 Ting Wang
Ting Wang Taichi Ishikawa
Taichi Ishikawa Minoru Sasaki
Minoru Sasaki Toshimi Chiba
Toshimi Chiba- 1Division of Internal Medicine, Department of Oral Medicine, Iwate Medical University, Morioka, Japan
- 2Division of Molecular Microbiology, Department of Microbiology, Iwate Medical University, Morioka, Japan
Gut microbiota play many important roles, such as the regulation of immunity and barrier function in the intestine, and are crucial for maintaining homeostasis in living organisms. The disruption in microbiota is called dysbiosis, which has been associated with various chronic inflammatory conditions, food allergies, colorectal cancer, etc. The gut microbiota is also affected by several other factors such as diet, antibiotics and other medications, or bacterial and viral infections. Moreover, there are some reports on the oral-gut-liver axis indicating that the disruption of oral microbiota affects the intestinal biota. Non-alcoholic fatty liver disease (NAFLD) is one of the systemic diseases caused due to the dysregulation of the oral-gut-liver axis. NAFLD is the most common liver disease reported in the developed countries. It includes liver damage ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis, and cancer. Recently, accumulating evidence supports an association between NAFLD and dysbiosis of oral and gut microbiota. Periodontopathic bacteria, especially Porphyromonas gingivalis, have been correlated with the pathogenesis and development of NAFLD based on the clinical and basic research, and immunology. P. gingivalis was detected in the liver, and lipopolysaccharide from this bacteria has been shown to be involved in the progression of NAFLD, thereby indicating a direct role of P. gingivalis in NAFLD. Moreover, P. gingivalis induces dysbiosis of gut microbiota, which promotes the progression of NAFLD, through disrupting both metabolic and immunologic pathways. Here, we review the roles of microbial dysbiosis in NAFLD. Focusing on P. gingivalis, we evaluate and summarize the most recent advances in our understanding of the relationship between oral-gut microbiome symbiosis and the pathogenesis and progression of non-alcoholic fatty liver disease, as well as discuss novel strategies targeting both P. gingivalis and microbial dysbiosis.
Introduction
Alteration of the normal microbial composition, known as microbial dysbiosis, has been widely studied owing to its pathologic impacts on the body. Numerous studies have provided evidence that gut dysbiosis is closely related to systemic diseases centered in various organs (1). Moreover, recent studies have revealed that periodontal disease, which is a typical dysbiosis disease of the oral cavity, also contributes to the pathogenesis of systemic diseases, such as metabolic syndrome, cardiovascular disease, kidney disease, and brain disease, and may be related to cancer (2, 3). The pathogenic mechanisms underlying the effect of microbial dysbiosis beyond its original location are complicated, including direct effects of pathologic bacteria translocated by the blood to a specific organ, and effects of bacteria-derived endotoxins, metabolites, and inflammatory immune-mediators (4–6). Specifically, dysbiosis of the oral microbiota can be pathogenic to the internal organs via gut dysbiosis (7).
Non-alcoholic fatty liver disease (NAFLD) is of interest because, as is typical of metabolic diseases closely related to metabolic syndromes such as obesity and type 2 diabetes, it is also a systemic disorder affecting not only the liver but also various extrahepatic organs throughout the body and is linked to various systemic diseases, such as cardiovascular complications, kidney disease, and in particular the increase of extrahepatic malignancies (8, 9) (Figure 1). Studies on NAFLD have been limited because of its unclear origin, pathogenesis, and development, difficulties in early diagnosis, and a lack of effective therapeutic options. In this review, we focus on the relationship between NAFLD and Porphyromonas gingivalis, which is a typical oral pathogenic bacteria in periodontal disease (10, 11).
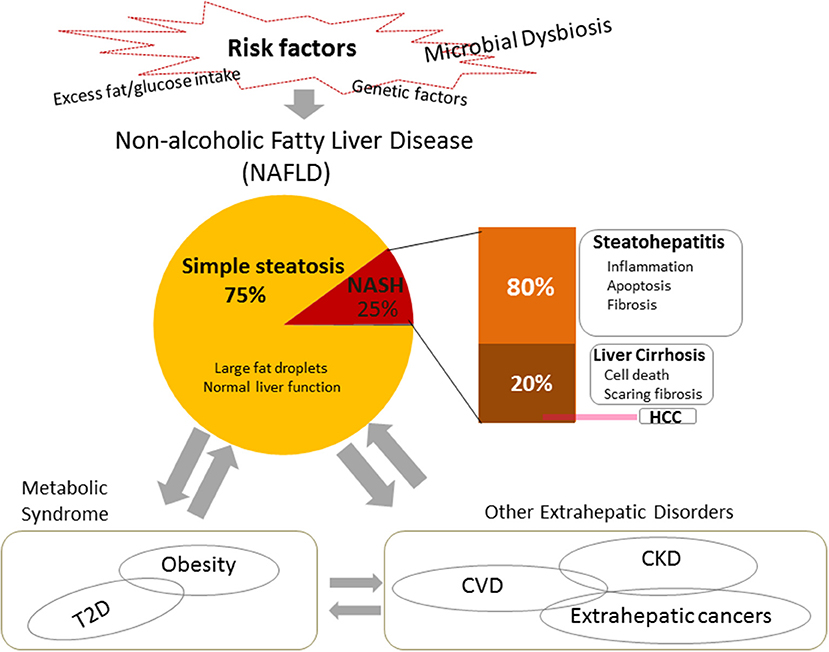
Figure 1. Risk factors, pathogenesis, and systemic impacts of non-alcoholic fatty liver disease (NAFLD). NAFLD is the most common chronic liver disease worldwide, usually presenting as fatty hepatocytes (simple steatosis). Non-alcoholic steatohepatitis (NASH) is a severe type of NAFLD. It is usually characterized by inflammation, cell damage, and apoptosis, and may include fibrosis. In turn, it can progress to cirrhosis or end-stage liver disease, i.e., hepatocellular carcinoma (HCC). In addition to heredity, one of the most widely accepted risk factors for NAFLD is excessive fat/glucose intake. Recent studies indicate that microbial dysbiosis takes an important role in the pathogenesis and the development of NAFLD. NAFLD leads not only to the development of hepatic diseases, but also connects with and contributes to systemic diseases, such as metabolic syndrome and extrahepatic disorders. NASH, non-alcoholic steatohepatitis; HCC, hepatocellular carcinoma; CDVs, cardiovascular diseases; CKD, chronic kidney disease, T2D, type 2 diabetes.
Human-Microorganism Symbiosis
In recent years, PCR, quantitative PCR, and metagenomic analyses have been used to investigate indigenous bacterial populations more deeply than can conventional culture methods and microscopic observations, and have made great progress. As a result, it has been found that there are more than 1,000 types of microorganisms in the human body, and that they have, collectively, 3.3 million genes, far exceeding the 25,000 genes in the human genome (12). The total amount of genomic information possessed by microorganisms in a certain environment is called a microbiome; Lederberg has written that “human beings are super-living organisms composed of symbiotic microorganisms and human beings, and symbiotic microorganisms are extremely important to human beings” (13). Even sophisticated microbiota analysis can determine only the bacterial biota, whereas microbiome analysis can determine both the bacterial biota (number of bacteria, composition ratio) and its component functions and metabolism. Bacteria, fungi, and protozoa are among the microorganisms that are present throughout the human body. Typical indigenous fungi include Candida albicans, and protozoans include Trichomonas and amoebas (14, 15). We focused on the bacteria that predominate among these indigenous microorganisms; they are present in healthy adults in or on the skin, nasopharynx, oral cavity, stomach, intestinal tract, vagina, etc. The number varies depending on the site, and the types vary from person to person (16–18). These bacteria are usually balanced to form a community (bacterial biota). Indigenous bacteria have beneficial effects on living organisms, such as acting against invasion and colonization by foreign pathogenic microorganisms, stimulating the immune system, enhancing host resistance and immune response, and synthesizing vitamins metabolites used by the living host body (19, 20). In contrast, such bacteria can also disadvantage the host by causing infectious disease in an easily infected host (opportunistic infection), or via pathogenic replacement, in which resident pathogenic microorganisms become predominant following reduction of the normal biota by antibacterial drugs. Moreover, resident bacteria that are not pathogenic in their original location can become pathogenic bacteria elsewhere (ectopic infection) (21). Indigenous bacteria are constantly exposed to the host's immune system, settle in the host tissues, and exist in a symbiotic, non-pathogenic state. It is thought that disruption of this interaction causes some infectious diseases (22).
The Intestinal Biota
Indigenous bacterial communities in the intestinal tract are composed of various bacterial species, such as Firmicutes, Bacteroidetes, and Actinobacteria (23). These are thought to invade mainly through the oral cavity (24). To date, there have been numerous reports of microbiome analyses (Table 1).
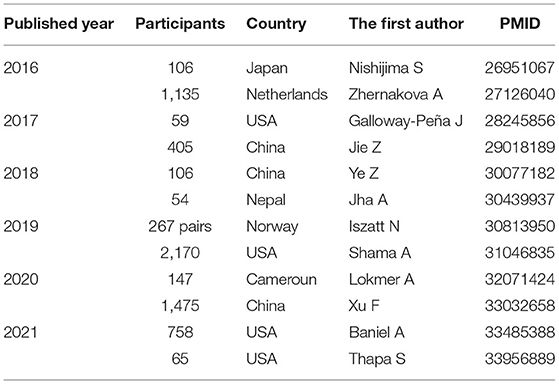
Table 1. Reports of the human intestinal microbiome by human genome analysis.
Dysbiosis changes in the intestinal biota is closely related to various pathological conditions, including intestinal-related diseases and systemic diseases (25–28). For example, inflammatory bowel diseases, including Crohn's disease, have been associated with increased mucosa-associated facultive anaerobes, Enterobacteriaceae, Pasteurellaceae, Veillonellaceae, Fusobacteriaceae, and decreased Faecalibacterium prausitzii, Erysipelotrichales, Bacteroidales, and Clostridiales (26). In obese mouse model, an increase in Firmicutes proportion and a decrease in Bacteroidetes proportion have been confirmed (29). Furthermore, the proportion of Akkermansia muciniphila, of the Verrucomicrobia, was reduced in diabetic patients (30), and the feces of children with autism spectrum disorders contained nine unknown species of Clostridium (28).
The Oral Biota
Previous reports suggest that the average adult has about 50–100 billion bacteria in the oral cavity, including as many as 500–700 bacterial species (31, 32). The oral biota varies across niches such as dental plaque, tongue, saliva, and gingival sulcus, and therefore exists as a wide variety of bacterial biota throughout the oral cavity (Table 2).

Table 2. Flora site and bacterial distribution in the human oral cavitya.
While these oral bacteria play a role as indigenous bacteria, some are thought to cause local infections such as tooth decay, periodontal disease, and endodontic infections (34). In addition, recent oral microbiome analysis shows that not only some pathogenic bacteria induce local disease, but also an abnormal bacterial biota (dysbiosis), in which the oral biota is disturbed by the growth of certain keystone species, may be the cause of various oral diseases (35). In addition, many studies have reported that dysbiosis of the oral bacteria is associated with many systemic diseases, such as aspiration pneumonia, bacterial endocarditis, preterm birth, diabetes, Alzheimer's disease, and atherosclerosis (36–38). Oral streptococci, including Streptococcus anginosus and S. intermedius, which are gram-positive facultative anaerobic cocci, and Prevotella, Fusobacterium, and Bacteroides, which are gram-negative obligate anaerobic cocci, are listed as the causative bacteria of aspiration pneumonia (39). It has been reported that bacterial endocarditis has a very high isolation frequency of oral streptococci and these are considered a causative agent (40). These are referred to as ectopic infections. In contrast, in preterm birth, diabetes, Alzheimer's disease, atherosclerosis, and one of the autoimmune diseases, rheumatism, the involvement of gram-negative obligate anaerobic bacilli, such as P. gingivalis and Treponema denticola, which are classified as periodontal pathogens, is strongly suspected; however, it is rare that the causative organism is specifically detected in the lesion. Various studies have been undertaken into the pathogenic mechanism of these bacteria, but there are many unclear points (41–43).
Mechanism by Which Oral Biota Induces Systemic Disease
Separate mechanisms underlying systemic disease caused by oral bacteria, differing from those involved in ectopic infection, have been considered. Conventionally, inflammatory cytokines and enzymes, present because of bacterial cell components in local lesions of the oral cavity, are transferred from the bloodstream to the whole body. More recently, it has been suggested that oral dysbiosis induces systemic disease by inducing intestinal dysbiosis, affecting the immune system, metabolic system, and intestinal barrier function (44, 45). Concerning this induction of intestinal dysbiosis by pre-existing oral dysbiosis, it has been reported that oral administration of Fusobacterium nucleatum, a gram-negative obligate anaerobic bacillus, significantly reduces the activity of natural killer cells (46). Additionally, P. gingivalis, whose proportion increases in periodontitis, may pass through the acid stomach and reach the intestines (47). In mice administered P. gingivalis orally, P. gingivalis does not colonize or proliferate in the intestinal tract, but changes the intestinal bacterial biota (48). The inside of the stomach is usually pH 1–2, and P. gingivalis cannot grow there. However, reports that P. gingivalis forms a biofilm and has a survival rate of 50% or more at pH 3, suggest that it can pass through the stomach at pH 4–5 immediately after eating (49). Changes in the intestinal biota may also decrease the expression of tight junction proteins involved in intestinal barrier function, increase inflammatory cytokine gene expression, and increase blood endotoxin levels because of suppression of the small intestinal alkaline phosphatase gene (50). As mentioned above, there are various reports on the mechanisms by which the oral biota can induce systemic diseases; further analysis is expected to contribute further details of these relationships, aiding the elucidation of the currently unclear etiology of systemic diseases.
Fatty Liver and Non-Alcoholic Fatty Liver Disease
As blood circulation is the key pathway through which pathogenic bacteria and their toxic metabolites can reach distant locations across the whole body, it is reasonable that those organs with abundant blood supply are more vulnerable to the above substances, and more easily develop diseases related to oral and gut microbial dysbiosis. Among them, the liver is the largest solid internal organ and is unique in having two sources of blood supply: 80% from the portal vein and 20% from the hepatic artery. The liver plays critical roles in the body, such as regulating metabolic processes, including synthesizing glucose and lipids, maintaining homeostasis, and protecting against toxic substances by means of considerable detoxification abilities (9, 51).
The liver has the potential to regenerate following tissue damage, although chronic inflammatory damage or potent drug-induced toxicity may lead to various pathologies, including excessive fat accumulation (fatty liver), inflammation, fibrosis, and cirrhosis, or even to end-stage liver disease, hepatocellular carcinoma (HCC) (52, 53). Among these conditions, simple steatosis of hepatocytes only rarely leads to clinical symptoms; it is also difficult to make a diagnosis, as the only effective method of determination so far is biopsy, which is invasive and thus generally not acceptable to patients who are suspected of having such a disease. However, fatty liver may be a precursor to all of the other, more severe states of liver disease, even cancer (54, 55). Understanding the mechanisms underlying the etiology and pathology of fatty liver diseases therefore has definite clinical significance in prevention, early prognosis, and treatment. It is known that alcohol can lead to fatty liver (56), although fatty liver has also been found in non-drinkers. The latter is therefore called NAFLD.
NAFLD is currently regarded as the most common chronic liver disease worldwide (9). Data have shown that approximately one quarter of the world's population is affected by it (57). Approximately 25% of NAFLD patients develop a severe clinical phenotype called non-alcoholic steatohepatitis (NASH), which is characterized by inflammation, cell apoptosis, and fibrosis (58). The prevalence of NAFLD and NASH in adults in the United States ranges from 30–40% to 3–12%, respectively (59). Furthermore, ~20% of NASH patients may progress to cirrhosis (60), and NASH-associated cirrhosis increases the risk of hepatocellular carcinoma (HCC) by 2.4–12.8% (61, 62). The pathogenesis of NAFLD has not yet been completely elucidated. A “two-hit” theory has been widely supported (63). Firstly, excess food intake causes metabolic disorders such as insulin resistance, which lead to excess fat accumulation, simple steatosis, in the liver. Secondly, lipotoxicity causes oxidative stress, mitochondrial dysfunction, and endoplasmic reticulum stress, which induces cell injury, inflammation, and fibrosis. However, research over the last decade supports a new “multiple-hits” hypothesis. In addition to various dietary components and genetic factors, microbial dysbiosis is a crucial pathogenic factor and plays important roles in the development of NAFLD (8, 58) (Figure 1).
Gut Microbial Dysbiosis and NAFLD: The Gut-Liver Axis
Risk factors such as long-term consumption of a high-fat diet, antibacterial drug use, and intestinal inflammation, cause alterations in gut microbial composition and functions (64–67). As a result, the gut barrier function is impaired, facilitating the entry of pathogenic bacteria, bacterial endotoxins, and other inflammatory cells and mediators to the portal vein, and thereby reaching the liver (62).
Numerous clinical and experimental studies have been conducted to reveal the relationships between gut dysbiosis and NAFLD, and the impact of gut dysbiosis on the etiology and pathology of NAFLD (65, 68, 69). Firstly, gut microbial alterations were detected in patients with NAFLD. For example, in the gut of the patients with NASH or cirrhosis, there was a decrease in health-related bacteria such as Bacteroidetes and an increase of pathogenic bacteria Proteobacteria and Enterobacteriaceae specie (70–72); The abnormal changes in the abundance of some bacterial phyla, such as Bacteroides, Prevotella, Proteobacteria, and Firmicutes, correlate with disease severity (73–76).
Secondly, microbiome signature profiles and metabolomics analysis have recently promoted wide discussion of the pathologic roles of bacteria-derived metabolites and products (77, 78). Such factors are known to contribute to hepatic steatosis, insulin resistance, and fibrosis (79–81). For example, the microbiome signature in NAFLD indicates that 3-(4-hydroxyphenyl), a metabolic material from Firmicutes, Bacteroidetes and Proteobacteria, is associated with liver fibrosis (82, 83), and Glycocholate is positively associated with advanced liver fibrosis (84). Moreover, metabolic studies have clarified that microbiome-generated secondary bile acid triggers NAFLD (85), and the gut microbiome of NAFLD patients shows abnormalities in carbon and amino acid metabolism (86), choline depletion, and increased production of certain short-chain fatty acids and alcohols (71). This suggests that gut dysbiosis is related to NAFLD by metabolic pathways.
Thirdly, endotoxemia resulted from increased gut permeability is also associated with NAFLD pathogenesis, indicating the effects of gut dysbiosis on immune system (55). Bacteria-derived endotoxins such as lipopolysaccharide (LPS) are closely related with intestinal immune function (73). Most reports give function of LPS as stimulation of the Toll-like receptor 4 (TLR4) (87, 88), which belongs to the Toll-like family of receptors that is crucial in host defense against invading pathogens (89). The localization of LPS has been confirmed in the livers of NALFD patients (58, 90, 91). Blocking LPS receptors is related to improved NAFLD in in-vivo studies (89, 92). Recent clinical and animal studies provide evidence that elevated concentrations of LPS and endotoxin-producing bacterial strains are positively related to the progression of NAFLD, via inflammation and oxidative stress, which finally leads to chronic inflammation (93) and insulin resistance (88, 94). LPS also triggers lipid peroxidation in the liver (95, 96). We will discuss LPS in the later part of this review.
It should be noted that although the above evidence indicates a promoter role for gut dysbiosis in the pathogenesis of NAFLD, the bilateral relationship between the disease and gut dysbiosis remains to be completely understood.
P. gingivalis and NAFLD
Recently, interest has been shown in the pathogenic role of oral, as well as gut, microbial dysbiosis. Chronic oral diseases such as periodontal disease typically involve such dysbiosis. Multiple reports from epidemiologic, in-vivo, and in-vitro studies indicate that periodontal disease is closely associated with NAFLD (93, 97). Oral pathologic bacteria related to oral diseases include P. gingivalis, Treponema denticola, Prevotella intermedia, Aggregatibacter actinomycetemcomitans, Tannerella forsythia, and Campylobacter rectus. Among them, P. gingivalis is regarded as a key pathogen leading to periodontitis and related systemic diseases (3, 10, 98). In this review, we emphasize the relationship between P. gingivalis and NAFLD, the effects of P. gingivalis on the pathogenesis of NAFLD, the mechanism underlying its pathogenic functions, and the therapeutic approaches targeting P. gingivalis and microbial dysbiosis.
P. gingivalis-Associated NAFLD
A potential link between P. gingivalis and NAFLD has been revealed. P. gingivalis and its DNA were detected in the oral cavities or livers of NAFLD patients at higher frequency than those of controls (99, 100). NASH patients with P. gingivalis infection had more severe fibrosis than those without infection (100). Furthermore, supportive data from various in-vivo studies showed that P. gingivalis infection can stimulate fat accumulation, increase the immune response, and result in insulin resistance, indicating the impact of P. gingivalis in NAFLD/NASH procession (97).
The spread of P. gingivalis to distant organs, such as the liver, occurs through two possible pathways. One is direct release into the blood circulation. During daily procedures such as brushing, or dental treatment, P. gingivalis and bacteria-associated factors such as LPS and cytokines spread into the blood via the microulceration in the periodontal pocket, and are transported to the liver through the hepatic artery. The indirect pathway involves swallowing P. gingivalis, which can translocate to the gastrointestinal tract and induce alteration of gut microbiota; this in turn negatively affects the liver through the portal vein system (93, 101). Therefore, the pathogenic action of the bacteria is related to both these pathways: directly through the oral-liver axis, and indirectly through the oral-gut-liver axis (Figure 2).
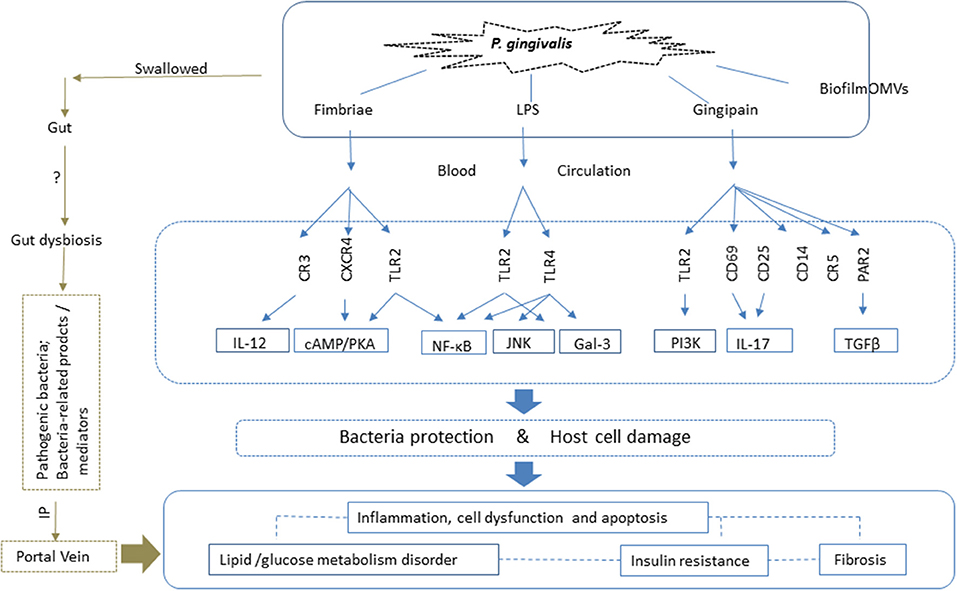
Figure 2. P. gingivalis impacts NAFLD via both the oral-liver and oral-gut axes. With respect to the oral-liver axis, P. gingivalis and its virulence factors can reach the liver directly via the blood circulation. Virulence factors such as fimbriae proteins, lipopolysaccharide (LPS) and gingipains elicit inflammatory immune reactions and activate intracellular signaling pathways by binding to the corresponding receptors. This results in inflammation, cell death, and dysfunctions, all of which are closely related to glucose and lipid metabolic disorders, insulin resistance, and fibrosis, which contribute to the pathogenesis of NAFLD. Biofilm and outer membrane vesicles (OMVs) play roles in the transfer of bacteria and virulence factors, such as gingipains, to the liver, the site of pathogenic action. P. gingivalis also affects NAFLD via the oral-gut-liver axis. It can enter the gastrointestinal tract via swallowing, where it can induce dysbiosis of the gut microbiota and enhance intestinal permeability. Thus, both the pathogenic bacteria, and related toxic products and mediators, can reach the liver via the portal vein, and play a role in NAFLD. LPS, lipopolysaccharide; TLR, Toll-like receptor; IL, interleukin; OMVs, outer membrane vesicles; CXCR, CXC-chemokine receptor; CR, complement receptor; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; NF-κB, nuclear factor kappa B; TGF-β, transforming growth factor β; PAR, proteinase-activated receptor; JNK, c-Jun-NH2-terminal kinase; IP, intestinal permeability; Gal, galectin; PI3K, phosphatidylinositol-3 kinase.
Direct Impacts of P. gingivalis in NAFLD: Structure-Related Functions
P. gingivalis is a gram-negative, obligate anaerobic bacteria. It is a common component of subgingival microbiomes that can colonize oral epithelial cells (102). P. gingivalis has unique structural components, known as virulence factors. These include some bacteria's own structural components such as fimbriae and LPS, and secretory components such as gingipains, and play critical roles in the survival and spread of P. gingivalis through cellular colonization, along with its pathogenic functions, such as inducing host immune response and inflammatory reactions (98, 103).
P. gingivalis fimbriae are filamentous structures on the surface of the bacterium that enable bacterial binding to host cells and tissues, enhance bacterial motility and invasiveness, and contribute to biofilm formation (104). There are two forms of P. gingivalis fimbriae: FimA, and the less important minor fimbriae. Studies of NAFLD patients showed that the invasive type II FimA constituted half of the total fimbriae detected (99). Type IV FimA, however, is closely associated with advanced liver fibrosis (105).
The mechanism underlying the impact of FimA on host cells is primarily related to the activation of adhesion and immuno-inflammatory pathways via interactions with various host receptors, which facilitates bacterial colonization and leads to host cell inflammation (104, 106, 107). In addition, FimA can also help the bacteria survive in the host cell over extended periods by triggering the complement system to protect the bacteria from host immune clearance (104).
FimA acts through three major types of receptor. Regarding its inflammatory effects, FimA binds to Toll-like receptor (TLR) 2, which activates the nuclear factor kappa B (NF-κB) system to induce the production of various pro-inflammatory cytokines (108). TLR2 has been shown to be associated with the pathogenesis of NAFLD (109). FimA also triggers the innate immune system by binding to complement receptor (CR) 3 and stimulating macrophages/monocytes (10). As a result, FimA inhibits the production of interleukin (IL) (12), which is related to bacterial clearance (110), thus enhancing P. gingivalis survival. CR3 is expressed in the liver (111), and the activation level of the complement system is related to NAFLD severity (112), suggesting the involvement of P. gingivalis fimbriae in the pathogenesis of NAFLD via immune response induction.
Furthermore, FimA activates CXC-chemokine receptor 4 (CXCR4) and TLR2 to induce cyclic adenosine monophosphate-dependent protein kinase A signaling, which not only results in disruption of phagocytosis by macrophages (113), but also inhibits host immune clearance of P. gingivalis (114). Dysfunctions of both macrophages and CXCR4 have been reported to be associated with NAFLD (115, 116).
Beyond FimA, P. gingivalis minor fimbriae can also induce inflammatory reactions in macrophages (117), and are reported to induce phosphorylation of FOXO1 (118), which plays a role in both lipid and glucose metabolism (119), indicating that both FimA and minor fimbriae in P. gingivalis may be factors leading to liver injury by this species.
LPS is a component of the outer cell membranes of gram-negative bacteria (120, 121). It critically contributes to the pathogenicity of microbes, mainly through lipid A, which is the biologically active region of LPS (122). LPS is structurally different in different bacterial species due to variations of fatty acid acyl chain composition of lipid A (10). P. gingivalis LPS contains multiple forms of lipid A, and the structural differences in lipid A may explain why P. gingivalis LPS can initiate differential signaling pathways and immune responses (10, 123). In addition to participating in biofilm formation (124), P. gingivalis LPS plays an important role in triggering host inflammatory responses via TLR activation; the specific type of TLR activation by LPS depends on the variation in the acylation of lipid A (10). However, it was recently reported that the activation of TLR2 by P. gingivalis is via other molecules on the surfaces of intact P. gingivalis cells rather than lipid A (125). Activation of TLR2 leads to an increase in the production of cytokines such as tumor necrosis factor α (TNF-α) and IL-6 by macrophages (126), while activation of TLR4 elevates the levels of IL-1β, IL-6, and IL-8 (127). In addition, NF-κB signaling pathway has also been reported to act downstream of TLRs, and to play a critical role in the inflammatory function of P. gingivalis LPS, including the production of cytokines (123). Several in-vivo and in-vitro studies have been performed to clarify the effects of P. gingivalis LPS on NAFLD. Injection of P. gingivalis LPS into the gingiva of animal models resulted in lipid deposition and inflammation in the liver (128–130), and in-vitro studies using HepG2 cells indicated that LPS may play a role in intracellular lipid accumulation and inflammation via both NF-κB and c-Jun-NH2-terminal kinase signaling (131). Moreover, P. gingivalis LPS can accelerate the progression of mild fatty liver to NASH (132), and studies using steatotic hepatocytes revealed that this activity may be related to increased TLR2 expression, inflammasome mRNA levels, and pro-inflammatory cytokines (100). Moreover, recent evidence shows that P. gingivalis LPS may contribute to hepatic fibrosis by activating hepatic stellate cells (HSCs); the mechanism involves triggering TLR4 to increase the production of galectin-3, which is critical for HSC activation (132). In addition, P. gingivalis LPS is possibly implicated in insulin resistance, either by stimulating the activation of pro-inflammatory cytokines such as TNF-α and IL-6, which play important roles in insulin resistance (133), or by directly inhibiting glucose incorporation into smooth muscle cells (134).
Kuraji et al. reported that P. gingivalis LPS accumulated predominantly in the liver, over other organs, and persisted in the livers of HFD-induced steatotic mice longer than in normal mouse liver. In addition, the diseased mice showed enhanced sensitivity to LPS and delayed clearance of LPS from the liver, indicating the potent role of P. gingivalis LPS in liver injury and NAFLD. The mechanism is possibly related to increased and activated hepatic macrophages (Kupffer cells) and TLR signaling (93).
As noted elsewhere in this review, gut bacteria-derived LPS also plays critical roles in the pathogenesis of NAFLD. The different functions and mechanisms involved in gut bacteria-derived LPS and P. gingivalis LPS have been investigated. For example, P. gingivalis LPS can activate both TLR2 and TLR4, whereas Escherichia coli LPS can only bind to TLR4 (135); although both LPS types activate TLR4, P. gingivalis LPS stimulates different pathways from those affected by E. coli LPS (136). Furthermore, P. gingivalis LPS shows a stronger ability to escape recognition by the host innate defense system than E. coli LPS (137), and P. gingivalis LPS induces more intracellular fat accumulation in HepG2 cells than E. coli LPS (131). In contrast to the substantial accumulated evidence that gut bacteria-derived LPS contributes to NAFLD by inducing oxidative stress (138), there is as yet little evidence (139) supporting direct oxidative stress stimulation by P. gingivalis LPS in NAFLD, which should be investigated further.
Gingipains are a family of secretory cysteine proteinases that are known to be the main virulence factors related to the pathogenicity of P. gingivalis. They consist of lysine and arginine gingipains (140, 141). As with fimbriae and LPS, gingipains also play roles in biofilm formation and produce immune-inflammatory responses by activating various immune cells (10, 142). Evidence shows that gingipains protect P. gingivalis from the host defense system in the following ways: they enhance the production of TNF-α by neutrophils by activating the TLR2/phosphatidylinositol-3 kinase (PI3K) pathway (143), thereby facilitating bacterial survival within host cells; they inhibit pathogen clearance by negatively regulating the production of neutrophil-derived molecules (144) and the macrophage immune receptor CD14 (145); and they promote the adaptability of P. gingivalis by triggering the complement system, for example by modulating the C5a receptor (C5aR) and its crosstalk with TLR2 signaling in macrophages (146).
Furthermore, gingipains may play a role in evading the host adaptive immune system by regulating T-cell immunity (147). Like LPS (148), they can induce the production by Th17 cells of the cell-specific cytokine, IL-17, by directly inducing the expression of CD69 and CD25 on T-cells (149). T-cell immunity, especially the Th17/IL-17 signaling pathway, plays a role in protecting pathogens and promoting inflammation, and was recently reported as important in the development of NAFLD (150–153). Moreover, recent work highlights a remarkable function of gingipains in NAFLD, revealing that they contribute to liver fibrosis by activating HSCs via the proteinase-activated receptor (PAR) 2 and TGF-β pathway (132). It should also be noted that gingipains can inactivate PI3K, protein kinase B (Akt), and Akt downstream proteins, including glycogen synthase kinase 3 (GSK3) and mammalian target of rapamycin (mTOR) (154). The PI3K/Akt signaling pathway plays multiple roles in various cell functions, including cell survival and glucose metabolism, and an in-vitro study has revealed that P. gingivalis suppresses glycogen synthesis in HepG2 cells by inhibiting the insulin receptor substrate 1/Akt/GSK3β pathway (155). This indicates that gingipains possibly participate in glucose metabolism impairment/insulin resistance, which needs to be investigated further.
Biofilms and outer membrane vesicles (OMVs) are two important microorganism-produced structures responsible for the survival, spread, and pathogenicity of microbes. Biofilms are composed of water, bacterial cells, and extracellular polymeric substances, which are responsible for microorganism protection and resistance to clinical treatments, such as antibiotics (156–158). P. gingivalis virulence factors, such as fimbriae, LPS, and gingipains, have been reported to contribute to biofilm formation, as mentioned previously, and the pathogenicity of P. gingivalis is enhanced by biofilm (159, 160). In addition to being a reservoir of pathogenic bacteria, biofilms also contribute to inflammation in many diseases, including inflammatory bowel disease and hepatobiliary carcinomas (161); however, the pathogenic effect of P. gingivalis biofilm, such as direct reactions of biofilm in NAFLD, remains unclear.
OMVs are small, spherical, bilayered membrane structures that are constantly released from the bacterial surface during growth. Each vesicle is composed of outer membrane proteins, lipoproteins, LPS, and some periplasmic components (162, 163). Like other bacteria such as Francisella and Pseudomonas putida, P. gingivalis can produce OMVs (164). P. gingivalis OMVs can concentrate virulence factors such as gingipains and LPS in the form of OMVs and discharge them to the environment to participate in bacteria-associated disorders (165, 166). P. gingivalis OMVs play roles in biofilm formation by binding to other periodontopathogens (167, 168), and facilitate bacterial adhesion and invasion in host cells, adaption to stress, and immune defense evasion (168). In addition to contributing to the destruction of periodontal tissues, P. gingivalis OMVs can migrate to the blood and play an important role in the pathogenesis of various systemic diseases, such as cardiovascular disease, rheumatoid arthritis, Alzheimer's disease, and carcinoma; the mechanism involved has been recently reviewed by Zhang et al. (169). Although the direct impact of P. gingivalis OMVs in NAFLD has not been completely elucidated, P. gingivalis OMVs carrying gingipains can transfer to the liver and impair hepatic glycogen synthesis in a mouse model; they can also inhibit insulin-induced Akt/GSK-3β signaling in a gingipain-dependent manner in HepG2 cells (170), indicating a potential role of these gingipain-carrying OMVs in the development of NAFLD through negative regulation of glucose metabolism and insulin sensitivity.
In summary, these bacteria-associated structures play roles in bacterial protection, as well as affecting the virulence of P. gingivalis, by activating various risk factors for NAFLD, such as fat accumulation, inflammation, insulin resistance/disturbance of glucose metabolism, and fibrosis. The mechanism involves immune cell-related inflammatory reactions and various intracellular signaling pathways (Figure 2). The scope of this review includes only the reported virulence mechanisms of P. gingivalis in relation to the pathogenesis of NAFLD; the functioning of these mechanisms beyond NAFLD and in other diseases also requires consideration. Moreover, other virulence factors, such as heat shock protein 60 (171), have been mentioned in relation to liver diseases (172). However, the majority of in-vitro studies have focused on circulating immune cells, while few studies have investigated the role of P. gingivalis in hepatic cells such as Kupffer cells and hepatocytes. Further studies are needed to elucidate the roles of the pathogenic components of P. gingivalis in NAFLD.
Indirect Impacts of P. gingivalis in NAFLD: Bacteria-Derived Gut Dysbiosis
The effects of P. gingivalis on the modulation of gut microbiota have been investigated in many studies. Both intravenous injection and oral administration of the bacteria in a mouse model produced alterations in the gut microbiota (50, 173, 174). Clinical studies have detected remarkably higher proportions of P. gingivalis in the guts of NAFLD patients than in those of non-NAFLD controls 99; patients with chronic periodontitis tended to have less diversity in their gut microbiomes (175); the major changes in the gut microbiota of patients with liver cirrhosis result from invasion by oral bacterial species (176). These results indicate a possible relationship between the bacteria, gut, and NAFLD (Figure 2). Studies involving oral administration of P. gingivalis to mouse models show that, in contrast to the increased blood endotoxin levels that are typical of gut dysbiosis/barrier dysfunction and lead to systemic inflammation contributing to liver injury, no P. gingivalis was detected in the blood system (174), or outgrowth of the bacteria in the gut (80). This strongly supports the speculation that oral P. gingivalis-induced endotoxemia-related liver injury indirectly, by inducing gut dysbiosis and barrier dysfunction.
However, despite the evidence that oral P. gingivalis can survive in the acidic conditions induced by gastric juice (49), enabling it to enter the gastrointestinal tract, and that dead bacteria from the mouth may stimulate several gut pathogens, upregulate bacterial virulence genes, and thereby increase cytotoxicity (177), the mechanism underlying bacteria-induced gut dysbiosis remains unclear. For example, P. gingivalis must inhabit the host cells and trigger immune-inflammatory reactions to initiate subsequent actions, and in-vitro studies have shown that intestinal inflammation can be detected following oral administration of P. gingivalis (50), but there is as yet no evidence to show whether and how P. gingivalis can localize in the gastrointestinal tract; in addition, the bilateral interactions between oral bacteria and gut microbiota should be considered. Furthermore, the lack of large-scale epidemiologic studies and some inconsistent results in different animal models make it difficult to define a clear mechanism underlying the effects of P. gingivalis in NAFLD via modulation of gut microbiota (93).
On the other hand, P. gingivalis is not the only periodontopathogen that can contribute to the pathogenesis of NAFLD. A. actinomycetemcomitans can induce gut dysbiosis and impairing glucose metabolism (178); the detection frequency of T. denticola in the NAFLD patients was significantly higher than that in the control subjects (99); in patients with liver cirrhosis, more than half (54%) of the patient-enriched, taxonomically assigned bacterial species were of oral origin (mostly veillonella and streptococci) (176, 179). Moreover, in comparison to growing evidence that supports P. gingivalis as a cause of gut dysbiosis, the precise mechanism through which P. gingivalis exerts its effects has yet to be determined. Further studies are needed to complete our knowledge of the roles of P. gingivalis in NAFLD via the oral-gut-liver axis.
Therapeutic Strategies in NAFLD: Targeting Oral Pathogens and Microbial Dysbiosis
Dietary factors have been accepted for over a decade as a critical risk factor for NAFLD. Food intake control (caloric restriction) and methods to increase energy consumption, such as physical activity and sports, have therefore been the major treatment options for NAFLD. Vitamin reagents, PPAR agonists, and other treatments are in use or undergoing clinical trials (48). However, most of them are symptomatic treatments, of uncertain efficacy, especially in severe progressive forms such as NASH or HCC. The newly established theory of bacteria-induced NAFLD provides guidance for the prevention, prediction, and treatment of the disease. Attenuating oral bacteria and improving gut dysbiosis are the main therapeutic approaches being studied (Figure 3, Table 3).
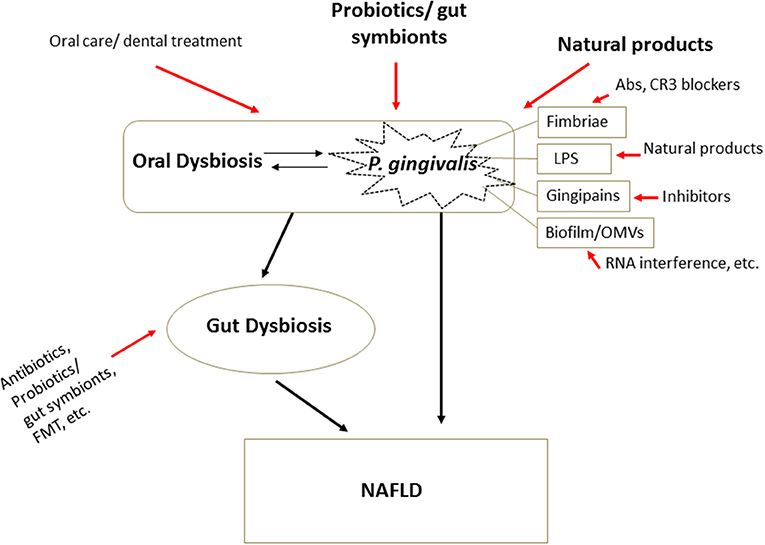
Figure 3. Therapeutic strategies in microbial dysbiosis-related NAFLD. Understanding the mechanism underlying microbial dysbiosis and its impacts on the pathogenesis of NAFLD through the gut-liver, oral-liver, and oral-gut-liver axes may contribute to the development of new options for treatment of the disease. Some very safe options, such as some probiotics, gut symbionts, and natural products, aimed at attenuating oral bacteria and correcting dysbiosis of the oral and gut microbiota, have received substantial attention. Additionally, treatments targeting the structures and virulence factors of P. gingivalis offer new treatment options specifically targeting bacteria. Abs, antibodies; FMT, fecal microbiota transplantation.
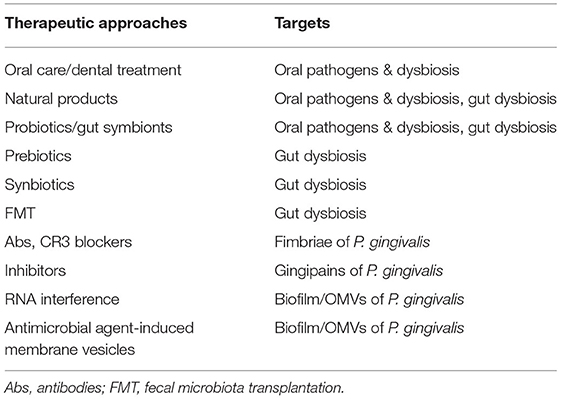
Table 3. Therapeutic approaches in NAFLD targeting oral pathogens and oral/gut microbial dysbiosis.
Treatment Based on Controlling Oral Disease
Given the remarkable impacts of periodontal disease in NAFLD, improving oral hygiene, and other approaches targeting pathogenic oral bacteria, are regarded as effective treatment strategies for bacteria-associated NAFLD. Administration of broad-spectrum antibiotics can effectively protect against various liver diseases, and some specific antibiotic treatments have produced improvement in the clinical symptoms of NAFLD, by means of eliminating harmful microbes, lowering circulating endotoxin and transaminase levels, and preventing lipid accumulation in the liver (180). Notably, despite their powerful positive effects, long-term administration of antibiotics may cause toxic side effects, eliciting antibiotic-resistant bacterial strains and itself generating gut dysbiosis (181). There is therefore increasing interest in the safety and specificity of therapeutic approaches targeting pathogenic oral bacteria, such as P. gingivalis.
One of the treatment modalities is the administration of probiotics. Two lactic acid bacteria, Lactococcus lactis and Lactobacillus reuteri, have millennia-long histories of use in the fermentation of foods and are generally accepted as safe for human consumption (“Generally Recognized As Safe,” or GRAS, in US Federal Drug Administration terminology) (182). It has been reported that L. lactis, together with one of its derivatives, nisin, was effective in periodontal disease, with potent antibacterial ability but low side effects. Its mechanism involves modulation of the formation, composition, and survival of oral bacterial biofilms (82). Moreover, L. lactis treatment reduced hepatic fat accumulation and showed anti-inflammatory effects in the liver, indicating its potential in the treatment of NAFLD (82, 182). Furthermore, antibacterial effects on P. gingivalis and other oral pathogens were seen not only from live L. reuteri cells but also supernatants from cell-free cultures and heat-killed L. reuteri (183), indicating a potent yet safe antibacterial activity of this probiotic.
With regard to the therapeutic approaches specifically targeting the structure of P. gingivalis, further methods for inhibiting biofilm formation by modulating OMVs have been investigated, in addition to the biofilm modulation approach mentioned above. These include RNA interference technology, antimicrobial agent-induced membrane vesicles, and some natural products, which have all been reviewed recently (120). Plant-derived products have attracted considerable attention in chronic disease therapy, given their safety. Natural compounds have been demonstrated to inhibit the growth and virulence factor activity of bacteria (184–186). We have reported that carnosic acid, extracted from rosemary plants, exerts potent effects, inhibiting lipid accumulation in both the adipose tissue and liver, improving glucose metabolism and liver functions in the ob-ob obesity mouse model (187), and inhibiting lipid accumulation in HepG2 cells (188). Moreover, our studies have clarified that carnosic acid can protect normal hepatocytes from H2O2-induced oxidative stress (189). Interestingly, carnosic acid has been reported to have remarkable antibacterial activities (190, 191). The mechanism is related to its inhibition of biofilm information (192); carnosic acid can also protect against LPS-induced liver injury (193), and is thus a potential new therapeutic approach for treating NAFLD. In our laboratory, we are currently investigating the roles of carnosic acid in NAFLD associated with P. gingivalis inoculation in both mouse and cell models.
Anti-fimbrial Ab has also been reported as effective in inhibiting the adherence of P. gingivalis to its host cells (194). Inhibition of or pre-immunity against potent virulence factors of P. gingivalis, such as gingipains, was effective against periodontitis and systemic diseases (195, 196). Moreover, as the CR3 receptor plays an important role in the P. gingivalis fimbriae-stimulated adhesion signal, as described above, CR3 attenuation has been considered as an approach for treating periodontitis and P. gingivalis-related systemic diseases (197). However, little is known about the significance of the above materials for the management of NAFLD associated with infection by oral pathogenic bacteria such as P. gingivalis, which should be investigated further.
Treatments Targeting Dysbiosis of Microbiota
Since the gut microbiota is closely related to the pathogenesis of NAFLD, various therapeutic approaches targeting gut microbiota have been developed. They include antibiotics, probiotics, prebiotics, synbiotics, and fecal microbiota transplantation, which have recently been reviewed (76, 180). In particular, dietary fiber is cited as useful: it may improve early-stage NAFLD by reducing calorie absorption and correcting the imbalance of gut microbiota (198); L. reuteri not only improves the dysbiosis of oral microbiota by targeting biofilm, but also shows the ability to correct gut dysbiosis and protect the liver from inflammatory damage (199). Moreover, a gut symbiont, Akkermansia muciniphila, and one of its membrane proteins has produced anti-inflammatory activity and improved gut permeability in animal models (30, 200, 201). Furthermore, human studies have shown that administration of A. muciniphila leads to decreased body weight, and the markers associated with inflammation and liver dysfunction in obese objects (202). However, it should be noted that while microbiome-based therapeutic approaches are usually regarded as safe, they cannot be considered risk-free, as they are still bioactive. Therefore, large-scale studies are required to evaluate their safety.
Conclusions
The close relationship between systemic disease and dysbiosis of both the oral and gut microbiota has been supported by substantial evidence from both basic and clinical studies. In this review, we emphasize one of the most commonly diagnosed such diseases, NAFLD, using it as a typical example of systemic disease, and P. gingivalis, which is the major pathogen of periodontitis, and typical of the pathogenic bacteria involved in systemic diseases via the oral-liver and oral-gut-liver axes. We focused on the structure-derived functions of P. gingivalis in the progression of NAFLD, such as hepatic steatosis, inflammation, and fibrosis, which cause conversion to NASH. NASH is associated with the pathogenesis of HCC, and it is therefore reasonable to suppose that P. gingivalis may contribute to HCC by promoting progression to NASH.
Currently, various therapeutic approaches are being developed, targeting P. gingivalis and dysbiosis of both oral and gut microbiota. Although most of these remain at the fundamental experimental level, limited to small-scale studies or experiencing difficulties in establishing ideal animal models, improving our understanding of the connections between NAFLD and oral/gut dysbiosis will definitely contribute to achieving the reasonable future goal of treating the disease successfully.
Author Contributions
TC, TW, TI, and MS conceptualized the review. TW and TI drafted the manuscript. TC and MS edited the manuscript. All the authors read and approved the final manuscript.
Funding
This study was supported by Grants-in-Aid for Scientific Research (JP 19K07737) from the Japan Society for the Promotion of Science.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gomaa EZ. Human gut microbiota/microbiome in health and diseases: a review. Antonie van Leeuwenhoek. (2020) 113:2019–40. doi: 10.1007/s10482-020-01474-7
2. Fi C, Wo W. Periodontal disease and systemic diseases: an overview on recent progresses. J Biol Regul Homeost Agents. (2021) 35 (1 Suppl. 1):1–9.
3. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X, et al. Porphyromonas gingivalis and its systemic impact: current status. Pathogens. (2020) 9:944. doi: 10.3390/pathogens9110944
4. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. (2020) 30:492–506. doi: 10.1038/s41422-020-0332-7
5. DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis. (2016) 22:1137–50. doi: 10.1097/MIB.0000000000000750
6. Singh R, Zogg H, Wei L, Bartlett A, Ghoshal UC, Rajender S, et al. Gut microbial dysbiosis in the pathogenesis of gastrointestinal dysmotility and metabolic disorders. J Neurogastroenterol Motil. (2021) 27:19–34. doi: 10.5056/jnm20149
7. Khor B, Snow M, Herrman E, Ray N, Mansukhani K, Patel KA, et al. Interconnections between the oral and gut microbiomes: reversal of microbial dysbiosis and the balance between systemic health and disease. Microorganisms. (2021) 9:496. doi: 10.3390/microorganisms9030496
8. Tilg H, Adolph TE, Moschen AR. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatology. (2021) 73:833–842. doi: 10.1002/hep.31518
9. Abdelmalek MF. Nonalcoholic fatty liver disease: another leap forward. Nat Rev Gastroenterol Hepatol. (2021) 18:85–6. doi: 10.1038/s41575-020-00406-0
10. Xu W, Zhou W, Wang H, Liang S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv Prot Chem Struct Biol. (2020) 120:45–84. doi: 10.1016/bs.apcsb.2019.12.001
11. Bregaint S, Boyer E, Fong SB, Meuric V, Bonnaure-Mallet M, Jolivet-Gougeon A. Porphyromonas gingivalis outside the oral cavity. Odontology. (2022) 110:1–19. doi: 10.1007/s10266-021-00647-8
12. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464:59–65. doi: 10.1038/nature08821
14. Alaalm L, Crunden JL, Butcher M, Obst U, Whealy R, Williamson C, et al. Identification and phenotypic characterization of hsp90 phosphorylation sites that modulate virulence traits in the major human fungal pathogen Candida albicans. Front Cell Infect Microbiol. (2021) 11:637836. doi: 10.3389/fcimb.2021.637836
15. Mach J, Sutak R. Iron in parasitic protists - from uptake to storage and where we can interfere. Metallomics. (2020) 12:1335–47. doi: 10.1039/d0mt00125b
16. Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA. (2008) 105:17994–9. doi: 10.1073/pnas.0807920105
17. Kim S, Jazwinski SM. The gut microbiota and healthy aging: a mini-review. Gerontology. (2018) 64:513–20. doi: 10.1159/000490615
18. Reid G. The need to focus on therapy instead of associations. Front Cell Infect Microbiol. (2019) 9:327. doi: 10.3389/fcimb.2019.00327
19. Suárez LJ, Arboleda S, Angelov N, Arce RM. Oral versus gastrointestinal mucosal immune niches in homeostasis and allostasis. Front Immunol. (2021) 12:705206. doi: 10.3389/fimmu.2021.705206
20. Ghattargi VC, Nimonkar YS, Burse SA, Davray D, Kumbhare SV, Shetty SA, et al. Genomic and physiological analyses of an indigenous strain, Enterococcus faecium 17OM39. Funct Integr Genomics. (2018) 18:385–99. doi: 10.1007/s10142-018-0596-x
21. Mu C, Zhu W. Antibiotic effects on gut microbiota, metabolism, and beyond. Appl Microbiol Biotechnol. (2019) 103:9277–85. doi: 10.1007/s00253-019-10165-x
22. Ballan R, Battistini C, Xavier-Santos D, Saad S. Interactions of probiotics and prebiotics with the gut microbiota. Prog Mol Biol Transl Sci. (2020) 171:265–300. doi: 10.1016/bs.pmbts.2020.03.008
23. Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. (2009) 11:2574–84. doi: 10.1111/j.1462-2920.2009.01982.x
24. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al. The human oral microbiome. J Bacteriol. (2010) 192:5002–17. doi: 10.1128/JB.00542-10
25. Kho ZY, Lal SK. The human gut microbiome - a potential controller of wellness and disease. Front Microbiol. (2018) 9:1835. doi: 10.3389/fmicb.2018.01835
26. Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
27. Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Specialized metabolites from the microbiome in health and disease. Cell Metab. (2014) 20:719–30. doi: 10.1016/j.cmet.2014.10.016
28. Kushak RI, Sengupta A, Winter HS. Interactions between the intestinal microbiota and epigenome in individuals with autism spectrum disorder. Dev Med Child Neurol. (2021) 63:1204–12. doi: 10.1111/dmcn.15052
29. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. (2006) 444:1022–3. doi: 10.1038/4441022a
30. Plovier H, Everard A, Druart C, Depommier C, Van Hul M, Geurts L, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. (2017) 23:107–13. doi: 10.1038/nm.4236
31. Krishnan K, Chen T, Paster BJ. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. (2017) 23:276–86. doi: 10.1111/odi.12509
32. Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. (2001) 183:3770–83. doi: 10.1128/JB.183.12.3770-3783.2001
33. Hamada S, Slade HD. Biology, Immunology, and cariogenicity of Streptococcus mutans. Microbiol Rev. (1980) 44:331–84. doi: 10.1128/mr.44.2.331-384.1980
34. Shi C, Cai L, Xun Z, Zheng S, Shao F, Wang B, et al. Metagenomic analysis of the salivary microbiota in patients with caries, periodontitis and comorbid diseases. J Dent Sci. (2021) 16:1264–73. doi: 10.1016/j.jds.2020.12.002
35. Álvarez S, Leiva-Sabadini C, Schuh C, Aguayo S. Bacterial adhesion to collagens: implications for biofilm formation and disease progression in the oral cavity. Crit Rev Microbiol. (2021) 48:1–13. doi: 10.1080/1040841X.2021.1944054
36. Ryder MI, Xenoudi P. Alzheimer disease and the periodontal patient: new insights, connections, and therapies. Periodontology. (2021) 87:32–42. doi: 10.1111/prd.12389
37. Koga S, Takazono T, Serita T, Ashizawa N, Hirayama T, Tashiro M, et al. Assessment of oral health in elderly patients with dementia by measuring volatile sulfur compounds and its relationship with pneumonia development: a pilot study. Respir Investig. (2021) 60:162–6. doi: 10.1016/j.resinv.2021.08.006
38. Pavlic V, Peric D, Kalezic IS, Madi M, Bhat SG, Brkic Z, et al. Identification of periopathogens in atheromatous plaques obtained from carotid and coronary arteries. BioMed Res Int. (2021) 2021:9986375. doi: 10.1155/2021/9986375
39. Hirai J, Sakanashi D, Haranaga S, Kinjo T, Hagihara M, Kato H, et al. Case-control study of pneumonia patients with Streptococcus anginosus group bacteria in their sputum. J Infect Chemother. (2016) 22:794–9. doi: 10.1016/j.jiac.2016.08.014
40. Sasaki M, Shimoyama Y, Kodama Y, Ishikawa T. Abiotrophia defectivaDnaK promotes fibronectin-mediated adherence to HUVECs and induces a proinflammatory response. Int J Mol Sci. (2021) 22:8528. doi: 10.3390/ijms22168528
41. Mahendra J, Mahendra L, Mugri MH, Sayed ME, Bhandi S, Alshahrani RT, et al. Role of periodontal bacteria, viruses, and placental mir155 in chronic periodontitis and preeclampsia-a genetic microbiological study. Curr Issues Mol Biol. (2021) 43:831–44. doi: 10.3390/cimb43020060
42. Gorabi AM, Kiaie N, Khosrojerdi A, Jamialahmadi T, Al-Rasadi K, Johnston TP, et al. Implications for the role of lipopolysaccharide in the development of atherosclerosis. Trends Cardiovasc Med. (2021) S1050-1738(21)00099-2. doi: 10.1016/j.tcm.2021.08.015. [Epub ahead of print].
43. Esberg A, Johansson L, Johansson I, Dahlqvist SR. Oral microbiota identifies patients in early onset rheumatoid arthritis. Microorganisms. (2021) 9:1657. doi: 10.3390/microorganisms9081657
44. Park SY, Hwang BO, Lim M, Ok SH, Lee SK, Chun KS, et al. Oral-Gut microbiome axis in gastrointestinal disease and cancer. Cancers. (2021) 13:2124. doi: 10.3390/cancers13092124
45. Fiorillo L, Cervino G, Laino L, D'Amico C, Mauceri R, Tozum TF, et al. Porphyromonas gingivalis, periodontal and systemic implications: a systematic review. Dent J. (2019) 7:114. doi: 10.3390/dj7040114
46. Kim YJ, Kim BK, Park SJ, Kim JH. Impact of Fusobacterium nucleatum in the gastrointestinal tract on natural killer cells. World J Gastroenterol. (2021) 27:4879–89. doi: 10.3748/wjg.v27.i29.4879
47. Tsuzuno T, Takahashi N, Yamada-Hara M, Yokoji-Takeuchi M, Sulijaya B, Aoki-Nonaka Y, et al. Ingestion of Porphyromonas gingivalis exacerbates colitis via intestinal epithelial barrier disruption in mice. J Periodontal Res. (2021) 56:275–88. doi: 10.1111/jre.12816
48. Kashiwagi Y, Aburaya S, Sugiyama N, Narukawa Y, Sakamoto Y, Takahashi M, et al. Porphyromonas gingivalis induces entero-hepatic metabolic derangements with alteration of gut microbiota in a type 2 diabetes mouse model. Sci Rep. (2021) 11:18398. doi: 10.1038/s41598-021-97868-2
49. Sato K, Takahashi N, Kato T, Matsuda Y, Yokoji M, Yamada M, et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci Rep. (2017) 7:6955. doi: 10.1038/s41598-017-07196-7
50. Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, et al. Oral Administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS ONE. (2015) 10:e0134234. doi: 10.1371/journal.pone.0134234
51. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. (2008) 47:729–36. doi: 10.1002/hep.22034
52. Shojaie L, Iorga A, Dara L. Cell death in liver diseases: a review. Int J Mol Sci. (2020) 21:9682. doi: 10.3390/ijms21249682
53. Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. (2021) 18:40–55. doi: 10.1038/s41575-020-0342-4
54. Antunes C, Azadfard M, Hoilat GJ, Gupta M. Fatty Liver. Treasure Island, FL: StatPearls Publishing (2021).
55. Kim JY, He F, Karin M. From liver fat to cancer: perils of the western diet. Cancers. (2021) 13:1095. doi: 10.3390/cancers13051095
57. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. (2020) 158:1851–64. doi: 10.1053/j.gastro.2020.01.052
58. Peng C, Stewart AG, Woodman OL, Ritchie RH, Qin CX. Non-Alcoholic steatohepatitis: a review of its mechanism, models and medical treatments. Front Pharmacol. (2020) 11:603926. doi: 10.3389/fphar.2020.603926
59. Sharma B, John S. Nonalcoholic Steatohepatitis (NASH). Treasure Island, FL: StatPearls Publishing (2021).
60. Khan F.Z, Perumpail RB, Wong RJ, Ahmed A. Advances in hepatocellular carcinoma: nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J Hepatol. (2015) 7:2155–61. doi: 10.4254/wjh.v7.i18.2155
61. White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. (2012) 10:1342–59.e2. doi: 10.1016/j.cgh.2012.10.001
62. Renne SL, Sarcognato S, Sacchi D, Guido M, Roncalli M, Terracciano L, et al. Hepatocellular carcinoma: a clinical and pathological overview. Pathologica. (2021) 113:203–17. doi: 10.32074/1591-951X-295
63. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. (1998) 114:842–5. doi: 10.1016/S0016-5085(98)70599-2
64. Ragonnaud E, Biragyn A. Gut microbiota as the key controllers of “healthy” aging of elderly people. Immunity Ageing. (2021) 18:2. doi: 10.1186/s12979-020-00213-w
65. Hrncir T, Hrncirova L, Kverka M, Hromadka R, Machova V, Trckova E, et al. Gut microbiota and NAFLD: pathogenetic mechanisms, microbiota signatures, and therapeutic interventions. Microorganisms. (2021) 9:957. doi: 10.3390/microorganisms9050957
66. Wieland A, Frank DN, Harnke B, Bambha K. Systematic review: microbial dysbiosis and nonalcoholic fatty liver disease. Aliment Pharmacol Ther. (2015) 42:1051–63. doi: 10.1111/apt.13376
67. Zhang YJ, Li S, Gan RY, Zhou T, Xu DP, Li HB. Impacts of gut bacteria on human health and diseases. Int J Mol Sci. (2015) 16:7493–519. doi: 10.3390/ijms16047493
68. Tilg H, Cani PD, Mayer EA. Gut microbiome and liver diseases. Gut. (2016) 65:2035–44. doi: 10.1136/gutjnl-2016-312729
69. Milosevic I, Vujovic A, Barac A, Djelic M, Korac M, Radovanovic Spurnic A, et al. Gut-Liver axis, gut microbiota, and its modulation in the management of liver diseases: a review of the literature. Int J Mol Sci. (2019) 20:395. doi: 10.3390/ijms20020395
70. Bashiardes S, Shapiro H, Rozin S, Shibolet O, Elinav E. Non-alcoholic fatty liver and the gut microbiota. Mol Metab. (2016) 5:782–94. doi: 10.1016/j.molmet.2016.06.003
71. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. (2016) 13:412–25. doi: 10.1038/nrgastro.2016.85
72. Qi X, Yang M, Stenberg J, Dey R, Fogwe L, Alam MS, et al. Gut microbiota mediated molecular events and therapy in liver diseases. World J Gastroenterol. (2020) 26:7603–18. doi: 10.3748/wjg.v26.i48.7603
73. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep. (2015) 3:8096. doi: 10.1038/srep08096
74. Oikonomou T, Papatheodoridis GV, Samarkos M, Goulis I, Cholongitas E. Clinical impact of microbiome in patients with decompensated cirrhosis. World J Gastroenterol. (2018) 24:3813–20. doi: 10.3748/wjg.v24.i34.3813
75. Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C, et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun. (2019) 10:1406. doi: 10.1038/s41467-019-09455-9
76. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. (2016) 63:764–75. doi: 10.1002/hep.28356
77. Ji Y, Yin Y, Li Z, Zhang W. Gut microbiota-derived components and metabolites in the progression of non-alcoholic fatty liver disease (NAFLD). Nutrients. (2019) 11:1712. doi: 10.3390/nu11081712
78. Zhou D, Fan JG. Microbial metabolites in non-alcoholic fatty liver disease. World J Gastroenterol. (2019) 25:2019–28. doi: 10.3748/wjg.v25.i17.2019
79. He FF, Li YM. Role of gut microbiota in the development of insulin resistance and the mechanism underlying polycystic ovary syndrome: a review. J Ovarian Res. (2020) 13:73. doi: 10.1186/s13048-020-00670-3
80. Wu J, Wang K, Wang X, Pang Y, Jiang C. The role of the gut microbiome and its metabolites in metabolic diseases. Protein Cell. (2021) 12:360–373. doi: 10.1007/s13238-020-00814-7
81. Delzenne NM, Bindels LB. Microbiome metabolomics reveals new drivers of human liver steatosis. Nat Med. (2018) 24:906–7. doi: 10.1038/s41591-018-0126-3
82. Sookoian S, Salatino A, Castaño GO, Landa MS, Fijalkowky C, Garaycoechea M, et al. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut. (2020) 69:1483–91. doi: 10.1136/gutjnl-2019-318811
83. Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, et al. Genetics of NAFLD in twins consortium. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology. (2018) 68:918–32. doi: 10.1002/hep.29892
84. Caussy C, Ajmera VH, Puri P, Hsu CL, Bassirian S, Mgdsyan M, et al. Serum metabolites detect the presence of advanced fibrosis in derivation and validation cohorts of patients with non-alcoholic fatty liver disease. Gut. (2019) 68:1884–92. doi: 10.1136/gutjnl-2018-317584
85. Zhou J, Tripathi M, Sinha RA, Singh BK, Yen PM. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res. (2021) 7:11. doi: 10.20517/2394-5079.2020.134
86. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. (2018) 15:397–411. doi: 10.1038/s41575-018-0011-z
87. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. (2009) 137:1716–24.e242. doi: 10.1053/j.gastro.2009.08.042
88. Ogawa Y, Imajo K, Honda Y, Kessoku T, Tomeno W, Kato S, et al. Palmitate-induced lipotoxicity is crucial for the pathogenesis of nonalcoholic fatty liver disease in cooperation with gut-derived endotoxin. Sci Rep. (2018) 8:11365. doi: 10.1038/s41598-018-29735-6
89. Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. (2009) 50:1094–104. doi: 10.1002/hep.23122
90. Dietrich P, Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. (2014) 28:637–53. doi: 10.1016/j.bpg.2014.07.008
91. Bertani B, Ruiz N. Function and biogenesis of lipopolysaccharides. EcoSal Plus. (2018) 8:1–19. doi: 10.1128/ecosalplus.ESP-0001-2018
92. Kuraji R, Sekino S, Kapila Y, Numabe Y. Periodontal disease-related nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an emerging concept of oral-liver axis. Periodontology. (2021) 87:204–40. doi: 10.1111/prd.12387
93. Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N. Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev. (2016) 2016:7432797. doi: 10.1155/2016/7432797
94. Fei N, Bruneau A, Zhang X, Wang R, Wang J, Rabot S, et al. Endotoxin producers overgrowing in human gut microbiota as the causative agents for nonalcoholic fatty liver disease. mBio. (2020) 11:e03263–19. doi: 10.1128/mBio.03263-19
95. Xu DX, Chen YH, Wang JP, Sun MF, Wang H, Wei LZ., et al. Perinatal lipopolysaccharide exposure downregulates pregnane X receptor and Cyp3a11 expression in fetal mouse liver. Toxicol Sci. (2005) 87:38–45. doi: 10.1093/toxsci/kfi239
96. Abdel-Salam OM, Youness ER, Mohammed NA, Morsy SM, Omara EA, Sleem AA. Citric acid effects on brain and liver oxidative stress in lipopolysaccharide-treated mice. J Med Food. (2014) 17:588–98. doi: 10.1089/jmf.2013.0065
97. Hatasa M, Yoshida S, Takahashi H, Tanaka K, Kubotsu Y, Ohsugi Y, et al. Relationship between NAFLD and periodontal disease from the view of clinical and basic research, and immunological response. Int J Mol Sci. (2021) 22:3728. doi: 10.3390/ijms22073728
98. How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. (2016) 7:53. doi: 10.3389/fmicb.2016.00053
99. Yoneda M, Naka S, Nakano K, Wada K, Endo H, Mawatari H, et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol. (2012) 12:16. doi: 10.1186/1471-230X-12-16
100. Furusho H, Miyauchi M, Hyogo H, Inubushi T, Ao M, Ouhara K, et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J Gastroenterol. (2013) 48:1259–70. doi: 10.1007/s00535-012-0738-1
101. Mulhall H, Huck O, Amar S. Porphyromonas gingivalis, a long-range pathogen: systemic impact and therapeutic implications. Microorganisms. (2020) 8:869. doi: 10.3390/microorganisms8060869
102. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. (2018) 16:745–59. doi: 10.1038/s41579-018-0089-x
103. Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of Porphyromonas gingivalis. Periodontology. (1999) 20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x
104. Enersen M, Nakano K, Amano A. Porphyromonas gingivalis fimbriae. J Oral Microbiology. (2013) 5:1–10. doi: 10.3402/jom.v5i0.20265
105. Nakahara T, Hyogo H, Ono A, Nagaoki Y, Kawaoka T, Miki D, et al. Involvement of Porphyromonas gingivalis in the progression of non-alcoholic fatty liver disease. J Gastroenterol. (2018) 53:269–80. doi: 10.1007/s00535-017-1368-4
106. Pathirana RD, O'Brien-Simpson NM, Reynolds EC. Host immune responses to Porphyromonas gingivalis antigens. Periodontology. (2010) 52:218–37. doi: 10.1111/j.1600-0757.2009.00330.x
107. Dashper SG, Mitchell HL, Seers CA, Gladman SL, Seemann T, Bulach DM, et al. Porphyromonas gingivalis uses specific domain rearrangements and allelic exchange to generate diversity in surface virulence factors. Front Microbiol. (2017) 8:48. doi: 10.3389/fmicb.2017.00048
108. Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama S, Ratti P, Schifferle RE, et al. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. (2006) 8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x
109. Miura K, Ohnishi H. Role of gut microbiota and toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. (2014) 20:7381–91. doi: 10.3748/wjg.v20.i23.7381
110. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. (2003) 3:133–46. doi: 10.1038/nri1001
111. Lima WG, Oliveira PS, Caliari MV, Gonçalves R, Michalick MS, Melo MN, et al. Histopathological and immunohistochemical study of type 3 complement receptors (CD11b/CD18) in livers and spleens of asymptomatic and symptomatic dogs naturally infected with leishmania (leishmania) chagasi. Vet Immunol Immunopathol. (2007) 117:129–36. doi: 10.1016/j.vetimm.2007.02.012
112. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, et al. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology. (2009) 50:1809–17. doi: 10.1002/hep.23228
113. Li L, Zhang YL, Liu XY, Meng X, Zhao RQ, Ou LL, et al. Periodontitis exacerbates and promotes the progression of chronic kidney disease through oral flora, cytokines, and oxidative stress. Front Microbiol. (2021) 12:656372. doi: 10.3389/fmicb.2021.656372
114. Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci USA. (2008) 105:13532–7. doi: 10.1073/pnas.0803852105
115. Zhang N, Qu Y, Qin B. Sodium butyrate ameliorates non-alcoholic fatty liver disease by upregulating miR-150 to suppress CXCR4 expression. Clin Exp Pharmacol Physiol. (2021) 48:1125–36. doi: 10.1111/1440-1681.13497
116. Boujedidi H, Robert O, Bignon A, Cassard-Doulcier AM, Renoud ML, Gary-Gouy H, et al. CXCR4 dysfunction in non-alcoholic steatohepatitis in mice and patients. Clin Sci. (2015) 128:257–67. doi: 10.1042/CS20130833
117. Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res. (2004) 39:136–42. doi: 10.1111/j.1600-0765.2004.00719.x
118. Meghil MM, Tawfik OK, Elashiry M, Rajendran M, Arce RM, Fulton DJ, et al. Disruption of immune homeostasis in human dendritic cells via regulation of autophagy and apoptosis by Porphyromonas gingivalis. Front Immunol. (2019) 10:2286. doi: 10.3389/fimmu.2019.02286
119. Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. (2008) 27:2320–36. doi: 10.1038/onc.2008.25
120. Płóciennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. (2015) 72:557–81. doi: 10.1007/s00018-014-1762-5
121. Darveau RP, Arbabi S, Garcia I, Bainbridge B, Maier RV. Porphyromonas gingivalis lipopolysaccharide is both agonist and antagonist for p38 mitogen-activated protein kinase activation. Infect Immunity. (2002) 70:1867–73. doi: 10.1128/IAI.70.4.1867-1873.2002
122. Marcano R, Rojo MÁ, Cordoba-Diaz D, Garrosa M. Pathological and therapeutic approach to endotoxin-secreting bacteria involved in periodontal disease. Toxins. (2021) 13:533. doi: 10.3390/toxins13080533
123. Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immunity. (2004) 72:5041–51. doi: 10.1128/IAI.72.9.5041-5051.2004
124. Li Y, Shi Z, Radauer-Preiml I, Andosch A, Casals E, Luetz-Meindl U, et al. Bacterial endotoxin (lipopolysaccharide) binds to the surface of gold nanoparticles, interferes with biocorona formation and induces human monocyte inflammatory activation. Nanotoxicology. (2017) 11:1157–75. doi: 10.1080/17435390.2017.1401142
125. Jain S, Coats SR, Chang AM, Darveau RP. A novel class of lipoprotein lipase-sensitive molecules mediates toll-like receptor 2 activation by Porphyromonas gingivalis. Infect Immunity. (2013) 81:1277–86. doi: 10.1128/IAI.01036-12
126. Holden JA, Attard TJ, Laughton KM, Mansell A, O'Brien-Simpson NM, Reynolds EC. Porphyromonas gingivalis lipopolysaccharide weakly activates M1 and M2 polarized mouse macrophages but induces inflammatory cytokines. Infect Immunity. (2014) 82:4190–203. doi: 10.1128/IAI.02325-14
127. Dahlen G, Basic A, Bylund J. Importance of virulence factors for the persistence of oral bacteria in the inflamed gingival crevice and in the pathogenesis of periodontal disease. J Clin Med. (2019) 8:1339. doi: 10.3390/jcm8091339
128. Fujita M, Kuraji R, Ito H, Hashimoto S, Toen T, Fukada T, et al. Histological effects and pharmacokinetics of lipopolysaccharide derived from Porphyromonas gingivalis on rat maxilla and liver concerning with progression into non-alcoholic steatohepatitis. J Periodontol. (2018) 89:1101–11. doi: 10.1002/JPER.17-0678
129. Kuraji R, Ito H, Fujita M, Ishiguro H, Hashimoto S, Numabe Y. Porphyromonas gingivalis induced periodontitis exacerbates progression of non-alcoholic steatohepatitis in rats. Clin Exp Dent Res. (2016) 2:216–25. doi: 10.1002/cre2.41
130. Varela-López A, Bullón P, Ramírez-Tortosa CL, Navarro-Hortal MD, Robles-Almazán M, Bullón B, et al. A diet rich in saturated fat and cholesterol aggravates the effect of bacterial lipopolysaccharide on alveolar bone loss in a rabbit model of periodontal disease. Nutrients. (2020) 12:1405. doi: 10.3390/nu12051405
131. Ding LY, Liang LZ, Zhao YX, Yang YN, Liu F, Ding QR, et al. Porphyromonas gingivalis-derived lipopolysaccharide causes excessive hepatic lipid accumulation via activating NF-κB and JNK signaling pathways. Oral Dis. (2019) 25:1789–97. doi: 10.1111/odi.13153
132. Nagasaki A, Sakamoto S, Chea C, Ishida E, Furusho H, Fujii M, et al. Odontogenic infection by Porphyromonas gingivalis exacerbates fibrosis in NASH via hepatic stellate cell activation. Sci Rep. (2020) 10:4134. doi: 10.1038/s41598-020-60904-8
133. Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. (2008) 14:222–31. doi: 10.2119/2007-00119.Tilg
134. Nagata T. [Relationhip between diabetes and periodontal disease]. Clin Calcium. (2009) 19:1291–8.
135. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. (1999) 11:443–51. doi: 10.1016/S1074-7613(00)80119-3
136. Ding PH, Wang CY, Darveau RP, Jin L. Porphyromonas gingivalis LPS stimulates the expression of LPS-binding protein in human oral keratinocytes in vitro. Innate Immunity. (2013) 19:66–75. doi: 10.1177/1753425912450348
137. Liu R, Desta T, Raptis M, Darveau RP, Graves DT., P. gingivalis, E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J Periodontol. (2008) 79:1241–7. doi: 10.1902/jop.2008.070575
138. Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: implications for prevention and therapy. Antioxidants. (2021) 10:174. doi: 10.3390/antiox10020174
139. Le Sage F, Meilhac O, Gonthier MP. Porphyromonas gingivalis lipopolysaccharide induces pro-inflammatory adipokine secretion and oxidative stress by regulating toll-like receptor-mediated signaling pathways and redox enzymes in adipocytes. Mol Cell Endocrinol. (2017) 446:102–10. doi: 10.1016/j.mce.2017.02.022
140. Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon's knife to a meat chopper-like brutal degradation of proteins. Periodontology. (2010) 54:15–44. doi: 10.1111/j.1600-0757.2010.00377.x
141. Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett. (2012) 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x
142. Grenier D, Tanabe S. Porphyromonas gingivalis gingipains trigger a proinflammatory response in human monocyte-derived macrophages through the p38α mitogen-activated protein kinase signal transduction pathway. Toxins. (2010) 2:341–52. doi: 10.3390/toxins2030341
143. Makkawi H, Hoch S, Burns E, Hosur K, Hajishengallis G, Kirschning CJ, et al. Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front Cell Infect Microbiol. (2017) 7:359. doi: 10.3389/fcimb.2017.00359
144. Maisetta G, Brancatisano FL, Esin S, Campa M, Batoni G. Gingipains produced by Porphyromonas gingivalis ATCC49417 degrade human-β-defensin 3 and affect peptide's antibacterial activity in vitro. Peptides. (2011) 32:1073–7. doi: 10.1016/j.peptides.2011.02.003
145. Wilensky A, Tzach-Nahman R, Potempa J, Shapira L, Nussbaum G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J Innate Immunity. (2015) 7:127–35. doi: 10.1159/000365970
146. Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. (2010) 3:ra11. doi: 10.1126/scisignal.2000697
147. Khalaf H, Bengtsson T. Altered T-cell responses by the periodontal pathogen Porphyromonas gingivalis. PLoS ONE. (2012) 7:e45192. doi: 10.1371/journal.pone.0045192
148. Park YD, Kim YS, Jung YM, Lee SI, Lee YM, Bang JB, et al. Porphyromonas gingivalis lipopolysaccharide regulates interleukin (IL)-17 and IL-23 expression via SIRT1 modulation in human periodontal ligament cells. Cytokine. (2012) 60:284–93. doi: 10.1016/j.cyto.2012.05.021
149. Yun LW, Decarlo AA, Hunter N. Blockade of protease-activated receptors on T cells correlates with altered proteolysis of CD27 by gingipains of Porphyromonas gingivalis. Clin Exp Immunol. (2007) 150:217–29. doi: 10.1111/j.1365-2249.2007.03488.x
150. Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology. (2013) 145:366–74.e743. doi: 10.1053/j.gastro.2013.04.010
151. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci. (2016) 130:193–203. doi: 10.1042/CS20150405
152. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The differential roles of t cells in non-alcoholic fatty liver disease and obesity. Front Immunol. (2019) 10:82. doi: 10.3389/fimmu.2019.00082
153. Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. (2011) 166:281–90. doi: 10.1111/j.1365-2249.2011.04471.x
154. Nakayama M, Ohara N. Molecular mechanisms of Porphyromonas gingivalis-host cell interaction on periodontal diseases. Jpn Dent Sci Rev. (2017) 53:134–40. doi: 10.1016/j.jdsr.2017.06.001
155. Ishikawa M, Yoshida K, Okamura H, Ochiai K, Takamura H, Fujiwara N, et al. Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the Akt/GSK-3β signaling pathway. Biochim Biophys Acta. (2013) 1832:2035–43. doi: 10.1016/j.bbadis.2013.07.012
156. Costerton JW, Geesey GG, Cheng KJ. How bacteria stick. Sci Am. (1978) 238:86–95. doi: 10.1038/scientificamerican0178-86
157. Yang L, Liu Y, Wu H, Song Z, Høiby N, Molin S, et al. Combating biofilms. FEMS Immunol Med Microbiol. (2012) 65:146–57. doi: 10.1111/j.1574-695X.2011.00858.x
158. Sharma D, Misba L, Khan AU. Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrobiol Resist Infect Control. (2019) 8:76. doi: 10.1186/s13756-019-0533-3
159. Kuboniwa M, Tribble GD, Hendrickson EL, Amano A, Lamont RJ, Hackett M. Insights into the virulence of oral biofilms: discoveries from proteomics. Exp Rev Proteomics. (2012) 9:311–23. doi: 10.1586/epr.12.16
160. Sakanaka A, Takeuchi H, Kuboniwa M, Amano A. Dual lifestyle of Porphyromonas gingivalis in biofilm and gingival cells. Microbiol Pathog. (2016) 94:42–7. doi: 10.1016/j.micpath.2015.10.003
161. Vestby LK, Grønseth T, Simm R, Nesse LL. Bacterial biofilm and its role in the pathogenesis of disease. Antibiotics. (2020) 9:59. doi: 10.3390/antibiotics9020059
162. Wang W, Chanda W, Zhong M. The relationship between biofilm and outer membrane vesicles: a novel therapy overview. FEMS Microbiol Lett. (2015) 362:fnv117. doi: 10.1093/femsle/fnv117
163. Pérez-Cruz C, Delgado L, López-Iglesias C, Mercade E. Outer-inner membrane vesicles naturally secreted by gram-negative pathogenic bacteria. PLoS ONE. (2015) 10:e0116896. doi: 10.1371/journal.pone.0116896
164. Williams GD, Holt SC. Characteristics of the outer membrane of selected oral bacteroides species. Can J Microbiol. (1985) 31:238–50. doi: 10.1139/m85-046
165. Nakao R, Takashiba S, Kosono S, Yoshida M, Watanabe H, Ohnishi M, et al. Effect of Porphyromonas gingivalis outer membrane vesicles on gingipain-mediated detachment of cultured oral epithelial cells and immune responses. Microbes Infect. (2014) 16:6–16. doi: 10.1016/j.micinf.2013.10.005
166. Bai D, Nakao R, Ito A, Uematsu H, Senpuku H. Immunoreactive antigens recognized in serum samples from mice intranasally immunized with Porphyromonas gingivalis outer membrane vesicles. Pathog Dis. (2015) 73:ftu006. doi: 10.1093/femspd/ftu006
167. Ho MH, Chen CH, Goodwin JS, Wang BY, Xie H. Functional advantages of Porphyromonas gingivalis vesicles. PLoS ONE. (2015) 10:e0123448. doi: 10.1371/journal.pone.0123448
168. Gui MJ, Dashper SG, Slakeski N, Chen YY, Reynolds EC. Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol Oral Microbiol. (2016) 31:365–78. doi: 10.1111/omi.12134
169. Zhang Z, Liu D, Liu S, Zhang S, Pan Y. The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol. (2021) 10:585917. doi: 10.3389/fcimb.2020.585917
170. Seyama M, Yoshida K, Yoshida K, Fujiwara N, Ono K, Eguchi T, et al. Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165731. doi: 10.1016/j.bbadis.2020.165731
171. Rajaiah R, Moudgil KD. Heat-shock proteins can promote as well as regulate autoimmunity. Autoimmun Rev. (2009) 8:388–93. doi: 10.1016/j.autrev.2008.12.004
172. Han P, Sun D, Yang J. Interaction between periodontitis and liver diseases. Biomed Rep. (2016) 5:267–76. doi: 10.3892/br.2016.718
173. Sasaki N, Katagiri S, Komazaki R, Watanabe K, Maekawa S, Shiba T, et al. Endotoxemia by Porphyromonas gingivalis injection aggravates non-alcoholic fatty liver disease, disrupts glucose/lipid metabolism, and alters gut microbiota in mice. Front Microbiol. (2018) 9:2470. doi: 10.3389/fmicb.2018.02470
174. Arimatsu K, Yamada H, Miyazawa H, Minagawa T, Nakajima M, Ryder MI., et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. (2014) 4:4828. doi: 10.1038/srep04828
175. Lourenςo T, Spencer SJ, Alm EJ, Colombo A. Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. J Oral Microbiol. (2018) 10:1487741. doi: 10.1080/20002297.2018.1487741
176. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. (2014) 513:59–64. doi: 10.1038/nature13568
177. Rodriguez Herrero E, Boon N, Pauwels M, Bernaerts K, Slomka V, Quirynen M, et al. Necrotrophic growth of periodontopathogens is a novel virulence factor in oral biofilms. Sci Rep. (2017) 7:1107. doi: 10.1038/s41598-017-01239-9
178. Komazaki R, Katagiri S, Takahashi H, Maekawa S, Shiba T, Takeuchi Y, et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci Rep. (2017) 7:13950. doi: 10.1038/s41598-017-14260-9
179. Olsen I, Yamazaki K. Can oral bacteria affect the microbiome of the gut?. J Oral Microbiol. (2019) 11:1586422. doi: 10.1080/20002297.2019.1586422
180. Chen HT, Huang HL, Li YQ, Xu HM, Zhou YJ. Therapeutic advances in non-alcoholic fatty liver disease: a microbiota-centered view. World J Gastroenterol. (2020) 26:1901–11. doi: 10.3748/wjg.v26.i16.1901
181. Shekhar S, Petersen FC. The dark side of antibiotics: adverse effects on the infant immune defense against infection. Front Pediatr. (2020) 8:544460. doi: 10.3389/fped.2020.544460
182. Van Pijkeren JP, Neoh KM, Sirias D, Findley AS, Britton RA. Exploring optimization parameters to increase ssDNA recombineering in Lactococcus lactis and Lactobacillus reuteri. Bioengineered. (2012) 3:209–17. doi: 10.4161/bioe.21049
183. Yang KM, Kim JS, Kim HS, et al. Lactobacillus reuteri AN417 cell-free culture supernatant as a novel antibacterial agent targeting oral pathogenic bacteria. Sci Rep. (2021) 11:1631. doi: 10.1038/s41598-020-80921-x
184. Murakami Y, Kawata A, Ito S, Katayama T, Fujisawa S. The radical scavenging activity and cytotoxicity of resveratrol, orcinol and 4-allylphenol and their inhibitory effects on cox-2 gene expression and Nf-κb activation in RAW264.7 cells stimulated with Porphyromonas gingivalis-FIMBRIAE. In Vivo. (2015) 29:341–349.
185. Choi GE, Hyun KY. Inhibitory effect of acer tegmentosum maxim extracts on P. gingivalis LPS-induced periodontitis. Arch Oral Biol. (2020) 109:104529. doi: 10.1016/j.archoralbio.2019.104529
186. Kariu T, Nakao R, Ikeda T, Nakashima K, Potempa J, Imamura T. Inhibition of gingipains and Porphyromonas gingivalis growth and biofilm formation by prenyl flavonoids. J Periodontal Res. (2017) 52:89–96. doi: 10.1111/jre.12372
187. Wang T, Takikawa Y, Satoh T, Yoshioka Y, Kosaka K, Tatemichi Y, et al. Carnosic acid prevents obesity and hepatic steatosis in ob/ob mice. Hepatol Res. (2011) 41:87–92. doi: 10.1111/j.1872-034X.2010.00747.x
188. Wang T, Takikawa Y, Tabuchi T, Satoh T, Kosaka K, Suzuki K. Carnosic acid (CA) prevents lipid accumulation in hepatocytes through the EGFR/MAPK pathway. J Gastroenterol. (2012) 47:805–13. doi: 10.1007/s00535-012-0546-7
189. Wang T, Takikawa Y. Carnosic acid protects normal mouse hepatocytes against H2 O2 -induced cytotoxicity via sirtuin 1-mediated signaling. Hepatol Res. (2016) 46:239–46. doi: 10.1111/hepr.12563
190. Del Campo J, Amiot MJ, Nguyen-The C. Antimicrobial effect of rosemary extracts. J Food Prot. (2000) 63:1359–68. doi: 10.4315/0362-028X-63.10.1359
191. Bernardes WA, Lucarini R, Tozatti MG, Souza MG, Silva ML, Filho AA, et al. Antimicrobial activity of Rosmarinus officinalis against oral pathogens: relevance of carnosic acid and carnosol. Chem Biodivers. (2010) 7:1835–40. doi: 10.1002/cbdv.200900301
192. Tsukatani T, Sakata F, Kuroda R, Akao T. Biofilm eradication activity of herb and spice extracts alone and in combination against oral and food-borne pathogenic bacteria. Curr Microbiol. (2020) 77:2486–95. doi: 10.1007/s00284-020-02017-z
193. Xiang Q, Liu Z, Wang Y, Xiao H, Wu W, Xiao C, et al. Carnosic acid attenuates lipopolysaccharide-induced liver injury in rats via fortifying cellular antioxidant defense system. Food Chem Toxicol. (2013) 53:1–9. doi: 10.1016/j.fct.2012.11.001
194. J Jeong SH, Nam Y, Jung H, Kim J, Rim YA, Park N, et al. Interrupting oral infection of Porphyromonas gingivalis with anti-FimA antibody attenuates bacterial dissemination to the arthritic joint and improves experimental arthritis. Exp Mol Med. (2018) 50:e460. doi: 10.1038/emm.2017.301
195. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:eaau3333. doi: 10.1126/sciadv.aau3333
196. Peng HY, Chen SY, Siao SH, Chang JT, Xue TY, Lee YH, et al. Targeting a cysteine protease from a pathobiont alleviates experimental arthritis. Arthritis Res Ther. (2020) 22:114. doi: 10.1186/s13075-020-02205-z
197. Cai J, Chen J, Guo H, Pan Y, Zhang Y, Zhao W, et al. Recombinant fimbriae protein of Porphyromonas gingivalis induces an inflammatory response via the TLR4/NF-κB signaling pathway in human peripheral blood mononuclear cells. Int J Mol Med. (2019) 43:1430–40. doi: 10.3892/ijmm.2019.4069
198. Pérez-Montes de Oca A, Julián MT, Ramos A, Puig-Domingo M, Alonso N. Microbiota, fiber, and NAFLD: is there any connection?. Nutrients. (2020) 12:3100. doi: 10.3390/nu12103100
199. Mu Q, Tavella VJ, Luo XM. Role of Lactobacillus reuteri in human health and diseases. Front Microbiol. (2018) 9:757. doi: 10.3389/fmicb.2018.00757
200. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. (2013) 110:9066–71. doi: 10.1073/pnas.1219451110
201. Ottman N, Reunanen J, Meijerink M, Pietilä TE, Kainulainen V, Klievink J, et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS ONE. (2017) 12:e0173004. doi: 10.1371/journal.pone.0173004
Keywords: oral microbiota, gut microbiota, microbial dysbiosis, NAFLD, P. gingivalis
Citation: Wang T, Ishikawa T, Sasaki M and Chiba T (2022) Oral and Gut Microbial Dysbiosis and Non-alcoholic Fatty Liver Disease: The Central Role of Porphyromonas gingivalis. Front. Med. 9:822190. doi: 10.3389/fmed.2022.822190
Received: 25 November 2021; Accepted: 19 January 2022;
Published: 02 March 2022.
Edited by:
Toshifumi Ohkusa, Juntendo University, JapanReviewed by:
Tomohisa Takagi, Kyoto Prefectural University of Medicine, JapanRaisa Catunda, University of Alberta, Canada
Copyright © 2022 Wang, Ishikawa, Sasaki and Chiba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshimi Chiba, dG9zY2hpYmFAaXdhdGUtbWVkLmFjLmpw; Ting Wang, dGluZ3dhbmdAaXdhdGUtbWVkLmFjLmpw