaga J, Bonaventura A, Mezzaroma E, Dhakal Y, Mauro AG, Abbate A, et al. Preservation of contractile reserve and diastolic function by inhibiting the NLRP3 inflammasome with OLT1177® (Dapansutrile) in a mouse model of severe ischemic cardiomyopathy due to non-reperfused anterior wall myocardial infarction. Molecules. (2021) 26:3534. doi: 10.3390/molecules26123534
291. Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset still's disease. Ann Rheum Dis. (2018) 77:840–847. doi: 10.1136/annrheumdis-2017-212608
292. Schnitzler L. Lesions Urticariennes Chroniques Permanentes (Erytheme Petaloide?). Case cliniques n. 46 B. Jounee Dermatologique d'Angers (1972). Available online at: https://ci.nii.ac.jp/naid/10016261259
293. Jain T, Offord CP, Kyle RA, Dingli D. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. (2013) 98:1581–5. doi: 10.3324/haematol.2013.084830
294. Greiner G, Gurbisz M, Ratzinger F, Witzeneder N, Simonitsch-Klupp I, Mitterbauer-Hohendanner G, et al. Digital PCR: a sensitive and precise method for KIT D816V quantification in mastocytosis. Clin Chem. (2018) 64:547–55. doi: 10.1373/clinchem.2017.277897
295. Krause K, Grattan CE, Bindslev-Jensen C, Gattorno M, Kallinich T, de Koning HD, et al. How not to miss autoinflammatory diseases masquerading as urticaria. Allergy. (2012) 67:1465–74. doi: 10.1111/all.12030
296. Bursztejn AC, Lipsker D. Delayed effort-induced swelling with myofasciitis and systemic manifestations: a so far unrecognized type of pressure-induced urticaria. Medicine. (2017) 96:e6112. doi: 10.1097/MD.0000000000006112
297. Gusdorf L, Asli B, Barbarot S, Néel A, Masseau A, Puéchal X, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy. (2017) 72:177–82. doi: 10.1111/all.13035
298. Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine. (2009) 88:23–31. doi: 10.1097/MD.0b013e3181943f5e
299. Bixio R, Rossini M, Giollo A. Efficacy of interleukin-1 blockade in Schnitzler's syndrome without detectable monoclonal gammopathy: a case-based review. Clin Rheumatol. (2021) 40:2973–7. doi: 10.1007/s10067-020-05501-w
300. Fujita Y, Asano T, Sakai A, Norikawa N, Yamamoto T, Matsumoto H, et al. A case of Schnitzler's syndrome without monoclonal gammopathy successfully treated with canakinumab. BMC Musculoskelet Disord. (2021) 22:257. doi: 10.1186/s12891-021-04120-z
301. Betrains A, Staels F, Vanderschueren S. Efficacy and safety of canakinumab treatment in schnitzler syndrome: a systematic literature review. Semin Arthritis Rheum. (2020) 50:636–42. doi: 10.1016/j.semarthrit.2020.05.002
302. Bonnekoh H, Frischbutter S, Roll S, Maurer M, Krause K. Tocilizumab treatment in patients with Schnitzler syndrome: an open-label study. J Allergy Clin Immunol Pract. (2021) 9:2486–9.e4. doi: 10.1016/j.jaip.2021.01.024
303. de Koning HD, Bodar EJ, van der Meer JW, Simon A, Schnitzler SSG. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. (2007) 37:137–48. doi: 10.1016/j.semarthrit.2007.04.001
304. Wechalekar AD, Lachmann HJ, Goodman HJ, Bradwell A, Hawkins PN, Gillmore JD. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood. (2008) 112:4009–16. doi: 10.1182/blood-2008-02-138156
305. Bodar EJ, Simon A, van der Meer JW. Effects of the histone deacetylase inhibitor ITF2357 in autoinflammatory syndromes. Mol Med. (2011) 17:363–8. doi: 10.2119/molmed.2011.00039
306. Paladini A, Vitale A, Frediani B, Cantarini L. Resolution of Schnitzler's syndrome after haematopoietic stem cell transplantation. Clin Exp Rheumatol. (2021) 39:704.
307. Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol. (2014) 32:3264–74. doi: 10.1200/JCO.2014.55.2018
308. Petersen MP, Möller S, Bygum A, Voss A, Bliddal M. Epidemiology of cutaneous lupus erythematosus and the associated risk of systemic lupus erythematosus: a nationwide cohort study in Denmark. Lupus. (2018) 27:1424–30. doi: 10.1177/0961203318777103
309. Klein R, Moghadam-Kia S, Taylor L, Coley C, Okawa J, LoMonico J, et al. Quality of life in cutaneous lupus erythematosus. J Am Acad Dermatol. (2011) 64:849–58. doi: 10.1016/j.jaad.2010.02.008
310. Little AJ, Vesely MD. Cutaneous lupus erythematosus: current and future pathogenesis-directed therapies. Yale J Biol Med. (2020) 93:81–95.
311. Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective janus kinase 1 inhibition is a promising therapeutic approach for lupus erythematosus skin lesions. Front Immunol. (2020) 11:344. doi: 10.3389/fimmu.2020.00344
312. Elman SA, Joyce C, Braudis K, Chong BF, Fernandez AP, Furukawa F, et al. Creation and validation of classification criteria for discoid lupus erythematosus. JAMA Dermatol. (2020) 156:901–6. doi: 10.1001/jamadermatol.2020.1698
313. Borucki R, Werth VP. Expert perspective: an evidence-based approach to refractory cutaneous lupus erythematosus. Arthritis Rheumatol. (2020) 72:1777–85. doi: 10.1002/art.41480
314. Concha JSS, Tarazi M, Kushner CJ, Gaffney RG, Werth VP. The diagnosis and classification of amyopathic dermatomyositis: a historical review and assessment of existing criteria. Br J Dermatol. (2019) 180:1001–8. doi: 10.1111/bjd.17536
315. Shiel WC, Jason M. The diagnostic associations of patients with antinuclear antibodies referred to a community rheumatologist. J Rheumatol. (1989) 16:782–5.
316. Tarazi M, Gaffney RG, Kushner CJ, Chakka S, Werth VP. Cutaneous lupus erythematosus patients with a negative antinuclear antibody meeting the american college of rheumatology and/or systemic lupus international collaborating clinics criteria for systemic lupus erythematosus. Arthritis Care Res. (2019) 71:1404–9. doi: 10.1002/acr.23916
317. Griesmacher A, Peichl P. Autoantibodies associated with rheumatic diseases. Clin Chem Lab Med. (2001) 39:189–208. doi: 10.1515/CCLM.2001.031
318. Egner W. The use of laboratory tests in the diagnosis of SLE. J Clin Pathol. (2000) 53:424–32. doi: 10.1136/jcp.53.6.424
319. Presto JK, Hejazi EZ, Werth VP. Biological therapies in the treatment of cutaneous lupus erythematosus. Lupus. (2017) 26:115–8. doi: 10.1177/0961203316670731
320. Rubio J, Kyttaris V. Journal club: efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomized, multicenter, placebo-controlled, phase 3 trial. ACR Open Rheumatol. (2021) 3:827–31. doi: 10.1002/acr2.11338
321. Chasset F, Bouaziz JD, Costedoat-Chalumeau N, Francès C, Arnaud L. Efficacy and comparison of antimalarials in cutaneous lupus erythematosus subtypes: a systematic review and meta-analysis. Br J Dermatol. (2017) 177:188–96. doi: 10.1111/bjd.15312
322. Gammon B, Hansen C, Costner MI. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. J Am Acad Dermatol. (2011) 65:717–21.e2. doi: 10.1016/j.jaad.2010.08.011
323. Islam MN, Hossain M, Haq SA, Alam MN, Ten Klooster PM, Rasker JJ. Efficacy and safety of methotrexate in articular and cutaneous manifestations of systemic lupus erythematosus. Int J Rheum Dis. (2012) 15:62–8. doi: 10.1111/j.1756-185X.2011.01665.x
324. Callen JP, Spencer LV, Burruss JB, Holtman J. Azathioprine. An effective, corticosteroid-sparing therapy for patients with recalcitrant cutaneous lupus erythematosus or with recalcitrant cutaneous leukocytoclastic vasculitis. Arch Dermatol. (1991) 127:515–22. doi: 10.1001/archderm.1991.04510010083008
325. Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? A retrospective analysis of 34 patients and a review of the literature. Dermatology. (2016) 232:91–6. doi: 10.1159/000441054
326. Ruzicka T, Sommerburg C, Goerz G, Kind P, Mensing H. Treatment of cutaneous lupus erythematosus with acitretin and hydroxychloroquine. Br J Dermatol. (1992) 127:513–8. doi: 10.1111/j.1365-2133.1992.tb14851.x
327. Fennira F, Chasset F, Soubrier M, Cordel N, Petit A, Francès C. Lenalidomide for refractory chronic and subacute cutaneous lupus erythematosus: 16 patients. J Am Acad Dermatol. (2016) 74:1248–51. doi: 10.1016/j.jaad.2016.01.054
328. Yuki EFN, Silva CA, Aikawa NE, Romiti R, Heise CO, Bonfa E, et al. Thalidomide and lenalidomide for refractory systemic/cutaneous lupus erythematosus Treatment: a narrative review of literature for clinical practice. J Clin Rheumatol. (2021) 27:248–59. doi: 10.1097/RHU.0000000000001160
329. Werth V, Merrill J, Furie R, van Vollenhoven R, Lipsky P, Weiswasser M, et al. Effect of Iberdomide on Cutaneous Manifestations in Systemic Lupus Erythematosus: Results of a 24-Week, Placebo-Controlled, Phase 2 Study. (2022). doi: 10.1136/annrheumdis-2021-eular.2181
330. Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med. (2021) 13:eabf8442. doi: 10.1126/scitranslmed.abf8442
331. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
332. Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. (2021) 12:3391. doi: 10.1038/s41467-021-23361-z
333. Werth V, Furie R, Romero-Diaz J, Navarra S, Kalunian K, Van Vollenhoven R, et al. OP0193 BIIB059, a Humanized Monoclonal Antibody Targeting BDCA2 on Plasmacytoid Dendritic Cells (PDC), Shows Dose-Related Efficacy in the Phase 2 LILAC Study in Patients (PTS) With Active Cutaneous Lupus Erythematosus (CLE) (2022). doi: 10.1136/annrheumdis-2020-eular.5743
334. Bogdanov I, Kazandjieva J, Darlenski R, Tsankov N. Dermatomyositis: current concepts. Clin Dermatol. (2018) 36:450–8. doi: 10.1016/j.clindermatol.2018.04.003
335. Mainetti C, Terziroli Beretta-Piccoli B, Selmi C. Cutaneous manifestations of dermatomyositis: a comprehensive review. Clin Rev Allergy Immunol. (2017) 53:337–56. doi: 10.1007/s12016-017-8652-1
336. Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. (2005) 17:701–6. doi: 10.1097/01.bor.0000179949.65895.53
337. Goreshi R, Chock M, Foering K, Feng R, Okawa J, Rose M, et al. Quality of life in dermatomyositis. J Am Acad Dermatol. (2011) 65:1107–16. doi: 10.1016/j.jaad.2010.10.016
338. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European league against rheumatism/American college of rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. (2017) 76:1955–64. doi: 10.1136/annrheumdis-2017-211468
339. Da Silva DM, Patel B, Werth VP. Dermatomyositis: a diagnostic dilemma. J Am Acad Dermatol. (2018) 79:371–3. doi: 10.1016/j.jaad.2017.12.074
340. Patel B, Khan N, Werth VP. Applicability of EULAR/ACR classification criteria for dermatomyositis to amyopathic disease. J Am Acad Dermatol. (2018) 79:77–83.e1. doi: 10.1016/j.jaad.2017.12.055
341. Callen JP. Collagen vascular diseases. J Am Acad Dermatol. (2004) 51:427–39. doi: 10.1016/j.jaad.2004.04.007
342. Marzecka M, Niemczyk A, Rudnicka L. Autoantibody markers of increased risk of malignancy in patients with dermatomyositis. Clin Rev Allergy Immunol. (2022). doi: 10.1007/s12016-022-08922-4. [Epub ahead of print].
343. Marasandra Ramesh H, Gude SS, Venugopal S, Peddi NC, Gude SS, Vuppalapati S. The role of myositis-specific autoantibodies in the dermatomyositis spectrum. Cureus. (2022) 14:e22978. doi: 10.7759/cureus.22978
344. Dawkins MA, Jorizzo JL, Walker FO, Albertson D, Sinal SH, Hinds A. Dermatomyositis: a dermatology-based case series. J Am Acad Dermatol. (1998) 38:397–404. doi: 10.1016/S0190-9622(98)70496-7
345. Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. (2013) 14:291–313. doi: 10.1007/s40257-013-0028-6
346. Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. (2002) 138:1231–3. doi: 10.1001/archderm.138.9.1231
347. Hollar CB, Jorizzo JL. Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis: a pilot study. J Dermatolog Treat. (2004) 15:35–9. doi: 10.1080/09546630310018509
348. Kim JE, Jeong MG, Lee HE, Ko JY, Ro YS. Successful treatment of cutaneous lesions of dermatomyositis with topical pimecrolimus. Ann Dermatol. (2011) 23:348–51. doi: 10.5021/ad.2011.23.3.348
349. Kurtzman DJ, Wright NA, Lin J, Femia AN, Merola JF, Patel M, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. (2016) 152:944–5. doi: 10.1001/jamadermatol.2016.0866
350. Cohen JB. Cutaneous involvement of dermatomyositis can respond to dapsone therapy. Int J Dermatol. (2002) 41:182–4. doi: 10.1046/j.1365-4362.2002.01409.x
351. Stirling DI. Thalidomide and its impact in dermatology. Semin Cutan Med Surg. (1998) 17:231–42. doi: 10.1016/S1085-5629(98)80019-9
352. Pharmaceuticals CORBUS. Corbus Pharmaceuticals Announces Topline Results From DETERMINE Phase 3 Study of Lenabasum for Treatment of Dermatomyositis. Pharmaceuticals CORBUS (2021). Available online at: https://www.corbuspharma.com/press-releases/detail/361/corbus-pharmaceuticals-announces-topline-results-from
353. Concha JSS, Pena S, Gaffney RG, Patel B, Tarazi M, Kushner CJ, et al. Developing classification criteria for skin-predominant dermatomyositis: the delphi process. Br J Dermatol. (2020) 182:410–417. doi: 10.1111/bjd.18096
354. Oldroyd AGS, Allard AB, Callen JP, Chinoy H, Chung L, Fiorentino D, et al. A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology. (2021) 60:2615–28. doi: 10.1093/rheumatology/keab166
355. Smith LN, Paik JJ. Promising and upcoming treatments in myositis. Curr Rheumatol Rep. (2020) 22:65. doi: 10.1007/s11926-020-00943-2
356. Nihtyanova SI, Tang EC, Coghlan JG, Wells AU, Black CM, Denton CP. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: a retrospective cohort study. QJM. (2010) 103:109–15. doi: 10.1093/qjmed/hcp174
357. Hao Y, Hudson M, Baron M, Carreira P, Stevens W, Rabusa C, et al. Early mortality in a multinational systemic sclerosis inception cohort. Arthritis Rheumatol. (2017) 69:1067–77. doi: 10.1002/art.40027
358. Li L, Cui Y, Chen S, Zhao Q, Fu T, Ji J, et al. The impact of systemic sclerosis on health-related quality of life assessed by SF-36: a systematic review and meta-analysis. Int J Rheum Dis. (2018) 21:1884–93. doi: 10.1111/1756-185X.13438
359. Park EH, Strand V, Oh YJ, Song YW, Lee EB. Health-related quality of life in systemic sclerosis compared with other rheumatic diseases: a cross-sectional study. Arthritis Res Ther. (2019) 21:61. doi: 10.1186/s13075-019-1842-x
360. van Leeuwen NM, Ciaffi J, Liem SIE, Huizinga TWJ, de Vries-Bouwstra JK. Health-related quality of life in patients with systemic sclerosis: evolution over time and main determinants. Rheumatology. (2021) 60:3646–55. doi: 10.1093/rheumatology/keaa827
361. Pavlov-Dolijanovic SR, Damjanov NS, Vujasinovic Stupar NZ, Baltic S, Babic DD. The value of pattern capillary changes and antibodies to predict the development of systemic sclerosis in patients with primary raynaud's phenomenon. Rheumatol Int. (2013) 33:2967–73. doi: 10.1007/s00296-013-2844-7
362. Bellando-Randone S, Matucci-Cerinic M. Very early systemic sclerosis. Best Pract Res Clin Rheumatol. (2019) 33:101428. doi: 10.1016/j.berh.2019.101428
363. Kill A, Tabeling C, Undeutsch R, Kühl AA, Günther J, Radic M, et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res Ther. (2014) 16:R29. doi: 10.1186/ar4457
364. Riemekasten G, Cabral-Marques O. Antibodies against angiotensin II type 1 receptor (AT1R) and endothelin receptor type A (ETAR) in systemic sclerosis (SSc)-response. Autoimmun Rev. (2016) 15:935. doi: 10.1016/j.autrev.2016.04.004
365. Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. (2017) 76:1327–39. doi: 10.1136/annrheumdis-2016-209909
366. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol. (2021) 3:e489–97. doi: 10.1016/S2665-9913(21)00107-7
367. Zamanian RT, Badesch D, Chung L, Domsic RT, Medsger T, Pinckney A, et al. Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension: a multicenter, double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. (2021) 204:209–21. doi: 10.1164/rccm.202009-3481OC
368. Rubenzik TT, Derk CT. Unmet patient needs in systemic sclerosis. J Clin Rheumatol. (2009) 15:106–10. doi: 10.1097/RHU.0b013e31819dbe83
369. Mouthon L, Alami S, Boisard AS, Chaigne B, Hachulla E, Poiraudeau S. Patients' views and needs about systemic sclerosis and its management: a qualitative interview study. BMC Musculoskelet Disord. (2017) 18:230. doi: 10.1186/s12891-017-1603-4
370. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
371. Langan SM, Seminara NM, Shin DB, Troxel AB, Kimmel SE, Mehta NN, et al. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol. (2012) 132:556–62. doi: 10.1038/jid.2011.365
372. Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA. (2006) 296:1735–41. doi: 10.1001/jama.296.14.1735
373. Ludwig RJ, Herzog C, Rostock A, Ochsendorf FR, Zollner TM, Thaci D, et al. Psoriasis: a possible risk factor for development of coronary artery calcification. Br J Dermatol. (2007) 156:271–6. doi: 10.1111/j.1365-2133.2006.07562.x
374. Kimball AB, Leonardi C, Stahle M, Gulliver W, Chevrier M, Fakharzadeh S, et al. Demography, baseline disease characteristics and treatment history of patients with psoriasis enrolled in a multicentre, prospective, disease-based registry (PSOLAR). Br J Dermatol. (2014) 171:137–47. doi: 10.1111/bjd.13013
375. Boehncke WH, Boehncke S, Tobin AM, Kirby B. The ‘psoriatic march': a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol. (2011) 20:303–7. doi: 10.1111/j.1600-0625.2011.01261.x
376. Vorobyev A, Gupta Y, Sezin T, Koga H, Bartsch YC, Belheouane M, et al. Gene-diet interactions associated with complex trait variation in an advanced intercross outbred mouse line. Nat Commun. (2019) 10:4097. doi: 10.1038/s41467-019-11952-w
377. Brunner PM, Silverberg JI, Guttman-Yassky E, Paller AS, Kabashima K, Amagai M, et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J Invest Dermatol. (2017) 137:18–25. doi: 10.1016/j.jid.2016.08.022
378. Lee HH, Gwillim E, Patel KR, Hua T, Rastogi S, Ibler E, et al. Epidemiology of alopecia areata, ophiasis, totalis, and universalis: a systematic review and meta-analysis. J Am Acad Dermatol. (2020) 82:675–82. doi: 10.1016/j.jaad.2019.08.032
379. Lee H, Jung SJ, Patel AB, Thompson JM, Qureshi A, Cho E. Racial characteristics of alopecia areata in the United States. J Am Acad Dermatol. (2020) 83:1064–70. doi: 10.1016/j.jaad.2019.06.1300
380. Boch K, Langan EA, Zillikens D, Ludwig RJ, Kridin K. Retrospective analysis of the clinical characteristics and patient-reported outcomes in vulval lichen planus: results from a single-center study. J Dermatol. (2021) 48:1913–7. doi: 10.1111/1346-8138.16191
381. Kridin K, Kridin M, Amber KT, Shalom G, Comaneshter D, Batat E, et al. the risk of pulmonary embolism in patients with pemphigus: a population-based large-scale longitudinal study. Front Immunol. (2019) 10:1559. doi: 10.3389/fimmu.2019.01559
382. van Beek N, Weidinger A, Schneider SW, Kleinheinz A, Gläser R, Holtsche MM, et al. Incidence of pemphigoid diseases in Northern Germany in 2016 - first data from the schleswig-holstein registry of autoimmune bullous diseases. J Eur Acad Dermatol Venereol. (2021) 35:1197–202. doi: 10.1111/jdv.17107
Keywords: medical need, skin, inflammation, atopic dermatitis, psoriasis, alopecia areata, chronic spontaneous urticaria, hidradenitis suppurativa
Citation: Ujiie H, Rosmarin D, Schön MP, Ständer S, Boch K, Metz M, Maurer M, Thaci D, Schmidt E, Cole C, Amber KT, Didona D, Hertl M, Recke A, Graßhoff H, Hackel A, Schumann A, Riemekasten G, Bieber K, Sprow G, Dan J, Zillikens D, Sezin T, Christiano AM, Wolk K, Sabat R, Kridin K, Werth VP and Ludwig RJ (2022) Unmet Medical Needs in Chronic, Non-communicable Inflammatory Skin Diseases. Front. Med. 9:875492. doi: 10.3389/fmed.2022.875492
Received: 14 February 2022; Accepted: 09 May 2022;
Published: 09 June 2022.
Edited by:
Devinder Mohan Thappa, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), IndiaReviewed by:
Hiroshi Koga, Kurume University School of Medicine, JapanCopyright © 2022 Ujiie, Rosmarin, Schön, Ständer, Boch, Metz, Maurer, Thaci, Schmidt, Cole, Amber, Didona, Hertl, Recke, Graßhoff, Hackel, Schumann, Riemekasten, Bieber, Sprow, Dan, Zillikens, Sezin, Christiano, Wolk, Sabat, Kridin, Werth and Ludwig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, cmFsZi5sdWR3aWdAdWtzaC5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.