 Nina Dwi Putri1†
Nina Dwi Putri1† Edison Johar2†
Edison Johar2† Yora Permata Dewi2
Yora Permata Dewi2 Nuri Dyah Indrasari3Dewi Wulandari3Merci Monica br Pasaribu3Teny Tjitra Sari1Fitri Prima Cakti1Madeline Ramdhani Jasin1
Nuri Dyah Indrasari3Dewi Wulandari3Merci Monica br Pasaribu3Teny Tjitra Sari1Fitri Prima Cakti1Madeline Ramdhani Jasin1 Tartila Tartila1
Tartila Tartila1 Frilasita Aisyah Yudhaputri2
Frilasita Aisyah Yudhaputri2 Safarina G. Malik2
Safarina G. Malik2 Khin Saw Aye Myint2*
Khin Saw Aye Myint2*- 1Department of Paediatrics, Dr. Cipto Mangunkusumo National Central Hospital, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
- 2Eijkman Institute for Molecular Biology, Jakarta, Indonesia
- 3Department of Clinical Pathology, Dr. Cipto Mangunkusumo National Central Hospital, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
Background: Thus far, Indonesia has recorded over 4,000,000 confirmed COVID-19 cases and 144,000 fatalities; 12.8% of cases have been in children under 18 years. Whole-genome viral sequencing (WGS) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been demonstrated to help differentiate hospital-acquired infection from community-acquired coronavirus disease 2019 (COVID-19) infection. Our study highlighted the use of WGS to investigate the origin of infection among pediatric oncology patients in Jakarta. The aim of our study was to evaluate clinical and laboratory characteristics and also the efficacy of using WGS to confirm hospital-acquired COVID-19 infection in a cluster of immunocompromised children within a single ward of a tertiary hospital in metropolitan Jakarta based on quasispecies, viral load, and admission dates.
Method: Real-time reverse-transcription polymerase chain reaction (RT-PCR) from nasopharyngeal (NP) swabs was used to diagnose the patients and also guardians and healthcare workers (HCWs) in the ward, followed by WGS of RT-PCR positive cases to establish their phylogenetic relationships.
Result: Using WGS, we showed that SARS-CoV-2 transmission in a cluster of children with underlying malignancy was characterized by high similarity of whole virus genome, which suggests nosocomial transmission.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly contagious virus with a potential for outbreaks in healthcare institutions (1). Hospital-acquired infections of SARS-CoV-2 are rarely reported in children (2), most of whom were infected from household contacts (3). Although COVID-19 has often been reported as asymptomatic or manifested as mild-to-moderate upper respiratory tract infections (4), children with significant comorbidities are more prone to severe infection and poor prognosis (5).
Although whole-genome sequencing (WGS) is an effective tool to incriminate hospital-acquired infections (6), there have been relatively few reports of it being used in hospital SARS-CoV-2 outbreaks, especially in developing countries, such as Indonesia, where comprehensive and systematic contact tracing is challenging.
Notably, RNA viruses, which have both high replication rates and limited genetic proofreading, mutate to suit the new environment of new hosts (7, 8). Consequently, a pool of highly related mutants within the host, termed quasispecies, is created (7). This phenomenon has been documented in SARS-CoV-2 infections (9–13), with quasispecies diversifying as the virus resides longer in the host (12, 13). Transmission of the virus from host to host brings not only dominant mutants, but also a pool of minor mutants. In the next host, the transmitted pool of mutants experiences a transmission bottleneck, with only a handful of mutants fitting the new host, as observed previously in household SARS-CoV-2 transmission (12), and in influenza A virus (14, 15).
Consensus sequences portray master or dominant sequences, which provide a picture of genetic relatedness within an infection cluster, but are unable to capture the dynamics of virus population or sequence of infection. On the other hand, quasispecies dynamics could be depicted by highlighting active mutation sites even though it is difficult to separate the pool of highly related mutants into individual virus entity. In our study, we highlighted the use of quasispecies approach which could provide more insight into the potential source and sequence of infection in a hospital outbreak of SARS-CoV-2 in a cluster of children considered to be immunocompromised due to treatment for malignancies.
Methods
This study was conducted in a general pediatric ward at Dr. Cipto Mangunkusomo Hospital, a tertiary care hospital in DKI Jakarta, Indonesia, from end December 2020 to March 2021. The study protocol was approved by the Eijkman Institute Research Ethics Committee (Approval Number 127) and FKUI-RSCM Health Research Ethics Committee (Ethical approval number KET-596/UN2.F1/ETIK/PPM.00.02/2020).
Nasopharyngeal (NP) swab specimens were collected on February 2 and 3, 2021 from the cluster of COVID-19 patients with malignancies, patients' guardians, and healthcare workers (HCWs). Samples were tested for RT-PCR in Clinical Pathology Laboratory of Integrated Laboratory Dr. Cipto Mangunkusumo Hospital. The second specimen was requested for WGS characterization on February 4, 2021. Subsequent swabs were not obtained to determine the duration of viral shedding due to the critical nature of the disease in the affected children. Data on clinical manifestations were obtained, together with routine hematology, homeostasis function, inflammatory markers, and chest X-ray (CXR) investigations from all positive cases.
Nucleic acid amplification test was performed on the specimens prior to sequencing (16). WGS was performed using ARTIC Network multiplex PCR method with version 3 primers (17). Library was prepared with Illumina TruSeq Nano DNA Library Prep kit and sequenced using Illumina MiSeq with 600 cycles kit. 30 bp were trimmed from 3′ and 5′ ends of each raw read using Geneious v2021.1.1 (https://www.geneious.com). The resulting reads were filtered using BBDuk v38.84 (18); only sequencing reads with length of ≥50 bp and Phred quality score of ≥30 were included in genome assembly using bowtie2 v2.3.0 (19). Consensus sequences were determined by representative bases with frequency ≥50%. Resulting sequences were deposited in GISAID (https://www.gisaid.org) with accession numbers of EPI_ISL_8540880, EPI_ISL_8540881, EPI_ISL_8540882, EPI_ISL_8540883, and EPI_ISL_8542984. These were aligned with sequences of PANGO lineage B.1.470 with high genome coverage available in GISAID (https://www.gisaid.org) and SARS-CoV-2 reference genome (Wuhan Hu-1, NC_045512.2, NCBI, https://www.ncbi.nlm.nih.gov/genome) using MAFFT v7.450 (20). A phylogenetic tree was generated using FastTree v2.1.12 GTR model with pseudocount (21).
To identify active mutation sites, 30 bp were trimmed from 3′ and 5′ ends of each raw read using Geneious v2021.1.1. The resulting reads were filtered using BBDuk v38.84; only sequencing reads with length of ≥50 bp and Phred quality score of ≥30 were mapped to SARS-CoV-2 reference genome using bowtie2 v2.3.0. Consensus sequences were generated by increasing representative base threshold to ≥95%. Any nucleotides with IUPAC ambiguity codes were defined as active mutation sites, and base composition was collected from the corresponding nucleotides using contig view of Geneious v2021.1.1. Only those with depth ≥100 reads were included in further active mutation site analysis. All bioinformatic tools used in this study were run in Geneious v2021.1.1.
Results
Five immunocompromised children admitted for hematology–oncology disorders sharing a 6-patient room in a general pediatric ward at the Dr. Cipto Mangunkusomo Hospital were diagnosed with COVID-19 infection in early February 2021. The 6 beds were only separated by curtains for privacy. They were primarily admitted for the relapse of their underlying conditions (four with acute myeloid leukemia and one with Ewing's sarcoma); during that period, patients were screened for COVID-19 based on the symptoms in the emergency department and rapid SARS-CoV-2 antigen test before admitted to the ward. Only patient with the clinical symptoms of COVID-19 were tested with SARS-CoV-2 RT-PCR and admitted to the isolation ward. Mask wearing was mandatory to guardians but not for children, and the guardians were also allowed to move freely in and out of hospital. There was no history of travel nor known contact with a positive case for any of the children for the 2 weeks prior onset of illness.
The first child (case 3) admitted with fever and abdominal pain. During follow-up, she developed COVID-19-related symptoms that include fever and cough and was confirmed by RT-PCR on day 8. Additional cases were detected following contact tracing with RT-PCR including the remaining four children and one guardian of case 5. HCWs and guardians attending the children were asymptomatic. All staff members were negative for SARS-CoV-2.
Demographics, clinical characteristics, admission and SARS-CoV-2 PCR confirmation, comorbidities, laboratory investigations including chest imaging, treatment, and outcome are detailed in Table 1 and Supplementary Table S1. Cases 1 and 4 were discharged and readmitted after 1 and 2 weeks, respectively. Peripheral blood evaluation revealed that leucopenia, the most common white cell abnormality associated with children with COVID-19 (22), was seen in three patients (cases 2, 4, and 5); lymphopenia (23) and neutrophilia (24, 25) possible markers of severity were seen in two patients each [(cases 3, 5) and (cases 1, 3), respectively]. High total white cell (leucocytosis), lymphocyte counts (lymphocytosis), and thrombocytopenia associated with hematological malignancies (26) were seen only in a single patient (case 3), two patients (cases 2, 4), and three patients (cases 2, 4, and 5), respectively. Hypercoagulability (as evidenced by raised levels of D-dimer) reported in severe COVID-19 and multisystem inflammatory syndrome in children (MIS-C) (27) was seen in all four patients with available data (cases 1, 2, 3, and 5). C-reactive protein (CRP), an inflammatory marker, documented to be significantly increased in hematological malignancies (28) was seen in three patients (cases 1, 2, and 3). CXR confirmed pneumonia in three patients (cases 2, 3, and 4); consolidation, ground glass opacities, interstitial infiltrate, and pleural effusion being the common CXR findings. Blood, urine, and sputum culture were performed per standard of care. One child (case 5) with sepsis and systemic fungal infection was also diagnosed with MIS-C with raised CRP, procalcitonin, troponin, D-dimers, and fibrinogen. All five children had critical SARS-CoV-2 infection, and four succumbed to the illness.
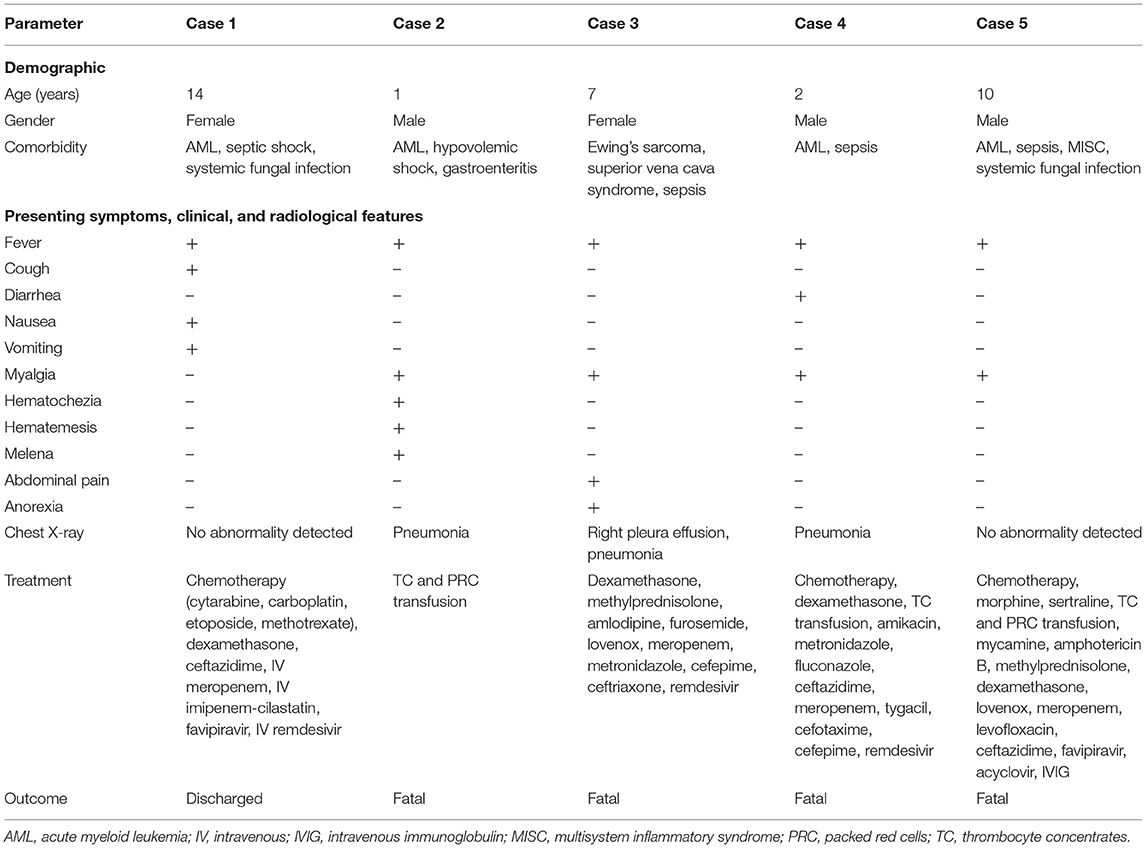
Table 1. Symptoms, clinical, radiological features, treatments, and outcomes of the pediatric patients.
Molecular Characterization
Samples from the five pediatric patients were subjected to SARS-CoV-2 WGS. A number of SARS-CoV-2-specific reads with Phred quality score of ≥30 in the 5 cases were 2,341,675 reads (mean coverage: 12,465 reads; range 6–47,663 reads), 1,163,149 (mean: 4,095; 6–21,364), 1,024,727 (mean: 3,436; 8–14,440), 2,168,464 (mean: 12,693; 0–112,052), and 2,543,718 (mean: 13,771; 22–57,918), respectively. Whole-genome sequences were successfully recovered from the specimens with genome coverage of 99.6, 99.1, 99.7, 90.0, and 99.6%, respectively. The sequences belonging to PANGO lineage B.1.470 were then aligned with 603 sequences of B.1.470 and SARS-CoV-2 reference genome (Wuhan-Hu1). The sequences formed a genetic clade with cases from Jakarta and immediate surrounding areas (in box, Figure 1). The first subcluster consisted of cases 1, 3, and 5; cases 2 and 4 formed another subcluster.
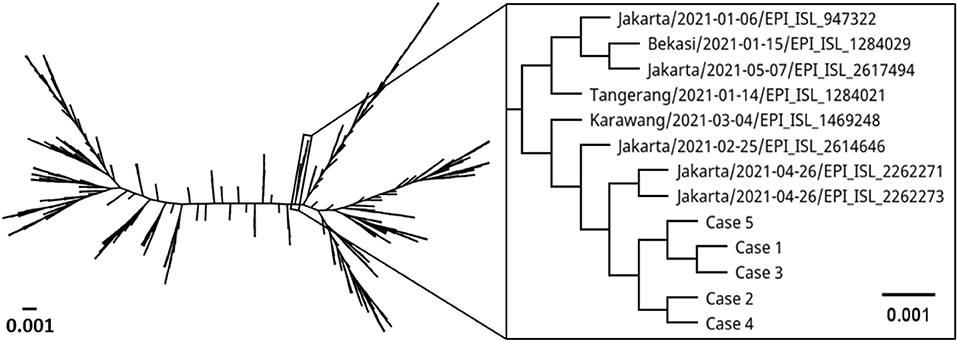
Figure 1. Phylogenetic tree comparing whole genome of SARS-CoV-2 from the five pediatric cases with SARS-CoV-2 lineage B.1.470 from around the world and reference genome (Wuhan Hu1). Label indicates collection site/collection date/GISAID accession number. The scale bar represents the number of nucleotide substitution per site.
We observed a few mutations in the genomes compared to Wuhan-Hu1 (Table 2). A total of 39 mutations were identified with 30 mutations found in all cases, and 9 mutations only found in some cases. The majority (53.9%) of the changes were a substitution to thymine base. Case 1 had one non-synonymous mutation each in envelope and spike, and one synonymous mutation in nsp4. Case 2 had one non-synonymous mutation in nsp2, and another in nsp4. Case 3 had one synonymous mutation each in nsp4 and spike. Case 4 was presented with one non-synonymous mutation each in nsp2 and spike, two synonymous mutations in nsp4, and a thymine base insertion in nsp3 gene. There was only one synonymous mutation in nsp4 observed in case 5. Moreover, there were two mutations shared among the cases; one synonymous mutation in nsp4 gene shared among cases 1, 3, 4, and 5 and a non-synonymous mutation in nsp2 gene of cases 2 and 4. In addition, we observed a rare C9565T substitution in nsp4 gene in four of the five cases. The mutation was reported in only 5.5% (33/603) of the analyzed B.1.470 sequences. None of the cases in the immediate genetic clade had the mutation (in box, Figure 1). High degree of similarity in mutations between cases is suggestive of hospital-acquired transmission.
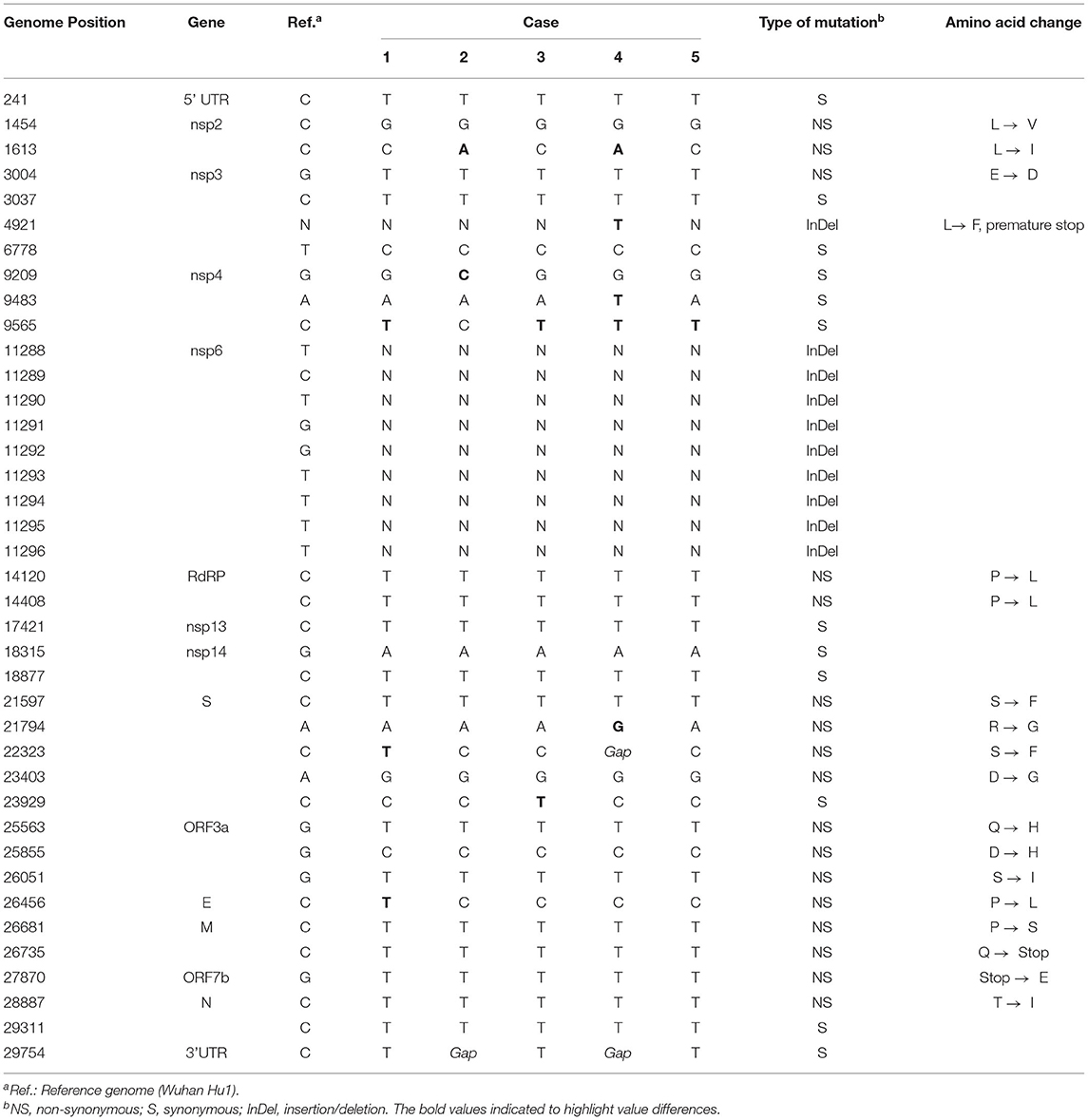
Table 2. List of mutations based on consensus sequences of the five pediatric cases.
Possible Source and Sequence of Infection
We attempted to reconstruct the sequence of infection based on active mutation sites, patient admission time, and viral load. To identify the sites, representative base threshold was increased to ≥95%, and bases with depth <100 reads were excluded from the analysis to avoid data distortion. Active mutation identified in the 5 cases were 7, 21, 14, 13, and 6 sites, respectively (Supplementary Table S2). The majority of sites (>70%) resided within genes encoding non-structural proteins. There were six shared active sites at genome position of 1613, 9565, 11286, 11287, 11511, and 28254, all in genes for non-structural proteins (Table 3).
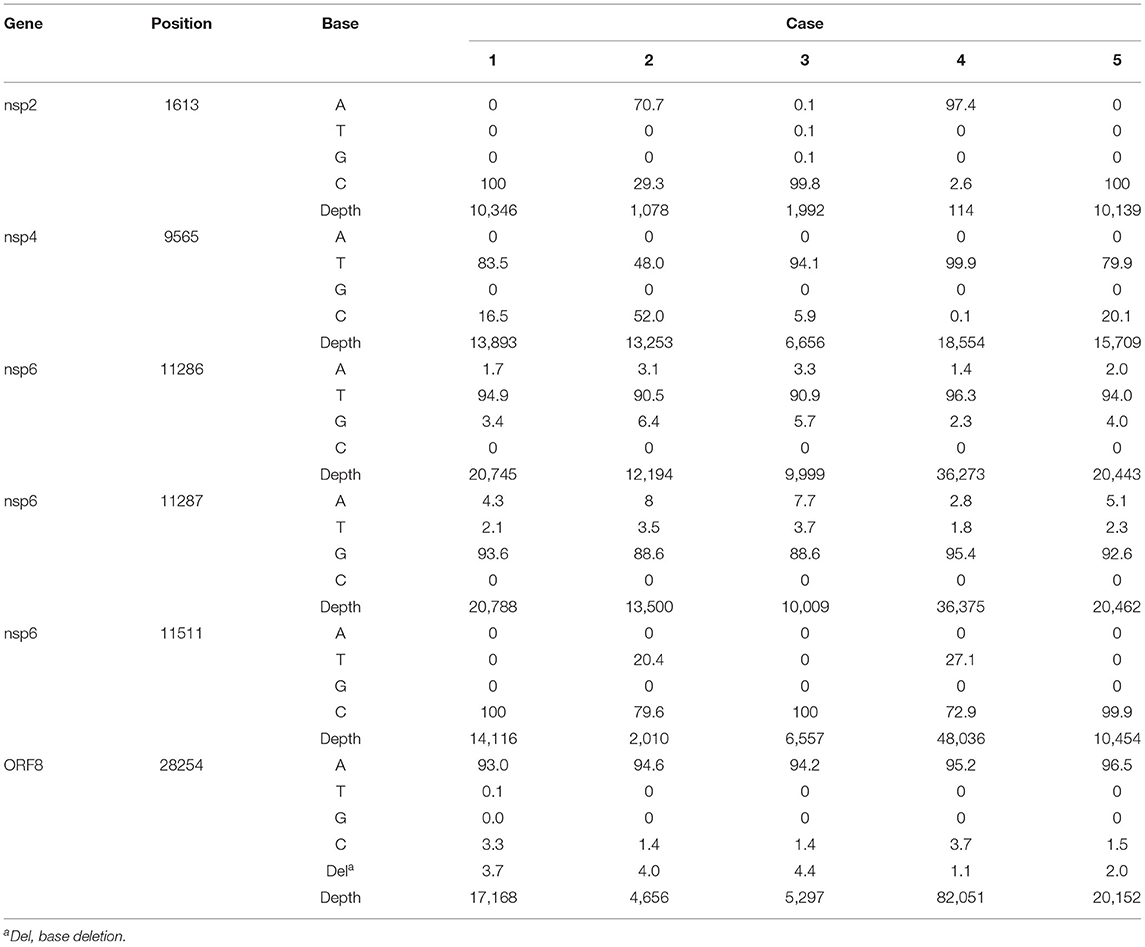
Table 3. List of shared active mutation sites of the five pediatric cases with base composition in percentage of total reads.
Case 2 had the most active mutation sites and thus presumed to be the source of the outbreak. This patient was admitted on January 18, 2021 for the underlying condition and likely was at that time infected with SARS-CoV-2. The virus might have infected case 5, who had been admitted on December 23. Case 1 was admitted into the room 2 days later (January 20, 2021) and could also have been infected by case 2. These speculations are supported by the similar viral load of case 1 (Ct value of 14.74) and case 5 (Ct value of 13.87), compared to a higher viral load (Ct value of 9.81) in case 2. Although case 4 was admitted on January 15, the patient appeared to be infected later, as indicated by much lower viral load at the time of sample collection (Ct value of 33.82). The C9565T mutation was observed in all cases, except case 2, in the consensus genome analysis. At the subconsensus sequence level, the substitution observed in case 2 had a proportion of 52.0% reads with cytosine and 48.0% with thymine. The other four cases exhibited low-base heterogeneity at the corresponding positions. We presume case 2 contributed the rare C9565T mutation. As the virus infected new hosts, that is, cases 1 and 5, a more favorable mutation could have prevailed. The mixture of high thymine (79.9–83.5%) and low cytosine (16.5–20.1%) in both cases demonstrated thymine gradually becoming a dominant base. In addition, thymine and cytosine in genome position 9565 were observed at similar proportion in cases 1 and 5, consistent with the conjecture of similar cases 1 and 5 infection time.
Case 3 was the last patient to be admitted on January 26, before the reported outbreak in the room. Assuming the average virus incubation period of 6 days (29), cases 1, 2, and 5 could be in the infectious stage at the time of case 3 admission. By taking into account the mutation spectra of ORF7b (position 28254), case 3 had base heterogeneity more similar to cases 1 and 2. We also observed the base heterogeneity in nsp6 (position 11286-7) in the cases. Case 3 had more similar profile to case 2 than case 1. Thus, case 3 was likely infected by virus from case 2.
Case 4 was likely infected later than case 3. Viral load in case 4 (Ct value of 33.82) was at least 10 times lower than case 3 (Ct value of 29.58), which indicates later infection. Cases 2 and 4 shared two unique mutation spectrum at genome position 1613 (nsp2) and 11511 (nsp6). It suggested that case 4 was likely infected by case 2, presumably gaining the mutations after cases 1, 3, and 5 infections. Furthermore, both cases 3 and 4 had low viral load at the time of sampling, which indicates that the cases were infected later than cases 1 and 5.
We observed similar level of base heterogeneity across all cases in genome position 11286 (nsp6), 11287 (nsp6), and 28254 (ORF8). A small subset (1.1–4.4%) of total reads in the ORF8 had one base deletion at the end of the gene, which results in amino acid change from isoleucine to serine. In addition, we noticed that case 4 had three sites with thymine insertion: genome position 4921 (nsp3, 62.2% reads with insertion), 11179 (nsp6, 41.0%), and 27821 (ORF7b, 11.9%). The insertions caused frameshift mutations, which results in truncated proteins.
Discussion
Our report described a linked cluster of B.1.470 infections, clinical manifestations, and outcome in a cohort of immunocompromised pediatric patients with oncological comorbidity in Jakarta during the second year of COVID-19 pandemic. The first case was diagnosed with hospital-associated SARS-CoV-2 eight days postadmission for her underlying condition. The commonest symptom of COVID-19 reported in pediatric cancer patients was fever followed by cough (30) the same as manifested in case 3. However, most of our study patients had masked symptoms of COVID-19, which was to be expected with the oncology comorbidity and therapy. Although children with cancer are vulnerable to COVID-19 infection due to immunosuppression associated with the disease and its treatment (24), the impact of SARS-CoV-2 on pediatric patients with hematological malignancies and solid tumors in low- and middle-income countries has been rarely reported. Pediatric cancer patients with SARS-CoV-2 infection were associated with milder infection than adults (31); however, they were reported to have a more severe disease and mortality compared to the general population (30). In our study, 4 of 5 in our cluster had succumbed to the illness likely due to complications of the underlying condition or opportunistic infection rather than the SARS-CoV-2 infection.
To prevent outbreaks of COVID-19 in a healthcare setting, it is important to investigate patients, HCWs, and close contacts with PCR test including asymptomatic individuals. Our study had undetermined source of infection as guardians and visitors were not vigorously screened during that period. Although the guardian of one immunocompromised child was positive by the onsite RT-PCR, the specimen was not saved for further genome sequencing. The majority of healthcare-associated SARS-CoV-2 infections were due to patient-to-patient or HCW-to-patient transmission (1). SARS-CoV-2 hospital outbreaks may often originate from HCWs (32); those attending the cluster were tested negative once and not serially checked, which did not provide enough evidence to exclude SARS-CoV-2 transmission from HCWs. With the strain from one positive guardian unavailable for WGS, genomic links between patients and other sources were also not thoroughly explored. Moreover, three confirmed patients had Ct ≤ 25, which suggests high viral loads, with a potential for patient-to-patient spread as studies have shown links between high viral loads and an increased transmission risk (33).
Symptoms of SARS-CoV-2 infection can overlap with exacerbation of the primary disorder, which suggests that routine screening is crucial in this vulnerable population for any viral respiratory outbreaks. In addition, testing of stool specimen in combination with chest CT is recommended to confirm COVID-19 infection in those with negative swabs as the infection can be masked by malignancy (34, 35). It has been suggested that immunodeficient individuals may have prolonged viral shedding and potentially be contagious for longer duration (36); however, in our study, subsequent respiratory specimens were not obtained due to the critical nature of the illness.
We successfully recovered SARS-CoV-2 genomes from the patients, assigned as PANGO lineage B.1.470. The variant, first identified a year ago in Indonesia, is now in circulation globally. More than 800 complete WGS of B.1.470 have been submitted to GISAID, mostly from Indonesia (>70%), including those from travelers visiting the country. Although all five immunocompromised children acquired the infection with high mortality (80%); transmissibility, severity, and neutralizing antibody response of this strain is not well studied. One study of 41 patients with B.1.470 infection showed 19.5% as asymptomatic, 31.7% as mild severity, and 48.8% as moderate severity (37). None were presented with severe clinical manifestations, but the patients were relatively young with median age of 31 years (range: 27.5–41.0). From our sequencing data, we identified 39 mutations compared to SARS-CoV-2 reference genome. Nine of the mutations were unique to the genetic clade of the five patients, which consisted of four synonymous mutations, four non-synonymous mutations, and one thymine insertion. A non-synonymous mutation of L270I in nsp2 has not been reported, and R78G in spike has been reported in GISAID, but without functional study. Another mutation was S254F in the N-terminal of spike for which no structural and antigenic changes were reported (38). Additional mutation was observed in envelope gene, which results in amino acid change from proline to leucine at position 71. The mutation has been reported to occur at a low frequency, but it appeared to produce no functional changes to the protein (39). Finally, thymine base insertion in nsp3 was observed in case 4, which results in premature protein translation. The truncated protein lost papain-like protease motif that has been described to involve in modulating host antiviral response (40). However, overall impact of the nine mutations remained unknown in regard to disease severity and virus fitness. Interestingly, we also observed that the majority of the mutations were substitution mutations, with a majority from cytosine to thymine base, similar to the analysis from the early stage of pandemic (41). It was hypothesized the changes fit mutational pattern mediated by the host APOBEC family proteins which are known to possess mRNA-editing activities (41, 42).
We also explored the use of quasispecies to determine possible source of infection. Specimens in this study were collected from the patients at the same day and in the early symptom onset. It has been documented in a study of one patient that intrahost SARS-CoV-2 virus quasispecies composition changed day by day (11). We also observed transmission bottleneck as certain variants became more dominant after jumping to other patients, as exemplified by C9565T variant in cases 1 and 5 following infection from case 2. Taken together with quasispecies dynamics, it allowed us to speculate on possible order of infection. We observed with interest that cases 1 and 5 did not develop pneumonia and had low quasispecies variants at 7 and 6, respectively. On the other hand, cases 2, 3, and 4 did develop pneumonia and had high quasispecies variants at 21, 14, and 13, respectively. Although it is difficult to rule out pneumonia caused by opportunistic pathogens due to the immunocompromised nature of the study patients, a link between disease severity and number of quasispecies variants has been established in SARS-CoV-2 (13, 43), and other RNA viruses (44). In addition, immunocompromised patients are at risk of prolonged infection, which allows the virus to develop detrimental mutations, which could lead to antibody evasion and potentially increased disease burden (45, 46).
Our study had a few limitations: 1. The study assumed a single introduction of one strain of virus into the shared ward; the possibility of multiple strains introduction could not be excluded as guardians' movements were not restricted prior to the outbreak. However, this was unlikely, as all five sequences from the patients formed one genetic clade, and the infection time presumably was not long enough to allow genome recombination from multiple virus strains. 2. Viral load comparison between samples may not represent true viral load due to challenges obtaining NP samples from young children. 3. Our study lacked systematic and comprehensive specimen collection that could strengthen the epidemiological link between patients. In addition, the cluster of immunocompromised children with malignancy was limited to five; the study should be expanded to include more patients to draw meaningful conclusions. 4. We could only investigate specimens at one-time point and did not have access to the subsequent specimens to study the dynamics of viral quasispecies. Gradual change of quasispecies variants in hosts infected by the same virus strain has been noted in HIV-1 infection, with high relatedness in early infection and progressively becoming less related (47). 5. Some quasispecies variants might be underrepresented or not visible in this study, as we did not perform ultra-deep sequencing.
Conclusion
In this study, WGS of a linked cluster of SARS-CoV-2 in immunocompromised children in a single ward demonstrated distinctive viral genomic mutations in the hospital cluster that indicated hospital-acquired transmission in a shared ward. Aggressive and routine contact tracing, and also widespread testing of patients and HCWs, including asymptomatic individuals, is essential to limit hospital-associated transmission of COVID-19 including the new variants. Our study also highlighted the use of viral quasi-species to establish epidemiological link between patients in a shared ward.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GISAID with accession numbers for the five cases: EPI_ISL_8540880, EPI_ISL_8540881, EPI_ISL_8540882, EPI_ISL_8540883, and EPI_ISL_8542984.
Ethics Statement
The studies involving human participants were reviewed and approved by Eijkman Institute Research Ethics Committee (Approval Number: 127) and FKUI-RSCM Health Research Ethics Committee (Ethical Approval Number: KET-596/UN2.F1/ETIK/PPM.00.02/2020). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
NP, NI, DW, MP, TS, FC, MJ, and TT collected the clinical data. EJ and NP performed data analysis and interpretation. EJ, KM, and YD wrote the first draft. NP, EJ, YD, NI, DW, MP, TS, FC, MJ, TT, FY, SM, and KM revised the final manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Ministry of Research and Technology/National Research and Innovation Agency, Republic of Indonesia and U.S. Centers for Disease Control and Prevention (US CDC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the patients and their families for agreeing to participate, Board of Directors, Head of Pediatric Department, Pediatric Residents, and Kiara Ultimate Medical Team of Dr. Cipto Mangunkusumo National Central Hospital for their support. We would also like to thank Ronald Rosenberg and Kyaw K. Saw for critically reviewing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.835998/full#supplementary-material
References
1. Abbas M, Robalo Nunes T, Martischang R, Zingg W, Iten A, Pittet D, et al. Nosocomial transmission and outbreaks of coronavirus disease 2019: the need to protect both patients and healthcare workers. Antimicrob Resist Infect Control. (2021) 10:7. doi: 10.1186/s13756-020-00875-7
2. Swann OV, Holden KA, Turtle L, Pollock L, Fairfield CJ, Drake TM, et al. Clinical characteristics of children and young people admitted to hospital with covid-19 in United Kingdom: prospective multicentre observational cohort study. BMJ. (2020) 2020:m3249. doi: 10.1136/bmj.m3249
3. Zimmermann P, Curtis N. Coronavirus infections in children including COVID-19: an Overview of the epidemiology, clinical features, diagnosis, treatment and prevention options in children. Pediat Infect Dis J. (2020) 39:355–68. doi: 10.1097/INF.0000000000002660
4. Qiu H, Wu J, Hong L, Luo Y, Song Q, Chen D. Clinical and epidemiological features of 36 children with coronavirus disease 2019 (COVID-19) in Zhejiang, China: an observational cohort study. Lancet Infect Dis. (2020) 20:689–96. doi: 10.1016/S1473-3099(20)30198-5
5. Tsankov BK, Allaire JM, Irvine MA, Lopez AA, Sauvé LJ, Vallance BA, et al. Severe COVID-19 infection and pediatric comorbidities: a systematic review and meta-analysis. Int J Infect Dis. (2021) 103:246–56. doi: 10.1016/j.ijid.2020.11.163
6. Lucey M, Macori G, Mullane N, Sutton-Fitzpatrick U, Gonzalez G, Coughlan S, et al. Whole-genome sequencing to track severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission in nosocomial outbreaks. Clin Infect Dis. (2021) 72:e727–35. doi: 10.1093/cid/ciaa1433
7. Domingo E, Perales C. Viral quasispecies. PLoS Genet. (2019) 15:e1008271. doi: 10.1371/journal.pgen.1008271
8. Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. (2011) 8:270–9. doi: 10.4161/rna.8.2.15013
9. Lau BT, Pavlichin D, Hooker AC, Almeda A, Shin G, Chen J, et al. Profiling SARS-CoV-2 mutation fingerprints that range from the viral pangenome to individual infection quasispecies. Genome Med. (2021) 13:62. doi: 10.1186/s13073-021-00882-2
10. Capobianchi MR, Rueca M, Messina F, Giombini E, Carletti F, Colavita F, et al. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin Microbiold Infect. (2020) 26:954–6. doi: 10.1016/j.cmi.2020.03.025
11. Jary A, Leducq V, Malet I, Marot S, Klement-Frutos E, Teyssou E, et al. Evolution of viral quasispecies during SARS-CoV-2 infection. Clin Microbiol Infect. (2020) 26:1560.e1-1560.e4. doi: 10.1016/j.cmi.2020.07.032
12. Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L, et al. Genomic diversity of severe acute respiratory syndrome–coronavirus 2 in patients with coronavirus disease 2019. Clin Infect Dis. (2020) 71:713–20. doi: 10.1093/cid/ciaa203
13. Gao R, Zu W, Liu Y, Li J, Li Z, Wen Y, et al. Quasispecies of SARS-CoV-2 revealed by single nucleotide polymorphisms (SNPs) analysis. Virulence. (2021) 12:1209–26. doi: 10.1080/21505594.2021.1911477
14. Stack JC, Murcia PR, Grenfell BT, Wood JLN, Holmes EC. Inferring the inter-host transmission of influenza A virus using patterns of intra-host genetic variation. Proc R Soc B. (2013) 280:20122173. doi: 10.1098/rspb.2012.2173
15. McCrone JT, Woods RJ, Martin ET, Malosh RE, Monto AS, Lauring AS. Stochastic processes constrain the within and between host evolution of influenza virus. Elife. (2018) 7:e35962. doi: 10.7554/eLife.35962
16. Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance. (2020) 25:45. doi: 10.2807/1560-7917.ES.2020.25.3.2000045
17. Tyson JR, James P, Stoddart D, Sparks N, Wickenhagen A, Hall G, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. BioRxiv [Preprint]. (2020). doi: 10.1101/2020.09.04.283077
18. Bushnell B BBTools (2014) Available online at: https://sourceforge.net/projects/bbmap/ (accessed January 25, 2022).
19. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. (2009) 10:R25. doi: 10.1186/gb-2009-10-3-r25
20. Katoh K, Standley DM. MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
21. Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. (2010) 5:e9490. doi: 10.1371/journal.pone.0009490
22. Kosmeri C, Koumpis E, Tsabouri S, Siomou E, Makis A. Hematological manifestations of SARS-CoV-2 in children. Pediatr Blood Cancer. (2020) 67: 28745.doi: 10.1002/pbc.28745
23. Liu J, Li H, Luo M, Liu J, Wu L, Lin X, et al. Lymphopenia predicted illness severity and recovery in patients with COVID-19: a single-center, retrospective study. PLoS ONE. (2020) 15:e0241659. doi: 10.1371/journal.pone.0241659
24. Yang K, Sheng Y, Huang C, Jin Y, Xiong N, Jiang K, et al. Clinical characteristics, outcomes, and risk factors for mortality in patients with cancer and COVID-19 in Hubei, China: a multicentre, retrospective, cohort study. Lancet Oncol. (2020) 21:904–13. doi: 10.1016/S1470-2045(20)30310-7
25. Zhang B, Yu Y, Hubert SM, Zhang Y, Lu J, Liu S, et al. Prognostic value of pro-inflammatory neutrophils and c-reactive protein in cancer patient with coronavirus disease 2019: a multi-center, retrospective study. Front Pharmacol. (2020) 11:576994. doi: 10.3389/fphar.2020.576994
26. Kim JS, Lee KH, Kim GE, Kim S, Yang JW Li H, Hong SH, et al. Clinical characteristics and mortality of patients with hematologic malignancies and COVID-19: a systematic review. Eur Rev Med Pharmacol Sci. (2020) 24:11926–33. doi: 10.26355/eurrev_202011_23852
27. Godfred-Cato S, Bryant B, Leung J, Oster ME, Conklin L, Abrams J, et al. COVID-19–associated multisystem inflammatory syndrome in children — United States, March–July 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:1074–80. doi: 10.15585/mmwr.mm6932e2
28. Russell B, Moss C, Papa S, Irshad S, Ross P, Spicer J, et al. Factors affecting COVID-19 outcomes in cancer patients: a first report from Guy's Cancer Center in London. Front Oncol. (2020) 10:e1279. doi: 10.3389/fonc.2020.01279
29. Elias C, Sekri A, Leblanc P, Cucherat M, Vanhems P. The incubation period of COVID-19: a meta-analysis. Int J Infect Dis. (2021) 104:708–10. doi: 10.1016/j.ijid.2021.01.069
30. Meena JP, Kumar Gupta A, Tanwar P, Ram Jat K, Mohan Pandey R, Seth R. Clinical presentations and outcomes of children with cancer and COVID-19: a systematic review. Pediatr Blood Cancer. (2021) 68:e290055. doi: 10.1002/pbc.29005
31. de Rojas T, Pérez-Martínez A, Cela E, Baragaño M, Galán V, Mata C, Peretó A, Madero L. COVID-19 infection in children and adolescents with cancer in Madrid. Pediatr Blood Cancer. (2020) 67: e28397. doi: 10.1002/pbc.28397
32. Sikkema RS, Pas SD, Nieuwenhuijse DF, O'Toole Á, Verweij J, van der Linden A, et al. COVID-19 in health-care workers in three hospitals in the south of the Netherlands: a cross-sectional study. Lancet Infect Dis. (2020) 20:1273–80. doi: 10.1016/S1473-3099(20)30527-2
33. Marks M, Millat-Martinez P, Ouchi D, Roberts C h, Alemany A, Corbacho-Monné M, Ubals M, Tobias A, Tebé C, Ballana E, et al. Transmission of COVID-19 in 282 clusters in Catalonia, Spain: a cohort study. Lancet Infect. Dis. (2021) 21:629–36. doi: 10.1016/S1473-3099(20)30985-3
34. Brogna B, Brogna C, Petrillo M, Conte AM, Benincasa G, Montano L, et al. SARS-CoV-2 detection in fecal sample from a patient with typical findings of COVID-19 pneumonia on CT but negative to multiple SARS-CoV-2 RT-PCR tests on oropharyngeal and nasopharyngeal swab samples. Medicina. (2021) 57:290. doi: 10.3390/medicina57030290
35. Brogna B, Bignardi E, Brogna C, Alberigo M, Grappone M, Megliola A, et al. Typical CT findings of COVID-19 pneumonia in patients presenting with repetitive negative RT-PCR. Radiography. (2021) 27:743–7. doi: 10.1016/j.radi.2020.09.012
36. Beran A, Zink E, Mhanna M, Abugharbyeh A, Hanrahan J, Duggan J, et al. Transmissibility and viral replication of SARS-COV-2 in immunocompromised patients. J Med Virol. (2021) 93:4156–60. doi: 10.1002/jmv.26970
37. Song Y, Ge Z, Cui S, Tian D, Wan G, Zhu S, et al. COVID-19 Cases from the first local outbreak of the SARS-CoV-2 B.1.1.7 variant in china may present more serious clinical features: a prospective, comparative cohort study. Microbiol Spectr. (2021) 9: e00273-21. doi: 10.1128/Spectrum.00273-21
38. McCallum M, De Marco A, Lempp FA, Tortorici MA, Pinto D, Walls AC, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell. (2021) 184:2332–47.e16. doi: 10.1016/j.cell.2021.03.028
39. Singh J, Samal J, Kumar V, Sharma J, Agrawal U, Ehtesham NZ, et al. Hasnain SE. Structure-function analyses of new SARS-CoV-2 variants B117, B1351 and B11281: clinical, diagnostic, therapeutic and public health implications. Viruses. (2021) 13:439. doi: 10.3390/v13030439
40. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. (2020) 587:657–62. doi: 10.1038/s41586-020-2601-5
41. Simmonds P. Rampant C → U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere. (2020) 5: e00408-20. doi: 10.1128/mSphere.00408-20
42. Mourier T, Sadykov M, Carr MJ, Gonzalez G, Hall WW, Pain A. Host-directed editing of the SARS-CoV-2 genome. Biochem Biophys Res Commun. (2021) 538:35–9. doi: 10.1016/j.bbrc.2020.10.092
43. Al Khatib HA, Benslimane FM, Elbashir IE, Coyle PV, Al Maslamani MA, Al-Khal A, et al. Within-host diversity of SARS-CoV-2 in COVID-19 patients with variable disease severities. Front Cell Infect Microbiol. (2020) 10:575613. doi: 10.3389/fcimb.2020.575613
44. Mandary MB, Masomian M, Poh CL. Impact of RNA virus evolution on quasispecies formation and virulence. IJMS. (2019) 20:4657. doi: 10.3390/ijms20184657
45. Corey L, Beyrer C, Cohen MS, Michael NL, Bedford T, Rolland M. SARS-CoV-2 variants in patients with immunosuppression. N Engl J Med. (2021) 385:562–6. doi: 10.1056/NEJMsb2104756
46. Chen L, Zody MC, Di Germanio C, Martinelli R, Mediavilla JR, Cunningham MH, et al. Emergence of multiple SARS-CoV-2 antibody escape variants in an immunocompromised host undergoing convalescent plasma treatment. mSphere (2021) 6:e00480–21. doi: 10.1128/mSphere.00480-21
Keywords: COVID-19, SARS-CoV-2, quasispecies, whole-genome sequencing, hospital-acquired infection, children, immunocompromised, Indonesia
Citation: Putri ND, Johar E, Dewi YP, Indrasari ND, Wulandari D, br Pasaribu MM, Sari TT, Cakti FP, Jasin MR, Tartila T, Yudhaputri FA, Malik SG and Myint KSA (2022) Whole-Genome Sequencing of SARS-CoV-2 Infection in a Cluster of Immunocompromised Children in Indonesia. Front. Med. 9:835998. doi: 10.3389/fmed.2022.835998
Received: 15 December 2021; Accepted: 26 January 2022;
Published: 04 March 2022.
Edited by:
Sanjay Kumar, Armed Forces Medical College, Pune, IndiaReviewed by:
Vikrant Madhukar Bhor, National Institute for Research in Reproductive Health (ICMR), IndiaMarina Piscopo, University of Naples Federico II, Italy
Copyright © 2022 Putri, Johar, Dewi, Indrasari, Wulandari, br Pasaribu, Sari, Cakti, Jasin, Tartila, Yudhaputri, Malik and Myint. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khin Saw Aye Myint, a2hpbnNhd3lpbmdAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship