 Daisuke Watanabe
Daisuke Watanabe Nobuhiko Kamada
Nobuhiko Kamada
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 07 February 2022
Sec. Gastroenterology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.826240
This article is part of the Research Topic Manipulation of Gut Microbiota as a Key Target to Intervene on the Onset and Progression of Digestive System Diseases View all 11 articles
In Crohn's disease (CD), intestinal fibrosis is a critical determinant of a patient's prognosis. Although inflammation may be a prerequisite for the initiation of intestinal fibrosis, research shows that the progression or continuation of intestinal fibrosis can occur independently of inflammation. Thus, once initiated, intestinal fibrosis may persist even if medical treatment controls inflammation. Clearly, an understanding of the pathophysiological mechanisms of intestinal fibrosis is required to diminish its occurrence. Accumulating evidence suggests that the gut microbiota contributes to the pathogenesis of intestinal fibrosis. For example, the presence of antibodies against gut microbes can predict which CD patients will have intestinal complications. In addition, microbial ligands can activate intestinal fibroblasts, thereby inducing the production of extracellular matrix. Moreover, in various animal models, bacterial infection can lead to the development of intestinal fibrosis. In this review, we summarize the current knowledge of the link between intestinal fibrosis in CD and the gut microbiota. We highlight basic science and clinical evidence that the gut microbiota can be causative for intestinal fibrosis in CD and provide valuable information about the animal models used to investigate intestinal fibrosis.
About a third of patients with Crohn's disease (CD) exhibit a distinct phenotype of intestinal fibrosis and stenosis over a period of 10 years (1). During the clinical course of their disease most of these patients undergo surgery or endoscopic dilation to relieve the symptoms of obstruction. Postoperative recurrence rates are high: 11–32% at 5 years, 20–44% at 10 years, and 46–55% at 20 years (2). This disease course prompted the development of several new biologics, such as vedolizumab, a monoclonal antibody against α4b7 integrin, and ustekinumab, an antibody against interleukin (IL)-12/23, in the field of CD therapy. However, mechanical treatments remain the only practical method to treat obstructive complications (3, 4). Therefore, elucidation of the cellular and molecular mechanisms of intestinal fibrosis in CD is required to improve patients' quality of life.
Why do CD patients develop intestinal fibrosis? It is a generally accepted that inflammatory bowel disease (IBD), consisting of CD and ulcerative colitis, is caused by a loss of tolerance to the gut resident bacteria, which evokes an excessive immune response in a genetically susceptible host (5). Although IBD is a multifactorial disease, human genetic studies support the association of the gut microbiota with the etiology of intestinal fibrosis. CD patients who carry mutations at the nucleotide-binding oligomerization domain 2 (NOD2) locus tend to display the fibrotic phenotype more frequently (6). The dysfunction of bacterial sensing caused by NOD2 mutations implies that intestinal fibrosis is due to the dysfunction of the recognition of the gut microbiota. In addition, several mouse models have shown that specific bacteria taxa (e.g., Salmonella spp.) directly induce a profibrogenic response in the gastrointestinal tract (7).
It has been shown that chronic tissue damage, impaired wound healing, and the expansion of mesenchymal cells are associated with the development of fibrosis. Multiple complex mechanisms involve several cellular components, including mesenchymal cells and immune cells. Physiologically, intestinal fibrosis is the result of an excessive accumulation of the extracellular matrix (ECM). Mesenchymal cells, such as myofibroblasts and stellate cells, serve as the main ECM producers and play a central role in the pathogenesis of fibrosis (8). Studies of cell biology have shown that microbial components affect mesenchymal cell differentiation (9), and that myofibroblasts proliferate at a faster pace in IBD patients compared to healthy individuals (10). These studies highlighted the importance of the gut microbiota in the pathophysiology of intestinal fibrosis.
On the other hand, chronic inflammation is known to be necessary for the initiation of fibrosis, based on the evidence that inflammation promotes mesenchymal differentiation, activation, and proliferation. However, an animal study using mice infected with Salmonella enterica serovar Typhimurium showed that eradication of the pathogen using antibiotics in the early phase of the fibrotic process did not prevent intestinal fibrosis formation (11). Despite major therapeutic advances that focus on the suppression of inflammation, the incidence rate of intestinal complications, including stricture and penetration, in CD patients has not markedly changed (12). These observations suggest that the suppression of inflammation does not simply change the clinical consequences of intestinal fibrosis in CD patients. In this context, the gut microbiota can directly activate pro-fibrotic process in myofibroblasts in addition to indirect activation of fibrotic processes through inducing inflammatory responses. However, the notion that inflammation-independent mechanisms may mediate a self-perpetuating intestinal fibrosis has not been elucidated well, leaving a knowledge gap in terms of the precise mechanisms by which the gut microbiota promotes the pathophysiology of intestinal fibrosis.
Herein, we review the insights of clinical and basic science research that link intestinal fibrosis and the gut microbiota. We highlight the cellular and molecular mechanisms by which the gut microbiota induces the formation of intestinal fibrosis.
Clinically, it is known that recurrent CD can be prevented by postoperative diversion of the fecal stream (13–17). Fecal diversion surgery such as ileostomy or colostomy is indicated for patients who have advanced perianal or colorectal CD as it promotes mucosal healing and resolution of perianal disease. Most individuals who undergo fecal diversion surgery exhibit striking clinical improvement within 3–6 months, and a substantial proportion of these patients achieve remission in the long term (13–17). It has also been shown that treatment with antibiotics confers notable benefits on the clinical course of CD (18–25). These studies suggest that the eradication of certain populations of bacteria has a beneficial influence on clinical outcome for patients with CD.
Human genetic studies revealed that individuals who carry variants of the NOD2 gene are more susceptible to CD (26–28). A detailed study including disease subphenotype analysis confirmed that NOD2 has the largest effect on the development of CD and is strongly associated with ileal disease (29). Biologically, NOD2 functions as an intracellular pattern recognition receptor (PRR) for muramyl dipeptide, which is derived from peptidoglycan of both gram-positive and gram-negative bacteria (30). After intracellular stimulation by bacterial products, NOD2 activates the nuclear factor kappa B (NF-κB) pathway and provides a defensive response to protect the host from bacterial infection. A study of mice revealed that variants of the NOD2 gene directly influence intestinal inflammation and bacterial translocation. Maeda and colleagues reported that mice carrying the homozygous NOD21007fs variant have an increased activation of NF-κB after exposure to muramyl dipeptide, which increases susceptibility to bacteria-induced intestinal inflammation, thereby compromising the integrity of the intestinal barrier (31). The study of intestinal biopsies from patients with CD revealed that the presence of NOD2 variants (especially R702W and 1007fs) is associated with increased NF-κB activation and altered epithelial cell–cell contacts, leading to higher intramucosal levels of endotoxin (32). Notably, these studies indicate that a dysfunctional bacterial sensing mechanism in the host can trigger the development of CD. In addition, given that NOD2 recruits ATG16L1 to the plasma membrane, the failure to do so, as occurs in the presence of NOD2 mutants, ultimately impairs autophagosomal encapsulation of invading bacteria in dendritic cells (33, 34). In line with this function, a single nucleotide polymorphism (SNP) in ATG16L1 (re2241880, Thr300Ala) appears to be associated with an increased risk of CD (35, 36).
The introduction of culture-independent techniques to analyze 16S rRNA gene sequences facilitated a more in-depth analysis of the composition of the gut microbiota (37). It was shown that CD is associated with gut dysbiosis, a condition characterized by an imbalance between protective and harmful bacteria (38). A consistent finding of 16S rRNA gene sequencing analysis was the increase in the abundance of members of the phylum Proteobacteria (gram-negative rods, including Escherichia spp.) in CD patients compared with non-IBD or healthy controls, and a decrease in members of the phylum Firmicutes (gram-positive bacteria, including Clostridium and Bacillus spp.) (39). Many microbiome studies recognized the adherent–invasive strains of Escherichia coli (AIEC) within the Enterobacteriaceae family, which were often found in ileal biopsies of the patients with active CD (40, 41). Some studies suggested that the decreased abundance of the phylum Firmicutes is directly associated with the pathogenesis of CD by modulating immune functions in the intestine. Animal studies revealed that 17 strains within Clostridia clusters IV, XIVa, and XVIII can induce regulatory T cells (Tregs) in the intestine (42, 43). In addition, Faecalibacterium prausnitzii, which belongs to Clostridium cluster IV, was identified as a key player in the dysbiosis associated with ileal CD (44), and shown to produce high amounts of butyrate that has beneficial effects on IBD (45). Intriguingly, a low abundance of F. prausnitzii is associated with an increased risk of future flares in CD (44). Moreover, F. prausnitzii appears to protect the host mucosa from inflammatory injury by favoring the production of antiinflammatory cytokines, such as IL-10 (44). These data support the notion that the composition of intestinal microbiota is one of the critical factors in the pathogenesis of CD.
It has been shown that CD patients carrying a NOD2 variant, such as Arg702Trp, Gly908Arg, or the frameshift mutation Leu1007insC, are at increased risk for complications and surgery (46, 47). A metaanalysis showed that carriage of at least one NOD2 variant increased the risk of stricture in CD patients (odds ratio 1.94; 95% confidence interval 1.61–2.34) (48). Further, the presence of two NOD2 mutations predicted a 41% increase in the risk of complicated disease (i.e., the stricturing or fistulizing subtype of CD) and a 58% increase in the risk of surgery (49). These data support the idea that dysfunction of bacterial sensing by NOD2 triggers intestinal fibrosis in CD.
The increased production of microbial antibodies in serum also supports the contribution of the gut microbiota toward the pathogenesis of intestinal fibrosis in CD. The serum antibody to flagellin anti-CBir1, which reflects aberrant adaptive immunity to luminal commensal bacteria, is significantly elevated in CD patients (50). Anti-CBir1 has been shown to react with flagellins from Clostridium species in the gut (50). It is known that flagellins are important molecules, located on the bacterial surface and involved in both adhesion and motility (51). In addition, flagellin interacts with its toll-like receptor TLR5, leading to the activation of NF-κB and the subsequent transcriptional induction of many proinflammatory cytokines (50, 52). Dubinsky and colleagues showed that children with CD who have anti-CBir1, anti-E. coli outer-membrane protein C antibodies (anti-OmpC), anti-Pseudomonas-associated sequence I2 antibodies (anti-I2), and anti-Saccharomyces cerevisiae antibodies (ASCA) are at an 11-fold higher risk of developing strictures and fistulas compared to those who are seronegative (53). Also, an Irish study reported a significant association between serum anti-CBir1 positivity and a complicated disease behavior as well as ileal location (54). These results suggest that immune responses against gut microbes may contribute to the development of intestinal fibrosis.
In addition, microbiome analysis of CD patients provides information about the relationship between the gut microbiota and intestinal fibrosis. As mentioned, Sokol and colleagues reported the association of a reduced abundance of F. prausnitzii with an increased risk of postoperative recurrence of CD (55). Another study showed that the increased risk of CD recurrence after bowel resection was associated with enriched diversity in members of the Enterobacteriaceae family, and the maintenance of remission was associated with increased diversity in members of the Lachnospiraceae family, which reside within Clostridium cluster XIVa (56). Therefore, although clinical evidence of the direct influence of the gut microbiota on intestinal fibrosis has been insufficient, several studies suggest that specific microbiota contribute to the pathogenesis of intestinal fibrosis in CD.
Moreover, it is reported that intestinal myofibroblasts display different functional capacities between normal individuals and patients with IBD, particularly CD (10). Myofibroblasts isolated from CD patients proliferated faster than those derived from normal individuals and UC (10). Also, the expression patterns of TGF-β isoforms differ in CD compared to normal or UC. In CD myofibroblasts, TGF-β3 is significantly reduced, while TGF-β2 is enhanced compared to normal or UC (10). These results indicate that the differential functional capacity of myofibroblasts in CD may lead to the development of intestinal fibrosis. However, the involvement of the gut microbiota in the regulation of TGF-β isoforms remains unclear.
Excessive ECM synthesis is a fundamental factor in the development of fibrostenosis. In IBD, myofibroblasts originate from numerous sources, including the cells of Cajal and subepithelial myofibroblasts. It is known that bacterial components directly activate intestinal myofibroblasts (Figure 1). Activated myofibroblasts are modified fibroblasts with smooth muscle–like features and considered to be responsible for the development of intestinal fibrosis. Once activated, myofibroblasts synthesize large quantities of ECM components; primarily collagen, glycosaminoglycans, tenascin, and fibronectin (57).
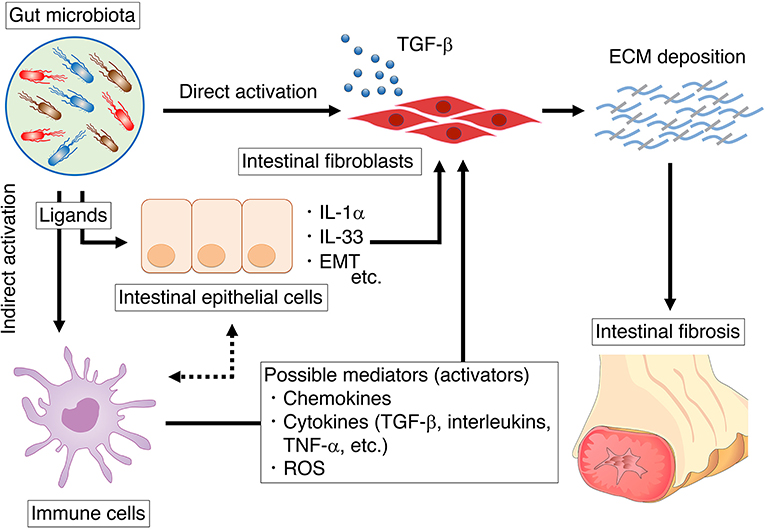
Figure 1. The summary of mechanisms by which the gut microbiota influences intestinal fibroblasts. We summarize the direct influence of microbiota on intestinal fibroblast (section direct activation of fibroblasts through microbial ligands). In addition, we also summarize the indirect influence of microbiota on intestinal fibroblast via epithelial cells or immune cells (section indirect activation of fibroblasts through the microbial ligands).
It is known that microbe-derived pathogen-associated molecular patterns (PAMPs) are sensed by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and Nod-like receptors (NLRs), expressed in intestinal immune and nonimmune cells (58). Likewise, mesenchymal cells in the intestine also express TLR1–9 and NOD1–2 (59). Among several receptors, TLR4 functions as the signaling receptor for lipopolysaccharide (LPS), the major component of the outer membrane of gram-negative bacteria (60), whereas TLR2 is activated by the cell wall components of gram-positive bacteria (61–63). Cultured intestinal myofibroblasts, once activated, secrete cytokine after TLR2 or TLR4 ligand stimulation (59). There is also evidence that intestinal fibroblasts respond to LPS by activating NF-κB signaling, which enhances collagen contraction (64).
As well as TLR2 and 4, TLR5 signaling is associated with the pathogenesis of intestinal fibrosis. Zhao et al. reported that a profibrogenic phenotype of intestinal fibroblasts is triggered exclusively by the TLR5 ligand flagellin (present in all flagellated bacteria), and this event is TGF-β1–independent and post-transcriptionally regulated (65) (Figure 2). In this study, the role of myofibroblasts to directly sense PAMPs in intestinal fibrosis was confirmed in vivo, as the selective deletion of MyD88 (the adaptor molecule for all TLRs except TLR3) in cells expressing α-smooth muscle actin (α-SMA) ameliorated intestinal fibrosis (65). Furthermore, the TLR5 ligand appears to promote cell cycle entry and proliferation of mesenchymal cells in vitro (67). Consistent with intestinal fibrosis, there is evidence that the gut microbiota promotes liver fibrosis. Elevated LPS levels have been measured in the systemic and portal circulation of patients with cirrhosis (68, 69). These studies suggest that the microbial ligand of LPS arriving from the portal vein or bacteria translocated to the liver promotes a liver fibrogenic response via TLR4 (68, 69). In accordance with clinical study, it has been shown that hepatic stellate cells, are activated by TLR4 ligands and mediate various fibrogenic effects (70, 71). In addition, TLR4 ligands indirectly contribute to liver fibrogenesis, rendering hepatic stellate cells more susceptible to TGF-β1 through downregulation of the TGF-β1 decoy receptor BAMBI (70). In line with the results of these studies, it has been shown that selective decontamination of the intestinal microbiota using an antibiotic agent inhibits experimental liver fibrosis with a decreased level of plasma LPS (70, 72).
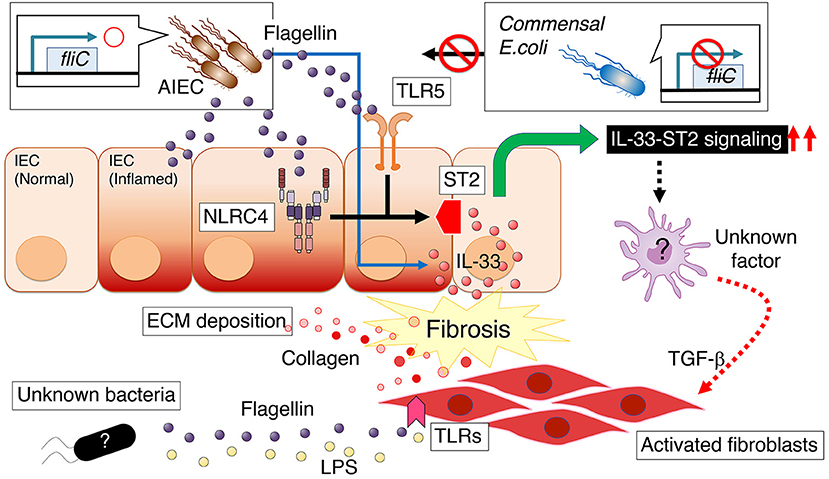
Figure 2. Persistent intestinal colonization of AIEC strain LF82 potentiates the development of intestinal fibrosis, a common and potentially severe complication of intestinal colitis (66). Flagellin produced by AIEC promotes the expression of interleukin 1 receptor–like 1 (IL1RL1, also known as ST2) in intestinal epithelial cells (IECs), which depends on flagellin ligands TLR5 and NLRC4 on IECs. ST2 expression augments IL-33 signaling, thereby promoting intestinal fibrosis. Conversely, there is a mechanism by which intestinal myofibroblasts directly respond to flagellin with enhanced fibronectin or collagen production in a MyD88-dependent manner (65). AIEC, adherent–invasive Escherichia coli; ECM, extracellular matrix; LPS, lipopolysaccharide; ST2, interleukin 1 receptor–like 1 (IL1RL1, also known as ST2).
In addition to intestinal fibroblasts, the gut microbiota influences immune cells and epithelial cells, which may also serve as possible cell mediators for the development of intestinal fibrosis (Figure 1). Microbial stimulation induces chemokines, cytokines, and reactive oxygen species (ROS) production by immune or epithelial cells, which in turn promote the activation of intestinal fibroblasts.
Cytokines are mediators that send a signal from a cell by binding to receptors on themselves or another cell surface. Several types of immune cells in lamina propria exert their function by producing specific cytokines, which can affect the intestinal fibroblasts. It is well known that TGF-β plays a crucial role in the machinery of intestinal mesenchymal cell activation and ECM production. The canonical TGF-β intracellular signal transduction pathway is mediated by Smad proteins as TGF-β receptor activation phosphorylates Smad2 and Smad3 and induces binding with Smad4 (73). The Smad2/3-Smad4 complex translocates into the nucleus, regulating TGF-β target genes. The Smad-dependent pro-fibrotic effects of TGF-β can result in myofibroblast activation and ECM accumulation (collagen production).
Several other cytokines are also involved in the formation of intestinal fibrosis. For instance, IL-1β can be the mediator to connect the microbiota-immune cells-intestinal fibroblasts interactions. In line with the previous studies, our laboratory showed that specific microbes accelerate IL-1β production by mononuclear cells in the lamina propria (74). It is also known that IL-1β is mainly produced by mononuclear phagocytes, acting as a pro-inflammatory effector cell of intestinal inflammation (75). In turn, IL-1β promotes the secretion of collagens I and IV, IL-8, monocyte chemoattractant protein (MCP)-1, and MMP-1 from colonic subepithelial myofibroblasts (76). These findings imply the possibility that the gut microbiota contributes to the development of intestinal fibrosis via induction of IL-1β from immune cells, albeit there is another contradictory report showing IL-1β inhibits collagen synthesis and induces collagenase and TIMP-1 production in intestinal smooth muscle cells (77, 78). Furthermore, TL1A, a protein encoded by TNFSF15, binds to death domain receptor 3 (DR3) and is expressed by various cell types, including immune cells. Primary intestinal myofibroblasts express DR3 and respond to TL1A, increasing collagen deposition (79). Consistently, it was reported that constitutive expression of TL1A in either lymphoid or myeloid cells leads to the acceleration of intestinal and colonic fibrosis (80, 81). Importantly, TL1A-mediated intestinal fibrosis and fibroblast activation are dependent on specific microbial populations (9). It is generally believed that Th1 cell-associated cytokines drive inflammation, whereas uncontrolled type 2 and type 17 cell responses might drive tissue fibrosis through the excessive deposition of ECM (75). IL-17 cytokines, primarily produced by Th17 cells, consists of six related proteins: IL-17A (also called IL-17), IL-17B, IL-17C, IL-17D, IL-17E (also called IL-25), and IL-17F, which signal through five receptor subunits IL-17RAIL17RE (82). It has been shown that pathogenic IL-17A-dependent immune responses are induced by microbial stimulation of DCs through NOD2, and therefore the deletion of NOD2 prevents the development of colitis (83). In addition, IL-17A enhances the production of collagen I and heat shock protein 47 (HSP47) in subepithelial myofibroblasts, which is significantly elevated in the intestinal tissues of patients with active CD (84). In line with these findings, the colonization by adherent-invasive Escherichia coli (AIEC) induces Th17 responses, heightens proinflammatory cytokines and fibrotic growth factors, with transmural inflammation and fibrosis (85). Previous studies have also shown that another mediator is chemokines that are leukocyte chemo-attractants that cooperate with profibrotic cytokines in fibrogenesis by recruiting myofibroblasts, macrophages, and other critical effector cells to sites of tissue injury (86). Blockade of CC- and CXC chemokine receptors decreases fibrosis in association with decreased IL-4 and IL-13 (86).
In addition to immune cells, intestinal epithelial cells also act upstream of intestinal fibroblasts, thereby contributing to intestinal fibrosis development. Epithelial cells are located at the interface of the inner lumen of the digestive tract and inside the intestinal wall. Like immune cells, epithelial cells also express various receptors for microbial ligands and produce several cytokines. For instance, IL-1α is constitutively expressed in epithelial cells, although it can be expressed by other cell types, such as macrophages, monocytes, and endothelial cells (87). Previously, it was reported that intestinal epithelial cell-derived IL-1α induces cytokine production by human intestinal fibroblasts (HIFs) (88). In addition, it has been known that IL-1α and TNF-α also increase TGF-β1 and TIMP-1 production by colonic epithelial cells (89). Also, IL-1α acts as a profibrotic cytokine in other organs, as IL-1α -deficient mice exhibit reduced collagen deposition in response to bleomycin treatment in lung fibroblasts (90).
In addition to these mechanisms, intestinal epithelial cells are involved in the pathogenesis of intestinal fibrosis via the machinery of epithelial-mesenchymal transition (EMT). It is well known that EMT is the primary mechanism in the development and progression of cancer and fibrosis (91). While EMT, epithelial cells change their morphology to spindle-shape, down-regulating the expression of epithelial molecules, such as E-cadherin, and gain mesenchymal characteristics, including vimentin and alpha-smooth muscle actin (α-SMA). Wang and colleagues have shown the role of the NLRP3 inflammasome in the EMT process. Interestingly, the EMT process is independent of the NLRP3 inflammasome complex formation but requires the presence of the NLRP3 protein (92).
Other mechanisms that cause intestinal fibrosis due to intestinal microbiota can be evoking oxidative stress. It has been known that polymorphonuclear leukocytes (PMNs) migrate to the site of infection or injury, engulf invading pathogens, and secrete ROS (93). ROSs are small molecules, including oxygen radicals (superoxide and hydroxyl) and non-radicals such as hypochlorous acid, singlet oxygen, and hydrogen peroxide. In the intestine, PMNs are the primary sources of ROS and reactive nitrogen species (RNS), as these cells express nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes (NOX/dual oxidase), the mitochondrial electron transport chain (mETC), and nitric oxide synthases (NOSs). It has also been shown that certain intestinal epithelial cells rapidly generate reactive oxygen species (ROS) in response to microbial signals (94), and subsequently, generated ROS promotes the production of several profibrogenic factors that stimulate the production or inhibit the degradation of ECM (95).
Clinically, it is known that radiation of the small bowel and colon induces severe intestinal fibrosis. A model of radiation-induced intestinal fibrosis in rats, and to a lesser extent in mice, has been widely used to study the mechanisms of intestinal fibrosis. This model reproduces the events responsible for the intestinal fibrosis in humans observed during radiation therapy. Morphological and pathological findings in this model include radiation-induced thickening of the bowel wall, accompanied by an enlarged submucosa, increased proliferation rates of fibroblasts and smooth muscle cells, as well as enhanced accumulation of collagen and other ECM components (96). Some genetically engineered animal models of IBD do not develop intestinal fibrosis when maintained in germ-free (GF) conditions (97). Mice colonized with only Bacteroides thetaiotaomicron and E. coli have similar outcomes to the GF mice (97). Consistent with GF mice, Zhao and colleagues showed that antibiotic cocktail pretreatment before radiation effectively reduces the content of LPS and inhibits the TLR4/MyD88/NF-κB signaling pathway in the ileum (98). Antibiotic treatment also significantly improves the survival rate and attenuates intestinal injury of the mice after radiation by reducing inflammation and preventing intestinal fibrosis (98). These results indicate that the intestinal microbiota plays an important role in this model (Table 1).
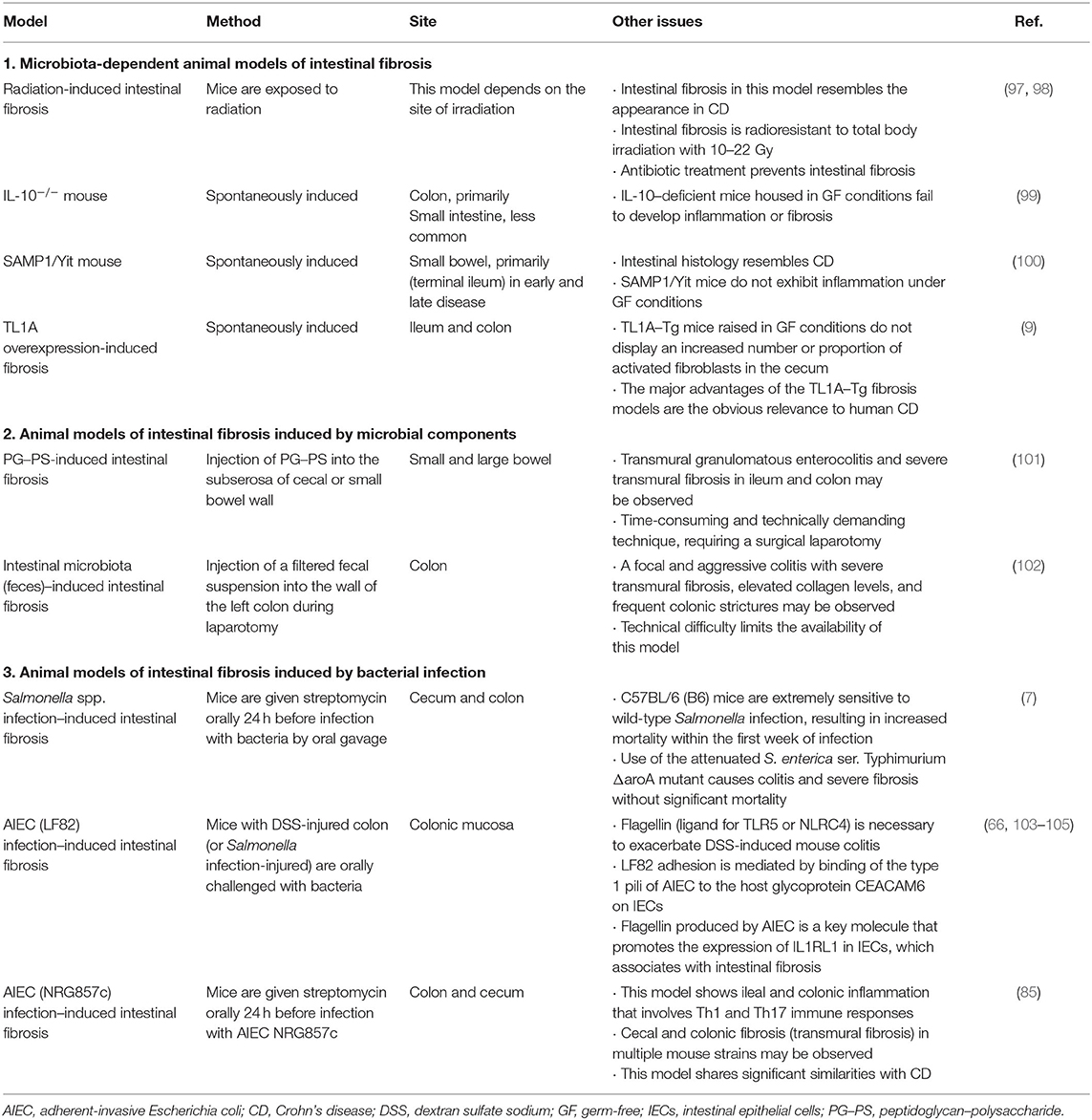
Table 1. Summary of animal models of intestinal fibrosis associated with the gut microbiota.
It was reported that interleukin 10 (IL-10)-deficiency aggravates intestinal (106) and renal fibrosis (107). It is well known that the interleukin 10 (IL-10)–deficient mouse shares several characteristics in common with CD (106). Nevertheless, this model has not been extensively adopted to study intestinal fibrosis. However, it has been shown that if IL-10–deficient mice undergo ileocecal resection, they develop postsurgical fibrosis in the small intestine, distant from the site of surgery, with an associated increase in procollagen-α1(I) (COL1A1) mRNA expression (99). In addition, consistent with CD patients, intestinal fibrosis develops on the proximal side of the anastomosis (108). Therefore, this model is clinically relevant and useful to study postoperative recurrence as occurs in CD. As for the machinery of intestinal fibrosis associated with IL-10 deficiency, the prohibition, which serves as a chaperone involved in stabilizing mitochondrial proteins (92), might be associated with the pathogenesis of intestinal fibrosis in IBD (109). It is well known that IL-10–deficient mice show no inflammation when housed in specific pathogen-free (SPF) conditions, but they develop intestinal inflammation on transfer to regular housing conditions (110). Interestingly, IL-10–deficient mice housed in GF conditions fail to develop inflammation or fibrosis even after ileocecal resection, suggesting that this response also depends on the presence of the gut microbiota (99) (Table 1).
Spontaneous models of intestinal fibrosis are particularly promising because they do not depend on exogenous stimulations. The senescence accelerated mouse (SAM) P1/Yit strain was originally generated by selective breeding of the SAMP1 line (100). The SAMP1/YitFc substrain (SAMP1/Fc) was developed in Fabio Cominelli's laboratory (111) and shown to share more histomorphological features in common with human CD than the SAMP1/Yit mouse line. The SAMP1/Yit mouse develops spontaneous enteric inflammation in the ileum within 10 weeks after birth and reveals a 100% penetrance of fibrosis by 30 weeks after birth (100). In addition, a fraction of these mice spontaneously develops perianal fistulas and accumulate ECM in the small bowel and colon with thickening of the muscularis mucosa, predominantly in the terminal ileum, a feature closely resembling intestinal stricture in CD patients (111). Like the two intestinal fibrosis models just described, the SAMP1/Yit mice do not exhibit inflammation in GF conditions (100). However, the GF SAMP1/Yit mice reconstituted by transfer of the gut microbiota from SPF SAMP1/Yit mice do develop intestinal disease (100). Evidently unknown host–microbial interactions amplify the severity of intestinal disease in this model (Table 1).
TL1A (a protein encoded by the TNFSF15 gene) is a member of the tumor necrosis factor (TNF) superfamily that can bind to death domain receptor 3 (DR3). A TNFSF15 haplotype appears to be associated with higher TL1A production, increased risk of CD, intestinal fibrostenosis, and greater need for surgery (112–114). In accordance with clinical data, constitutive TL1A expression in mice increases collagen deposition in the colon without detectable histologic colitis, whereas the ileum exhibits increased collagen deposition with spontaneous ileitis (80, 81, 115, 116). In addition, colitogenic conditions induced by chronic dextran sulfate sodium (DSS) treatment or adoptive T-cell transfer increase collagen deposition with fibrostenotic lesions that cause intestinal obstruction in this model (117) (Table 1).
Jacob and colleagues showed that the profibrotic and inflammatory phenotype resulting from constitutive TL1A expression is abrogated in the absence of the resident microbiota (9). Although an increased proportion of intestinal myofibroblasts can be observed in TL1A–transgenic (Tg) mice raised in conventional SPF conditions (79), TL1A–Tg mice raised in GF conditions do not display an increased number of activated fibroblasts in the cecum (9). Colonic fibroblasts isolated from the TL1A–Tg mice also displayed a significantly higher migratory capacity compared with those isolated from wild-type mice in a scratch cell migration assay; however, the enhanced rate of fibroblast gap-closure observed in TL1A–Tg mice raised in native conditions was eliminated in GF conditions (9). Furthermore, reconstitution with intestinal microbiota from SPF mice, but not human donor microbiota, resulted in increased intestinal collagen deposition and fibroblast activation in TL1A–Tg mice (9). Thus, these results indicate that TL1A-mediated intestinal fibrosis and fibroblast activation are dependent on specific microbial populations (Table 1).
It has been well known that mice drinking the sugar polymer of DSS for several days develop highly reproducible colitis with bloody diarrhea, ulcerations, and weight loss (118). In the same way, chronic administration of DSS for several cycles results in intestinal fibrosis in certain strains (119). It was reported that DSS administration induces colitis in GF mice to the same extent or even more severely compared with conventionally housed mice (120), indicating that resident gut microbiota is not required for DSS-induced colitis.
However, mice develop more severe intestinal fibrosis when colonized with a pathobiont AIEC (66), suggesting that specific, most likely pathobiont-type microbiota contributes to the development of fibrosis in this model. Consistently, deletion of MyD88 results in the amelioration of intestinal fibrosis in this model (65). In contrast, a probiotic Lactobacillus acidophilus strain reduces the severity of DSS-induced intestinal fibrosis (121).
Likewise, the trinitrobenzene sulfonic (TNBS) acid-induced intestinal fibrosis model is one of the most commonly applied chemically-induced intestinal fibrosis models (122). Repetitive rectal TNBS application results in chronic colitis accompanied by intestinal fibrosis with luminal stenosis and bowel dilatation. TNBS administration disrupts the epithelial barrier, thereby leading to the invasion of luminal bacteria into the colonic wall in conventionally-housed animals (123). In contrast, no colitis occurs when TNBS is administered after eradication of the colonic microbiota by antibiotics (124), and some bacteria, including Lactobacillus casei (DN 114-001 strain), may even have protective properties in TNBS mouse model (123). Thus, the gut microbiota regulates the pathogenesis of TNBS-induced colitis and intestinal fibrosis.
Peptidoglycan–polysaccharide (PG–PS) is a polymer composed of sugars and amino acids that is found in the bacterial cell wall. Transmural enterocolitis in rats can be observed after injection of purified sterile PG–PS derived from bacteria (e.g., Streptococcus pyogenes) into the subserosa of the cecal or small bowel wall during laparotomy (101). In this model, the initial insult is characterized by intense transmural inflammation and avid infiltration of acute inflammatory cells, including polymorphonuclear leukocytes. After several weeks, the acute inflammatory response becomes a patchy, chronic granulomatous inflammation, which has similarities to the chronic inflammation in CD. The affected intestinal wall becomes thickened and intraabdominal adhesions can develop (125), while areas of granulomatous inflammation express increased levels of collagen-α1 (COL1A1), TGF-β1, and IL-6 mRNA (126). Moreover, significant fibrosis and abundant mesenchymal cells surround the granulomas in this model. The mesenchymal cells have morphological and immunostaining patterns consistent with myofibroblasts, which are the key effector cells in intestinal fibrosis (126). The PG–PS model shows that the infiltration of nonviable bacterial components into the intestinal wall is sufficient to trigger inflammation and initiate intestinal fibrosis, and this infiltration can be enacted by bacterial components in the healthy intestinal lumen (Table 1).
This model shares many technical similarities to the PG–PS model. An injection of a filtered fecal suspension into the wall of the left colon of rats during laparotomy causes a focal and aggressive colitis with severe transmural fibrosis, elevated collagen levels, and frequent colonic strictures (102). A subserosal injection of a single organism suspension of intestinal anaerobes, but not aerobes, reproduces similar findings (102). The treated animals show signs of chronic inflammation and fibrosis with stricture development, significantly elevated levels of mucosal and serum TGF-β and increased collagen deposition. In this model, the increased production of TGF-β1 stimulates Smad2/3 phosphorylation and enhanced ALK5, TIMP-1, and COL1A2 gene expression (127). In addition, it has been shown that use of anti-TGF-β antibodies significantly abrogate collagen deposition in this model (102). These observations emphasize the impact of commensal intestinal bacteria on TGF-β1, collagen production, and intestinal fibrogenesis (Table 1).
Nontyphoidal Salmonella enterica spp., such as Salmonella enterica serovar Typhimurium, are intestinal pathogens that can infect a wide range of animals, including humans (128, 129). It is known that certain Salmonella serovars are host restricted, whereas others have a broad host range. In humans, S. enterica ser. Typhimurium and Paratyphi can cause typhoid characterized by systemic infection, fever, and often, gastrointestinal symptoms such as diarrhea. In contrast, S. enterica ser. Typhimurium causes enterocolitis in humans and cattle, but systemic infection in mice (Table 1).
Serovars of S. enterica spp. are widely used in laboratory studies to gain an understanding of the basis of mucosal immune responses and intestinal diseases such as gastroenteritis and typhoid. It is known that oral infection with S. enterica ser. Typhimurium leads to spread via the gut-associated lymphoid tissue (GALT) to systemic sites in genetically susceptible mice. The bacteremia and lesions in the systemic organs of these mice are akin to typhoidal salmonellosis in humans; hence, this phenotype is known as mouse typhoid (130). As for Salmonella colonization in the mouse intestine, it is known that strain-dependent genetic susceptibility affects the host response to Salmonella infection. It has been shown that S. enterica ser. Typhimurium can cause chronic infection of systemic organs in some genetically resistant inbred mouse strains (e.g., 129SvEv, Nramp1+/+) (131). Consequently, these mice are useful animal models of persistent Salmonella systemic infection (131). On the other hand, several studies have aimed to improve Salmonella colonization in the mouse intestine. It is known that the intestinal tract of conventional SPF mice is poorly colonized by S. enterica ser. Typhimurium (~104 CFU/g of contents) (132, 133). However, pretreatment of SPF mice with an antibiotic agent results in abundant colonization of S. enterica ser. Typhimurium in the cecum and colon, and susceptibility to colitis (132); a technique that is now widely used in this murine model of Salmonella spp. infection (Table 1).
In addition to studies of enterocolitis, oral administration of live S. enterica ser. Typhimurium has been used to study the pathogenesis of intestinal fibrosis. The severity of disease and intestinal fibrosis has been shown to depend on the genetics of the mouse strain (7). 129sv/J mice pretreated with antibiotics and chronically infected with S. enterica ser. Typhimurium strain SL1344 can serve as a robust model of intestinal fibrosis (7). In contrast, the C57BL/6 mouse strain is extremely sensitive to wild-type Salmonella infection, resulting in increased mortality within the first week of infection (7). Grassl and colleagues reported that to use C57BL/6 mice for an intestinal fibrosis model using Salmonella infection, the attenuated S. enterica ser. Typhimurium mutant strain ΔaroA (attenuated by a block in the synthesis of aromatic amino acids) can be used. These C57BL/6 mice have severe fibrosis without significant mortality (7). Thus, infection with the S. enterica ser. Typhimurium ΔaroA mutant allows analysis of the mechanisms that contribute to intestinal fibrosis in knockout mouse models maintained on a C57BL/6 background. So, which mouse strain and bacterial strain are the best to study intestinal fibrosis? Johnson and colleagues reported that although the severity of fibrosis in the Salmonella infection models varies depending on the host and bacterial strain, CBA/J mice infected with the S. enterica ser. Typhimurium SL1344 strain may be the optimal model for intestinal fibrosis (134) (Table 1).
This model has provided several insights that attribute the pathogenesis of intestinal fibrosis to the intestinal microbiota. First, the Salmonella virulence factors such as Salmonella pathogenicity islands (SPI)-1 and−2 are essential for the induction of intestinal fibrosis in this model (7). The resulting extensive transmural inflammation, primarily evident in the cecum but also in the colon, is accompanied by an upregulation of T helper 1 (Th1) cytokines, fibrotic growth factors, and procollagen type I. These profibrotic profiles are consistent with CD, which is associated with strong Th1 immune responses, including elevations in proinflammatory cytokine TNF-α expression. Further, early blockade of inflammation by eradicating the S. enterica ser. Typhimurium infection with levofloxacin ameliorates intestinal fibrosis, but does not abolish subsequent fibrosis, suggesting that once initiated, intestinal fibrosis in this model is self-propagating (11). Finally, an animal study using Rorasg/sg BMT mice (i.e., group 2 innate lymphoid cell (ILC2)–deficient mice) showed that collagen deposition is associated with IL-17A and RORα-dependent innate lymphoid cells (ILCs) (135), affirming ILC involvement in intestinal fibrosis in this model (Table 1).
Members of the E. coli family constitute a normal component of the healthy intestinal microbiota. It is known that E. coli strains can acquire virulence factors to adapt to harsh circumstances in the host. Many studies have reported that some E. coli strains isolated from the ileal lesions of CD patients exhibit adherent and invasive capabilities in gastrointestinal epithelial cells and macrophages (136–141); hence, termed adherent–invasive Escherichia coli (AIEC) (136). Cell biological studies showed that AIEC phagocytosed by macrophages are more resistant to xenophagy and capable of inducing a persistent inflammatory response by releasing large amounts of TNF-α (141, 142). Interestingly, monocytes from CD patients who carry homozygous or heterozygous NOD2 polymorphisms display reduced secretion of IL-1β, IL-6, and IL-10 after AIEC infection in vitro compared with monocytes from CD patients without NOD2 polymorphisms (143). In addition, clinical studies showed that AIEC strains are preferentially observed in ileal CD (40, 137, 144).
To date, there are two well-characterized prototypic AIEC strains: LF82 and NRG857c (85, 136, 145). The prototype AIEC strain LF82 colonizes the intestinal mucosa and induces proinflammatory cytokines during acute DSS-induced colitis (103–105). In this model, flagellin (ligand for TLR5 and NLRC4) is necessary for the AIEC strain LF82 to exacerbate DSS-induced mouse colitis, while the nonflagellated LF82 mutant strain behaves like the nonpathogenic E. coli strain K12 (103). Mechanistically, it has been shown that its adhesion is mediated by binding of the type 1 pili of AIEC to the host glycoprotein carcinoembryonic antigen–related cell adhesion molecule (CEACAM) 6 on the intestinal epithelial cells (IECs) (104, 146). Barnich and colleagues reported that CD patients with ileal disease have an abnormal ileal expression of CEACAM5 and 6, and that only CEACAM6 acts as a receptor for AIEC (146). Intriguingly, in vitro studies demonstrated that CEACAM6 expression is increased in cultured IECs after infection with AIEC, indicating that AIEC promotes its own colonization through induction of CEACAM6 expression in the host (146). In addition to these mechanisms, it has been shown that bacterial adhesion to IECs is mediated via chitin-binding domains in bacteria, encoded by bacterial chitinase ChiA, that interact with human chitinase CHI3L1 expressed on IECs in inflammatory conditions (105) (Table 1).
As well as LF82, a human CD isolate of AIEC strain NRG857c was used to develop a chronic AIEC infection mouse model to study intestinal inflammation and fibrosis (85). Like the S. enterica ser. Typhimurium infection model, mice were pretreated with oral streptomycin prior to infection with NRG857c (85). After NRG857c infection, this model showed that ileal and colonic inflammation involves Th1 and Th17 immune responses (85). The resulting inflammation leads to cecal and colonic fibrosis in multiple mouse strains, in varying degrees, and progresses to transmural fibrosis (85). This model shares significant similarities with CD (Table 1).
Our laboratory has shown that persistent intestinal colonization of AIEC strain LF82 potentiates the development of intestinal fibrosis in conditions of Salmonella-induced or DSS-induced colitis (66) (Figure 2). In this model, flagellin produced by AIEC, a principal component of bacterial flagella, is a key molecule that promotes the expression of interleukin 1 receptor–like 1 (IL1RL1, also known as ST2) in intestinal epithelial cells (IECs), which depends on flagellin ligands TLR5 and NLRC4 on IECs (66). Further, it has been shown that ST2 expression in IECs augments IL-33 signaling, thereby promoting intestinal fibrosis, as the blockade of IL-33–ST2 signaling by anti-ST2 antibody significantly ameliorates intestinal fibrosis (66). Therefore, therapeutic approaches that target AIEC or its downstream IL-33–ST2 signaling pathway would benefit CD patients with intestinal fibrosis (Table 1).
Accumulating evidence suggests that interventions against the gut microbiota may regulate the prognosis of intestinal fibrosis in CD. However, we have not had any therapeutical options using microbiota-targeted interventions that specifically treat intestinal fibrosis (147). Although we have various strategies that modulate the gut microbiota (e.g., antibiotics, probiotics, fecal microbiota transplantation [FMT]), it is challenging to evaluate the effects of interventions on intestinal fibrosis. This is due to the unavailability of quantification methods for intestinal fibrosis. Albeit several diagnostic tools, such as ultrasound, computer tomography, magnetic resonance, and gastrointestinal endoscopy, are available to estimate the developmental status of intestinal fibrosis in CD, there are no modalities to quantify the degree of intestinal fibrosis without conducting the surgical resection of the affected intestine (147). In addition, because intestinal fibrosis gradually progresses in CD in several decades, a much longer time should be required to certificate its effectiveness of action. Another possible reason is that it is also challenging to target pathobionts selectively without affecting other bacteria in the healthy intestine. Although antibiotics have provided significant advances in therapies for infectious diseases, several bacterial species susceptible to the agents will be affected by the treatment.
On the other, there are some success stories of “microbiota-targeted therapy” in fields other than intestinal fibrosis. For instance, several researchers reported that FMT could be a promising treatment to induce remission in UC with active disease (148, 149). In addition, probiotics may be the candidate for the treatment because it has suppressed UC relapse (150). These data might indicate that microbiota-targeted therapy is promising, and therefore we hope that practical application of microbiota-targeted therapies for intestinal fibrosis come on the stage in the near future.
The cellular and molecular mechanisms of intestinal fibrosis are the focus of intense investigation. Clearly, patients who suffer from intestinal fibrosis need novel and more effective treatments that target this common, often severe, complication of CD. Compared to the enormous advances in the development of new therapies to control intestinal inflammation, such as anti-TNFs, anti-integrins, and kinase inhibitors, progress to develop therapeutic modalities that may prevent or reverse intestinal fibrosis in CD is limited. As reviewed, the gut microbiota may have a considerable impact on the pathophysiology of intestinal fibrosis in CD. Of note, several animal models enable the investigation of the precise role of the gut microbiota in the development of intestinal fibrosis. Also, technical advances provide access to the global data associated with the alterations of gene expression and gut microbial composition during the process of intestinal fibrosis. These research tools may identify specific microbes or microbial components and virulence factors that affect intestinal fibrosis development. A rational identification of microbes and microbial factors could lead to effective therapies for preventing and attenuating intestinal fibrosis.
DW and NK wrote the manuscript and approved it for publication.
This work was supported an Overseas Postdoctoral Fellowship from the Uehara Memorial Foundation and a Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science (to DW) and National Institutes of Health grants DK108901, DK119219, AI142047, and DK125087 (to NK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn's disease according to the Vienna classification: changing pattern over the course of the disease. Gut. (2001) 49:777–82. doi: 10.1136/gut.49.6.777
2. Nos P, Domenech E. Postoperative Crohn's disease recurrence: a practical approach. World J Gastroenterol. (2008) 14:5540–8. doi: 10.3748/wjg.14.5540
3. Peyrin-Biroulet L, Loftus EV. Jr., Colombel JF, Sandborn WJ. The natural history of adult Crohn's disease in population-based cohorts. Am J Gastroenterol. (2010) 105:289–97. doi: 10.1038/ajg.2009.579
4. Pittet V, Rogler G, Michetti P, Fournier N, Vader JP, Schoepfer A, et al. Penetrating or stricturing diseases are the major determinants of time to first and repeat resection surgery in Crohn's disease. Digestion. (2013) 87:212–21. doi: 10.1159/000350954
5. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
6. Abreu MT, Taylor KD, Lin YC, Hang T, Gaiennie J, Landers CJ, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn's disease. Gastroenterology. (2002) 123:679–88. doi: 10.1053/gast.2002.35393
7. Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. (2008) 134:768–80. doi: 10.1053/j.gastro.2007.12.043
8. Regan MC, Flavin BM, Fitzpatrick JM, O'Connell PR. Stricture formation in Crohn's disease: the role of intestinal fibroblasts. Ann Surg. (2000) 231:46–50. doi: 10.1097/00000658-200001000-00007
9. Jacob N, Jacobs JP, Kumagai K, Ha CWY, Kanazawa Y, Lagishetty V, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. (2018) 11:1466–76. doi: 10.1038/s41385-018-0055-y
10. McKaig BC, Hughes K, Tighe PJ, Mahida YR. Differential expression of TGF-beta isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Physiol Cell Physiol. (2002) 282:C172–82. doi: 10.1152/ajpcell.00048.2001
11. Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis. (2012) 18:460–71. doi: 10.1002/ibd.21812
12. Cosnes J, Nion-Larmurier I, Beaugerie L, Afchain P, Tiret E, Gendre JP. Impact of the increasing use of immunosuppressants in Crohn's disease on the need for intestinal surgery. Gut. (2005) 54:237–41. doi: 10.1136/gut.2004.045294
13. Yamamoto T, Allan RN, Keighley MR. Effect of fecal diversion alone on perianal Crohn's disease. World J Surg. (2000) 24:1258–62; discussion 62–3. doi: 10.1007/s002680010250
14. Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R, et al. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet. (1991) 338:771–4. doi: 10.1016/0140-6736(91)90663-A
15. Zelas P, Jagelman DG. Loop illeostomy in the management of Crohn's colitis in the debilitated patient. Ann Surg. (1980) 191:164–8. doi: 10.1097/00000658-198002000-00006
16. McIlrath DC. Diverting ileostomy or colostomy in the management of Crohn's disease of the colon. Arch Surg. (1971) 103:308–10. doi: 10.1001/archsurg.1971.01350080224035
17. D'Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn's disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. (1998) 114:262–7. doi: 10.1016/S0016-5085(98)70476-7
18. Shafran I, Burgunder P. Adjunctive antibiotic therapy with rifaximin may help reduce Crohn's disease activity. Dig Dis Sci. (2010) 55:1079–84. doi: 10.1007/s10620-009-1111-y
19. Prantera C, Lochs H, Campieri M, Scribano ML, Sturniolo GC, Castiglione F, et al. Antibiotic treatment of Crohn's disease: results of a multicentre, double blind, randomized, placebo-controlled trial with rifaximin. Aliment Pharmacol Ther. (2006) 23:1117–25. doi: 10.1111/j.1365-2036.2006.02879.x
20. Arnold GL, Beaves MR, Pryjdun VO, Mook WJ. Preliminary study of ciprofloxacin in active Crohn's disease. Inflamm Bowel Dis. (2002) 8:10–5. doi: 10.1097/00054725-200201000-00002
21. Colombel JF, Lémann M, Cassagnou M, Bouhnik Y, Duclos B, Dupas JL, et al. A controlled trial comparing ciprofloxacin with mesalazine for the treatment of active Crohn's disease. Groupe d'Etudes Thérapeutiques des Affections Inflammatoires Digestives (GETAID). Am J Gastroenterol. (1999) 94:674–8. doi: 10.1111/j.1572-0241.1999.935_q.x
22. Prantera C, Zannoni F, Scribano ML, Berto E, Andreoli A, Kohn A, et al. An antibiotic regimen for the treatment of active Crohn's disease: a randomized, controlled clinical trial of metronidazole plus ciprofloxacin. Am J Gastroenterol. (1996) 91:328–32.
23. Greenbloom SL, Steinhart AH, Greenberg GR. Combination ciprofloxacin and metronidazole for active Crohn's disease. Can J Gastroenterol. (1998) 12:53–6. doi: 10.1155/1998/349460
24. Sutherland L, Singleton J, Sessions J, Hanauer S, Krawitt E, Rankin G, et al. Double blind, placebo controlled trial of metronidazole in Crohn's disease. Gut. (1991) 32:1071–5. doi: 10.1136/gut.32.9.1071
25. Leiper K, Morris AI, Rhodes JM. Open label trial of oral clarithromycin in active Crohn's disease. Aliment Pharmacol Ther. (2000) 14:801–6. doi: 10.1046/j.1365-2036.2000.00753.x
26. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. (2001) 411:603–6. doi: 10.1038/35079114
27. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. (2001) 411:599–603. doi: 10.1038/35079107
28. Hampe J, Cuthbert A, Croucher PJP, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. (2001) 357:1925–8. doi: 10.1016/S0140-6736(00)05063-7
29. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited determinants of Crohn's disease and ulcerative colitis phenotypes: a genetic association study. Lancet. (2016) 387:156–67. doi: 10.1016/S0140-6736(15)00465-1
30. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. (2003) 278:8869–72. doi: 10.1074/jbc.C200651200
31. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. (2005) 307:734–8. doi: 10.1126/science.1103685
32. Kosovac K, Brenmoehl J, Holler E, Falk W, Schoelmerich J, Hausmann M, et al. Association of the NOD2 genotype with bacterial translocation via altered cell-cell contacts in Crohn's disease patients. Inflamm Bowel Dis. (2010) 16:1311–21. doi: 10.1002/ibd.21223
33. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. (2010) 16:90–7. doi: 10.1038/nm.2069
34. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. (2010) 11:55–62. doi: 10.1038/ni.1823
35. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. (2007) 39:207–11. doi: 10.1038/ng1954
36. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. (2007) 39:596–604. doi: 10.1038/ng2032
37. Zoetendal EG, Rajilic-Stojanovic M, de Vos WM. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut. (2008) 57:1605–15. doi: 10.1136/gut.2007.133603
38. Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. (2004) 53:1–4. doi: 10.1136/gut.53.1.1
39. Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn's disease. Nat Rev Gastroenterol Hepatol. (2011) 8:152–68. doi: 10.1038/nrgastro.2011.3
40. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. (2004) 127:412–21. doi: 10.1053/j.gastro.2004.04.061
41. Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT AIEC. pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut. (2014) 63:1069–80. doi: 10.1136/gutjnl-2013-304909
42. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. (2013) 500:232–6. doi: 10.1038/nature12331
43. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. (2011) 331:337–41. doi: 10.1126/science.1198469
44. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
45. Van Immerseel F, Ducatelle R, De Vos M, Boon N, Van De Wiele T, Verbeke K, et al. Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. J Med Microbiol. (2010) 59:141–3. doi: 10.1099/jmm.0.017541-0
46. Weersma RK, Stokkers PC, Cleynen I, Wolfkamp SC, Henckaerts L, Schreiber S, et al. Confirmation of multiple Crohn's disease susceptibility loci in a large Dutch-Belgian cohort. Am J Gastroenterol. (2009) 104:630–8. doi: 10.1038/ajg.2008.112
47. Buning C, Genschel J, Buhner S, Kruger S, Kling K, Dignass A, et al. Mutations in the NOD2/CARD15 gene in Crohn's disease are associated with ileocecal resection and are a risk factor for reoperation. Aliment Pharmacol Ther. (2004) 19:1073–8. doi: 10.1111/j.1365-2036.2004.01967.x
48. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. (2004) 99:2393–404. doi: 10.1111/j.1572-0241.2004.40304.x
49. Adler J, Rangwalla SC, Dwamena BA, Higgins PD. The prognostic power of the NOD2 genotype for complicated Crohn's disease: a meta-analysis. Am J Gastroenterol. (2011) 106:699–712. doi: 10.1038/ajg.2011.19
50. Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. (2004) 113:1296–306. doi: 10.1172/JCI200420295
51. Winstanley C, Morgan JA. The bacterial flagellin gene as a biomarker for detection, population genetics and epidemiological analysis. Microbiology (Reading). (1997) 143:3071–84. doi: 10.1099/00221287-143-10-3071
52. Hayashi F, Smith KD, Ozinsky A, Hawn TR Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. (2001) 410:1099–103. doi: 10.1038/35074106
53. Dubinsky MC, Lin YC, Dutridge D, Picornell Y, Landers CJ, Farrior S, et al. Serum immune responses predict rapid disease progression among children with Crohn's disease: immune responses predict disease progression. Am J Gastroenterol. (2006) 101:360–7. doi: 10.1111/j.1572-0241.2006.00456.x
54. O'Donnell S, O'Sullivan M, O'Morain CA, Ryan BM. The clinical significance of antimicrobial serologic responses within an Irish Crohn's disease population. Eur J Gastroenterol Hepatol. (2013) 25:1464–9. doi: 10.1097/MEG.0b013e328365a364
55. Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. (2009) 15:1183–9. doi: 10.1002/ibd.20903
56. Hamilton AL, Kamm MA, De Cruz P, Wright EK, Feng H, Wagner J, et al. Luminal microbiota related to Crohn's disease recurrence after surgery. Gut Microbes. (2020) 11:1713–28. doi: 10.1080/19490976.2020.1778262
57. Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I Paracrine cells important in health and disease. Am J Physiol. (1999) 277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1
58. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
59. Otte J-M, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. (2003) 124:1866–78. doi: 10.1016/S0016-5085(03)00403-7
60. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. (1999) 274:10689–92.
61. Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. (1998) 395:284–8. doi: 10.1074/jbc.274.16.10689
62. Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. (1999) 274:17406–9. doi: 10.1074/jbc.274.25.17406
63. Morath S, Stadelmaier A, Geyer A, Schmidt RR, Hartung T. Synthetic lipoteichoic acid from Staphylococcus aureus is a potent stimulus of cytokine release. J Exp Med. (2002) 195:1635–40. doi: 10.1084/jem.20020322
64. Burke JP, Cunningham MF, Watson RW, Docherty NG, Coffey JC, O'Connell PR. Bacterial lipopolysaccharide promotes profibrotic activation of intestinal fibroblasts. Br J Surg. (2010) 97:1126–34. doi: 10.1002/bjs.7045
65. Zhao S, Dejanovic D, Yao P, Bhilocha S, Sadler T, Schirbel A, et al. Selective deletion of MyD88 signaling in alpha-SMA positive cells ameliorates experimental intestinal fibrosis via post-transcriptional regulation. Mucosal Immunol. (2020) 13:665–78. doi: 10.1038/s41385-020-0259-9
66. Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. (2019) 12:632–43. doi: 10.1038/s41385-019-0138-4
67. Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem. (2005) 280:20620–7. doi: 10.1074/jbc.M500877200
68. Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol. (1995) 22:165–72. doi: 10.1016/0168-8278(95)80424-2
69. Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY Li CP, et al. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol. (1997) 32:942–6. doi: 10.3109/00365529709011206
70. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. (2007) 13:1324–32. doi: 10.1038/nm1663
71. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. (2003) 37:1043–55. doi: 10.1053/jhep.2003.50182
72. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. (2004) 118:229–41. doi: 10.1016/j.cell.2004.07.002
73. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
74. Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG, Hayashi A, Imai J, et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell. (2020). doi: 10.1016/j.cell.2020.05.048
75. Friedrich M, Pohin M, Powrie F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity. (2019) 50:992–1006. doi: 10.1016/j.immuni.2019.03.017
76. Okuno T, Andoh A, Bamba S, Araki Y, Fujiyama Y, Fujiyama M, et al. Interleukin-1beta and tumor necrosis factor-alpha induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol. (2002) 37:317–24. doi: 10.1080/003655202317284228
77. Graham MF, Willey A, Adams J, Yager D, Diegelmann RF. Interleukin 1 beta down-regulates collagen and augments collagenase expression in human intestinal smooth muscle cells. Gastroenterology. (1996) 110:344–50. doi: 10.1053/gast.1996.v110.pm8566579
78. Graham MF, Willey A, Zhu YN, Yager DR, Sugerman HJ, Diegelmann RF. Corticosteroids repress the interleukin 1 beta-induced secretion of collagenase in human intestinal smooth muscle cells. Gastroenterology. (1997) 113:1924–9. doi: 10.1016/S0016-5085(97)70012-X
79. Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. (2014) 7:1492–503. doi: 10.1038/mi.2014.37
80. Shih DQ, Barrett R, Zhang X, Yeager N, Koon HW, Phaosawasdi P, et al. Constitutive TL1A (TNFSF15) expression on lymphoid or myeloid cells leads to mild intestinal inflammation and fibrosis. PLoS ONE. (2011) 6:e16090. doi: 10.1371/journal.pone.0016090
81. Zheng L, Zhang X, Chen J, Ichikawa R, Wallace K, Pothoulakis C, et al. Sustained Tl1a (Tnfsf15) expression on both lymphoid and myeloid cells leads to mild spontaneous intestinal inflammation and fibrosis. Eur J Microbiol Immunol (Bp). (2013) 3:11–20. doi: 10.1556/EuJMI.3.2013.1.2
82. Ramani K, Biswas PS. Interleukin-17: Friend or foe in organ fibrosis. Cytokine. (2019) 120:282–8. doi: 10.1016/j.cyto.2018.11.003
83. Ermann J, Staton T, Glickman JN, de Waal Malefyt R. Glimcher LH. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-Rag2-/- (TRUC) mice. Proc Natl Acad Sci USA. (2014) 111:E2559–66. doi: 10.1073/pnas.1408540111
84. Honzawa Y, Nakase H, Shiokawa M, Yoshino T, Imaeda H, Matsuura M, et al. Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn's disease. Gut. (2014) 63:1902–12. doi: 10.1136/gutjnl-2013-305632
85. Small CL, Reid-Yu SA, McPhee JB, Coombes BK. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun. (2013) 4:1957. doi: 10.1038/ncomms2957
86. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
87. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. (2016) 38:517–34. doi: 10.1007/s00281-016-0559-z
88. Scarpa M, Kessler S, Sadler T, West G, Homer C, McDonald C, et al. The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol. (2015) 185:1624–37. doi: 10.1016/j.ajpath.2015.02.018
89. Drygiannakis I, Valatas V, Sfakianaki O, Bourikas L, Manousou P, Kambas K, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis. (2013) 7:286–300. doi: 10.1016/j.crohns.2012.04.008
90. Suwara MI, Green NJ, Borthwick LA, Mann J, Mayer-Barber KD, Barron L, et al. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. (2014) 7:684–93. doi: 10.1038/mi.2013.87
91. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. (2003) 112:1776–84. doi: 10.1172/JCI200320530
92. Berger KH, Yaffe MP. Prohibitin family members interact genetically with mitochondrial inheritance components in Saccharomyces cerevisiae. Mol Cell Biol. (1998) 18:4043–52. doi: 10.1128/MCB.18.7.4043
93. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
94. Kumar A, Wu H, Collier-Hyams LS, Hansen JM Li T, Yamoah K, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. (2007) 26:4457–66. doi: 10.1038/sj.emboj.7601867
95. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. (2015) 6:565–77. doi: 10.1016/j.redox.2015.09.009
96. Langberg CW, Sauer T, Reitan JB, Hauer-Jensen M. Relationship between intestinal fibrosis and histopathologic and morphometric changes in consequential and late radiation enteropathy. Acta Oncol. (1996) 35:81–7. doi: 10.3109/02841869609098484
97. Crawford PA, Gordon JI. Microbial regulation of intestinal radiosensitivity. Proc Natl Acad Sci USA. (2005) 102:13254–9. doi: 10.1073/pnas.0504830102
98. Zhao Z, Cheng W, Qu W, Shao G, Liu S. Antibiotic Alleviates Radiation-Induced Intestinal Injury by Remodeling Microbiota, Reducing Inflammation, and Inhibiting Fibrosis. ACS Omega. (2020) 5:2967–77. doi: 10.1021/acsomega.9b03906
99. Rigby RJ, Hunt MR, Scull BP, Simmons JG, Speck KE, Helmrath MA, et al. A new animal model of postsurgical bowel inflammation and fibrosis: the effect of commensal microflora. Gut. (2009) 58:1104–12. doi: 10.1136/gut.2008.157636
100. Matsumoto S, Okabe Y, Setoyama H, Takayama K, Ohtsuka J, Funahashi H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. (1998) 43:71–8. doi: 10.1136/gut.43.1.71
101. Sartor RB, Cromartie WJ, Powell DW, Schwab JH. Granulomatous enterocolitis induced in rats by purified bacterial cell wall fragments. Gastroenterology. (1985) 89:587–95. doi: 10.1016/0016-5085(85)90455-X
102. Mourelle M, Salas A, Guarner F, Crespo E, García-Lafuente A, Malagelada JR. Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology. (1998) 114:519–26. doi: 10.1016/S0016-5085(98)70535-9
103. Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A. Crohn's disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis. (2008) 14:1051–60. doi: 10.1002/ibd.20423
104. Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, et al. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. (2009) 206:2179–89. doi: 10.1084/jem.20090741
105. Low D, Tran HT, Lee IA, Dreux N, Kamba A, Reinecker HC, et al. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology. (2013) 145:602–12 e9. doi: 10.1053/j.gastro.2013.05.017
106. Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol Rev. (2005) 206:260–76. doi: 10.1111/j.0105-2896.2005.00291.x
107. Jin Y, Liu R, Xie J, Xiong H, He JC, Chen N. Interleukin-10 deficiency aggravates kidney inflammation and fibrosis in the unilateral ureteral obstruction mouse model. Lab Invest. (2013) 93:801–11. doi: 10.1038/labinvest.2013.64
108. Fichera A, Lovadina S, Rubin M, Cimino F, Hurst RD, Michelassi F. Patterns and operative treatment of recurrent Crohn's disease: a prospective longitudinal study. Surgery. (2006) 140:649–54. doi: 10.1016/j.surg.2006.07.011
109. Yuan C, Chen WX, Zhu JS, Chen NW, Lu YM, Ou YX, et al. IL-10 treatment is associated with prohibitin expression in the Crohn's disease intestinal fibrosis mouse model. Mediators Inflamm. (2013) 2013:617145. doi: 10.1155/2013/617145
110. Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. (1993) 75:263–74. doi: 10.1016/0092-8674(93)80068-P
111. Rivera-Nieves J, Bamias G, Vidrich A, Marini M, Pizarro TT, McDuffie MJ, et al. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. (2003) 124:972–82. doi: 10.1053/gast.2003.50148
112. Picornell Y, Mei L, Taylor K, Yang H, Targan SR, Rotter JI. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. (2007) 13:1333–8. doi: 10.1002/ibd.20223
113. Michelsen KS, Thomas LS, Taylor KD Yu QT, Mei L, Landers CJ, et al. IBD-associated TL1A gene (TNFSF15) haplotypes determine increased expression of TL1A protein. PLoS ONE. (2009) 4:e4719. doi: 10.1371/journal.pone.0004719
114. Hirano A, Yamazaki K, Umeno J, Ashikawa K, Aoki M, Matsumoto T, et al. Association study of 71 European Crohn's disease susceptibility loci in a Japanese population. Inflamm Bowel Dis. (2013) 19:526–33. doi: 10.1097/MIB.0b013e31828075e7
115. Meylan F, Song YJ, Fuss I, Villarreal S, Kahle E, Malm IJ, et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol. (2011) 4:172–85. doi: 10.1038/mi.2010.67
116. Taraban VY, Slebioda TJ, Willoughby JE, Buchan SL, James S, Sheth B, et al. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. (2011) 4:186–96. doi: 10.1038/mi.2010.70
117. Barrett R, Zhang X, Koon HW, Vu M, Chang JY, Yeager N, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. (2012) 180:636–49. doi: 10.1016/j.ajpath.2011.10.026
118. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R, et al. novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. (1990) 98:694–702. doi: 10.1016/0016-5085(90)90290-H
119. Lund PK, Zuniga CC. Intestinal fibrosis in human and experimental inflammatory bowel disease. Curr Opin Gastroenterol. (2001) 17:318–23. doi: 10.1097/00001574-200107000-00004
120. Kitajima S, Morimoto M, Sagara E, Shimizu C, Ikeda Y. Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Exp Anim. (2001) 50:387–95. doi: 10.1538/expanim.50.387
121. Park JS, Choi JW, Jhun J, Kwon JY, Lee BI, Yang CW, et al. Lactobacillus acidophilus Improves Intestinal Inflammation in an Acute Colitis Mouse Model by Regulation of Th17 and Treg Cell Balance and Fibrosis Development. J Med Food. (2018) 21:215–24. doi: 10.1089/jmf.2017.3990
122. Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S, et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc. (2017) 12:1295–309. doi: 10.1038/nprot.2017.044
123. Llopis M, Antolin M, Guarner F, Salas A, Malagelada JR. Mucosal colonisation with Lactobacillus casei mitigates barrier injury induced by exposure to trinitronbenzene sulphonic acid. Gut. (2005) 54:955–9. doi: 10.1136/gut.2004.056101
124. García-Lafuente A, Antolín M, Guarner F, Crespo E, Salas A, Forcada P, et al. Incrimination of anaerobic bacteria in the induction of experimental colitis. Am J Physiol. (1997) 272:G10–5. doi: 10.1152/ajpgi.1997.272.1.G10
125. Sartor RB, Bond TM, Schwab JH. Systemic uptake and intestinal inflammatory effects of luminal bacterial cell wall polymers in rats with acute colonic injury. Infect Immun. (1988) 56:2101–8. doi: 10.1128/iai.56.8.2101-2108.1988
126. van Tol EA, Holt L, Li FL, Kong FM, Rippe R, Yamauchi M, et al. Bacterial cell wall polymers promote intestinal fibrosis by direct stimulation of myofibroblasts. Am J Physiol. (1999) 277:G245–55. doi: 10.1152/ajpgi.1999.277.1.G245
127. Medina C, Santos-Martinez MJ, Santana A, Paz-Cabrera MC, Johnston MJ, Mourelle M, et al. Transforming growth factor-beta type 1 receptor (ALK5) and Smad proteins mediate TIMP-1 and collagen synthesis in experimental intestinal fibrosis. J Pathol. (2011) 224:461–72. doi: 10.1002/path.2870
128. Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol. (2008) 6:53–66. doi: 10.1038/nrmicro1788
129. Grassl GA, Finlay BB. Pathogenesis of enteric Salmonella infections. Curr Opin Gastroenterol. (2008) 24:22–6. doi: 10.1097/MOG.0b013e3282f21388
130. Carter PB, Collins FM. The route of enteric infection in normal mice. J Exp Med. (1974) 139:1189–203. doi: 10.1084/jem.139.5.1189
131. Monack DM, Bouley DM, Falkow S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J Exp Med. (2004) 199:231–41. doi: 10.1084/jem.20031319
132. Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. (2003) 71:2839–58. doi: 10.1128/IAI.71.5.2839-2858.2003
133. Collins FM, Carter PB. Growth of salmonellae in orally infected germfree mice. Infect Immun. (1978) 21:41–7. doi: 10.1128/iai.21.1.41-47.1978
134. Johnson LA, Rodansky ES, Moons DS, Larsen SD, Neubig RR, Higgins PDR. Optimisation of Intestinal Fibrosis and Survival in the Mouse S. Typhimurium model for anti-fibrotic drug discovery and preclinical applications. J Crohns Colitis. (2017) 11:724–36. doi: 10.1093/ecco-jcc/jjw210
135. Lo BC, Gold MJ, Hughes MR, Antignano F, Valdez Y, Zaph C, et al. The orphan nuclear receptor ROR alpha and group 3 innate lymphoid cells drive fibrosis in a mouse model of Crohn's disease. Sci Immunol. (2016) 1:eaaf8864. doi: 10.1126/sciimmunol.aaf8864
136. Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn's disease. Infect Immun. (1999) 67:4499–509. doi: 10.1128/IAI.67.9.4499-4509.1999
137. Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology. (1998) 115:1405–13. doi: 10.1016/S0016-5085(98)70019-8
138. Eaves-Pyles T, Allen CA, Taormina J, Swidsinski A, Tutt CB, Jezek GE, et al. Escherichia coli isolated from a Crohn's disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol. (2008) 298:397–409. doi: 10.1016/j.ijmm.2007.05.011
139. Subramanian S, Roberts CL, Hart CA, Martin HM, Edwards SW, Rhodes JM, et al. Replication of Colonic Crohn's Disease Mucosal Escherichia coli Isolates within Macrophages and Their Susceptibility to Antibiotics. Antimicrob Agents Chemother. (2008) 52:427–34. doi: 10.1128/AAC.00375-07
140. De la Fuente M, Franchi L, Araya D, Diaz-Jimenez D, Olivares M, Alvarez-Lobos M, et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int J Med Microbiol. (2014) 304:384–92. doi: 10.1016/j.ijmm.2014.01.002
141. Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun. (2001) 69:5529–37. doi: 10.1128/IAI.69.9.5529-5537.2001
142. Bringer MA, Glasser AL, Tung CH, Meresse S, Darfeuille-Michaud A. The Crohn's disease-associated adherent-invasive Escherichia coli strain LF82 replicates in mature phagolysosomes within J774 macrophages. Cell Microbiol. (2006) 8:471–84. doi: 10.1111/j.1462-5822.2005.00639.x
143. Peeters H, Bogaert S, Laukens D, Rottiers P, De Keyser F, Darfeuille-Michaud A, et al. CARD15 variants determine a disturbed early response of monocytes to adherent-invasive Escherichia coli strain LF82 in Crohn's disease. Int J Immunogenet. (2007) 34:181–91. doi: 10.1111/j.1744-313X.2007.00670.x
144. Martinez-Medina M, Aldeguer X, Lopez-Siles M, Gonzalez-Huix F, Lopez-Oliu C, Dahbi G, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn's disease. Inflamm Bowel Dis. (2009) 15:872–82. doi: 10.1002/ibd.20860
145. Nash JH, Villegas A, Kropinski AM, Aguilar-Valenzuela R, Konczy P, Mascarenhas M, et al. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genom. (2010) 11:667. doi: 10.1186/1471-2164-11-667
146. Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. (2007) 117:1566–74. doi: 10.1172/JCI30504
147. Lenti MV, Di Sabatino A. Intestinal fibrosis. Mol Aspects Med. (2019) 65:100–9. doi: 10.1016/j.mam.2018.10.003
148. Narula N, Kassam Z, Yuan Y, Colombel JF, Ponsioen C, Reinisch W, et al. Systematic review and meta-analysis: fecal microbiota transplantation for treatment of active ulcerative colitis. Inflamm Bowel Dis. (2017) 23:1702–9. doi: 10.1097/MIB.0000000000001228
149. Costello SP, Soo W, Bryant RV, Jairath V, Hart AL, Andrews JM. Systematic review with meta-analysis: faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment Pharmacol Ther. (2017) 46:213–24. doi: 10.1111/apt.14173
Keywords: gut microbiota, Crohn's disease, animal model, adherent-invasive Escherichia coli (AIEC), intestinal fibrosis
Citation: Watanabe D and Kamada N (2022) Contribution of the Gut Microbiota to Intestinal Fibrosis in Crohn's Disease. Front. Med. 9:826240. doi: 10.3389/fmed.2022.826240
Received: 30 November 2021; Accepted: 13 January 2022;
Published: 07 February 2022.
Edited by:
Wenrui Wu, Zhejiang University, ChinaReviewed by:
Xu Shu, The First Affiliated Hospital of Nanchang University, ChinaCopyright © 2022 Watanabe and Kamada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuhiko Kamada, bmthbWFkYUB1bWljaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.