 Érico Murilo Monteiro Cutrim1
Érico Murilo Monteiro Cutrim1 Precil Diego Miranda de Meneses Neves2Marcos Adriano Garcia Campos3Davi Campos Wanderley4
Precil Diego Miranda de Meneses Neves2Marcos Adriano Garcia Campos3Davi Campos Wanderley4 Antonio Augusto Lima Teixeira-Júnior5
Antonio Augusto Lima Teixeira-Júnior5 Monique Pereira Rêgo Muniz1Francisco Rasiah Ladchumananandasivam6
Monique Pereira Rêgo Muniz1Francisco Rasiah Ladchumananandasivam6 Orlando Vieira Gomes7Rafael Fernandes Vanderlei Vasco8Dyego José de Araújo Brito1Joyce Santos Lages1
Orlando Vieira Gomes7Rafael Fernandes Vanderlei Vasco8Dyego José de Araújo Brito1Joyce Santos Lages1 Natalino Salgado-Filho1Felipe Leite Guedes9
Natalino Salgado-Filho1Felipe Leite Guedes9 José Bruno de Almeida9
José Bruno de Almeida9 Marcelo Magalhães10Stanley de Almeida Araújo4
Marcelo Magalhães10Stanley de Almeida Araújo4 Gyl Eanes Barros Silva1*
Gyl Eanes Barros Silva1*
- 1University Hospital, Federal University of Maranhão, São Luís, Brazil
- 2Clinical Hospital, University of São Paulo, São Paulo, Brazil
- 3Clinical Hospital, Botucatu Faculty of Medicine of São Paulo State University, Botucatu, Brazil
- 4Nephropathology Institute, Federal University of Minas Gerais, Belo Horizonte, Brazil
- 5Department of Genetics and Postgraduate Program in Genetics, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
- 6University Hospital, Federal University of Paraíba, João Pessoa, Brazil
- 7University Hospital, Federal University of Vale do São Francisco, Petrolina, Brazil
- 8University Hospital, Federal University of Alagoas, Maceió, Brazil
- 9University Hospital, Federal University of Rio Grande do Norte, Natal, Brazil
- 10Laboratory of Genomic and Histocompatibility Studies, University Hospital, Federal University of Maranhão, São Luís, Brazil
Collapsing glomerulopathy (CG) is a clinicopathologic entity characterized by segmentar or global collapse of the glomerulus and hypertrophy and hyperplasia of podocytes. The Columbia classification of 2004 classified CG as a histological subtype of focal segmental glomerulosclerosis (FSGS). A growing number of studies have demonstrated a high prevalence of CG in many countries, especially among populations with a higher proportion of people with African descent. The present study is a narrative review of articles extracted from PubMed, Medline, and Scielo databases from September 1, 2020 to December 31, 2021. We have focused on populational studies (specially cross-sectional and cohort articles). CG is defined as a podocytopathy with a distinct pathogenesis characterized by strong podocyte proliferative activity. The most significant risk factors for CG include APOL1 gene mutations and infections with human immunodeficiency virus and severe acute respiratory syndrome coronavirus 2. CG typically presents with more severe symptoms and greater renal damage. The prognosis is notably worse than that of other FSGS subtypes.
Introduction
In many countries, focal segmental glomerulosclerosis (FSGS) comprises the main histological diagnosis in patients with nephrotic syndrome (NS) (1). However, FSGS is nowadays understood as a heterogeneously entity. Cases with more marked glomerular collapse patterns have been described since 1974 (2). In 1986, a notable article described six African-American patients with NS and severe renal damage. Histological evaluation demonstrated glomerular collapse and significant tubulointerstitial damage (3), and the follow-up was marked by rapid progression requiring renal replacement therapy (RRT). Weiss followed this series of patients—one of whom was later diagnosed with human immunodeficiency virus (HIV). In 1988, the renal biopsy findings of nine patients supported the association of HIV with glomerular collapse (4). Since then, collapsing glomerulopathy (CG) has been recognized as one of the main histological forms of HIV-associated nephropathy (5).
Further studies have reported a strong association between CG and APOL1 gene mutations, especially among the black population of sub-Saharan Africa (6). In 2020, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection showed a strong association with this histological pattern (7), which highlighted the need to improve our understanding about this disease.
Epidemiology
The prevalence of CG varies widely between countries and may be difficult to assess because CG and FSGS are commonly analyzed together (8). Population-based studies in the United States, India, Pakistan, Macedonia, and Portugal proposed prevalence rates of 1.7, 0.75, 0.38, 1.7, and 0.29%, respectively (Table 1) (9–13). To the best of our knowledge, no population-based prevalence studies have been conducted in Brazil; however, the São Paulo Registry of Glomerulopathies indicates that 36% of FSGS cases comprise patients with CG (14).
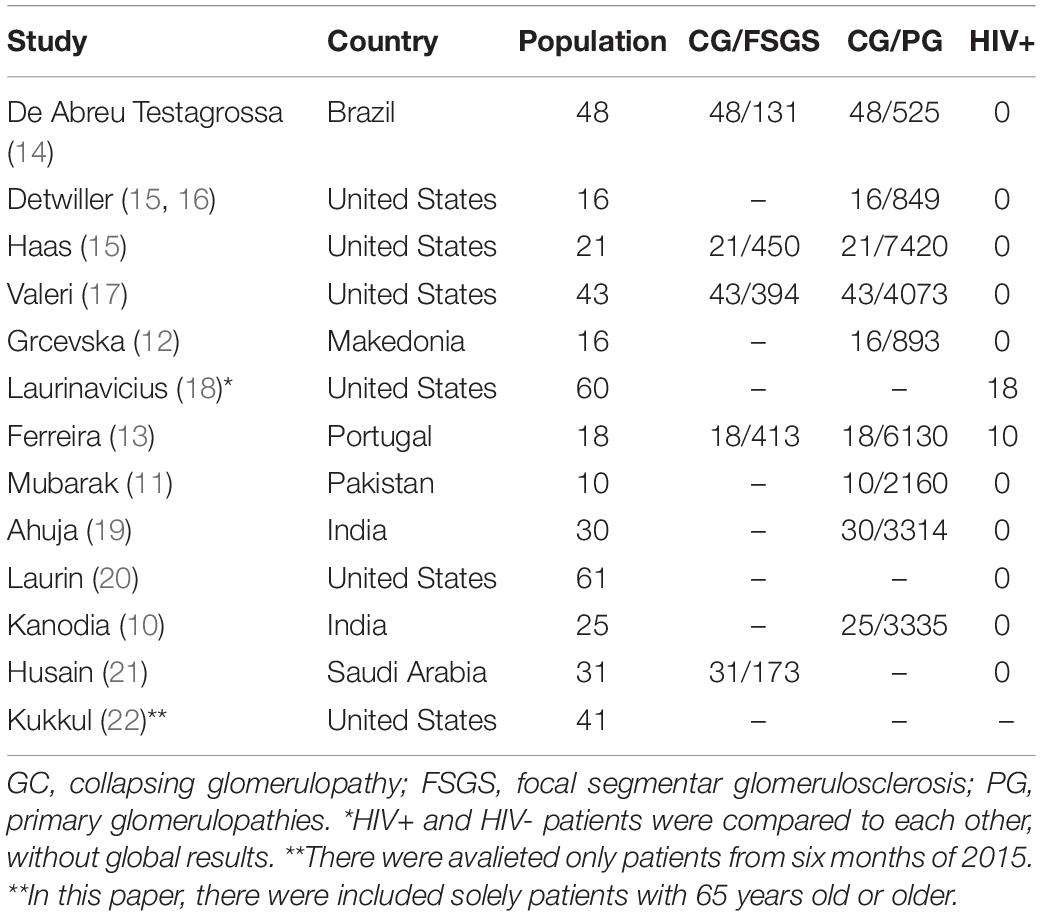
Table 1. CG prevalence in populational studies.
A survey conducted between 1979 and 1993 in the renal pathology center of Chicago suggested a significant increase in the proportion of CG among primary FSGS cases. In particular, the proportion of CG increased by 10, 20, and 24% in 1979–1985, 1985–1989, 1989–1993, respectively (15), which may be attributed to improvements in histopathologic diagnoses or changes in the epidemiological behavior of conditions such as HIV.
Pathogenesis
Cell Behavior
From a histopathologic perspective, CG falls within the spectrum of podocytopathies (Figure 1). Podocytes are terminal cells that constitute an essential part of the glomerular structure. They consist of a cell body, primary, secondary, and tertiary podocyte processes, and interdigitations between these processes (8). The spaces between pedicels are filled with slit diaphragm proteins, which function as mechanical and electrical barriers for a plasma ultrafiltrate (23–25). The proteins that constitute the diaphragm are strongly anchored to the cytoskeleton of the podocytes (23) and comprise a strong functional part of the glomerular filtration barrier.
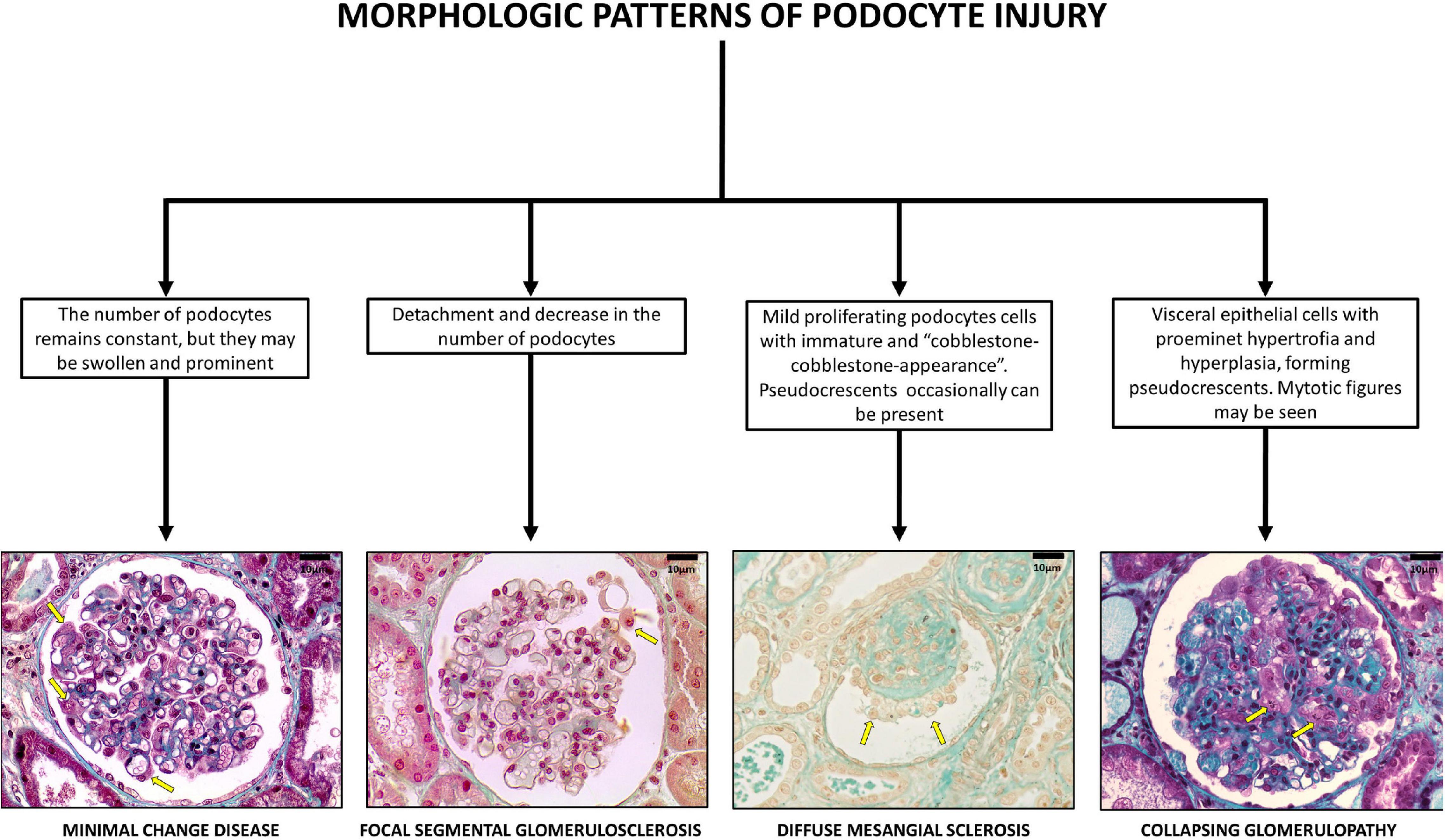
Figure 1. Glomerular pattern and podocytes morphological changes in human podocytopathies (bar = 10 μm).
Barisoni (26) classified podocytopathies based on their morphological characteristics and pathogenesis. Minimal change disease presents with slit diaphragm involvement instead of podocyte damage, whereas FSGS presents with marked podocytopenia (27). In contrast, diffuse mesangial sclerosis (DMS) and CG are characterized by high proliferation rates (9). The proliferation rate is higher in CG than in DMS (26).
Although podocytes are usually considered as terminal differentiation cells, they have a marked loss of differentiation and high mitotic rates in CG. Shkeri utilized the western blot technique to analyze the gene expressions of several glomerulopathies and demonstrated that CG was associated with increased cell proliferation markers, such as T-telomerase, Ki-67 protein, and beta-cadherin 1; however, these findings were not observed in the other types of FSGS (28).
The human Wilms tumor 1 (WT1) gene is a tumor suppressor gene that spans ∼50 kb and consists of 10 exons. It encodes a protein that shares a high degree of structural homology with the early growth response family of transcription factors. Several lines of evidence suggest that WT1 is important for normal podocyte function (29). Overexpression of the Wilms tumor 1 (WT1) protein results in an increased telomerase function, which promotes podocyte cell differentiation and renal histological organization. Inhibition of the WT1 protein is associated with terminal phenotype loss and podocyte hyperplasia (28). In CG, WT1 inhibition results in podocytes that have similar gene expressions patterns with those of the glomerular parietal epithelium, which may represent a phenotypic return to the original cells that yielded the Bowman’s capsule epithelium (28). WT1 has been demonstrated to activate transcription of the podocalyxin gene. The integral membrane protein podocalyxin connects to the cytoskeleton of the podocytes and is implicated in maintaining the complex three-dimensional shape of the cells (29). Gene inhibition of podocalyxin and other proteins that contribute to the structure of podocytes and slit diaphragms by determining protein binding and local electrostatic forces as synaptopodin, glomerular epithelial protein 1, common acute lymphoblastic leukemia antigen, and the C3b receptor has also been documented in CG (24).
Other cells may also be associated with the pathophysiology of CG. Under hypoxic conditions, glomerular endothelial cells have been reported to secrete paracrine factors that modify podocyte structure (30). HIV-affected T lymphocytes are also involved in cell proliferation (31).
Genetics
APOL1, which is located on the long arm of chromosome 22 (22q12 region), contributes to the pathogenesis of CG. It is responsible for forming high-density lipoproteins in different cell membranes and is associated with innate immunity. APOL1 confers resistance to Trypanosoma brucei rhodesiense, the etiologic agent for African sleeping sickness. APOL1 improves gene function and fights against infections through G1 (missense) and G2 (deletion of two amino acids) mutations. APOL1 is also associated with several cell damage mechanisms, such as mitochondrial damage, lysosomal degranulation, and cell pore formation (6). Lysosomal degranulation plays a particular role in destroying the cell membranes of foreign pathogens (32).
APOL1 mutations are correlated with the distribution of sleeping sickness. As such, these mutations are quite prevalent in sub-Saharan Africa, reaching over 40% in countries such as Ghana and Nigeria (6). In several countries including Brazil, APOL1 mutations are considered as risk factors and determinants of chronic kidney disease (CKD). A national case-control study demonstrated that these mutations are associated with an odds ratio of 10.95 for progression to CKD compared to controls. APOL1 mutations have also been associated with an increased indication for hemodialysis within a mean period of 12 years or earlier (33).
Other genes have been implicated in the pathogenesis of CG. The COQ2 gene (4q21.23) encodes the mitochondrial protein CoQ1, which plays a role in some neurologic, muscle, and renal syndromes. Mitochondrial gene mutations contribute to the proliferation of a poorly differentiated podocyte profile, which results in CG (34, 35). In contrast, the MHY9 gene is responsible for synthesizing myosin microfilaments that maintain the podocyte structure and filtration barrier. Among patients with HIV, MHY9 gene mutations increase the risk of progression to CG by 4–8-fold (36).
Etiologies
Barisoni proposed a classification scheme for podocytopathies and characterized CG as idiopathic/primary, genetic, and reactive/secondary (26) (Figure 2). The primary conditions that cause CG are highlighted here and classified according to the nature of injury, as follows:
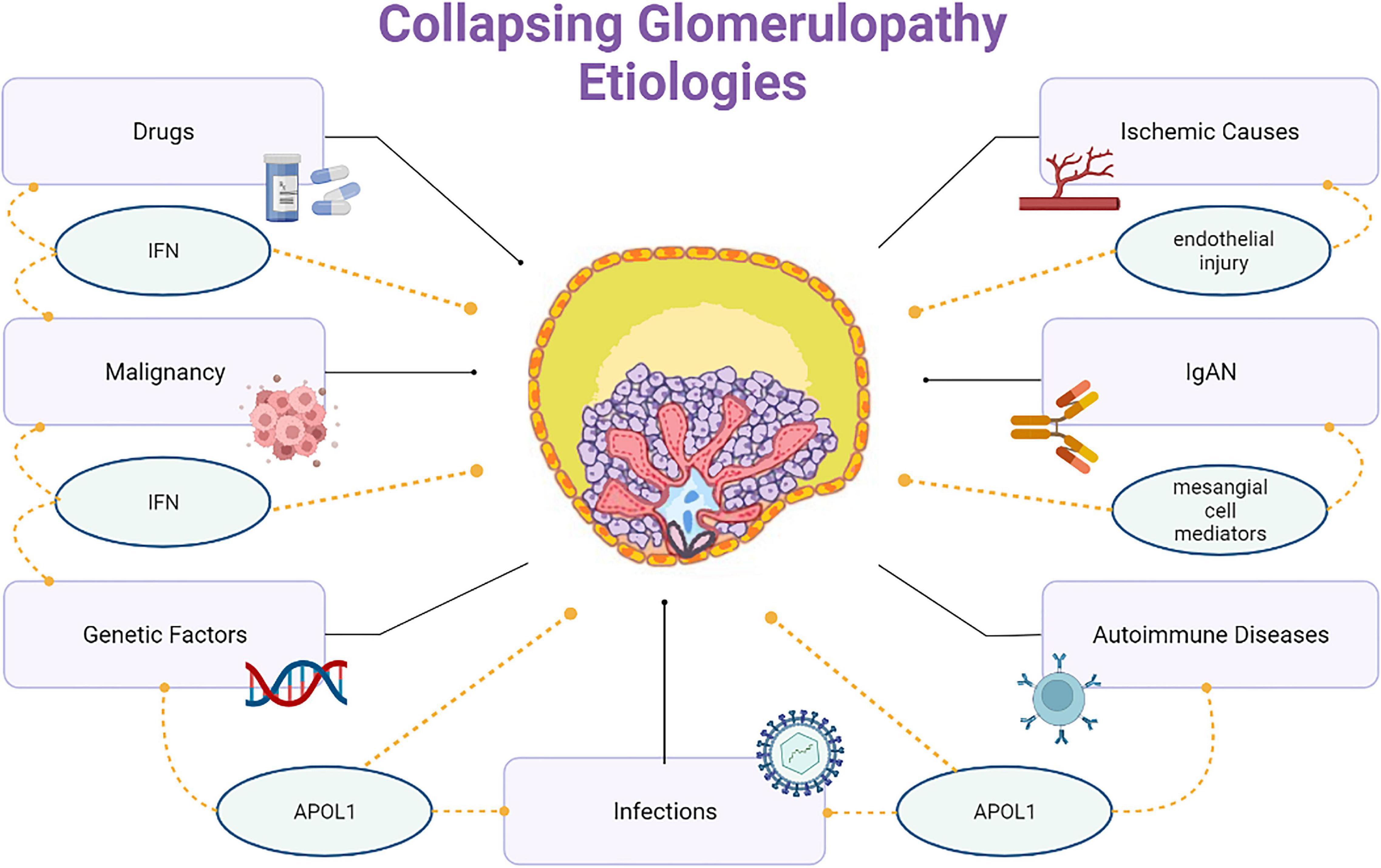
Figure 2. Collapsing glomerulopathy etiologies.
(1) Infectious etiologies like HIV, T-cell lymphotropic virus, hepatitis B virus (HBV), hepatitis C virus (HCV), Epstein-Barr virus, cytomegalovirus (CMV), parvovirus B19, dengue, Zika virus, Chikungunya, tuberculosis, visceral leishmaniasis, filariasis, and SARS-COV-2.
(2) Autoimmune etiologies including systemic lupus erythematosus (SLE), adult Still’s disease, temporal arteritis, mixed connective tissue disease, and Behçet’s disease.
(3) Neoplastic etiologies, such as multiple myeloma, acute monoblastic leukemia, natural killer cell leukemia, and acquired hemophagocytic syndrome.
(4) Drug-related etiologies like bisphosphonates, interferons, anabolic steroids, heroin, valproic acid, and anthracyclines.
(5) Ischemic etiologies, that include thrombotic microangiopathy (TMA), atheroembolic disease, sickle cell anemia, and thromboembolism by hydrophilic polymers.
Collapsing glomerulopathy can also coexist with other nephropathies, such as immunoglobulin A (IgA) nephropathy, membranous glomerulopathy, and other histological types of FSGS and DMS.
Several case reports are published annually that link CG to infectious diseases. Chandra (37) proposed the following criteria to assess the causal link between infection and CG: (1) CG demonstration in multiple cases of viral disease; (2) clear demonstration of CG lesions, including glomerular tuft collapse and changed podocyte phenotype; (3) demonstration of viral proteins or nucleic acids in glomerular cells, especially in podocytes; and (4) experimental demonstration (in animal models) of some (or all) CG findings.
Collapsing glomerulopathy is the most common morphological pattern in HIV nephropathy (38). The increased incidence of HIV infection has been identified as a cause for the proportionate increase in the cases of CG (17). Disease activity characterized by a high viral load and low CD4 lymphocyte count results in greater renal damage (5); however, the virus has also been detected in the renal tissue of patients with undetectable viral loads (31). Non-structural HIV proteins, such as viral protein R and negative factor, promote cell cycle dysregulation, which stimulates podocyte proliferation. Other factors (such as chronic inflammation) are associated with the genetic predisposition for APOL1 and MHY9 mutations, which result in CG (31). In some studies, the prevalence of HIV infection in patients with CG ranged from 30 to 55% (13, 18).
Cytomegalovirus infection is associated with immunosuppression, but it can also occur in immunocompetent patients and progress to CG, even in the acute phase of the disease. Specific treatment is associated with several benefits for prognosis in this population (37, 39–41).
Arboviruses were recently identified as an important causative factor for CG, particularly in Brazil. Eight of 13 CG biopsy samples obtained from the first half of 2016 from a large Brazilian kidney biopsy center were positive for arbovirus, wherein six samples were positive for dengue, one for Zika, and one for a concomitant infection; only one case had APOL1 mutations. These findings suggested that direct viral action in tissues may be associated with other risk factors, such as G1 and G2 mutations (42, 43).
The association between SARS-CoV-2 infection and CG was initially demonstrated in a series of autopsies (7). A recent systematic review of 59 studies reporting COVID-19 related histopathological diagnoses from kidney biopsy identified CG as the most common finding, followed by acute tubular injury and trombotic microangiopathy (44). Various mechanisms of AKI secondary to COVID-19 have been proposed—from direct intrarenal infection to dysregulation of the renin-angiotensin-aldosterone system, to altered hemodynamic control, coagulation and cytokine homeostasis (44). Although the association is multifactorial, it has been emphasized the influence of the hyperactive inflammatory process and participation of circulating interferons (44) and direct infection appears highly unlikely to play a significant pathogenic role.
APOL1 mutations have been detected in many patients with COVID-19, which suggested that the virus is a potential secondary trigger for glomerular damage (45). In fact, studies have confirmed the strong association of CG and non-collapsing podocytopathies with concurrent or recent COVID-19 in patients with APOL1 high-risk alleles (44–46).
Systemic lupus erythematosus can trigger CG as an extreme form of lupus podocytopathy in the absence of other lupus nephritis patterns (47). CG may also result from the association of SLE with other risk factors, such as black ethnicity and APOL1 mutations. CG is occasionally present during the diagnosis of SLE (47), with low levels of therapeutic response.
Collapsing glomerulopathy can be drug-induced; bisphosphonates, especially pamidronate and zoledronic acid, inhibit the mevalonate synthesis pathways, which are essential for cell differentiation. This triggers podocyte proliferation and progression to CG (48). Synthetic interferons (which are used to treat some infectious, autoimmune, and neoplastic diseases) can result in APOL1 overexpression in the glomerular epithelium, which triggers podocyte damage. This effect is evidenced by the presence of tubuloreticular inclusions on electron microscopy (48). Illicit drugs, such as cocaine and heroin, are also associated with CG. The most probable mechanism involves ischemic glomerular damage from oxidative endothelial damage, accelerated atheromatosis, and direct vasoconstriction (49).
Acquired Hemophagocytic Syndrome results from a hyperactive immune system that develops secondary to infectious diseases or lymphatic hematological neoplasms. Excessive T lymphocyte and circulating cytokine activation can promote podocyte proliferation and progression to CG (50). Monoclonal gammopathies can also encourage progression toward CG. Histological analysis demonstrates diffuse Ig chain deposits, with glomerular collapse resulting from the deposition of extracellular elements instead of from podocyte proliferation (51). Once triggered, CG progresses independently of the underlying disease. Further, while the underlying gammopathy may go into remission, CG can continue to worsen until RRT becomes necessary (52).
Collapsing glomerulopathy is also associated with diseases characterized by microvascular damage, such as sickle cell anemia, intravascular hemolysis syndromes, drug reactions (calcineurin inhibitors), malignant arterial hypertension, and antiphospholipid syndrome. A total of 53 histological samples that demonstrated TMA were evaluated. These samples were acquired from 33 patients with FSGS, 19 of whom had CG (30). Glomerular ischemia resulted in the loss of podocyte differentiation and reduced cell proliferation. The population in this study was predominantly of white ethnicity and was characterized by a lower incidence of nephrotic proteinuria.
Other conditions, such as hypertensive disorders of pregnancy (HDP), have also been implicated as a trigger for CG. HDP are characterized by diffuse endothelial damage and possible glomerular ischemia. Kidney biopsy often shows association between CG and TMA (46).
Histology
Although initially treated as a single entity, FSGS encompasses several distinct histological patterns. FSGS is heterogenous in terms of etiology, histological characteristics, clinical presentation, treatment response, and prognosis. For this reason, a classification of FSGS histological subtypes was proposed in 2004 (53). The diagnosis of CG requires the presence of at least one glomerulus with segmental or global collapse and podocyte hypertrophy and hyperplasia (Figure 3). CG can occur with other morphological subtypes, and a single characteristic lesion is adequate for a diagnosis of CG.
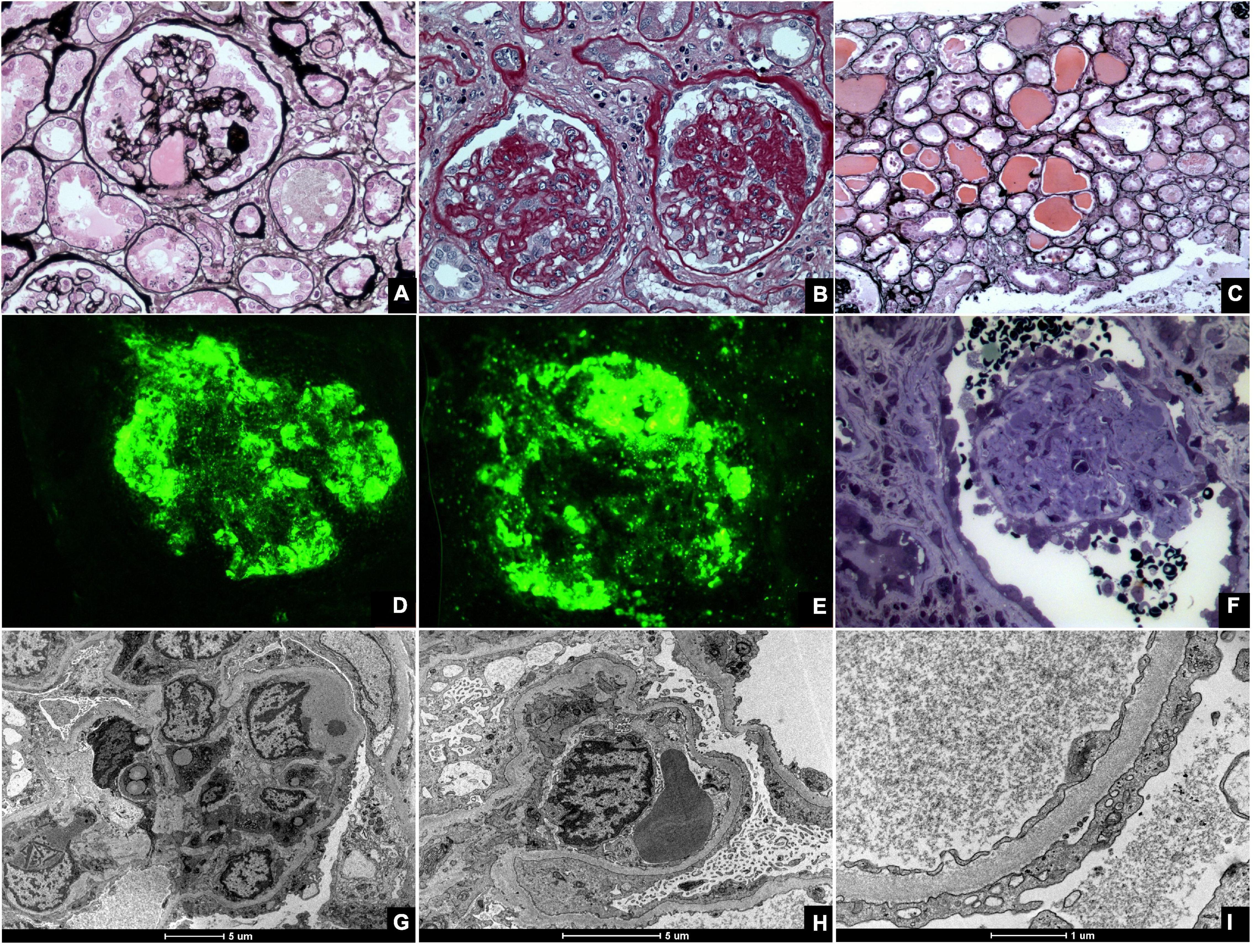
Figure 3. Kidney biopsies of collapsing glomerulopathy. (A,B) Periodic Acid Schiff (PAS) and Jones Methenamine Silver (JMS) (40×), respectively show intense podocyte hyperplasia and glomerular tuft collapse. (C) JMS (20×) exhibits microcytic transformation of distal convoluted tubules with accumulations of hyaline material inside of those. (D,E) Fluorescence microscopy (40×) shows, respectively, IgM and C3 trapping in areas of collapse/sclerosis. (F) Semi-fine stained in Toluidine Blue (63×) with collapse of the entire glomerular tuft and hyperplasia of podocytes and dilated Bowman’s space. (G,H) Transmission electron microscopy contrasted with Osmium Tetroxide, Lead Citrate and Uranyl in block shows capillary loop collapse with hyalinosis in addition to diffuse fusion and flattening of the pedicels associated with microvillous transformation. (I) Electron microscopy tubes contrasted with osmium tetroxide, lead citrate, and uranyl in block with detail of disorganization of the cytoskeleton in the podocyte cytoplasm, with extensive effacement of the pedicels.
Light microscope shoes partial or total obliteration of the lumen of the glomerular capillaries secondary to podocyte hypertrophy and hyperplasia (53, 54). Podocyte crowns may contain intracytoplasmic deposits, which denote protein reabsorption. Pseudocrescents, which resemble glomerular crescents, may temporally precede glomerular sclerosis (27). Other findings, such as glomerulomegaly, hyalinosis, hypercellularity, and adhesions, are unusual but more common in the final stages of the disease (55).
As a rule, tubulointerstitial damage in CG is more intense than in other FSGS subtypes (17). Important specific findings include tubular dilatation with molding and the formation of tubular microcysts (10, 17). Tissue and lymphatic macrophages, specifically CD4 and CD8, can also be found in interstitial infiltrates (10).
Immunofluorescence (IF) findings are non-specific and, in many cases, negative. If present, they usually comprise granular or mesangial IgM and C3 deposits. IF may suggest associated conditions such as Berger’s disease, which is characterized by mesangial IgA deposits (54), or SLE, which is associated with several antibody deposits.
Electron microscopy (EM) is mostly unnecessary; however, it can demonstrate collapsed and ruptured capillary membranes and swollen, hypertrophic, and/or hyperplastic podocytes (54). EM may also identify intracellular inclusions that are often associated with HIV or SLE. In these conditions, the podocyte processes are greatly compromised with loss of glomerular filtration barrier integrity.
Clinical Presentation
The most frequent clinical presentation of CG is massive, severe proteinuria associated with pure NS. Patients may present with hypertension, lipiduria, and hematuria on urinalysis. A significantly high number of patients present with renal failure on admission and rapidly progress to end-stage renal disease (ESRD) (13, 17, 20, 27, 56).
Collapsing glomerulopathy demonstrates no sex predilection, but preferentially affects patients of African descent. The disease also often affects young adult patients and has a varying predominance in the pediatric age group (10, 13, 17, 19, 20, 57).
The normal kidney dimensions are usually preserved in CG and presents as in other conditions, such as diabetic nephropathy, polycystic kidney disease, and amyloidosis. Ultrasonography may demonstrate normal or increased renal size and hyperechogenicity, which may be due to edema, fibrosis, and tubulointerstitial infiltrates resulting from the rapid progression to ESRD and absence of renal parenchymal contraction (17).
Treatment
The treatment of CG consists of the following: (1) targeted therapy for disorders resulting from NS, such as dyslipidemia, hypertension, and edema; (2) treatment of the underlying disease when CG is associated with other conditions; and (3) immunosuppressive therapy.
According to the Kidney Disease Improving Global Outcomes guidelines on the management of glomerulopathies (58, 59), the measures to control complications associated with NS consist of edema control through a low-sodium diet (<2 g/day), fluid restriction, diuretic therapy, and, if necessary, hemodialysis or ultrafiltration. Systolic blood pressure should be maintained at <120 mmHg with angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, unless renal function worsens on these medications (60).
The severity of dyslipidemia (DLP) in patients with nephrotic syndrome (NP) is proportional to the degree of proteinuria. Once the proteinuria in patients with GC can be massive, the serum levels of cholesterol and its fractions as well as lipoproteins can reach high elevated values. The potential contributors to DLP genesis in patients with nephrotic syndrome are the patient’s diet, use of drugs (such as corticosteroids, calcineurin inhibitors, and mTOR inhibitors), in addition to genetic predisposition (61, 62). According to the new KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases (60) in patients with NP the DLP should be controlled, especially in those ones with other cardiovascular risk factors (e.g., hypertension and diabetes) and/or diseases that respond poorly to stabilized therapy.
As non-pharmacological measures, lifestyle changes (diet, smoking cessation, weight loss, and physical activity) must be encouraged in all patients. The tratment with statins as a first-line treatment should be indicated in patients at increased risk of atherosclerotic cardiovascular disease, such as those with GFR <60 ml/min/1.73m2 and/or albuminuria >30 mg/g, in addition to treating infectious/inflammatory diseases that contribute to increased cardiovascular risk. For patients who do not tolerate the use of statins or cannot achieve the recommended lipid levels targets, other drugs as bile acid chelators, fibrates, nicotinic acid, ezetimibe, PCSK9 inhibitors and lipid apheresis can be used.
Special attention should also be given to the risk of thrombotic events; prophylactic anticoagulation should be considered for susceptible populations. Immunocompromised patients should be given full immunizations; tested for HIV, HBV, HCV, tuberculosis, and syphilis; and initiated on strongyloidiasis and pneumocystis prophylaxis.
When an underlying etiology is identified, such as drug-related, infectious, genetic, autoimmune, and other causes, the patient should undergo specific therapy, when available. In drug-induced cases, the offending drug must be discontinued whenever possible (63). In cases associated with infections, specific antimicrobial treatments can reduce CG progression or even promote remission. Some HIV-associated cases demonstrate 38% delay in the progression to ESRD following the initiation of antiretroviral therapy (ART) (64); however, other studies proposed that up to 50% of patients progress toward the initiation of RRT despite adequate treatment (65). Nevertheless, HIV-associated nephropathy should always be treated with ART because it is very effective at controlling the disease (66).
Intravenous Ig and cidofovir can be used to treat parvovirus B19 infection, which is particularly important from the perspective of renal transplantation (67). CMV infections should be treated with ganciclovir, which also promotes CG remission and renal function improvement (39).
Immunosuppressant therapy was administered to most patients. While there are no specific recommendations for CG, the immunotherapy guidelines for CG are extrapolated from the FSGS protocol (20). Several types of immunosuppressants have been proposed (16–18, 20), but the initial therapeutic regimen consists of high-dose of oral corticosteroids for 4–16 weeks or until remission is achieved. Calcineurin inhibitors or cyclophosphamide are considered second-line medications for patients that exhibit corticosteroid resistance, corticosteroid dependence, or frequent relapses; they should be given for at least 12 weeks. Other immunosuppressants (such as rituximab and mycophenolate mofetil) are indicated for patients who do not respond to other immunosuppressive regimens. Patients with extensive kidney damage may choose to forego treatment for CG because of the lack of benefits and risks associated with immunosuppression therapy.
Prognosis
Collapsing glomerulopathy has a poor prognosis. Most cases present with refractory proteinuria, severe loss of renal function, and progression to permanent RRT (15, 53, 54). While other FSGS subtypes have a renal survival time of 62.5 months, CG has a renal survival time of 13 months (17). Table 2 summarizes the treatment data and prognoses collected by the studies that examined patients with CG.
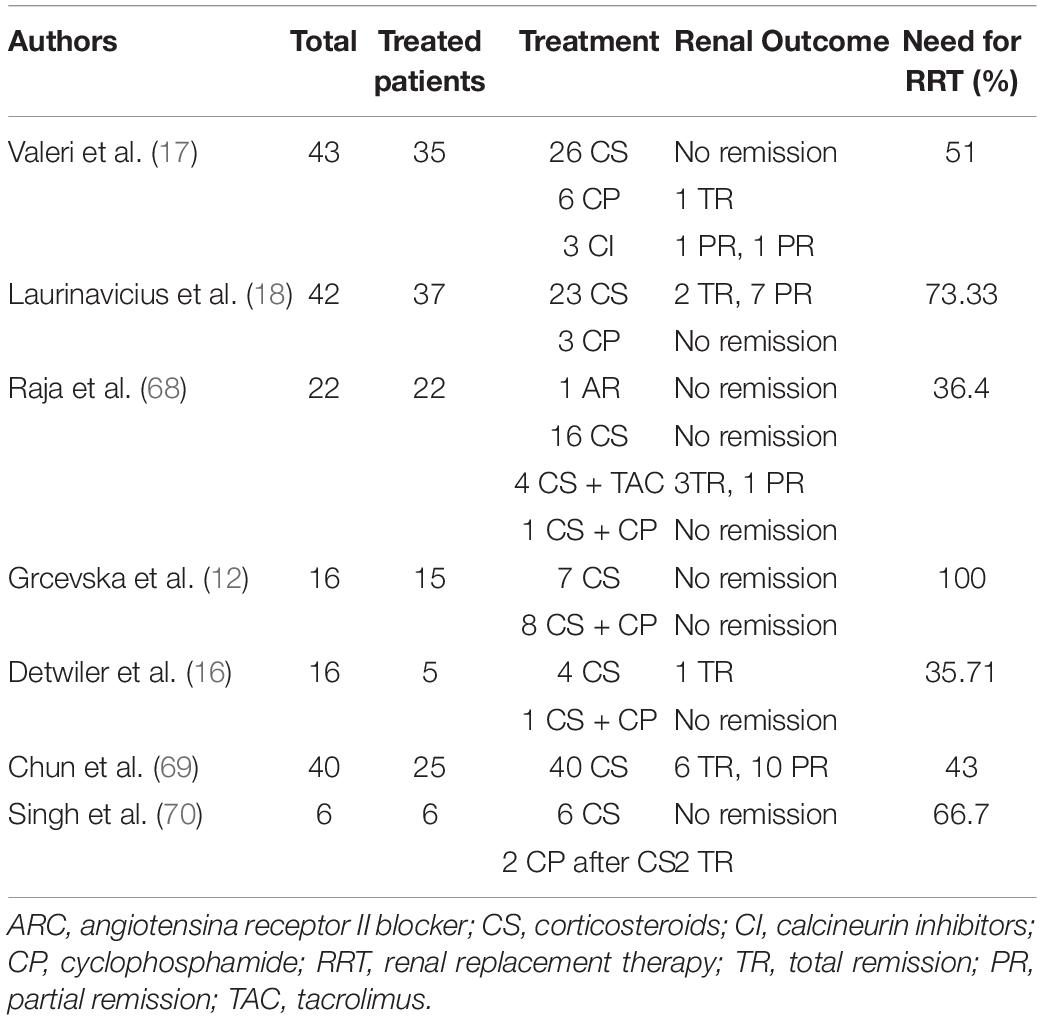
Table 2. CG patients’ treatment and follow-up data.
Conclusion
The current literature demonstrates that CG is a distinct clinicopathological entity that results from complex interactions between intrinsic (genetic mutations) and extrinsic (especially infections) factors. CG presents at more advanced stages and with poorer renal function, which emphasizes that it must be evaluated properly, especially in developing countries where risk factors, such as APOL1 mutations and viral infections, are more common.
The prognosis of CG is notably worse than that of the other FSGS subtypes. A large number of young patients with CG progress to permanent RRT. Larger longitudinal studies are needed to assess the factors associated with CG in different populations and determine the clinical and histological elements that contribute to the final prognosis.
This article was written by the Collapsing Brazilian Consortium, an initiative in Brazil that brings nephrologists and pathologists together to study CG.
Author Contributions
ÉC, PN, MC, and GS conceived and designed the analysis, performed the analysis, and wrote the manuscript. DW, MMu, FL, OG, RV, DB, JL, NS-F, JA, MMa, and SA contributed data or analysis tools and performed the analysis. AT-J contributed data or analysis tools, performed the analysis, and wrote the manuscript. FG contributed data or analysis tools, performed the analysis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001, and by the University Hospital of Federal University of Maranhão (HUUFMA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Special thanks to all collaborators from Collapsing Brazilian Consortium (COBRAZ).
References
1. Polito MG, De Moura LAR, Kirsztajn GM. An overview on frequency of renal biopsy diagnosis in Brazil: clinical and pathological patterns based on 9617 native kidney biopsies. Nephrol Dial Transplant. (2010) 25:490–6. doi: 10.1093/ndt/gfp355
2. Brown CB, Cameron JS, Turner DR, Chantler C, Ogg CS, Willians DG, et al. Focal segmental glomerulosclerosis with rapid decline in renal function (‘malignant FSGS’). Clin Nephrol. (1978) 10:51–61.
3. Weiss MA, Daquioag E, Margolin EG, Pollak VE. Nephrotic syndrome, progressive irreversible renal failure, and glomerular “collapse”: a new clinicopathologic entity? Am J Kidney Dis. (1986) 7:20–8. doi: 10.1016/s0272-6386(86)80052-x
4. Cohen AH, Nast CC. HIV-associated nephropathy. A unique combined glomerular, tubular, and interstitial lesion. Mod Pathol. (1988) 1:87–97.
5. Wyatt CM, Klotman PE, D’Agati VD. HIV-associated nephropathy: clinical presentation, pathology, and epidemiology in the era of antiretroviral therapy. Semin Nephrol. (2008) 28:513–22. doi: 10.1016/j.semnephrol.2008.08.005
6. Freedman BI, Limou S, Ma L, Kopp JB. APOL1-associated nephropathy: a key contributor to racial disparities in CKD. Am J Kidney Dis. (2018) 72(5 Suppl. 1):S8–16. doi: 10.1053/j.ajkd.2018.06.020
7. Nasr SH, Kopp JB. COVID-19–associated collapsing glomerulopathy: an emerging entity. Kidney Int Rep. (2020) 5:759–61. doi: 10.1016/j.ekir.2020.04.030
8. Kopp JB, Anders HJ, Susztak K, Posteda MA, Remuzzi G, Friedhelm H, et al. Podocytopathies. Nat Rev Dis Primers. (2020) 6:1–24. doi: 10.1038/s41572-020-0196-7
9. Cossey NL, Larsen CP, Liapis H. Collapsing glomerulopathy: a 30-year perspective and single, large center experience. Clin Kidney J. (2017) 10:443–9. doi: 10.1093/ckj/sfx029
10. Kanodia KV, Vanikar AV, Patel RD, Shutar KS, Nigan LK, Patel HV, et al. Collapsing glomerulopathy: a single centre clinicopathologic study of seven years. J Clin Diagn Res. (2016) 10:EC15–7. doi: 10.7860/JCDR/2016/17297.7646
11. Mubarak M, Kazi JI. Collapsing FSGS: a clinicopathologic study of 10 cases from Pakistan. Clin Exp Nephrol. (2010) 14:222–7. doi: 10.1007/s10157-010-0275-2
12. Grcevska L, Polenakovik M. Collapsing glomerulopathy: clinical characteristics and follow-up. Am J Kidney Dis. (1999) 33:652–7. doi: 10.1016/s0272-6386(99)70215-5
13. Ferreira AC, Carvalho D, Carvalho F, Galvo MJ, Nolasco F. Collapsing glomerulopathy in Portugal: a review of the histological and clinical findings in HIV and non-HIV patients. Nephrol Dial Transplant. (2011) 26:2209–15. doi: 10.1093/ndt/gfq686
14. De Abreu Testagrossa L, Malheiros DMAC. Estudo brasileiro das variantes morfológicas da glomerulosclerose segmentar e focal. J Bras Patol Med Lab. (2012) 48:211–5. doi: 10.1590/S1676-24442012000300009
15. Haas M, Spargo BH, Coventry S. Increasing incidence of focal-segmental glomerulosclerosis among adult nephropathies: a 20-year renal biopsy study. Am J Kidney Dis. (1995) 26:740–50. doi: 10.1016/0272-6386(95)90437-9
16. Detwiler RK, Falk RJ, Hogan SL, Jennette JC. Collapsing glomerulopathy: a clinically and pathologically distinct variant of focal segmental glomeruloscierosis. Kidney Int. (1994) 45:1416–24. doi: 10.1038/ki.1994.185
17. Valeri A, Barisoni L, Appel GB, Seigle R, D’Agati V. Idiopathic collapsing focal segmental glomeruloscierosis: a clinicopathologic study. Kidney Int. (1996) 50:1734–46. doi: 10.1038/ki.1996.493
18. Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. (1999) 56:2203–13. doi: 10.1046/j.1523-1755.1999.00769
19. Ahuja A, Gupta R, Sharma A, Bagga A, Bohwmik DN, Agarwal SK, et al. Idiopathic collapsing glomerulopathy: a clinicopathologic analysis of 30 cases. Indian J Nephrol. (2014) 24:239–42. doi: 10.4103/0971-4065.133009
20. Laurin LP, Gasim AM, Derebail VK, McGregor JG, Kidd JM, Hogan SL, et al. Renal survival in patients with collapsing compared with not otherwise specified FSGS. Clin J Am Soc Nephrol. (2016) 11:1752–9. doi: 10.2215/CJN.13091215
21. Husain S. Collapsing glomerulopathy, the Saudi Arabian scenario. A study of 31 cases and a review of literature. Saudi Med J. (2017) 38:509–16. doi: 10.15537/smj.2017.5.19299
22. Kukkul B, Rupali SA, Smith KD, Houghton SD, Troxell ML, Andeen NK. Collapsing glomerulopathy in older adults. Mod Pathol. (2019) 32:532–8. doi: 10.1038/s41379-018-0154-z
23. Cheng H, Harris RC. The glomerulus – a view from the outside - the podocyte. Int J Biochem Cell Biol. (2010) 42:1380–7. doi: 10.1016/j.biocel.2010.05.014
24. Barisoni L, Kriz W, Mundel P, D’agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (1999) 10:51–61. doi: 10.1681/ASN.V10151
25. Chugh SS, Clement LC. Telomerase at the center of collapsing glomerulopathy. Nat Med. (2012) 18:26–7. doi: 10.1038/nm.2602
26. Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. (2007) 2:529–42. doi: 10.2215/CJN.04121206
27. Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing glomerulopathy. J Am Soc Nephrol. (2006) 17:2854–63. doi: 10.1681/ASN.2006030225
28. Shkreli M, Sarin KY, Pech MF, Papeta N, Chang W, Brockmann SA, et al. Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat Med. (2012) 18:111–9. doi: 10.1038/nm.2550
29. Scholz H, Kirschner KMA. Role for the Wilms’ tumor protein WT1 in organ development. Physiology. (2005) 20:54–9. doi: 10.1152/physiol.00048.2004
30. Buob D, Decambron M, Gnemmi V, Frimat M, Hoffmann M, Azar R, et al. Collapsing glomerulopathy is common in the setting of thrombotic microangiopathy of the native kidney. Kidney Int. (2016) 90:1321–31. doi: 10.1016/j.kint.2016.07.021
31. Ross MJ. Advances in the pathogenesis of HIV-associated kidney diseases. Kidney Int. (2014) 86:266–74. doi: 10.1038/ki.2014.167
32. Siemens TA, Riella MC, Moraes TP, Riella CV. APOL1 risk variants and kidney disease: what we know so far. J Braz Nephrol. (2018) 40:388–402. doi: 10.1590/2175-8239-JBN-2017-0033
33. Riella C, Siemens TA, Wang M, Campos RP, Moraes TP, Riella LV, et al. APOL1-associated kidney disease in Brazil. Kidney Int Rep. (2019) 4:923–9. doi: 10.1016/j.ekir.2019.03.006
34. Barisoni L, Diomedi-camassei F, Santorelli FM, Caridi G, Thomas DB, Emma F, et al. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int. (2008) 74:237–43. doi: 10.1038/sj.ki.5002767
35. Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, et al. COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol. (2018) 33:1257–61. doi: 10.1007/s00467-018-3937-z
36. Winkler CA, Nelson G, Oleksyk TK, Nava MB, Kopp JB. Genetics of focal segmental glomerulosclerosis and human immunodeficiency virus-associated collapsing glomerulopathy: the role of MYH9 genetic variation. Semin Nephrol. (2010) 30:111–25. doi: 10.1016/j.semnephrol.2010.01.003
37. Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. (2013) 6:1–5. doi: 10.1093/ckj/sft002
38. Husain NE, Ahmed MH, Almobarak AO, Noor SK, Elmadhoun WM, Awadalla H, et al. HIV-associated nephropathy in Africa: pathology, clinical presentation and strategy for prevention. J Clin Med Res. (2018) 10:1–8. doi: 10.14740/jocmr3235w
39. Grover V, Gaiki MR, DeVita MV, Schwimmer JA. Cytomegalovirus-induced collapsing focal segmental glomerulosclerosis. Clin Kidney J. (2013) 6:71–3. doi: 10.1093/ckj/sfs097
40. Freitas GRR, Praxedes MRG, Malheiros D, Testagrossa L, Dias CB, Woronik V. Collapsing variant of focal segmental glomerulosclerosis by parvovirus B19: case report. J Bras Nefrol. (2015) 37:121–6. doi: 10.5935/0101-2800.20150017
41. Besse W, Mansour S, Jatwani K, Nast CC, Brewster UC. Collapsing glomerulopathy in a young woman with APOL1 risk alleles following acute parvovirus B19 infection: a case report investigation. BMC Nephrol. (2016) 17:125. doi: 10.1186/s12882-016-0330-7
42. Araújo SDA, Cordeiro TM, Belisário AR, Araújo RFA, Marinho PES, Kroon EG, et al. First report of collapsing variant of focal segmental glomerulosclerosis triggered by arbovirus: dengue and zika virus infection. Clin Kidney J. (2019) 12:355–61. doi: 10.1093/ckj/sfy104
43. Queiroz PC, Jorge AES, Mourão PHV, Penido MGMG. Collapsing focal segmental glomerulosclerosis probably triggered by dengue virus infection – two case reports. J Bras Nefrol. (2020) 42:489–93. doi: 10.1590/2175-8239-JBN-2019-0237
44. Oliveira P, Cunha K, Neves P, Muniz M, Gatto G, Salgado-Filho N, et al. Renal morphology in coronavirus disease: a literature review. Medicina (Kaunas). (2021) 57:258. doi: 10.3390/medicina57030258
45. Teixeira Júnior AAL, Neves PDMM, Lages JS, Cunha KA, Muniz MRP, Brito DJA, et al. Brazilian consortium for the study on renal diseases associated with COVID-19: a multicentric effort to understand SARS-CoV-2-related nephropathy. Front Med. (2020) 7:584235. doi: 10.3389/fmed.2020.584235
46. Gopalakrishnan N, Dhanapriya J, Padmakumar C, Dineshkumar T, Kurien AA, Sakthirajan R, et al. Collapsing glomerulopathy and thrombotic microangiopathy in postpartum period: two case reports. Indian J Nephrol. (2018) 28:157–9. doi: 10.4103/ijn.IJN_242_16
47. Salvatore SP, Barisoni LMC, Herzenberg AM, Chander PN, Nickeleit V, Seshan SV. Collapsing glomerulopathy in 19 patients with systemic lupus erythematosus or lupus-like disease. Clin J Am Soc Nephrol. (2012) 7:914–25. doi: 10.2215/CJN.11751111
48. Markowitz GS, Bomback AS, Perazella MA. Drug-induced glomerular disease: direct cellular injury. Clin J Am Soc Nephrol. (2015) 10:1291–9. doi: 10.2215/CJN.00860115
49. Jaffe JA, Kimmel PL. Chronic nephropathies of cocaine and heroin abuse: a critical review. Clin J Am Soc Nephrol. (2006) 1:655–67. doi: 10.2215/CJN.00300106
50. Gebregeorgis W, Patel I, Thakur M, Bhutani D, Woldie I. Collapsing glomerulopathy associated with hemophagocytic syndrome in a patient with NK/T cell lymphoma. Clin Nephrol Case Stud. (2016) 4:11. doi: 10.5414/CNCS108586
51. Korbet SM, Schwartz MM. Multiple myeloma. J Am Soc Nephrol. (2006) 17:2533–45. doi: 10.1681/ASN.2006020139
52. Bhowmik D, Dinda AK, Gupta S, Agarwal SK, Tiwari SC, Dash SC. Multiple myeloma presenting as collapsing glomerulopathy. Indian J Pathol Microbiol. (2003) 46:233–4.
53. D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. (2004) 43:368–82. doi: 10.1053/j.ajkd.2003.10.024
54. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of renal pathology: collapsing glomerulopathy. Am J Kidney Dis. (2015) 66:e3–4. doi: 10.1053/j.ajkd.2015.04.009
55. Nieto Ríos JF, Milena Brand S, Serna Higuita LM, Fernando Arias L. The collapsing variant of focal segmental glomerulosclerosis in children. Iatreia. (2013) 26:481–6.
56. Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol. (2008) 19:1276–81. doi: 10.1681/ASN.2007080926
57. Watanabe A, Guaragna MS, Belangero VMS, Casimiro FMS, Pesquero JB, Feltran LS, et al. APOL1 in an ethnically diverse pediatric population with nephrotic syndrome: implications in focal segmental glomerulosclerosis and other diagnoses. Pediatr Nephrol. (2021) 36:1–10. doi: 10.1007/s00467-021-04960-w
58. Floege J, Barbour SJ, Cattran DC, Hogan JJ, Nachman PH, Tang SCW, et al. Management and treatment of glomerular diseases (part 1): conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2019) 95:268–80. doi: 10.1016/j.kint.2018.10.018
59. Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, et al. Management and treatment of glomerular diseases (part 2): conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2019) 95:268–80. doi: 10.1016/j.kint.2018.11.008
60. Cheung AK, Chang TI, Cushman WC, Furth SL, Hou FF, Ix JH, et al. Executive summary of the KDIGO 2021 clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int. (2021) 99:559–69. doi: 10.1016/j.kint.2020.10.026
61. Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int. (2016) 90:41–52. doi: 10.1016/j.kint.2016.02.026
62. Agrawal S, Zaritsky JJ, Fornoni A, Smoyer WE. Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat Rev Nephrol. (2018) 14:57–70. doi: 10.1038/nrneph.2017.155
63. Morales E, Alonso M, Gutiérrez E. Collapsing glomerulopathy: update. Med Clin (Barc). (2019) 152:361–7. doi: 10.1016/j.medcli.2018.10.021
64. Schwartz EJ, Szczech LA, Ross MJ, Klotman ME, Winston JA, Klotman PE. Highly active antiretroviral therapy and the epidemic of HIV end-stage renal disease. J Am Soc Nephrol. (2005) 16:2412–20. doi: 10.1681/ASN.2005040340
65. Szczech LA, Gupta SK, Habash R, Guasch A, Kalayjian R, Appel A, et al. The clinical epidemiology and course of the spectrum of renal diseases associated with HIV infection. Kidney Int. (2004) 66:1145–52. doi: 10.1111/j.1523-1755.2004.00865.x
66. Atta MG, Gallant JE, Rahman MH, Nagajothi N, Racusen LC, Scheel PL, et al. Antiretroviral therapy in the treatment of HIV-associated nephropathy. Nephrol Dial Transplant. (2006) 21:2809–13. doi: 10.1093/ndt/gfl337
67. Nair V, Jandovitz N, Jhaveri KD, Hirschwerk D, Grodstein L, Bijol V, et al. Treatment of parvovirus B19 viremia to facilitate kidney transplantation in a patient with collapsing glomerulopathy. Clin Nephrol Case Stud. (2020) 8:41–5. doi: 10.5414/CNCS110113
68. Ramachandran R, Rajakumar V, Duseja R, Sakhuja V, Jha V. Successful treatment of adult-onset collapsing focal segmental glomerulosclerosis with rituximab. Clin Kidney J. (2013) 6:500–2. doi: 10.1093/ckj/sft102
69. Chun MJ, Korbet SM, Schwartz MM, Lewis EJ. Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol. (2004) 15:2169–77. doi: 10.1097/01.ASN.0000135051.62500.97
Keywords: segmental and focal glomerulosclerosis, nephrotic syndrome (NS), renal biopsy, podocytes, glomerulopathy
Citation: Cutrim ÉMM, Neves PDMdM, Campos MAG, Wanderley DC, Teixeira-Júnior AAL, Muniz MPR, Ladchumananandasivam FR, Gomes OV, Vasco RFV, Brito DJdA, Lages JS, Salgado-Filho N, Guedes FL, de Almeida JB, Magalhães M, Araújo SdA and Silva GEB (2022) Collapsing Glomerulopathy: A Review by the Collapsing Brazilian Consortium. Front. Med. 9:846173. doi: 10.3389/fmed.2022.846173
Received: 30 December 2021; Accepted: 08 February 2022;
Published: 03 March 2022.
Edited by:
Roberta Fenoglio, Ospedale San Giovanni Bosco, ItalyReviewed by:
Claudio Ponticelli, Retired, Milan, ItalyMarco Quaglia, Università del Piemonte Orientale, Italy
Copyright © 2022 Cutrim, Neves, Campos, Wanderley, Teixeira-Júnior, Muniz, Ladchumananandasivam, Gomes, Vasco, Brito, Lages, Salgado-Filho, Guedes, de Almeida, Magalhães, Araújo and Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gyl Eanes Barros Silva, gyl.silva@ufma.br