 Cristina Egido-Turrión1,2†
Cristina Egido-Turrión1,2† Elisa Rossi3†
Elisa Rossi3† Claudia Ollauri-Ibáñez1,2
Claudia Ollauri-Ibáñez1,2 María L. Pérez-García2,4María A. Sevilla1,2
María L. Pérez-García2,4María A. Sevilla1,2 José María Bastida1,2,5José Ramón González-Porras2,5
José María Bastida1,2,5José Ramón González-Porras2,5 Alicia Rodríguez-Barbero1,2
Alicia Rodríguez-Barbero1,2 Carmelo Bernabeu6
Carmelo Bernabeu6 José M. Lopez-Novoa1,2
José M. Lopez-Novoa1,2 Miguel Pericacho1,2*
Miguel Pericacho1,2*- 1Department of Physiology and Pharmacology, Universidad de Salamanca, Salamanca, Spain
- 2Institute for Biomedical Research of Salamanca (IBSAL), Salamanca, Spain
- 3Université de Paris, Innovative Therapies in Haemostasis, INSERM, Paris, France
- 4Department of Internal Medicine, Complejo Asistencial Universitario de Salamanca (CAUSA)-SACYL, Salamanca, Spain
- 5Department of Hematology, Complejo Asistencial Universitario de Salamanca (CAUSA)-SACYL, Salamanca, Spain
- 6Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
Hereditary Hemorrhagic Telangiectasia (HHT) is an autosomal-dominant genetic disorder involving defects in two predominant genes known as endoglin (ENG; HHT-1) and activin receptor-like kinase 1 (ACVRL1/ALK1; HHT-2). It is characterized by mucocutaneous telangiectases that, due to their fragility, frequently break causing recurrent epistaxis and gastrointestinal bleeding. Because of the severity of hemorrhages, the study of the hemostasis involved in these vascular ruptures is critical to find therapies for this disease. Our results demonstrate that HHT patients with high bleeding, as determined by a high Epistaxis Severity Score (ESS), do not have prolonged clotting times or alterations in clotting factors. Considering that coagulation is only one of the processes involved in hemostasis, the main objective of this study was to investigate the overall mechanisms of hemostasis in HHT-1 (Eng+/−) and HHT-2 (Alk1+/−) mouse models, which do not show HHT vascular phenotypes in the meaning of spontaneous bleeding. In Eng+/− mice, the results of in vivo and in vitro assays suggest deficient platelet-endothelium interactions that impair a robust and stable thrombus formation. Consequently, the thrombus could be torn off and dragged by the mechanical force exerted by the bloodstream, leading to the reappearance of hemorrhages. In Alk1+/− mice, an overactivation of the fibrinolysis system was observed. These results support the idea that endoglin and Alk1 haploinsufficiency leads to a common phenotype of impaired hemostasis, but through different mechanisms. This contribution opens new therapeutic approaches to HHT patients' epistaxis.
Introduction
Hereditary Hemorrhagic Telangiectasia (HHT), also known as Osler-Weber-Rendu disease, is an autosomal-dominant genetic disorder involving genetic defects in two predominant genes: ENG, codifying for endoglin (HHT-1 type) and ACVRL1/ALK1, codifying for activin receptor-like kinase 1 (HHT-2 type); and, much less frequently, in two other genes, such as MADH4/SMAD4 (JP-HHT) or BMP9/GDF2 (HHT-5) (1). HHT has an estimated prevalence of at least 1/5,000 and is characterized by the presence of multiple dermal, mucous and visceral arteriovenous malformations (AVMs) in lungs, liver, gastrointestinal tract and nervous system (2–4), as well as recurrent and severe hemorrhages (5, 6).
Generally, nosebleeds, also known as epistaxis, are not only the first symptom to manifest the pathology, but also the most frequent, since patients usually bleed more than once a week. This frequency increases with age and 12% of diagnosed cases have more than one bleeding per day (7). The intensity can vary from a few drops to events that last up to 10 min, and although nasal packing is often used to control acute bleeding, even though it may cause re-bleeding, there are no well-designed studies on the first-line treatment of acute epistaxis (8, 9). The combination of the high frequency linked to the severity of the bleeding leads 50% of patients to develop iron deficiency anemia (10). Usually, bleeds are so uncontrolled that administration of iron supplement is not enough to reverse anemia, so it is not uncommon to treat patients with repeated blood transfusions (11, 12). Both bevacizumab, an antiangiogenic monoclonal antibody that selectively targets Vascular Endothelial Growth Factor (VEGF) (13), and thalidomide, a potent immunosuppressive and antiangiogenic agent (14) could be used to treat HHT patient hemorrhages but with long-term side effects (15). For these reasons the study of hemostasis in HHT mouse models could contribute to better understand the pathology and improve treatments.
It is widely recognized that the origin of HHT bleeding is the fragility of the mucocutaneous telangiectases which tend to break, causing recurrent epistaxis and gastrointestinal bleeding. However, it is unclear why the hemostatic system does not properly control these bleeds. Intriguingly, despite the severity of the bleeds, HHT has not been described as a disease that presents deficiencies in any of the clotting factors (16) and no systematic studies on the rest of the processes related to the control of bleeding have been performed. A previous pioneer study demonstrated that endoglin (Eng), a protein expressed on endothelial cells and in heterozygous condition in HHT-1 patients, is able to bind the integrin αIIbβ3 present on platelets via its RGD motif (17). This binding contributes to platelet interaction with activated endothelial cells and increases platelet resistance to shear, whereas loss of Eng seems to interfere with these platelets-endothelium interactions (17), confirming that Eng can display a role as an adhesion molecule in addition to its well-known function as TGFβ co-receptor (18).
In this study, after confirming with HHT-1 and HHT-2 patients that increased bleeding is not associated with alterations in prothrombin time (PT), activated partial thromboplastin time (aPTT) or coagulation factors, we used the most common genetic models of HHT, namely Eng+/− for HHT-1 and Alk1+/− for HHT-2 (19), as well as their Wildtype (WT) littermates (Eng+/+ and Alk1+/+, respectively) as controls to analyze the overall process of hemostasis. Our hypothesis is that, while the high frequency of bleeds seems clearly due to rupture of fragile vessels resulting from impaired angiogenesis, the severity of bleeds may be due to alterations in the hemostatic process. Taking into account that Eng and ALK1 are mostly expressed in the endothelium, we have focused on the processes where the endothelium is most relevant, especially in thrombus stabilization, where endoglin may play an important role.
Materials and Methods
Materials
If not specifically mentioned, all the reagents and vasoactive substances were from Sigma-Aldrich (Merck Life Science S.L.U, Darmstadt, Germany).
Patients and Epistaxis Severity Score
In total, 36 HHT patients, 5 with HHT-1 and 31 with HHT-2, participated in the study. Relatives not affected by the disease were recruited as controls for the study. All of them signed an informed consent form. The study was approved by the Local Ethical Committee. Epistaxis Severity Score were calculated for all participants in the study as previously described (20).
Coagulation Studies
Venous blood samples were drawn into commercial buffered 3.2% sodium citrate for coagulation studies. Coagulation basic studies consisted of prothrombin time (PT) and activated partial thromboplastin time (APTT) and were measured according to international recommendations (21). Factor coagulant activity (Factor:C) was measured on a BCS XP System (Siemens, Healthcare Diagnostics, Erlangen, Germany) by modification of the one-stage clotting method with the use of factor deficient or depleted plasmas. Coagulation time by BCS XP was calibrated with standard human plasma. The normal ranges were considered to be 60–120%. Von Willebrand (vW) Factor activity was analyzed using latex particles surface-coated with recombinant GPIb (Siemens, Healthcare Diagnostics, Erlangen, Germany) (22, 23).
Animal Models: Eng+/− (HHT-1), Alk1+/− (HHT-2), and ENG+
All animal protocols were performed with approval of the Committee for the Care and Use of Animals of the University of Salamanca (Spain), project #305 and complied with the current guides of the European Union and the US Department of Health and Human Services for the Care and Use of Laboratory Animals. Generation of Eng+/− and Alk1+/− mice has been described previously (24, 25). The animals were a generous gift from Michelle Letarte (Hospital for Sick Children, Toronto, Canada) and Peter ten Dijke (Leiden University, Netherlands), respectively. Both transgenic lines were backcrossed with C57BL/6J mice for more than ten generations. Of note, the genetic background of these HHT animal models do not show the characteristic bleeding phenotype due to fragile telangiectases as in other strains (26). Routine genotyping of DNA isolated from mouse tail biopsies was performed by PCR using the primers previously reported (25, 27). The generation and characteristics of the transgenic mice ubiquitously overexpressing human endoglin (ENG+) was performed by microinjection of a pCAGGS vector containing the complementary DNA sequence (cDNA) of endoglin in the fertilized egg of CBAxC57BL/6J mice, as previously described (28). WT littermates for each model were used as controls.
For animal anesthesia, 2% isoflurane in oxygen was used. During recovery from the anesthesia, heat was provided and, when necessary, a dose of buprenorphine (0.05 mg/kg) was subcutaneously administered. Animals were sacrificed by CO2 inhalation or cervical dislocation, depending on the experiment requirements.
Tail-Bleeding Assay
Isoflurane-anesthetized mice were placed in prone position. Their tails were transected at ~3 mm from the tip (29, 30) (constant tail diameter 1.4 mm) and immediately immersed in PBS at 37°C. Each animal was monitored for 30 min even if bleeding ceased, in order to detect any re-bleeding. However, when mice bled continuously for more than 10 min, the experiment was interrupted so that the life of the animal was not threatened. Three parameters were analyzed: (i) first bleeding time, the period between the beginning of the hemorrhage and the first time it stopped; (ii) re-bleeding time, defined as the time of the rest of periods where the hemorrhage starts again; and (iii) total bleeding time, the sum of the time in which the bleeding is active.
Vascular Reactivity of Thoracic Aorta Rings
To analyze the capacity for reflex vasoconstriction of the vessels of mice models, we conducted studies of vasomotor functionality in thoracic aorta arteries obtained from mice of the different genotypes, as previously described (31). Briefly, arteries were removed and placed in chilled Krebs solution (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, and 11 mM glucose). The pH of the solution after saturation with carbogen was 7.4. The arteries were carefully cleaned, cut into rings (2 mm) and aortic rings were mounted, with Krebs solution, onto a 4-channel myograph (Multiwire Myograph System, DMT, Denmark) After an equilibration period all rings were normalized and set at a resting tension of 5 mN and allowed to equilibrate for 30 min. After that, the vessels were tested for responsiveness to a hyperpotassium solution (KPSS; KCl 120 mM) and cumulative concentration–response curves to norepinephrine, serotonin and thromboxane analog U-46619 were generated in separate rings until the maximal response was consistent. Vasoconstrictor responses were expressed as a percentage of KCl-induced contractions. The cumulative concentration–response curves, were fitted to a logistic equation and analyzed with GraphPad Prism 9.0 software. We carried out statistical analysis of concentration–response curves using the extra sum-of-squares F test principle.
Induction of Thrombogenic Event
Mice were anesthetized as previously described and a catheter was implanted in the jugular vein. Collagen (150 ng/g body weight; Chrono-Log, Havertown, PA, USA) and epinephrine (15 ng/g body weight) were administered intravenously and blood samples were collected from jugular vein into EDTA tubes before and after 3 min of the injection of these compounds, that lead to platelet activation, aggregation and adhesion and, consequently, a decrease in platelet count. Platelet count in these samples was performed in an automated complete blood count analyzer (AdviaTM 120 hematology; Bayer, Leverkusen, Germany). At the end of experiment, anesthetized animals were euthanized by cervical dislocation.
Platelet Activation and Aggregation Analysis by Flow Cytometry
Murine blood samples were obtained by retroorbital puncture using citrated capillary. Whole blood samples were centrifuged at 1,300 g for 5 min and resuspended in 1 mL of Tyrode's buffer (134 mM NaCl; 20 mM HEPES; 12 mM NaHCO3; 5 mM glucose; 2.9 mM KCl; 1 mM MgCl2; 0.34 mM Na2HPO4; pH 7.4). Due to the limited volume of blood samples obtained, flow cytometry was used to test platelets activation and aggregation as previously reported (32). Aliquots of diluted blood (30 μL) were incubated for 15 min at room temperature with the appropriate anti-mouse monoclonal antibody: APC-conjugated rat anti-integrin alpha IIb (GPIIb, CD41) (6.7 μg/mL; clone MWReg30; eBioscience, Thermo Fisher Scientific, Waltham, MA, USA) and PE-labeled rat anti-mouse integrin αIIbβ3 (GPIIbIIIa, CD41/61) (25 μg/mL; clone JON/A; Emfret Analytics, Würzburg, Germany) were used to detect platelets activation. PE-labeled CD9 and FITC-labeled CD9 (5 μg/mL; clone MZ3; BioLegend, San Diego, CA, USA) were used to visualize aggregation and the double positive population was considered the aggregated platelets. Thrombin (1 U/mL) was added when necessary. To remove excess antibody, aliquots were centrifuged (2,250 g for 5 min) and resuspended in 150 μL of Tyrode's buffer. Analysis was immediately performed in a BD Accury™ C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Primary Culture of Mouse Lung Endothelial Cells (MLEC)
Mouse Lung Endothelial Cells (MLEC) were isolated from the different mice described above (33). MLECs were grown in Advanced-DMEM F-12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 20% FBS, 50 U/mL penicillin–streptomycin, 2 mM glutamine, 30 μg/mL endothelial cell growth supplement (ECGS) (Generon, Slough, UK) and 100 μg/mL bovine heparin in Petri dishes coated with 0.1% gelatin and supplemented with 0.01% collagen (Corning, Corning, NY, USA) and 0.01% fibronectin. Cells were maintained in 90% RH, 5% CO2 atmosphere at 37°C. MLECs between passages 3 and 6 were used for the experiments.
Obtention of Platelets From Human Blood Samples
Citrated blood samples were obtained from healthy donors, who had not taken any medication for at least 10 days. Whole blood was centrifuged at 100 g for 20 min to obtain platelet-rich plasma (PRP). PRP was washed twice by diluting with washing buffer (103 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose and 36 mM citric acid; pH 6.5) supplemented with platelet activation inhibitor PGE1 (2 × 10−7 M) and the ADP scavenger apyrase (1 U/mL), followed by centrifugation for 12 min at 1,240 g. Platelets were finally resuspended at 2.5 × 108 platelets/mL in Walsh's buffer (137 mM NaCl, 20 mM PIPES, 5.6 mM dextrose, 1 g/L BSA, 1 mM MgCl2, 2.7 mM KCl, 3.3 mM NaH2PO4; pH 7.4). Finally, washed platelets were stained with 1 μM calcein (Thermo Fisher Scientific, Waltham, MA, USA). Prior to any experimental procedure, platelets were typically left at room temperature for 30 min. Under resting conditions, platelets did not show activation.
Static Platelet Adhesion Assay
MLECs were seeded in slides (Millicell EZ slide; Millipore, Burlington, MA, USA) and grown to reach a monolayer. Human-stained-platelets (107 platelets/250 μL) were added on the MLECs monolayer. After an incubation for 30 min in the presence of CXCL12 (200 ng/mL; R&D Systems, Minneapolis, MN, USA), chamber wells were washed twice with PBS. Platelet adhesion was observed under a fluorescent microscope (Observer D1; Zeiss, Jena, Germany) coupled to a CCD camera (QIclick FCLR-12; Qimaging, Surrey, Canada) and quantifications were performed with ImageJ software.
Dynamic Platelet Adhesion Assays
Microfluid devices were used to perform flow assays as previously described (34). Briefly, the channels were coated with a monolayer of MLECs. Calcein-stained platelets (2.5 × 108 platelets/ml) were perfused through the channels at 2 dynes/cm2, allowed to adhere in the absence of flow for 10 min in presence of CXCL12. Then, the chambers were subjected to 2 dynes/cm2 flow for 2 min. Real-time platelet adhesion was monitored under fluorescent microscope (Observer D1; Zeiss, Jena, Germany) coupled to a CCD camera (QIclick FCLR-12; Qimaging, Surrey, Canada). Then, images were quantified by ImageJ software.
Carotid Artery Occlusion Induced by FeCl3
Carotid arteries of isoflurane-anesthetized animals were exposed by a surgical opening in the ventral face of the neck. Before causing the vascular injury, a flow probe was placed around the carotid artery and carotid blood flow was recorded for 3 min using a Transonic Model TS420 flowmeter (Transonic Systems, Ithaca, NY, USA) in order to determine basal blood flow. After causing vascular damage by applying a filter paper saturated with 7.5% FeCl3 on top of the vessel for 1.5 min, carotid blood flow was recorded for 30 min and represented as the percentage of carotid basal blood flow. The time to carotid occlusion was determined as the time required to achieve a flow rate of <5% that remains stable for more than 5 min.
Study of Fibrinolysis
Murine EDTA-blood samples were collected before and after 30 min, 6 h or 24 h since the intravenous administration of collagen (150 ng/g body weight; Chrono-Log, Havertown, PA, USA) and epinephrine (15 ng/g body weight) to generate a thrombogenic event and fibrinolysis system activation. Anticoagulated EDTA-blood was centrifuged and stored according to ELISA kits instructions. Plasminogen, t-PA, PAI-1 and D-Dimer levels were analyzed, respectively, by Mouse Plasminogen (PLG) ELISA kit (Cusabio, Houston, TX, USA), Mouse t-PA (Tissue-type Plasminogen Activator) ELISA kit (Elabscience, Houston, TX, USA), RayBio® Mouse PAI-1 ELISA kit (RayBiotech, Norcross, GA, USA) and Mouse D-Dimer, D2D ELISA kit (Cusabio, Houston, TX, USA) strictly following datasheet instructions. In all cases, we analyzed statistically, by two-way ANOVA, whether in each mouse model there were significant changes in the levels of the factor studied after the thrombotic event.
Statistical Analysis
Data are expressed as the mean + standard error of the mean. All results are from at least 3 independent experiments and subjected to statistical test. Data were checked for Gaussian distribution using the D'Agostino-Pearson normality test. For normally distributed datasets, Student's t-test, ANOVA and two-way ANOVA was used, followed by Sidak's test for multiple comparisons. The statistical analysis of non-parametric datatests was made by the Mann-Whitney test (for two datasets) and Kruskal-Wallis test (more than two datasets). Fisher's exact test was used for the comparison of proportions when necessary. Results were considered statistical significant if p < 0.05 (ns: non-significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Graphs and statistical analysis were performed using GraphPad Prism 9 software (GraphPad software, San Diego, CA, USA).
Results
Increased Bleeding Is Not Associated With Alterations of Coagulation Pathways in HHT Patients
Epistaxis Severity Score (ESS) was used to measure the severity and frequency of epistaxis in a group of HHT-1 and HHT-2 patients, as well as in relatives not affected by the disease who are the controls of the study. The distribution of controls and patients in age and gender is shown in Figure 1A. In the patient's group there is a slightly more number of men than women and a higher mean age compared to controls, but no significant statistical differences between the two groups were found.
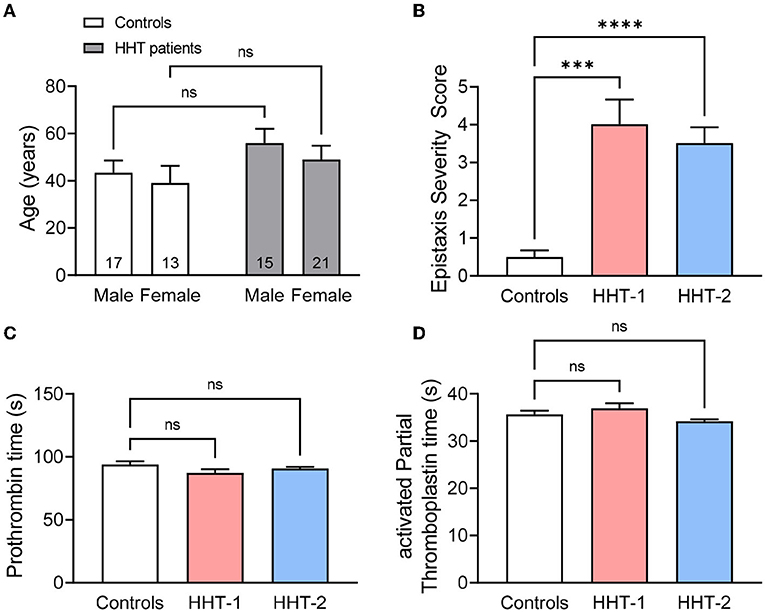
Figure 1. (A) Mean age of HHT patients (n = 15 males and 21 females) and its relatives unaffected by the disease that are the controls (n = 17 males and 13 females) of the study, disaggregated by gender. (B) Epistaxis Severity Score of controls (n = 30), HHT-1 (n = 5) and HHT-2 (n = 31) patients. (C,D) Prothrombin time (C) and activated Partial Thromboplastin time (D) of controls, HHT-1 and HHT-2 patients. Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
As expected, the ESS of HHT patients is significantly higher than that of their healthy relatives (Figure 1B). In line with previous data in the literature and Rendu's statements when first describing the disease (35), there are no differences between control subjetcs and HHT-1 or HHT-2 patients in the coagulation basic studies, neither coagulation factor activity of the intrinsic or extrinsic coagulation pathways (Figures 1C,D, Supplementary Figure S1).
Endoglin or Alk1 Deficiency Leads to Longer Tail Bleeding Time in Mice
Having confirmed that increased bleeding in HHT patients is not due to coagulation alterations, and because dissecting other components of the hemostasis process in patients is not feasible due to ethical reasons, we decided to investigate it in murine genetic models of the disease: endoglin heterozygous mice (Eng+/−) for HHT-1 and Alk1 heterozygous mice (Alk1+/−) for HHT-2. As indicated in the methods section, the genetic background of these models do not present a bleeding phenotype due to fragile telangiectases as in other strains (26).
First, we aimed to analyze whether HHT mouse models exhibit increased bleeding when the lesion occurs in a normal vessel, rather than in a fragile vascular lesion as an HHT telangiectasia. For this purpose, we measured the tail bleeding time, which allows the analysis not only of the bleeding time of mice after wounding, but also the presence and duration of any spontaneous rebleeding.
Although, our data show that there is no difference in the first bleeding (Figures 2A,B), when rebleeding is analyzed, the number of mice with intense rebleeds (longer than 120 s) is significantly higher in both Eng+/− and Alk1+/− mice with respect to their controls (Figures 2C,D). Actually, rebleeding can become so severe that in some Eng+/− and Alk1+/− animals the bleeding may not stop spontaneously after 30 min from injury and, thus, it must be stopped by the investigator (Figures 2E,F). For these reasons, the rebleeding time, i.e., the sum of the times of the subsequent spontaneous bleedings, is significantly longer in both Eng+/− or Alk1+/− mice with respect to their controls (Figures 2G,H). In conclusion, both murine models of the disease have a prolonged bleeding time suggesting an impaired hemostasis.
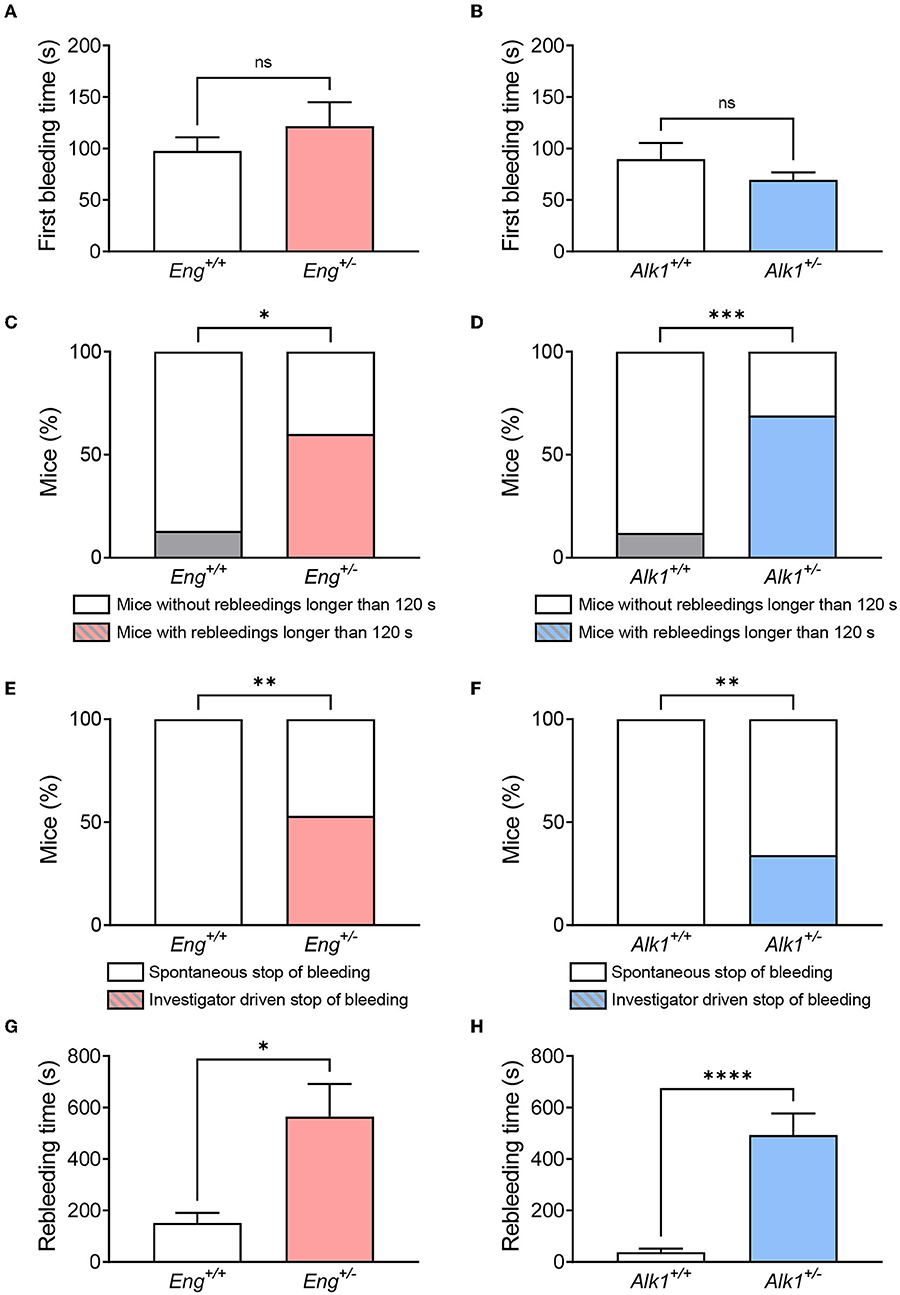
Figure 2. (A,B) Time of first bleeding after cutting the tail tip in the HHT-1 murine model (A), Eng+/− mice (n = 15) and its respective controls Eng+/+ (n = 15); and in the HHT-2 murine model (B), Alk1+/− mice (n = 16) and its respective controls Alk1+/+ (n = 29). (C,D) Percentage of mice showing rebleeds longer than 2 min in the mouse model of HHT-1 (C) and HHT-2 (D). (E,F) Proportion of mice that are able to stop rebleeding on their own compared to those that need investigator intervention to stop rebleeding in the animal model of HHT-1 (E) or HHT-2 (F). (G,H) Rebleeding time, calculated as the sum of the time taken for the different spontaneous rebleeding times that a mouse may have during the experiment, for Eng-deficient mice and their controls (G), as well as for Alk1-deficient mice and their controls (H). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Vascular Contractile Response Is Not Reduced in Eng+/− or Alk1+/− Mice
Hemostasis is firstly dependent on a vascular spasm that reduces blood flow and increments the efficacy of the following steps of hemostasis, such as platelet activation and aggregation. Thus, we analyzed in artery rings obtained from thoracic aorta of HHT-1 and HHT-2 animal models whether they presented a lower response to different vasoconstrictor agents involved in vascular spasm, such as norepinephrine, serotonin and thromboxane analog U-46619, which could explain the increased bleeding in these animals. As shown in Supplementary Figure S2, significant differences were only found with respect to the control in the response of Eng+/– mice to serotonin (Supplementary Figure S2C). However, these mice showed an increase in the contraction response, rather than a reduction. Taken together, our results support the conclusion that the areterial contractile response is not reduced in both HHT animal models.
Platelet Activation and Aggregation Is Not Altered in Animal Models of HHT
Primary hemostasis depends on the ability of platelets to activate and aggregate in response to different stimuli. For in vivo analysis of these processes we studied the effect of intravenous injection of a sublethal mixture of collagen with epinephrine, which causes the activation and aggregation of circulating platelets forming thrombi throughout the vessels. Thus, after treatment, a decrease in non-aggregated platelets is observed, which correspond to those platelets that have formed aggregates. We found a significant difference between control and collagen/epinephrime treatment but no significant differences between WT and Eng+/− or Alk1+/− mice (Figures 3A,B). Thus, our results suggest that Eng or Alk1 deficiency does not alter the ability of platelets to activate or form platelet aggregates.
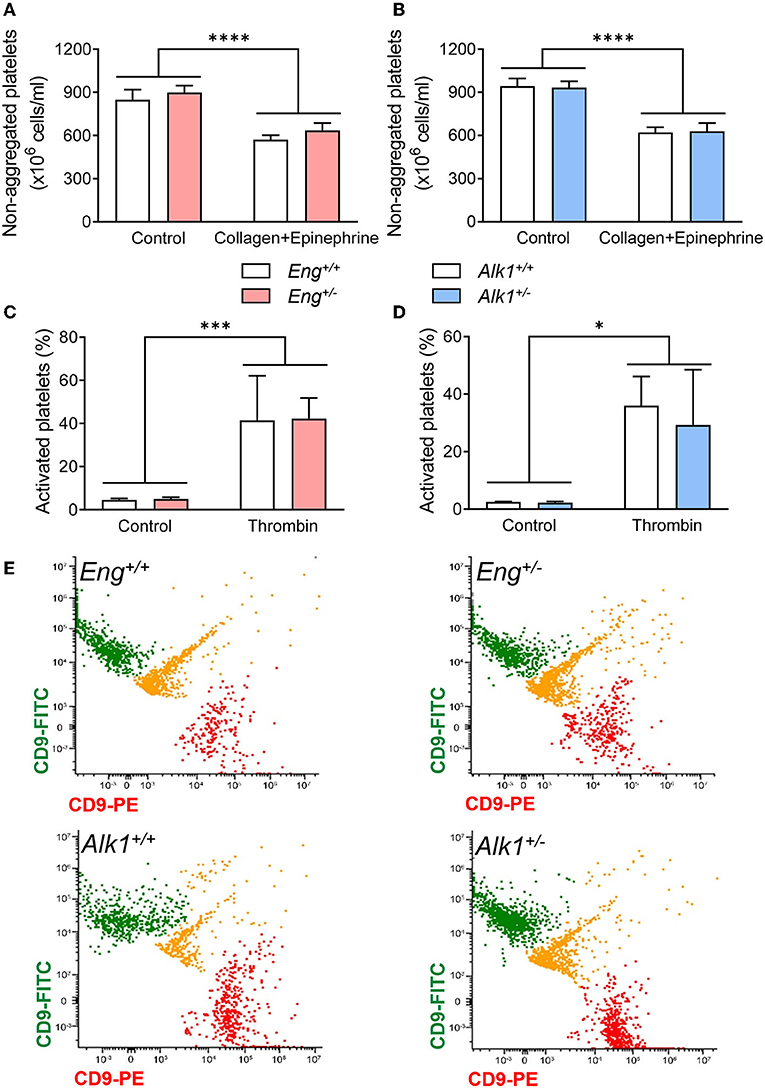
Figure 3. (A,B) Number of non-aggregated platelets per mL before and after the induction of a thrombogenic event with a bolus of collagen and epinephrine in the HHT-1 murine model (A), Eng+/− mice (n = 8) and their respective controls Eng+/+ (n = 9); and in the HHT-2 murine model (B), Alk1+/− mice (n = 10) and their respective controls Alk1+/+ (n = 14). (C,D) Percentage of activated platelets, determined by flow cytometry, after treatment with thrombin of diluted blood from Eng+/− mice (n = 7 mice) and their respective controls Eng+/+ (n = 6 mice) (C) or from Alk1+/− mice (n = 6 mice) and their controls Alk1+/+ (n = 5 mice) (D). (E) Representative images, from 3 independent experiments, of flow cytometry analysis of platelet aggregation after thrombin stimulation of diluted blood from Eng+/+, Eng+/−, Alk1+/+, and Alk1+/−. Platelets are divided into 2 populations that are labeled with PE or with FITC bound in both cases to CD9. If the thrombin stimulus stimulates aggregation, green and red colocalization events are observed, labeled in yellow. Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
We confirmed these results by in vitro experiments performed with platelets isolated from Eng+/− or Alk1+/− mice. As shown in Figure 3, platelets from both mice are able to activate (Figures 3C,D) or aggregate (Figures 3E,F) in response to thrombin, with no differences in these processes between Eng- or Alk1-deficient mice and their respective controls.
Because the excessive bleeding in Eng+/− and Alk1+/− mice does not appear to be due to a defect in coagulation, a defective contractile response or defects in platelet function, we posit that HHT proteins, predominantly expressed in endothelial cells, could be involved in later stage endothelium-dependent processes of hemostasis, namely thrombus stabilization or fibrinolysis.
Endoglin Deficiency Impairs Platelet-Endothelium Adhesion and Thrombus Stabilization
Although the quiescent endothelium is remarkably refractory to platelet adhesion, platelet-endothelium adhesion can occur after endothelial stimulation and this mechanism is important for proper thrombus stabilization. Recently, we have described that endoglin may participate in this interaction between platelets and endothelium through integrin binding (17). We therefore analyzed whether Eng, or Alk1, heterozygosity in the endothelium was causing reduced platelet adhesion. To this end, monolayers of MLECs from Eng+/− and Alk1+/− animals and their respective control mice, were incubated for 30 min with calcein-labeled platelets and the integrin activator CXCL12 and binding of platelets was measured. We found that platelets displayed a lower adhesion to Eng+/− MLECs compared to controls (Eng+/+ MLECs) (Figures 4A,B). In addition, to confirm that the effect is dependent on endoglin expression, we analyzed platelet adhesion to MLECs from ENG+ mice, that constitutively and ubiquitously express high levels of endoglin. Our results show that adhesion to ENG+ MLECs was significantly higher than to WT MLECs (Figures 4C,D). Interestingly, this result does not hold in Alk1-deficient cells, since no significant differences were found when we compared platelet adhesion to Alk1+/− vs. Alk1+/+ MLECs (Figures 4E,F).
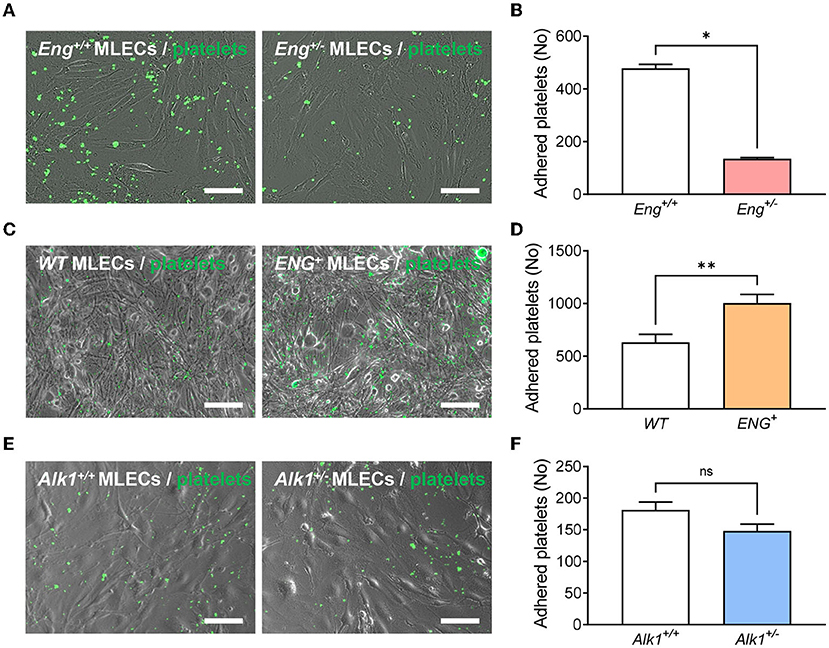
Figure 4. (A) Representative images of calcein-labeled platelets (green fluorescence) adhered on a monolayer of endothelial cells (phase contrast image), isolated from Eng+/+ or Eng+/− mice, treated with CXCL12. (B) Number of adherent platelets after 30 min of incubation of platelets on CXCL12-activated endothelial cells isolated from Eng+/+ or Eng+/− mice (n = 4). (C) Representative images of calcein-labeled platelets (green fluorescence) adhered on a monolayer of endothelial cells (phase contrast image), isolated from WT or ENG+ mice, treated with CXCL12. (D) Number of adherent platelets after 30 min of incubation of platelets on CXCL12-activated endothelial cells isolated from WT or ENG+ mice (n = 9). (E) Representative images of calcein-labeled platelets (green fluorescence) adhered on a monolayer of endothelial cells (phase contrast image), isolated from Alk1+/+ or Alk1+/− mice, treated with CXCL12. (F) Number of adherent platelets after 30 min of incubation of platelets on CXCL12-activated endothelial cells isolated from Alk1+/+ or Alk1+/− mice (n = 5). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Next, we assessed the role of Eng in resistance to shear under flow, using microfluidic devices. Platelets were allowed to adhere to endothelial cells, previously stimulated for 10 min with the integrin activator CXCL12, and the resistance to perfusion at 2 dynes/cm2 for 2 min was measured (Supplementary Figures S3A,B, Supplementary Videos 1, 2). We found that platelets resistance to flow was strongly reduced in Eng+/− compared to WT endothelial monolayers. Similar to the static adhesion results, the resistance to shear flow of platelets attached to the Alk1+/− MLECs monolayer is comparable to that of WT MLECs (Supplementary Figures S3C,D and Supplementary Videos 3, 4). These results suggest that the presence of Eng in the monolayer of MLECs contributes to guarantee a platelets' firm adhesion and this may be critical to ensure proper thrombus formation and stabilization.
Next, we analyzed thrombus formation and stabilization by measuring blood flow in the carotid artery after FeCl3 administration on the external carotid wall. FeCl3 diffuses to the endothelial layer and generates a damage capable of activating the hemostasis response and generating a thrombus that reduces and even blocks blood flow. Our results show that stable occlusion, determined as the reduction of carotid blood flow to <5% of baseline blood flow, was delayed in Eng+/− compared to WT mice (Figures 5A,B). This is in agreement with the above results on platelet adhesion to the endothelium. In order to further assess the role of endoglin in these hemostasis functions, we analyzed thrombus formation in the mouse model overexpressing human endoglin (ENG+). We found that wound closure was significantly faster in ENG+ mice, which present constitutively increased levels of endoglin, than in control animals (Figures 5C,D), suggesting that endoglin is directly related to the formation of a stable thrombus and positively contributes to hemostasis. To rule out that this effect on stable thrombus formation was not due to endoglin overexpression leading to changes in platelet activity, we analyzed in vivo platelet activation and aggregation by measuring (i) non-aggregated platelets after the induction of a thrombogenic event in ENG+ mice and (ii) in vitro platelet aggregation in response to thrombin. Our results show that endoglin overexpression did not alter platelet activation or aggregation either in vivo (Supplementary Figure S4A) or in vitro (Supplementary Figures S4B,C). In addition, we analyzed the effect of endoglin overexpression on the bleeding time of these mice and also observed no difference between ENG+ and control mice (Supplementary Figure S4D).
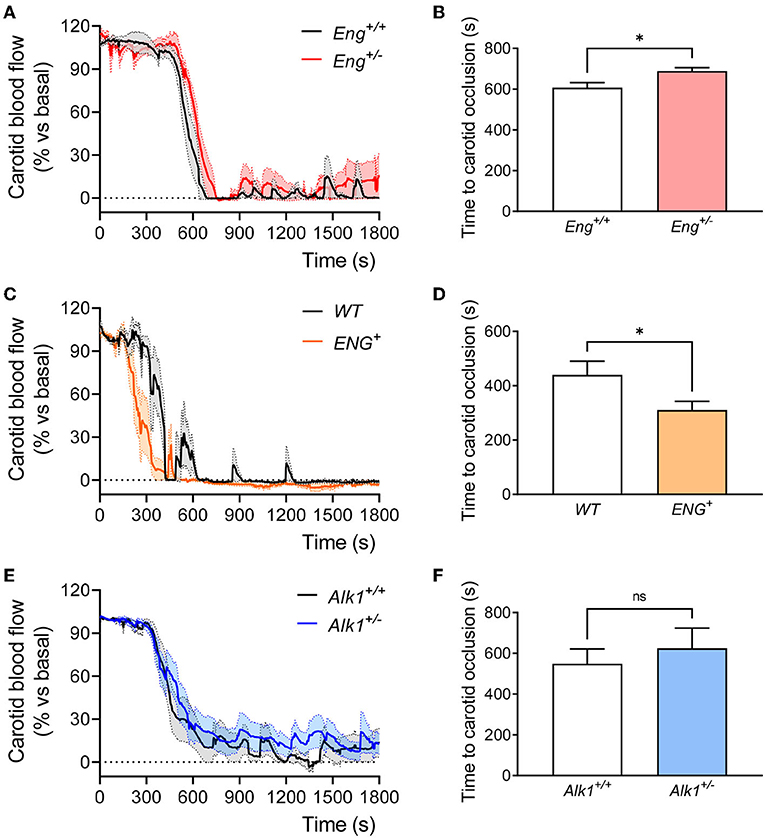
Figure 5. (A) Evolution of carotid blood flow after application of FeCl3 in carotid arteries of Eng+/+ (n = 5) and Eng+/− (n = 5) mice. Percentage of baseline blood flow is represented as the mean of all mice (thick line) and the standard error of the mean (dotted line and shaded area). (B) Time to complete a stable carotid occlusion after FeCl3 administration in carotids of Eng+/− mice (n = 5) and their controls (n = 5). (C) Evolution of carotid blood flow after application of FeCl3 in carotid arteries of WT (n = 5) and ENG+ (n = 6) mice. Percentage of baseline blood flow is represented as the mean of all mice (thick line) and the standard error of the mean (dotted line and shaded area). (D) Time to complete a stable carotid occlusion after FeCl3 administration in carotids of ENG+ mice (n = 6) and their WT controls (n = 5). (E) Evolution of carotid blood flow after application of FeCl3 in carotid arteries of Alk1+/+ (n = 10) and Alk1+/− (n = 12) mice. Percentage of baseline blood flow is represented as the mean of all mice (thick line) and the standard error of the mean (dotted line and shaded area). (F) Time to complete a stable carotid occlusion after FeCl3 administration in carotids of Alk1+/− mice (n = 12) and their controls (n = 10). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
At variance with the Eng+/− mice, no significant differences between Alk1+/− mice and controls were observed regarding thrombus formation and carotid closure (Figures 5E,F). This is consistent with the absence of differences in platelet adhesion to endothelial cells when comparing Alk1+/− vs. Alk1+/+ MLECs (Figures 4E,F and Supplementary Figures S3C,D).
These results suggest that endoglin haploinsufficiency, but not Alk1 haploinsufficiency, leads to unstable thrombus formation that may explain the occurrence of rebleeding, whereas in the Alk1-deficient condition, the difficulty in stopping bleedings seems to be due to a different mechanism.
Alk1 Deficiency Stimulates Thrombus Fibrinolysis
Fibrinolysis is involved in the process of removing the clot formed upon hemostasis activation. We analyzed the main components of the fibrinolysis cascade in plasma samples from Eng+/−, Alk1+/− and their control mice, collected immediately before (basal levels) and 30 min, 6 or 24 h after an acute thrombotic event: the intravenous administration of collagen and epinephrine as above described. These collection times were selected by choosing the periods where the highest concentrations of each of the components are expected to be found.
The basal plasminogen concentration in plasma was similar in Eng+/− and WT mice, and no significant changes were observed in both groups 30 min after the induction of the thrombotic event (Figure 6A). However, 6 h after the thrombotic event, tissue plasminogen activator (t-PA) (Figure 6B) and its inhibitor PAI-1 (Figure 6C) levels significantly increased in Eng+/− mice while in the WT this increment is lower and not statistically significant. Finally, we analyzed the plasmatic levels of D-dimer, a breakdown product of the blood clot fibrinolysis. Our results show that 24 h after the thrombotic event, D-dimer levels do not increase significantly in either WT or Eng+/−mice (Figure 6D). Therefore, Eng deficiency causes a greater release not only of t-PA, but also of its inhibitor PAI-1 after the occurrence of a thrombotic event; these changes being associated with no differences in in the plasma levels of D-dimer.
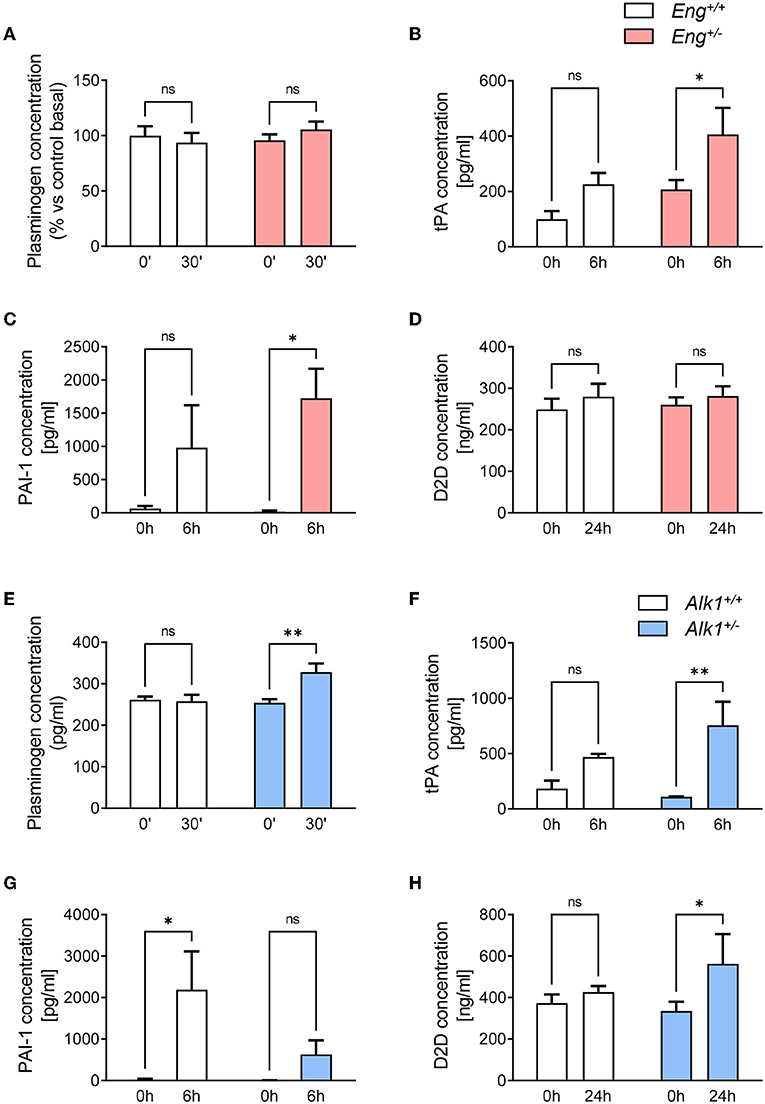
Figure 6. (A) Plasma levels of plasminogen at baseline and 30 min after a thrombogenic event in Eng+/+ (n = 5) and Eng+/− (n = 5) mice. (B) Plasma levels of t-PA at baseline and 6 h after a thrombogenic event in Eng+/+ (n = 5) and Eng+/− (n = 5) mice. (C) Plasma levels of PAI-1 at baseline and 6 h after a thrombogenic event in Eng+/+ (n = 5) and Eng+/− (n = 5) mice. (D) Plasma levels of D-dimer at baseline and 24 h after a thrombogenic event in Eng+/+ (n = 5) and Eng+/− (n = 5) mice. (E) Plasma levels of plasminogen at baseline and 30 min after a thrombogenic event in Alk1+/+ (n = 5) and Alk1+/− (n = 5) mice. (F) Plasma levels of t-PA at baseline and 6 h after a thrombogenic event in Alk1+/+ (n = 5) and Alk1+/− (n = 5) mice. (G) Plasma levels of PAI-1 at baseline and 6 h after a thrombogenic event in Alk1+/+ (n = 5) and Alk1+/− (n = 5) mice. (H) Plasma levels of D-dimer at baseline and 24 h after a thrombogenic event in Alk1+/+ (n = 5) and Alk1+/− (n = 5) mice. Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Interestingly, the fibrinolytic response of the HHT-2 animal model was substantially different from that of HHT-1 mice. Thus, after the thrombotic event induced in Alk1+/− mice, a significant increase in plasminogen levels at 30 min was observed, whereas the control group remained unresponsive (Figure 6E). Similar to the HHT-1 model, 6 h after activating thrombogenesis, there was a statistically significant increase in plasma levels of t-PA in Alk1+/− mice, but not in WT mice (Figure 6F). However, contrary to what was observed in Eng-deficient mice, in Alk1+/− mice the increase in PAI-1 levels did not reach statistical significance and was clearly much lower than the significant increase that occurred in control mice (Figure 6G). Analysis of D-dimer levels 24 h after the thrombotic event showed a significant increase in plasma levels of Alk1+/− mice with no statistical differences in control mice (Figure 6H). In summary, the generation of a thrombotic event in the murine model of HHT-2 results in a significant increase in plasminogen and t-PA. However, the increment in PAI-1 levels is much lower than in controls. Finally, an increase in D-dimer levels is detected that is not observed in controls. Together, these results support the existence of a greater fibrinolysis activity in Alk1+/− mice.
Discussion
The patterns of presentation of HHT are highly variable, but spontaneous and recurrent nosebleeds is the most common clinical manifestation, affecting more than 90% of patients, and is usually the first to appear; thus, it is one of the established Curaçao Criteria for the diagnosis of HHT. In addition, over the progression of the disease, 15–20% of individuals eventually develop recurrent bleeding from the gastrointestinal tract (36). Nosebleeds, GI bleeds, or both, cause a large proportion of HHT patients to suffer chronic anemia, and the need to receive periodic blood transfusions in those patients unresponsive to iron supplementation treatments (37).
Currently it is widely considered that the increased bleeding of HHT patients is due to the abnormalities and fragility of the vasculature (35, 38). For this reason, most of HHT-related studies have focused on angiogenesis and the defective structure of telangiectases. However, whereas the fragility of telangiectases could explain the higher frequency of hemorrhages, it is unclear why bleeds are so intense, especially considering that other pathologies that also present telangiectases, lack the intense bleeding characteristic of HHT (39–44). It should be noted that most other pathologies which present telangiectases rarely display them in nasal and GI mucosa, as skin telangiectases are more usually frequent, which may explain this difference. However, also among HHT patients they show a wide variation in bleeding severity, which is not completely explained by the well-known factors, such as number and size of telangiectases. Therefore, one can assume that, in addition to the fragility of the vascular lesions, an alteration in hemostasis could also participate in the intense bleeding observed in HHT patients.
Some authors have already proposed that there may be alterations in phases of primary and secondary hemostasis associated with HHT even increasing the risk of thrombosis, but most of them are old studies and there is considerable controversy among them (5, 45–50). In this regard, our results demonstrate that the presence of increased bleeding in HHT patients is not associated with lengthened clotting times or lower clotting factor activity, as suggested in the classical description of the disease (35). In that context, to elucidate if the abnormally increased bleeding intensity of HHT patients may be due to an alteration of hemostasis in non-coagulation related phases, here we have analyzed in detail the hemostasis system in two murine models of the disease. We have used mice that are haploinsufficient for the endoglin gene (Eng+/−) or for the Alk1 gene (Alk1+/−). These mice are the closest experimental models of HHT-1 and HHT-2 patients in terms of genotype (19). Of note, the HHT phenotype of these animals is highly dependent on the mouse strain. Thus, Eng+/− and Alk1+/− animals with 129/Ola background develop highly penetrant and frequent clinical manifestations similar to those of HHT, such as dilated vessels, nosebleeds, telangiectases and vascular lesions in the liver, the nail bed, ears, intestine and skin (24, 51, 52). By contrast, in Eng+/− and Alk1+/− animals with a C57BL/6J background the penetrance of the HHT phenotype is incomplete and its frequency is negligible (24, 51). Therefore, Eng+/− and Alk1+/− animals in the C57BL/6J background, as used in this work, represent a useful model to analyze the hemostasis system in the absence of vascular lesions that could affect the bleeding. Regardless, the findings obtained using the C57BL/6J genetic background should be confirmed in future studies using other genetic backgrounds.
When analyzing in the HHT animal models the bleeding from a normal vessel, that does not involve telangiectases or arteriovenous malformations, we observe that both murine models (Eng+/− and Alk1+/−) have more abundant rebleeding than controls (Figure 2). These results suggest an alteration in the mechanisms of hemostasis that may account for the difficulty in stopping the bleeding in HHT patients, prompting us to analyze the different phases involved in hemostasis, e.g., vessel spasm, formation of platelet plug, thrombus stabilization and thrombus dissolution (fibrinolysis).
Previous studies have described the role of endoglin or ALK1 in vascular tone, where the endothelium has a main role in the secretion of vasoactive molecules (53). Thus, Eng+/− mice present reduced nitric oxide-dependent vasodilation (27) and Alk1+/− mice show increased arterial pressure due to sympathetic activation (54). Moreover, heterozygous mutations in either ENG or ALK1 in humans are associated, in a few cases, with pulmonary arterial hypertension due their functional link with the BMP type II receptor, the main target responsible for most family cases of pulmonary arterial hypertension (46, 55). Because these findings suggest that a deficient expression of endoglin or ALK1 contributes to the deterioration of vascular tone, we analyzed whether the vasoconstrictor response of aortic rings was impaired as a consequence of the genetic alteration in Eng+/− and Alk1+/− mice. Our results, using three vasoactive agents present in the hemostatic response, only exhibited an increased contraction in response to serotonin in the aortic rings from Eng+/− mice when compared with the control, whereas no significant impairment in the contractile response of aortic rings from the two HHT mouse models with respect to their controls was observed. These results support that the greater bleeding of Eng+/− and Alk1+/− mice is not due to less vascular spasm. They also reinforce the idea that the effect of these genes on the regulation of vascular tone is primarily due to alterations in vasodilation rather than vascular contraction, although it might be interesting to investigate the outcome in response to serotonin in endoglin-deficient mice in case it contributes to the relationship between endoglin and pulmonary hypertension.
With regard to thrombus formation, we show that, compared to control animals, Eng+/− mice take longer time to occlude the carotid artery after FeCl3-induced local thrombotic response. This effect does not seem to depend on the ability of platelets to activate and form aggregates, since no differences in these processes are observed between Eng+/− mice and controls. Therefore, as previously hypothesized (17), Eng deficiency seems to be involved in thrombus instability. This is corroborated by platelet adhesion assays on monolayers of Eng+/+ and Eng+/− MLECs, which show that Eng deficiency in endothelial cells compromises platelet adhesion and their resistance to shear stress, whereas endoglin-overexpressing MLECs show increased platelet adhesion to these endothelial cells, supporting the adhesive role of endoglin in the interaction between platelets and activated endothelium, as previously proposed (17). This conclusion is reflected in the fact that mice with overexpression of endoglin are able to generate a thrombus stable enough to occlude the carotid in a shorter time than WT mice. Interestingly, this does not translate into a shorter bleeding time, so it seems that the physiological levels of Eng are sufficient to ensure a correct hemostasis.
Surprisingly, a different situation was found in Alk1+/− mice, the murine model for HHT-2, since, in this case the excessive bleeding does not appear to be due to alterations in platelet-endothelium adhesion. Similarly to Eng-deficient mice, the rebleeds of Alk1-deficient mice are more severe than those in controls, but in this case, in addition to finding no differences in platelet activation or aggregation, we also found no differences in platelet adhesion to activated endothelium or carotid occlusion time after FeCl3 application. Interestingly, when analyzing the fibrinolysis system, we found that the stimulation of a thrombotic event in Alk1+/− mice result in higher plasma levels of plasminogen and its activator t-PA, and lower levels of PAI-1, the inhibitor of t-PA. These results suggest that Alk1 deficiency may result in an increased fibrinolysis and higher degradation of the thrombus that may account for the increased levels of D-Dimer found in these animals and severe rebleedings. Of note, a relationship between ALK1 deficiency and increased t-PA has been previously reported (25). Thus, in line with our results, abnormally high levels of t-PA have been shown in Alk1 knock-out mice (25) and in telangiectases of HHT patients (56, 57). However, although there is sufficient evidence that ALK1 deficiency leads to an increase in PAI-1 (25, 58), our results point in the opposite direction. The studies that relate ALK1 deficiency with PAI-1 are studies on TGFβ1 signaling pathways in which 2 pathways are described with opposite effects depending on whether it signals through ALK1 or ALK5, both TGFβ1 type I receptors. Thus, if there is ALK1 deficiency, signaling would be mainly through ALK5, giving rise to different cellular responses. In this context, PAI-1 has traditionally been used as a reporter of the activity of the TGFβ1-ALK5 pathway, so it seems that ALK1 deficiency would have to cause an increase in PAI-1. However, these studies did not analyze the accumulation of PAI-1 in plasma but its cellular expression in response to treatments and therefore we consider that our results do not contradict the literature but may be a consequence of the excessively high levels of t-PA. The way PAI-1 inhibits t-PA is by forming covalent bonds that are rapidly degraded (59, 60). Thus, if t-PA levels are very high, as they are in ALK1-deficient mice, the rise in PAI-1 would be masked by its increased binding to t-PA and its rapid elimination.
Taken together, our findings allow us to suggest the model presented in Figure 7. In a condition of endoglin deficiency, as in HHT-1 patients or Eng+/− mice, platelets interact with each other and with the sub-endothelium to generate an apparently correct platelet plug. However, the deficient expression of endoglin in the endothelium results in suboptimal platelet-endothelium interactions, impairing the formation of a robust and stable thrombus. This unstable thrombus could be torn off and dragged by the mechanical force exerted by the bloodstream, which would result in the reappearance of the hemorrhage (Figure 7A). After the onset of a thrombogenic event, the fibrinolysis cascade is set in motion, which allows fibrin to be degraded by activation of plasminogen to plasmin by t-PA. Under physiological conditions a significant increase in the t-PA inhibitor, PAI-1, prevents excessive fibrinolysis that could compromise the integrity of the newly formed thrombus. However, ALK1 deficiency results in a much greater increase in t-PA that PAI-1 is unable to effectively control. This results in increased fibrin degradation leading to instability of the newly formed thrombus and increased risk of rebleeding (Figure 7B).
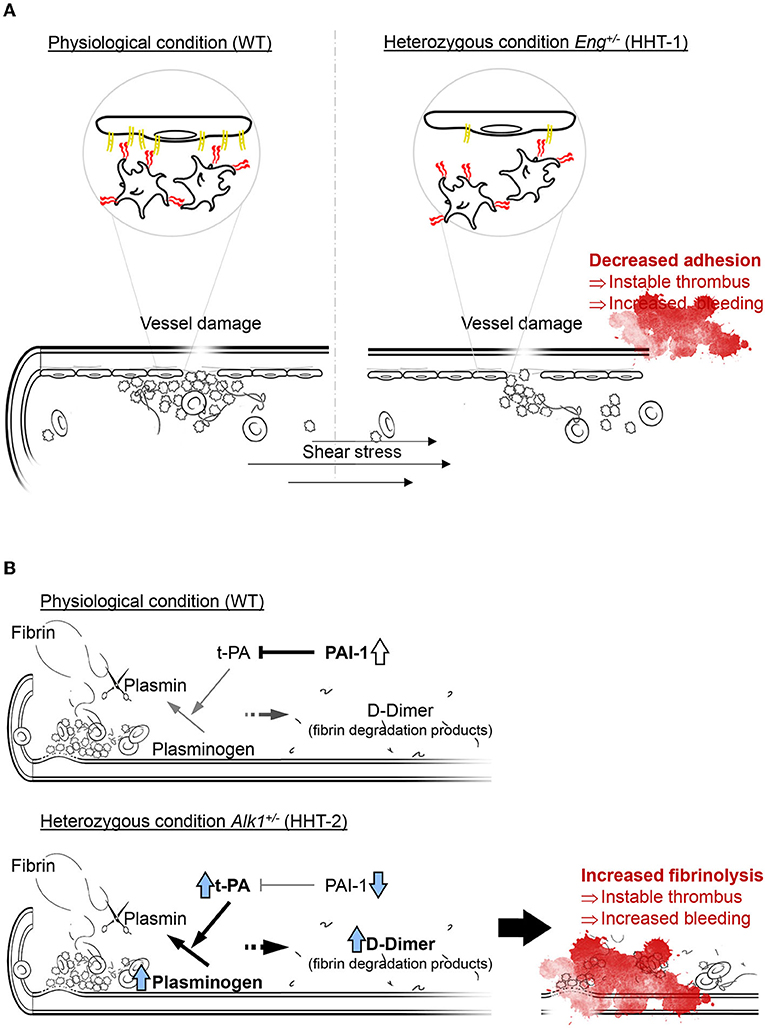
Figure 7. (A) In normal conditions platelets rolling on endothelial cells is effective and the thrombus generated is stable, while a loss on endoglin in the endothelium mean less effective platelets adhesion and an instable thrombus that can be carried away by the bloodstream, compromising thrombosis and increasing bleeding. Thus, endoglin-deficient mice need more time to generate a thrombus large and stable enough not to be entrained. (B) Fibrinolysis depends on the activity of t-PA catalyzing the conversion of plasminogen to plasmin, which is the enzyme responsible for fibrin degradation and thrombus disruption. In physiological conditions, there is an increase in PAI-1 that inhibits the function of t-PA and allows stabilization of the newly formed thrombus. However, ALK1 deficiency causes plasminogen and mainly t-PA levels to be much higher, so that PAI-1 cannot inhibit it and fibrinolysis is increased, which we can detect by the increase in D-dimer.
In summary, our results support the hypothesis that in HHT-1 and HHT-2 mouse models, Eng and Alk1 haploinsufficiency leads to a common phenotype of impaired hemostasis but through two different mechanisms: Eng haploinsufficiency affects thrombus stabilization due to deficient interactions between platelets and endothelial cells, while Alk1 deficiency generates an upregulation of the fibrinolysis system. This contribution could open new therapeutic approaches not only in the management of nosebleeds and gastrointestinal bleeding in HHT patients, but also to the understanding of the effect of other endothelium- or thrombosis-related pathologies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité de Ética del Complejo Asistencial Universitario de Salamanca. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Comité de Bioética de la Universidad de Salamanca.
Author Contributions
CE-T, ER, CO-I, MP-G, JMB, JG-P, AR-B, CB, JL-N, and MP: conceptualization. CE-T, ER, MS, JMB, JG-P, and MP: methodology. CE-T, ER, MP-G, and MP: formal analysis. CE-T, ER, CO-I, MP-G, MS, and MP: investigation. ER, MP-G, MS, JG-P, AR-B, CB, JL-N, and MP: resources. ER and MP: writing—original draft preparation. CE-T, ER, and MP: visualization. MP: supervision and project administration. ER, CB, and MP: funding acquisition. All authors: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Junta de Castilla y León (BIO/SA70/14, GRS2135/A/2020, and GRS2314/A/2021), Fundación Mutua Madrileña (FMM AP172142019), the Instituto de Salud Carlos III (PI16/00460 and PI19/01630, co-funded by FEDER), and Consejo Superior de Investigaciones Científicas (CSIC; 201920E022 to CB). CO-I was supported by a contract from the Ministerio de Economía y Competitividad of Spain. CE-T was a fellow from the Fundación Miguel Casado San José.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.871903/full#supplementary-material
Supplementary Figure S1. (A–H) Plasma activity of factors involved in coagulation cascade in controls (n = 10), HHT-1 (n = 5), and HHT-2 (n = 21) patients. The factors analyzed were Factor II (A), Factor V (B), Factor VII (C), Factor VIII (D), Factor IX (E), Factor X (F), Factor XI (G), and Factor vW (H). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Supplementary Figure S2. (A–F) Vascular contraction in response to increasing concentrations of vasoconstrictor agents, expressed as % of contraction compared with KPSS-induced contraction, of aortic rings isolated from HHT murine models and its controls. Mean ± SEM for each concentration and nonlin fit curve, calculated with the model: log(agonist) vs. response (three parameters) of Graphpad 9, are represented. (A,B) Vascular contraction in response to norepinephrine of aortic rings isolated from Eng+/− mice (n = 20 aortic rings from 5 mice) and its respective controls Eng+/+ (n = 20 aortic rings from 5 mice) (A) or from Alk1+/− mice (n = 10 aortic rings from 5 mice) and its controls Alk1+/+ (n = 10 aortic rings from 5 mice) (B). (C,D) Vascular contraction in response to serotonin of aortic rings isolated from Eng+/− mice (n = 10 aortic rings from 5 mice) and its respective controls Eng+/+ (n = 10 aortic rings from 5 mice) (C) or from Alk1+/− mice (n = 12 aortic rings from 5 mice) and its controls Alk1+/+ (n = 12 aortic rings from 5 mice) (D). (E,F) Vascular contraction in response to U-46619 (thromboxane analog) of aortic rings isolated from Eng+/− mice (n = 10 aortic rings from 5 mice) and its respective controls Eng+/+ (n = 10 aortic rings from 5 mice) (E) or from Alk1+/− mice (n = 8 aortic rings from 5 mice) and its controls Alk1+/+ (n = 8 aortic rings from 5 mice) (F). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Supplementary Figure S3. (A) Representative images of calcein-labeled platelets (green fluorescence) adhered on a monolayer of endothelial cells, isolated from Eng+/+ or Eng+/− mice, treated with CXCL12 before (left panel) or after (right panel) the application of a flow of 2 dyne/cm2 (yellow arrow: flow direction). After applying shear stress most platelets are removed, but some withstand the drag and remain attached to the endothelium (white arrowheads). (B) Fluorescence intensity of adhered platelets on CXCL12-activated endothelial cells isolated from Eng+/+ or Eng+/− mice after the application of a flow of 2 dyne/cm2 for 2 min (n = 3). (C) Representative images of calcein-labeled platelets (green fluorescence) adhered on a monolayer of endothelial cells, isolated from Alk1+/+ or Alk1+/− mice, treated with CXCL12 before (left panel) or after (right panel) the application of a flow of 2 dyne/cm2 (yellow arrow: flow direction). After applying shear stress most platelets are removed, but some withstand the drag and remain attached to the endothelium (white arrowheads). (D) Fluorescence intensity of adhered platelets on CXCL12-activated endothelial cells isolated from Alk1+/+ or Alk1+/− mice after the application of a flow of 2 dyne/cm2 for 2 min (n = 3). Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
Supplementary Figure S4. (A) Number of non-aggregated platelets per mL before and after the induction of a thrombogenic event with a bolus of collagen and epinephrine in ENG+ mice (n = 17) and its respective WT controls (n = 17). (B) Total bleeding time, calculated as the sum of first bleeding time and rebleeding time, in WT (n = 25) and ENG+ mice (n = 25). (C) Representative images, from 3 independent experiments, of flow cytometry analysis of platelet aggregation after thrombin stimulation of diluted blood from WT and ENG+ mice. Results were considered statistically significant if p < 0.05 (ns: non-significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
References
1. Begbie ME, Wallace GMF, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J. (2003) 79:18–24. doi: 10.1136/pmj.79.927.18
2. Cottin V, Chinet T, Lavolé A, Corre R, Marchand E, Reynaud-Gaubert M, et al. Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia. Medicine. (2007) 86:1–17. doi: 10.1097/MD.0b013e31802f8da1
3. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. (2011) 48:73–87. doi: 10.1136/jmg.2009.069013
4. Brinjikji W, Iyer VN, Wood CP, Lanzino G. Prevalence and characteristics of brain arteriovenous malformations in hereditary hemorrhagic telangiectasia: a systematic review and meta-analysis. J Neurosurg. (2017) 127:302–10. doi: 10.3171/2016.7.JNS16847
5. Sureda A, César J, García Frade LJ, García Avello A, Fernández Fuentes I, Navarro JL. Hereditary hemorrhagic telangiectasia: analysis of platelet aggregation and fibrinolytic system in seven patients. Acta Haematol. (1991) 85:119–23. doi: 10.1159/000204872
6. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. (2009) 17:860–71. doi: 10.1038/ejhg.2009.35
7. Dheyauldeen S, Abdelnoor M, Bachmann-Harildstad G. The natural history of epistaxis in patients with hereditary hemorrhagic telangiectasia in the norwegian population: a cross-sectional study. Am J Rhinol Allergy. (2011) 25:214–8. doi: 10.2500/ajra.2011.25.3616
8. Plauchu H, De Chadarévian J-P, Bideau A, Robert J-M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. (1989) 32:291–7. doi: 10.1002/ajmg.1320320302
9. Silva BM, Hosman AE, Devlin HL, Shovlin CL. Lifestyle and dietary influences on nosebleed severity in hereditary hemorrhagic telangiectasia. Laryngoscope. (2013) 123:1092–9. doi: 10.1002/lary.23893
10. Pahl K, Choudhury A, Kasthuri RS. Causes and severity of anemia in hereditary hemorrhagic telangiectasia. Blood. (2016) 128:3776. doi: 10.1182/blood.V128.22.3776.3776
11. Weingarten TN, Hanson JW, Anusionwu KO, Moncrief ML, Opdhl TJ, Schneider DD, et al. Management of patients with hereditary hemorrhagic telangiectasia undergoing general anesthesia: a cohort from a single academic center's experience. J Anesth. (2013) 27:705–11. doi: 10.1007/s00540-013-1601-0
12. Jackson SB, Villano NP, Benhammou JN, Lewis M, Pisegna JR, Padua D. Gastrointestinal manifestations of hereditary hemorrhagic telangiectasia (HHT): a systematic review of the literature. Dig Dis Sci. (2017) 62:2623–30. doi: 10.1007/s10620-017-4719-3
13. Rosenberg T, Fialla A, Kjeldsen J, Kjeldsen A. Does severe bleeding in HHT patients respond to intravenous bevacizumab? Review of the literature and case series. Rhinology. (2019) 57:242–51. doi: 10.4193/Rhin18.289
14. Lebrin F, Srun S, Raymond K, Martin S, Van der Brink S, Freitas C, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. (2010) 16:420–8. doi: 10.1038/nm.2131
15. Buscarini E, Botella LM, Geisthoff U, Kjeldsen AD, Mager HJ, Pagella F, et al. Safety of thalidomide and bevacizumab in patients with hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. (2019) 14:1–14. doi: 10.1186/s13023-018-0982-4
16. Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist's perspective. Haematologica. (2018) 103:1433–43. doi: 10.3324/haematol.2018.193003
17. Rossi E, Pericacho M, Bachelot-Loza C, Pidard D, Gaussem P, Poirault-Chassac S, et al. Human endoglin as a potential new partner involved in platelet–endothelium interactions. Cell Mol Life Sci. (2018) 75:1269–84. doi: 10.1007/s00018-017-2694-7
18. Rossi E, Bernabeu C, Smadja DM. Endoglin as an adhesion molecule in mature and progenitor endothelial cells: a function beyond TGF-β. Front Med. (2019) 6:10. doi: 10.3389/fmed.2019.00010
19. Tual-Chalot S, Oh SP, Arthur HM. Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front Genet. (2015) 6:25. doi: 10.3389/fgene.2015.00025
20. Hoag JB, Terry P, Mitchell S, Reh D, Merlo CA. An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope. (2010) 120:838–43. doi: 10.1002/lary.20818
21. Baker P, Platton S, Gibson C, Gray E, Jennings I, Murphy P, et al. Guidelines on the laboratory aspects of assays used in haemostasis and thrombosis. Br J Haematol. (2020) 191:347–62. doi: 10.1111/bjh.16776
22. Bastida JM, del Rey M, Lozano ML, Sarasquete ME, Benito R, Fontecha ME, et al. Design and application of a 23-gene panel by next-generation sequencing for inherited coagulation bleeding disorders. Haemophilia. (2016) 22:590–7. doi: 10.1111/hae.12908
23. Bastida Bermejo JM, González-Porras JR, Jiménez C, Benito R, Ordoñez GR, Álvarez-Román MT, et al. Application of a molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes. Thromb Haemost. (2017) 117:66–74. doi: 10.1160/TH16-05-0375
24. Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. (1999) 104:1343–51. doi: 10.1172/JCI8088
25. Oh SP, Seki T, Goss KA, Imamura T, Donahoe PK, Li L, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. (2000) 97:2626–31. doi: 10.1073/pnas.97.6.2626
26. Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. (2010) 5:1518–34. doi: 10.1038/nprot.2010.113
27. Jerkic M, Rivas-Elena JV, Prieto M, Carrón R, Sanz-Rodríguez F, Pérez-Barriocanal F, et al. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. (2004) 18:609–11. doi: 10.1096/fj.03-0197fje
28. Oujo B, Muñoz-Félix JM, Arévalo M, Núñez-Gómez E, Pérez-Roque L, Pericacho M, et al. L-Endoglin overexpression increases renal fibrosis after unilateral ureteral obstruction. PLoS ONE. (2014) 9:e110365. doi: 10.1371/journal.pone.0110365
29. Muczynski V, Casari C, Moreau F, Aymé G, Kawecki C, Legendre P, et al. A factor VIII–nanobody fusion protein forming an ultrastable complex with VWF: effect on clearance and antibody formation. Blood. (2018) 132:1193–7. doi: 10.1182/blood-2018-01-829523
30. Laurent PA, Hechler B, Solinhac R, Ragab A, Cabou C, Anquetil T, et al. Impact of PI3Kα (phosphoinositide 3-kinase alpha) inhibition on hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. (2018) 38:2041–53. doi: 10.1161/ATVBAHA.118.311410
31. De Cárcer G, Wachowicz P, Martínez-Martínez S, Oller J, Méndez-Barbero N, Escobar B, et al. Plk1 regulates contraction of postmitotic smooth muscle cells and is required for vascular homeostasis. Nat Med. (2017) 23:964–74. doi: 10.1038/nm.4364
32. van Asten I, Schutgens R, Urbanus R. Toward flow cytometry based platelet function diagnostics. Semin Thromb Hemost. (2018) 44:197–205. doi: 10.1055/s-0038-1636901
33. Ollauri-Ibáñez C, Núñez-Gómez E, Egido-Turrión C, Silva-Sousa L, Díaz-Rodríguez E, Rodríguez-Barbero A, et al. Continuous endoglin (CD105) overexpression disrupts angiogenesis and facilitates tumor cell metastasis. Angiogenesis. (2020) 23:231–47. doi: 10.1007/s10456-019-09703-y
34. Rossi E, Sanz-Rodriguez F, Eleno N, Duwell A, Blanco FJ, Langa C, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. (2013) 121:403–15. doi: 10.1182/blood-2012-06-435347
35. Rendu H. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muqueux. Bull Soc Medicale des Hop Paris. (1896) 13:731–3.
36. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. (2000) 91:66–7. doi: 10.1002/(SICI)1096-8628(20000306)91:1<66::AID-AJMG12>3.0.CO;2-P
37. Geisthoff UW, Nguyen H-L, Röth A, Seyfert U. How to manage patients with hereditary haemorrhagic telangiectasia. Br J Haematol. (2015) 171:443–52. doi: 10.1111/bjh.13606
38. Guttmacher A, Marchuk A, White R. Hereditary hemorrhagic telangiectasia. (1995) 333:918–24. doi: 10.1056/NEJM199510053331407
39. Braverman IM, Keh A, Jacobson BS. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J Invest Dermatol. (1990) 95:422–7. doi: 10.1111/1523-1747.ep12555569
40. Nabatian A, Suchter MF, Milgraum S. Palmar telangiectases as a manifestation of graves disease. Cutis. (2012) 89:84–88. Available online at: https://www.mdedge.com/dermatology/article/68284/palmar-telangiectases-manifestation-graves-disease
41. Wenson SF, Jan F, Sepehr A. Unilateral nevoid telangiectasia syndrome: a case report and review of the literature. Dermatol Online J. (2011) 17:1–2. doi: 10.5070/D39MB4458K
42. Smith PC. Management of reticular veins and telangiectases. Phlebology. (2015) 30:46–52. doi: 10.1177/0268355515592770
43. Yiannakopoulou E. Safety concerns for sclerotherapy of telangiectases, reticular and varicose veins. Pharmacology. (2016) 98:62–9. doi: 10.1159/000445436
44. Casadio R, Santi V, Mirici-Cappa F, Magini G, Cacciari M, Bernardi M, et al. Telangiectasia as a presenting sign of Graves' disease. Horm Res. (2008) 69:189–92. doi: 10.1159/000112593
45. Shovlin CL, Chamali B, Santhirapala V, Livesey JA, Angus G, Manning R, et al. Ischaemic strokes in patients with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia: associations with iron deficiency and platelets. PLoS ONE. (2014) 9:e88812. doi: 10.1371/journal.pone.0088812
46. Hodgson CH, Burchell HB, Good CA, Clagett OT. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous fistula: survey of a large family. N Engl J Med. (1959) 261:625–36. doi: 10.1056/NEJM195909242611301
47. Muckle T. Low in-vivo adhesive-platelet count in Hereditary Haemorrhagic Telangiectasia. Lancet. (1965) 285:169. doi: 10.1016/S0140-6736(65)91133-5
48. Macfarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature. (1964) 202:498–9. doi: 10.1038/202498a0
49. Shovlin CL, Sulaiman NL, Govani FS, Jackson JK, Begbie ME. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): association with venous thromboembolism. Thromb Haemost. (2007) 98:1031–9. doi: 10.1160/TH07-01-0064
50. Livesey J, Manning R, Meek JH, Jackson JE, Kulinskaya E, Laffan MA, et al. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax. (2012) 67:328–33. doi: 10.1136/thoraxjnl-2011-201076
51. Srinivasan S, Hanes MA, Dickens T, Porteous EM, Oh SP, Hale LP, et al. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. (2003) 12:473–82. doi: 10.1093/hmg/ddg050
52. Torsney E, Charlton R, Diamond AG, Burn J, Soames JV, Arthur HM. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation. (2003) 107:1653–7. doi: 10.1161/01.CIR.0000058170.92267.00
53. González-Núñez M, Muñoz-Félix JM, López-Novoa JM. The ALK-1/Smad1 pathway in cardiovascular physiopathology. A new target for therapy? Biochim Biophys Acta Mol Basis Dis. (2013) 1832:1492–510. doi: 10.1016/j.bbadis.2013.05.016
54. Gonzalez-Nunez M, Riolobos AS, Castellano O, Fuentes-Calvo I, Sevilla MA, Oujo B, et al. Heterozygous disruption of activin receptor-like kinase 1 is associated with increased arterial pressure in mice. Dis Model Mech. (2015) 8:1427–39. doi: 10.1242/dmm.019695
55. Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, et al. Mutations of the TGF-β type II receptorBMPR2 in pulmonary arterial hypertension. Hum Mutat. (2006) 27:121–32. doi: 10.1002/humu.20285
56. Kwaan HC, Silverman S. Fibrinolytic activity in lesions of hereditary hemorrhagic telangiectasia. Arch Dermatol. (1973) 107:571–3. doi: 10.1001/archderm.107.4.571
57. Watanabe M, Hanawa S, Morishima T. Fibrinolytic activity in cutaneous lesions of hereditary hemorrhagic telangiectasia. Nihon Hifuka Gakkai Zasshi. (1985) 95:11–6.
58. Goumans M-J, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Djke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. (2002) 21:1743–53. doi: 10.1093/emboj/21.7.1743
59. Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. (2008) 88:887–918. doi: 10.1152/physrev.00033.2007
Keywords: bleeding, hemostasia, Hereditary Hemorrhagic Telangiectasia (HHT), ALK1 (ACVRL1), endoglin (CD105)
Citation: Egido-Turrión C, Rossi E, Ollauri-Ibáñez C, Pérez-García ML, Sevilla MA, Bastida JM, González-Porras JR, Rodríguez-Barbero A, Bernabeu C, Lopez-Novoa JM and Pericacho M (2022) Functional Alterations Involved in Increased Bleeding in Hereditary Hemorrhagic Telangiectasia Mouse Models. Front. Med. 9:871903. doi: 10.3389/fmed.2022.871903
Received: 08 February 2022; Accepted: 06 April 2022;
Published: 19 May 2022.
Edited by:
Giancarlo Castaman, University of Florence, ItalyReviewed by:
Mirjana Jerkic, St Michael's Hospital, CanadaCarlo Sabbà, University of Bari Medical School, Italy
Copyright © 2022 Egido-Turrión, Rossi, Ollauri-Ibáñez, Pérez-García, Sevilla, Bastida, González-Porras, Rodríguez-Barbero, Bernabeu, Lopez-Novoa and Pericacho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Pericacho, cGVyaWNhY2hvQHVzYWwuZXM=
†These authors share first authorship