
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 25 April 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.864554
 Nina O'Connell1
Nina O'Connell1 Jun Oh2
Jun Oh2 Klaus Arbeiter3
Klaus Arbeiter3 Anja Büscher4
Anja Büscher4 Dieter Haffner1
Dieter Haffner1 Jessica Kaufeld5Christine Kurschat6Christoph Mache7
Jessica Kaufeld5Christine Kurschat6Christoph Mache7 Dominik Müller8Ludwig Patzer9
Dominik Müller8Ludwig Patzer9 Lutz T. Weber10
Lutz T. Weber10 Burkhard Tönshoff11Marcus Weitz12
Burkhard Tönshoff11Marcus Weitz12 Katharina Hohenfellner13†
Katharina Hohenfellner13† Lars Pape1,4*†
Lars Pape1,4*†Background: Infantile nephropathic cystinosis (INC) is a rare lysosomal storage disorder resulting in progressive chronic kidney disease (CKD) and a variety of extrarenal manifestations. This orphan disease remains a challenge for patients, their families and health care providers. There is currently no comprehensive study on patients' clinical course in Germany and Austria.
Methods: A retrospective cohort study including 74 patients at eleven centers of care was conducted. Data on time of diagnosis, CKD stage, leukocyte cystine levels (LCL), extrarenal manifestations, and treatment was collected from medical charts and subsequently analyzed using explorative statistics. Age at initiation of kidney replacement therapy (KRT) was evaluated by Kaplan–Meier analyses for different groups of patients.
Results: Patients were diagnosed at a median age of 15 months (IQR: 10–29, range: 0–110), more recent year of birth was not associated with earlier diagnosis. Oral cystine-depleting therapy (i.e., cysteamine) was prescribed at a median dose of 1.26 g/m2 per day (IQR: 1.03–1.48, range: 0.22–1.99). 69.2% of all 198 LCL measurements of 67 patients were within the desired target range (≤ 1 nmol cystine/mg protein). Median time-averaged LCLs per patient (n = 65) amounted to 0.57 nmol cystine/mg protein (IQR: 0.33–0.98, range: 0.07–3.13) when considering only values at least 1 year after initiation of therapy. The overall median height of 242 measurements of 68 patients was at the 7th percentile (IQR: 1–25, range: 1–99). 40.5% of the values were ≤ the 3rd percentile. Patient sex and year of birth were not associated with age at initiation of KRT, but patients diagnosed before the age of 18 months required KRT significantly later than those patients diagnosed at the age of ≥ 18 months (p = 0.033): median renal survival was 21 years (95% CI: 16, -) vs. 13 years (95% CI, 10, -), respectively.
Conclusion: Early diagnosis and initiation of cystine depleting therapy is important for renal survival in children with INC. Cysteamine doses and LCL showed that treatment in this cohort met international standards although there is great interindividual variety. Patient growth and other aspects of the disease should be managed more effectively in the future.
Infantile nephropathic cystinosis (INC), MIM 219800, is a lysosomal storage disorder due to loss of function of the proton/cystine symporter cystinosin on lysosomal membranes. Accounting for 95% of all cases, it is the most common but also the most severe form of cystinosis (1).
It is due to bi-allelic mutations of the 12-exon CTNS gene on chromosome region 17p13 (2, 3). Up to now, over 140 different types of mutations have been described, the most common one in Northern Europeans being the 57kb-deletion of the promotor region and the first 10 exons of the CTNS gene together with two upstream genes (4, 5).
In Northern European studies conducted during the twentieth century, the prevalence of INC was estimated as 0.3 to 0.9 per 100,000 live births (6). In 2018/19, a screening study of 292,000 German newborns which used multiplex PCR and next-generation sequencing detected 2 patients (7, 8). According to the Interdisciplinary Cystinosis Clinic in Rosenheim, there are currently approximately 130 pediatric and adult INC patients in Germany. At this clinic, all German INC patients have the opportunity to receive standardized and coordinated interdisciplinary care at yearly intervals (9).
In INC, several mechanisms account for the phenotype (10). However, despite many efforts, it is still not fully known how these mechanisms are interlinked or how they impact upon the clinical course of the disease.
Lysosomal efflux of cystine into the cytoplasm is mostly cystinosin-dependent. Following loss of function of cystinosin in INC, the amino-acid accumulates and crystallizes inside the lysosome (11). Alterations in mitochondrial metabolism, lysosomal dynamics, autophagy, apoptosis, the mTORC1-pathway and inflammatory responses have also been explored in INC (10).
A diagnosis of INC is confirmed by measurement of elevated LCL (levels of healthy subjects are below 0.2 nmol cystine/mg protein), slit lamp examination of corneal cystine crystals which become apparent by the age of 18 months at the latest and CTNS gene sequencing (1, 12).
Although cystinosin is expressed in all tissues, manifestations of INC vary in degree of severity and time of onset: Children mostly present with renal Fanconi syndrome before their first birthday, which manifests as failure to thrive, polyuria, polydipsia, vomiting, dehydration, electrolyte imbalance, and hypophosphatemic rickets. These symptoms are caused by the failure of renal proximal tubular cells to reabsorb water, glucose, amino acids, uric acid, carnitine, phosphate, bicarbonate, and other small molecules (1). In later life, glomerular function also deteriorates, a phenomenon which has been linked to pathognomonic podocyte injury (13, 14). When treated adequately, the need for kidney replacement therapy (KRT) can be delayed until the second or even third decade of life (15), historical cohorts reached end stage kidney failure at approximately 10 years of age (16).
There are many extrarenal complications which accompany INC and strongly impact a patient's quality of life: Ophthalmological manifestations can affect every part of the eye—an early and frequent symptom is photophobia due to corneal crystal deposition (17). Gastrointestinal symptoms of INC are caused by cysteamine treatment and/or by cystine deposition in the gut wall (18). The most common endocrinological complications of the disease are hypothyroidism, diabetes mellitus (1), pubertal delay (19, 20), and male infertility (21). Muscle impairment is not only of concern for bone metabolism and strength, but may lead to swallowing difficulties and respiratory insufficiency (16, 18). Short stature, skeletal pain and deformities, as well as a higher risk of fractures are more common in INC and growth also seems to follow a different pattern than in other nosologic entities of chronic kidney disease (CKD) (22, 23). A primary defect in cystinotic osteoblasts and osteoclasts as well as secondary implications of INC account for the so-called cystinosis-associated metabolic bone disease (22, 24, 25). Varying individually, neurological (26), hematological, dermatological, cardiac, and psychosocial implications of the disease have also been reported (27).
If left untreated, morbidity and mortality in INC are high (1, 16, 28)—due to the development of adequate specific and supporting therapy, patients with the former solely pediatric disease today might live past the age of 40 years (29): In end-stage kidney disease, dialysis and kidney transplantation are provided. Extra-renal manifestations of INC are treated according to the symptoms, e.g. through administration of thyroxine or growth hormones (12). The only currently available specific therapy for INC is the cystine-depleting agent cysteamine, which is administered orally. Although cysteamine is not a rescue treatment for Fanconi syndrome—and cannot cure INC—it can delay progression of the disease (16, 30). At present, an immediate-release cysteamine bitartrate (IRC, to be administered strictly every 6 h) and an extended-release formulation (ERC, for twice-daily dosing) are available (31). Topical cysteamine application is needed to address the avascular cornea (32).
Improved cysteamine formulations and new approaches based on recently discovered mechanisms are being investigated (10). A Phase I/II clinical trial is ongoing (NCT03897361) to examine safety and efficacy of autologous transplantation of hematopoietic stem cells expressing the CTNS gene after lentiviral modification ex vivo.
The present study assesses the clinical course of INC in a large German-Austrian cohort, characterizing factors associated with the progression of the disease and exploring possibilities for improvement in the quality and structure of health care.
To participate, patients had to be diagnosed with INC and to have received treatment in Germany or Austria within the last 10 years. Written informed consent from the patients themselves and/or their parents/guardians was mandatory. A superordinate ethics committee approval was received by the ethics committee of Hannover Medical School. The study was conducted in accordance with the Declaration of Helsinki.
Baseline data included date of birth, sex, time of initial diagnosis and initiation of systemic cystine-depleting therapy, dialysis and kidney transplantation.
Data was collected retrospectively from the patients' health records in the cooperating hospitals: These included the most recent routine examination along with examination results at approximately 12-monthly intervals from that point until up to 10 years earlier and at the time of the initial diagnosis.
Patients' records on routine examinations were searched for anthropometric data, laboratory parameters, medication and clinical symptoms. Percentiles were calculated for patient height and weight (33). Laboratory parameters to calculate estimated glomerular filtration rate (eGFR) according to Schwartz 2009 [0.413 x (height in cm/serum-creatinine in mg/dl)] (34) and CKD stage according to KDIGO 2012 (35) were documented, as well as the cystine level in mixed white blood cells (nmol cystine/mg protein) to monitor therapy with cysteamine. A leucocyte fraction was isolated via density gradient centrifugation. Cystine content was analyzed via GC-MS and normalized to protein content, our results were multiplied by a factor of 2 to get 1/2 cystine-results. Cut-off values were 0.1–0.5 nmol cystine/mg protein for carriers and sufficient therapy control in INC patients was defined as LCL below 1.00 nmol cystine/mg protein.
Additional data included information on medication: cysteamine in g/m2 per day calculated according to DuBois (36) with overdosing defined at the recommended maximum of 1.95 g/m2 per day (12), type of cysteamine (IRC vs. ERC), use of growth hormone therapy and further supporting medication. Manifestation of anemia, hypothyroidism, rickets, skeletal deformities, gastrointestinal, ophthalmological, neurological, and muscular symptoms, as well as the need for tube feeding, were recorded as binary variables.
For statistical analysis, R (version: 4.0.5) was used (37). All documented values for laboratory parameters and cysteamine dosage were included. Continuous data was reported using arithmetic mean and standard deviation (SD) or median, interquartile range (IQR), and range when data did not follow a normal distribution according to the Shapiro–Wilk test. Categorical data was expressed using counts and percentages; relative frequencies always referred to the total number of patients regardless of missing values. The main focus was on Kaplan–Meier-estimators including 95%-confidence intervals (95% CI) and log-rank tests for p-value. The level of significance was set at p = 0.05. Time until KRT was analyzed (age in years at first dialysis or kidney transplantation) and stratified for age at initial diagnosis, sex and year of birth to investigate whether treatment was related to era. To examine possible correlations of data which did not follow normal distribution, scatterplots, and Spearman's correlation were used. The nearer the absolute value of Spearman's coefficient ρ is to 1, the more the data correlates. Here, p was set to 0.02 to increase the probability of rejecting the null hypothesis.
A total of 74 patients from eleven healthcare centers was included in this cohort study. Thirty-nine (52.7%) of the participants were male, 35 (47.3%) were female. Half of the patients were born until 2002 (IQR: 1995–2009, range: 1975–2019). Health records were available for the years 1986 to 2020 (median: 2016, IQR: 2013–2016). Median survey duration was 2 years (IQR: 1–5, range: 1–17). The median age patients had at the recorded examinations was 11 years (IQR: 6–15, range: 0–42). The study cohort mostly represented underage patients at a total of 240 points in time; only 36 observations were made for patients at the age of 18 years and older.
Median age at diagnosis of INC (n = 67) was 15 months (IQR: 10–29, range: 0–110). In one case the date of initial diagnosis was unknown, while in six cases only the year was given. Spearman's correlation of birth year and age at diagnosis, as well as a scatterplot, showed no correlation (ρ = −0.17, p = 0.16).
Systemic cystine-depleting therapy was initiated at a median age of 16.5 months (IQR: 11–30, range: 0–111). Data was missing on 12 patients born between 1975 and 2003. In 53 patients, cysteamine therapy started within 1 month after initial diagnosis, in seven cases within 4 months, whilst two patients (both born in 1981) started treatment 3 and 7 years after initial diagnosis.
For 72 patients, the treatment with systemic cysteamine was documented, data was missing for two patients. The use of IRC was recorded in 54 cases, with 17 patients changing to ERC at a median age of 11 years (IQR: 6–14, range: 2–16). Two patients changed back to IRC after 1 and 2 years, one of them due to increased vomiting. The use of ERC alone was reported for two patients, whilst two more patients took IRC twice during the daytime and ERC once at night-time.
The exact cysteamine dosage was available for 67 patients, with the median of the 235 documented values being 1.26 g/m2 per day (IQR: 1.03–1.48, range: 0.22–1.99). 84 values of 35 patients were >1.35 g/m2 per day with a median of 1.57 (IQR: 1.46–1.67, range: 1.36–1.99) at a median age of 12 years (IQR: 7–16, range: 1–35). Two patients received a dose slightly above the recommended maximum of 1.95 g/m2 per day, with 1.99 each at the age of 12 and 14 years. There was no documentation on side effects available.
The median dose of 171 values of the patients on IRC was 1.31 (IQR: 1.16–1.51, range: 0.22–1.99). The median dose of 71 values of the patients on ERC was 1.03 (IQR: 0.95–1.21, range: 0.27–1.86).
The median time-averaged LCL values based on 65 patients amounted to 0.57 (IQR: 0.33–0.98, range: 0.07–3.13). Here, only measurements at least 1 year after initiation of therapy were considered. 69.2% of all 198 LCL measurements of 67 patients were within the desired target range ≤ 1 nmol cystine/mg protein. An overview of all documented LCLs is given in Table 1.
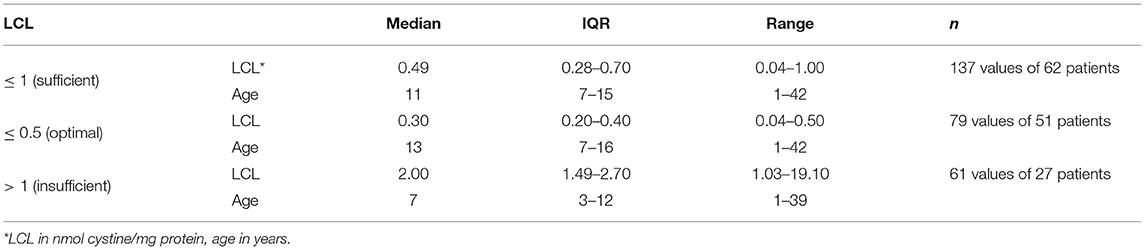
Table 1. Leukocyte cystine levels of 67 patients.
There was no association between cysteamine dosage and age (ρ = 0.06, p = 0.38) or LCLs (ρ = (−0.06), p = 0.44).
For 52 patients, 195 values of eGFR could be calculated before KRT and assigned to CKD stages (Figure 1).
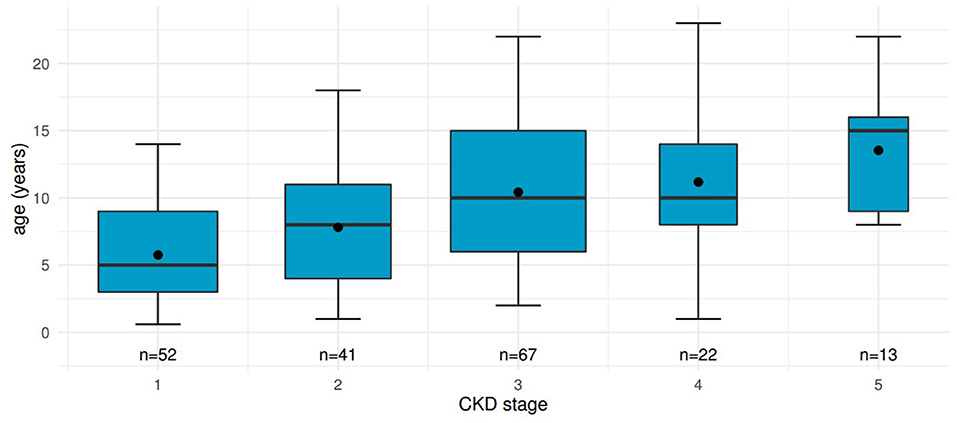
Figure 1. Distribution of age at different CKD stages in patients before kidney transplantation. Boxplots including mean values show renal function declining with increasing age. “n”, number of measurements contributing to each boxplot.
Thirty-one patients (41.9%), 17 female and 14 male, underwent KRT (either dialysis or pre-emptive kidney transplantation), data was missing in four cases. The median age at initiation of KRT was 10 years (IQR: 9–15, range: 7–25).
Thirty patients underwent kidney transplantation at least once. 18 of those patients received their first transplant after a period of dialysis therapy, 4 kidneys were transplanted pre-emptively whereas in 8 cases data on preceding dialysis was missing. One patient on chronic dialysis therapy had not yet been listed for transplantation.
Kaplan–Meier analysis was used to evaluate time from birth to KRT (Figure 2). Data was stratified for age at initial diagnosis in months (Figure 3). Thirty-four patients (45.9%) were each assigned to one of two groups (initial diagnosis before 18 months of life vs. at 18 months or older), for which a log-rank test showed a significant difference in renal survival.
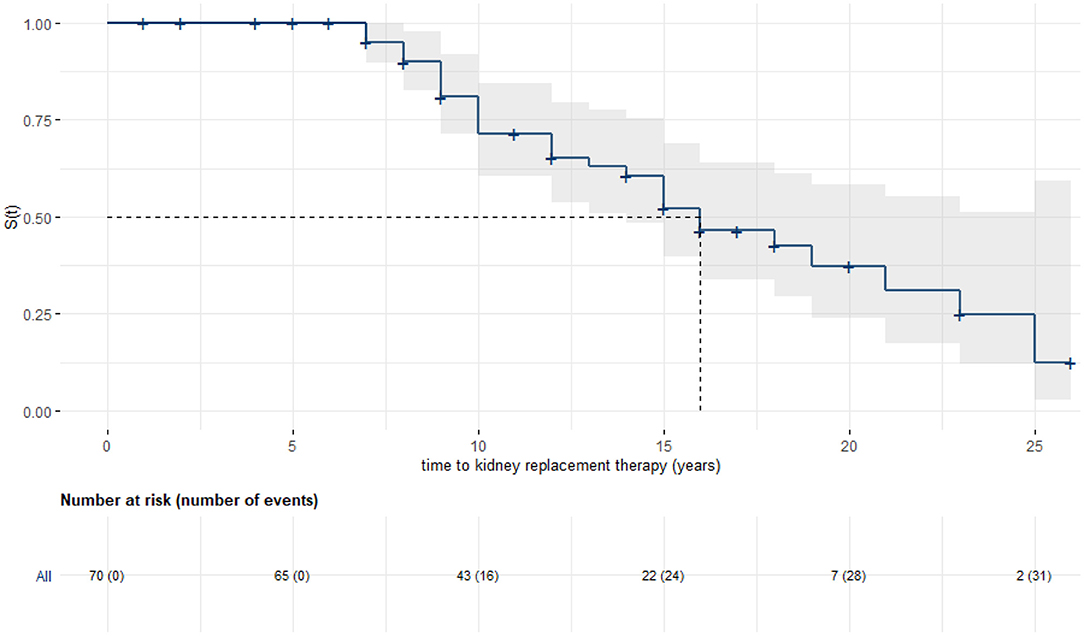
Figure 2. Kaplan–Meier analysis with 95% confidence band: Kidney survival in years. Seventy patients could be included, median renal survival was 16 years (95% CI: 14, -).

Figure 3. Stratified Kaplan–Meier analysis with 95% confidence bands and log-rank test: Kidney survival in years. Groups were stratified for age at initial diagnosis (cut-off point at 18 months). The 34 patients each showed a median renal survival of 13 years (95% CI: 10, -) in the group diagnosed at an older age vs. 21 years (95% CI: 16, -) in the group with earlier diagnosis.
When stratified for year of birth (1975 to 2001 vs. 2002 to 2019) or sex, with crossing hazards and therefore failing log-rank test, no difference in renal survival was detected.
Percentiles for height were calculated for 68 patients with a total of 242 documented values available. The overall median lay at the 7th percentile (IQR: 1–25, range: 1–99). Detailed boxplots are shown in Figure 4, displaying all age groups from 1 year to adulthood and differentiating two subgroups (before and after initiation of KRT).
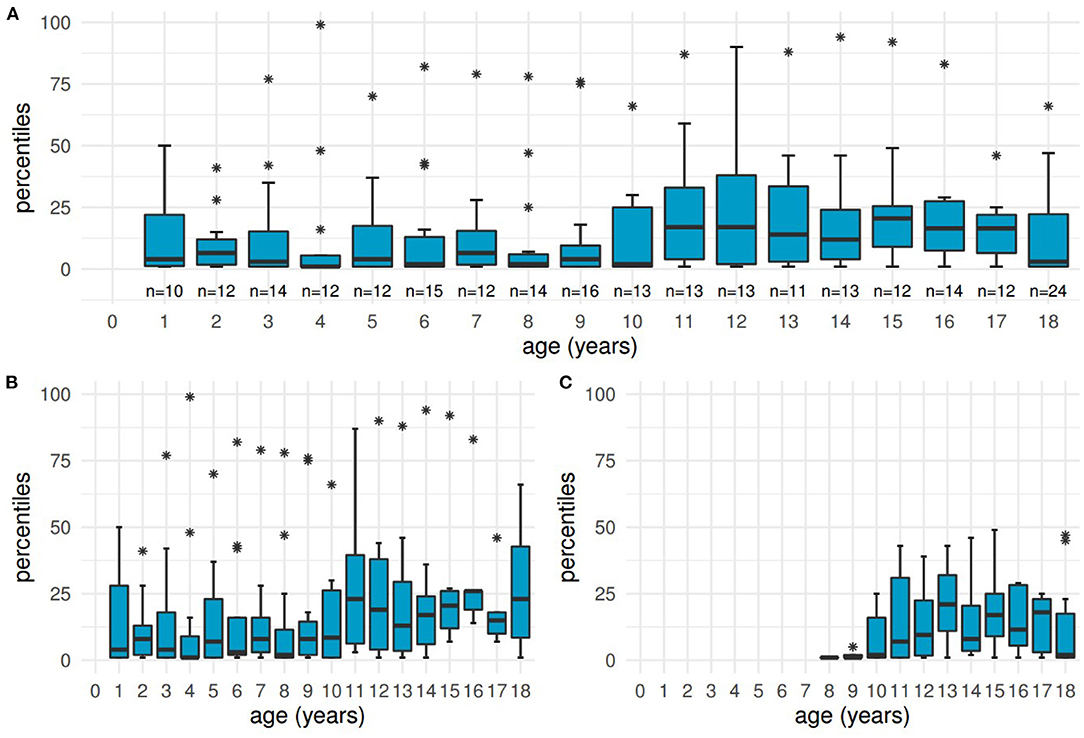
Figure 4. Distribution of height at different ages. Boxplots showing low percentiles at the ages of 1 year to adulthood, with the age of “18” including the heights of all adult patients. (A) all patients, (B) subgroup of 41 patients before initiation of kidney replacement therapy, (C) subgroup of 27 patients receiving kidney replacement therapy.
Ninety-eight values (40.5%) from 43 patients were within the defined pathological range of ≤ 3rd percentile at a median age of 8 years (IQR: 5–13, range: 1–18 and older). Pathological heights were mostly documented for younger children and adults born between 1975 and 2001. Only 3 patients reached the 50th percentile or higher at least once.
The subgroup not yet in need of KRT (41 patients, 164 measurements) had a median percentile of 8 (IQR: 1–26, range: 1–99), the median of 27 patients after initiation of KRT lay at the 6th percentile (IQR: 1–23, range: 1–49, including 66 values).
Percentiles for weight were calculated for 71 patients with a total of 248 documented values available. The overall median lay at percentile 15.50 (IQR: 5.00–34.25, range: 1.00–97.00). Fifty-three values (21.4%) from 28 patients were ≤ 3rd percentile at a median age of 8 years (IQR: 4–13, range: 1–18 and older). Only 16 patients reached the 50th percentile or higher at least once.
Data on tube feeding was available for 25 patients (33.8%). Median age when beginning tube feeding was 1 year (IQR: 1–3, range: 1–10). For 11 patients, the median duration was 6 years (IQR: 2–13, range: 0.04–23), whilst for 13 patients the median duration was at least 4 years (IQR: 2–5, range:1–11), and was still ongoing through the period of observation.
Thirty-nine (52.7%) of the patients received recombinant growth hormone therapy at some point. In 10 cases, the exact duration was known (mean: 7.70 years, SD: 3.53, range: 1.00–13.00); in those cases the therapy started at a mean age of 8.00 years (SD: 3.62, range: 1–12). In 26 cases, the documented duration was a median of at least 4.00 years (IQR: 1.25–7.50, range: 1.00–15.00), but the exact starting and/or ending points remained unclear due to insufficient documentation.
Observed extrarenal manifestations are listed in Table 2. The available documents did not distinguish the absence of those symptoms from missing information.
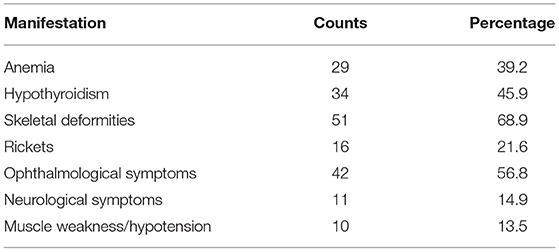
Table 2. Extrarenal manifestations.
The most reported skeletal deformities were genu valgum (33 cases), pes planovalgus (10 cases), and scoliosis (7 cases). Where further details on ophthalmological symptoms were given, photophobia and corneal discomfort were reported to be the main concern. Regarding neurological symptoms, 7 patients with seizures were reported, and there was one case each of movement disorder, dyslalia, motor retardation and stroke with speech impediments. There were no entries on diabetes, but one patient was reported as suffering from exocrine pancreatic insufficiency. Detailed information on the context of these observations was not available.
Gastrointestinal symptoms are presented in Table 3.
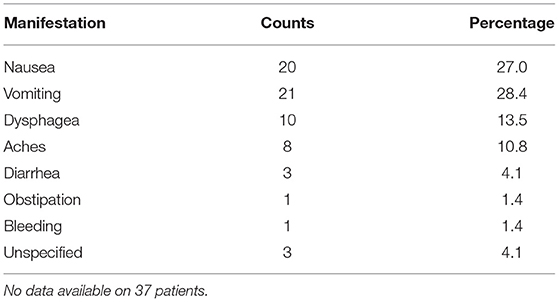
Table 3. Gastrointestinal symptoms.
Our results indicate that cystine-depleting treatment of INC in Germany and Austria is comparable to other recently reported international data with the length of renal survival being determined by age at diagnosis and subsequent initiation of cysteamine therapy.
Despite the tremendous progress treatment has made within the last decades, INC continues to be a severe chronic condition (16, 28, 29).
Frequently, the first sign of INC noticed by parents and clinicians is failure to thrive (38). We observed that children often needed the support of tube feeding at a young age, continuing for several years. Growth has been an important outcome parameter in previous studies as it reflects the multi-factorial influences of different aspects of the disease as well as therapeutic success (29, 38). We were able to show that even though medication in Germany and Austria started at an early age, patients remained very short in stature. Our results are in concordance with another study focusing on patient's body growth before initiation of KRT (23). However, our data was not sufficient to explain the great interindividual variety, with several patients having fewer difficulties than others. These results might indicate an improvement around length of therapy and medical developments in recent years, as pathological percentiles were mostly concentrated at younger ages and adults born before 2001. While thorough analyses of its impact on growth in INC patients are still necessary, we recommend an early start of GH treatment in infancy for children with INC based on the suggestion directing the general population of infants with CKD (39).
Our study also demonstrated that there was subsequent kidney failure with advancing age. Overall median renal survival in German and Austrian patients corresponded to the large European-Turkish cohort analyzed recently by Emma et al. (29). We also showed that in German-Austrian patients, time of initial diagnosis accounted for a great difference in median renal survival with our cut-off point at the age of 1.5 years (arbitrarily chosen at a younger age than in previous studies (15, 30), oriented to age at initial diagnosis in our cohort).
Information on extrarenal symptoms and their onset could not be determined as thoroughly as planned, but our results still provided an insight into the greatest challenges regarding this disease. Corresponding to international studies (30, 40), hypothyroidism, anemia, ophthalmological, skeletal and gastrointestinal complications were very common manifestations of INC. Considering the incomplete health records on this topic, it is likely that more patients were affected than was actually documented in the present study.
We aimed to characterize the clinical course for German-Austrian patients with INC, with a main focus on assessing the quality of their healthcare using data on initial diagnosis and treatment as well as undertaking an analysis of the dosage and monitoring of specific therapies.
According to the findings of Bertholet-Thomas et al. (15), our cohort was representative for management of INC in developed nations. We showed that systemic application of cysteamine was offered to patients very soon after initial diagnosis, addressing the importance of cystine-depleting therapy. Prescribed doses of systemic cysteamine were at an optimal level according to international standards (12), regardless of whether IRC or ERC was used. No relevant overdoses were documented.
As the measurement of LCLs provides an indirect insight into patients' adherence and overall success of cysteamine treatment, analyzing this aspect thoroughly was of importance in this study. The aim of the therapy is to remain ≤ 1 nmol cystine/mg protein, with an optimal outcome being a level ≤ 0.5 (31). We found our cohort to have overall satisfying LCLs when calculating the time-averaged mean for each patient as well as when considering all measurements individually. The therapy in our cohort seemed to be particularly efficient after a few years of treatment which is indicated by the observation that those values above 1 nmol cystine/mg protein are found at younger ages than the sufficient LCLs. The comparison of our findings with international data (15, 29) is remarkable as it shows that, though there may have been room for improvement on an individual level, this cohort had exceptional overall therapy control based upon LCL. However, the assay to detect LCL, the current gold standard to monitor cysteamine therapy, is technically demanding and not always available. Biomarkers of macrophage activation are promising monitoring candidates, reflecting the macrophage-mediated inflammation in cystinosis. Among them, chitotriosidase enzyme activity showed to be a good predictor of LCL and was significantly correlated to extrarenal complications (41). Concentration of ceroid in macrophages (a product of lysosomal oxidation of LDL that plays an important role in the development of atherosclerosis) might become a future marker to monitor the antioxidative properties of cysteamine (42). Monitoring of therapy adherence should not be restricted to the analysis of direct cysteamine effects, but should also include biomarker tracking of disease complications, such as those of bone status (43), thyroid function and glucose metabolism. The most relevant clinical biomarker correlated to cysteamine effect doubtlessly remains preservation of glomerular filtration rate (44) and might be the best motivation for therapy adherence.
Age at initial diagnosis in our patient cohort was comparable to recent experiences in other countries (15, 29). Interestingly, in our cohort spanning four decades, a more recent date of birth was not associated with earlier diagnosis or relevantly improved renal survival, leading to the hypothesis that the awareness for diagnosis of cystinosis in the first year of live has not increased and an optimal therapeutic approach might have already been reached. Thus, new initiatives, such as new-born screening to enable earlier diagnosis, need to be implemented to achieve even more favorable outcomes. It has to be noted, though, that it is currently unclear whether cysteamine treatment bears more advantages than risks when initiated before children experience symptoms of INC.
Limitations of our study were closely linked to its retrospective nature, and mainly consist of incomplete records on some patients as well as heterogeneous distribution of duration of individual follow-up.
There was no information available on the underlying genetic defect.
Particularly in cases where there was no reporting on extrarenal manifestations, it remained unclear whether the information was missing or whether the symptoms did not occur, so more patients might have been affected. Our numbers on those manifestations are therefore to be understood as minimal counts. Also, patient ages at onset remained uncertain. Concerning all extrarenal manifestations and especially our data on growth, important information could not be evaluated: Data on supporting medication, nutritional intervention, acidosis, bone disease and other disease-related as well as therapeutic factors was not available.
For Kaplan–Meier-analyses, data on eight patients was included although it was not known whether these persons had received dialysis before their documented transplantation date. In our opinion, however, it was deemed important to include data on these patients nevertheless to avoid loss of valuable information.
Additionally, despite being a large cohort for national studies on this orphan disease, statistical analysis was still flawed because of its relatively small sample size.
A certain level of bias might have also played a role as patients' dates of birth varied greatly and health care has changed fundamentally for patients born more recently. Changes in awareness and profound knowledge of INC, pharmaceutical developments such as the availability of cysteamine and growth hormones, advances in KRT but also individual patients' characteristics such as adherence and psychosocial aspects in general may have had an impact on our observations that we cannot differentiate nor quantify.
Furthermore, there may have been sampling bias as there are rarely coordinated programs for transitioning care to specialized centers in adulthood and it was therefore very difficult to include adult patients in this study. Adult nephrologists were very widely contacted but did not report back. The majority of all pediatric nephrology centers in Germany that treat patients with cystinosis have included their data in the registry of the German Society for Pediatric Nephrology. In our view, establishing national and international registries will be the key to understanding and managing this orphan disease in the future.
This study is the first to extensively characterize a large cohort of patients with INC in Germany and Austria, with a focus on the previous decade. Our findings underline that continuous monitoring of the effects of treatment in rare diseases by international initiatives is needed. Our results confirm recent international reports that treatment with cysteamine is adequate, whereas other disease aspects, such as growth and treatment of extrarenal manifestations, should be optimized in the future.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee Hannover Medical School. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
LPe and JO designed the study. NO'C collected the data and performed the statistical analyses. LPe and NO'C wrote the first draft of the manuscript. LPe, JO, KA, AB, DH, JK, CK, CM, DM, LW, BT, MW, and KH provided patient data. All authors contributed to manuscript revision, read, and approved the submitted version.
LPe received an unrestrictive grant from Chiesi Germany to conduct this study. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
DH received speaker fees and research support, CK and BT received speaker and advisory board fees from Chiesi. LPe, LW, and JO received speaker fees from Chiesi and Orphan Europe.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.864554/full#supplementary-material
CKD, chronic kidney disease; 95% CI, 95% confidence interval; eGFR, estimated glomerular filtration rate; ERC, extended-release cysteamine; IRC, immediate-release cysteamine; IQR, interquartile range; LCL, leukocyte cystine level; INC, infantile nephropathic cystinosis; KRT, kidney replacement therapy; SD, standard deviation.
1. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. (2002) 347:111–21. doi: 10.1056/NEJMra020552
2. The Cystinosis Collaborative Research Group. Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat Genet. (1995) 10:246–8. doi: 10.1038/ng0695-246
3. Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. (1998) 18:319–24. doi: 10.1038/ng0498-319
4. David D, Berlingerio SP, Elmonem MA, Arcolino FO, Soliman N, van den Heuvel B, et al. Molecular basis of cystinosis: geographic distribution, functional consequences of mutations in the CTNS gene, and potential for repair. Nephron. (2019) 141:133–46. doi: 10.1159/000495270
5. Freed KA, Blangero J, Howard T, Johnson MP, Curran JE, Garcia YR, et al. The 57 kb deletion in cystinosis patients extends into TRPV1 causing dysregulation of transcription in peripheral blood mononuclear cells. J Med Genet. (2011) 48:563–6. doi: 10.1136/jmg.2010.083303
6. Levy M, Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. (2000) 58:925–43. doi: 10.1046/j.1523-1755.2000.00250.x
7. Fleige T, Burggraf S, Czibere L, Haering J, Glück B, Keitel LM, et al. Next generation sequencing as second-tier test in high-throughput newborn screening for nephropathic cystinosis. Eur J Hum Genet. (2020) 28:193–201. doi: 10.1038/s41431-019-0521-3
8. Hohenfellner K, Bergmann C, Fleige T, Janzen N, Burggraf S, Olgemoeller B, et al. Molecular based newborn screening in Germany: follow-up for cystinosis. Mol Genet Metab Rep. (2019) 21:100514. doi: 10.1016/j.ymgmr.2019.100514
9. Hohenfellner K, Deerberg-Wittram J. Coordinated, cost-effctive care for rare disease: the cystinosis outpatient consultation program at RoMed. NEJM Catal Innov Care Deliv. (2020) 1. doi: 10.1056/CAT.19.1116
10. Jamalpoor A, Othman A, Levtchenko EN, Masereeuw R, Janssen MJ. Molecular mechanisms and treatment options of nephropathic cystinosis. Trends in Mol Med. (2021) 27:673–86. doi: 10.1016/j.molmed.2021.04.004
11. Thoene JG, Lemons RM. Cystine accumulation in cystinotic fibroblasts from free and protein-linked cystine but not cysteine. Biochem J. (1982) 208:823–30. doi: 10.1042/bj2080823
12. Emma F, Nesterova G, Langman C, Labbe A, Cherqui S, Goodyer P, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. (2014) 29(Suppl 4):iv87. doi: 10.1093/ndt/gfu090
13. Elmonem MA, Khalil R, Khodaparast L, Khodaparast L, Arcolino FO, Morgan J, et al. Cystinosis (CTNS) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Sci Rep. (2017) 7:42583. doi: 10.1038/srep42583
14. Ivanova EA, Arcolino FO, Elmonem MA, Rastaldi MP, Giardino L, Cornelissen EM, et al. Cystinosin deficiency causes podocyte damage and loss associated with increased cell motility. Kidney Int. (2016) 89:1037–48. doi: 10.1016/j.kint.2016.01.013
15. Bertholet-Thomas A, Berthiller J, Tasic V, Kassai B, Otukesh H, Greco M, et al. Worldwide view of nephropathic cystinosis: results from a survey from 30 countries. BMC Nephrol. (2017) 18:210. doi: 10.1186/s12882-017-0633-3
16. Gahl W, Balog J, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med. (2007) 147:242–50. doi: 10.7326/0003-4819-147-4-200708210-00006
17. Pinxten AM, Hua MT, Simpson J, Hohenfellner K, Levtchenko E, Casteels I. Clinical practice: a proposed standardized ophthalmological assessment for patients with cystinosis. Ophthalmol Ther. (2017) 6:93–104. doi: 10.1007/s40123-017-0089-3
18. Topaloglu R, Gültekingil-Keser A, Gülhan B, Ozaltin F, Demir H, Ciftci T, et al. Cystinosis beyond kidneys: gastrointestinal system and muscle involvement. BMC Gastroenterol. (2020) 20:242. doi: 10.1186/s12876-020-01385-x
19. Gültekingil-Keser A, Topaloglu R, Bilginer Y, Besbas N. Long-term endocrinologic complications of cystinosis. Minerva Pediatr. (2014) 66:123–130.
20. Winkler L, Offner G, Krull F, Brodehl J. Growth and pubertal development in nephropathic cystinosis. Eur J Pediatr. (1993) 152:244–9. doi: 10.1007/BF01956154
21. Rohayem J, Haffner D, Cremers JF, Huss S, Wistuba J, Weitzel D, et al. Testicular function in males with infantile nephropathic cystinosis. Hum Reprod. (2021) 36:1191–204. doi: 10.1093/humrep/deab030
22. Ewert A, Leifheit-Nestler M, Hohenfellner K, Büscher A, Kemper M, Oh J, et al. Bone and mineral metabolism in children with nephropathic cystinosis compared with other CKD entities. J Clin Endocrinol Metab. (2020) 105:dgaa267. doi: 10.1210/clinem/dgaa267
23. Kluck R, Müller S, Jagodzinski C, Hohenfellner K, Bascher A, Kemper M, et al. Body growth, upper arm fat area, and clinical parameters in children with nephropathic cystinosis compared with other pediatric chronic kidney disease entities. J Inherit Metab Dis. (2022) 11:170. doi: 10.1002/jimd.12473
24. Hohenfellner K, Rauch F, Ariceta G, Awan A, Bacchetta J, Bergmann C, et al. Management of bone disease in cystinosis: statement from an international conference. J Inherit Metab Dis. (2019) 42:1019–29. doi: 10.1002/jimd.12134
25. Battafarano G, Rossi M, Rega L, Di Giovamberardino G, Pastore A, D'Agostini M, et al. Intrinsic bone defects in cystinotic mice. Am J Pathol. (2019) 189:1053–64. doi: 10.1016/j.ajpath.2019.01.015
26. Curie A, Touil N, Gaillard S, Galanaud D, Leboucq N, Deschênes G, et al. Neuropsychological and neuroanatomical phenotype in 17 patients with cystinosis. Orphanet J Rare Dis. (2020) 15:59. doi: 10.1186/s13023-019-1271-6
27. Kasimer RN, Langman CB. Adult complications of nephropathic cystinosis: a systematic review. Pediatr Nephrol. (2021) 36:223–236. doi: 10.1007/s00467-020-04487-6
28. Manz F, Gretz N. Progression of chronic renal failure in a historical group of patients with nephropathic cystinosis. European Collaborative Study on Cystinosis. Pediatr Nephrol. (1994) 8:466–71. doi: 10.1007/BF00856532
29. Emma F, Van't Hoff W, Hohenfellner K, Topaloglu R, Greco M, Ariceta G, et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int. (2021) 100:1112–23. doi: 10.1016/j.kint.2021.06.019
30. Brodin-Sartorius A, Tete M, Niaudet P, Antignac C, Guest G, Ottolenghi C, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. (2012) 81:179–89. doi: 10.1038/ki.2011.277
31. Ahlenstiel-Grunow T, Kanzelmeyer N, Froede K, Kreuzer M, Drube J, Lerch C, et al. Switching from immediate- to extended-release cysteamine in nephropathic cystinosis patients: a retrospective real-life single-center study. Pediatric Nephrol. (2017) 32:91–7. doi: 10.1007/s00467-016-3438-x
32. Castro-Balado A, Mondelo-Garcia C, Varela-Rey I, Moreda-Vizcaino B, Sierra-Sanchez JF, Rodriguez-Ares MT, et al. Recent research in ocular cystinosis: drug delivery systems, cysteamine detection methods and future perspectives. Pharmaceutics. (2020) 12:1177. doi: 10.3390/pharmaceutics12121177
33. Kromeyer-Hauschild K, Wabitsch M, Kunze D, Geller F, Geiß H, Hesse V, et al. Perzentile für den Body-mass-Index für das Kindes- und Jugendalter unter Heranziehung verschiedener deutscher Stichproben. Monatsschr Kinderheilk. (2001) 149:807–18. doi: 10.1007/s001120170107
34. Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. (2009) 20:629–37. doi: 10.1681/ASN.2008030287
35. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. (2013) 3:1–150. doi: 10.1038/kisup.2012.76
36. DuBois D, DuBois E. A formula to estimate the approximate surface area if height and weight be known. Arch Intern Med. (1916) 17:863–71. doi: 10.1001/archinte.1916.00080130010002
37. R Core Team. R: A Language Environment for Statistical Computing. Vienna (2021). Available online at: https://www.R-project.org/
38. Greco M, Brugnara M, Zaffanello M, Taranta A, Pastore A, Emma F. Long-term outcome of nephropathic cystinosis: a 20-year single-center experience. Pediatr Nephrol. (2010) 25:2459–67. doi: 10.1007/s00467-010-1641-8
39. Drube J, Wan M, Bonthuis M, Wühl E, Bacchetta J, Santos F, et al. Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat Rev Nephrol. (2019) 15:577–89. doi: 10.1038/s41581-019-0161-4
40. Topaloglu R, Gülhan B, Inözü M, Canpolat N, Yilmaz A, Noyan A, et al. The clinical and mutational spectrum of Turkish patients with cystinosis. Clin J Am Soc Nephrol. (2017) 12:1634–41. doi: 10.2215/CJN.00180117
41. Veys KRP, Elmonem MA, Dyck MV, Janssen MC, Cornelissen EAM, Hohenfellner K, et al. Chitotriosidase as a Novel Biomarker for Therapeutic Monitoring of Nephropathic Cystinosis. J Am Soc Nephrol. (2020) 31:1092–106. doi: 10.1681/ASN.2019080774
42. Wen Y, Ahmada F, Mohrib Z, Weinberg PD, Leake DS. Cysteamine inhibits lysosomal oxidation of low density lipoprotein in human macrophages and reduces atherosclerosis in mice. Atherosclerosis. (2019) 291:9–18. doi: 10.1016/j.atherosclerosis.2019.09.019
43. Bertholet-Thomas A, Claramunt-Taberner D, Gaillard S, Deschanes G, Sornay-Rendu E, Szulc P, et al. Teenagers and young adults with nephropathic cystinosis display significant bone disease and cortical impairment. Pediatr Nephrol. (2018) 33:1165–72. doi: 10.1007/s00467-018-3902-x
Keywords: cystinosis, dialysis, kidney transplantation, growth, metabolic disease, rare disease, chronic kidney disease
Citation: O'Connell N, Oh J, Arbeiter K, Büscher A, Haffner D, Kaufeld J, Kurschat C, Mache C, Müller D, Patzer L, Weber LT, Tönshoff B, Weitz M, Hohenfellner K and Pape L (2022) Patients With Infantile Nephropathic Cystinosis in Germany and Austria: A Retrospective Cohort Study. Front. Med. 9:864554. doi: 10.3389/fmed.2022.864554
Received: 28 January 2022; Accepted: 30 March 2022;
Published: 25 April 2022.
Edited by:
Simone Baldovino, University of Turin, ItalyReviewed by:
Changli Wei, Rush University, United StatesCopyright © 2022 O'Connell, Oh, Arbeiter, Büscher, Haffner, Kaufeld, Kurschat, Mache, Müller, Patzer, Weber, Tönshoff, Weitz, Hohenfellner and Pape. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Pape, bGFycy5wYXBlQHVrLWVzc2VuLmRl
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.