 Miquel Blasco
Miquel Blasco Elena Guillén-Olmos1†
Elena Guillén-Olmos1† Maribel Diaz-Ricart
Maribel Diaz-Ricart Marta Palomo
Marta Palomo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 25 April 2022
Sec. Hematology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.811504
This article is part of the Research TopicEndotheliopathies: Current Concepts and Importance in Clinical PracticeView all 12 articles
Thrombotic microangiopathies (TMA) constitute a group of different disorders that have a common underlying mechanism: the endothelial damage. These disorders may exhibit different mechanisms of endothelial injury depending on the pathological trigger. However, over the last decades, the potential role of the complement system (CS) has gained prominence in their pathogenesis. This is partly due to the great efficacy of complement-inhibitors in atypical hemolytic syndrome (aHUS), a TMA form where the primary defect is an alternative complement pathway dysregulation over endothelial cells (genetic and/or adquired). Complement involvement has also been demonstrated in other forms of TMA, such as thrombotic thrombocytopenic purpura (TTP) and in Shiga toxin-producing Escherichia coli hemolytic uremic syndrome (STEC-HUS), as well as in secondary TMAs, in which complement activation occurs in the context of other diseases. However, at present, there is scarce evidence about the efficacy of complement-targeted therapies in these entities. The relationship between complement dysregulation and endothelial damage as the main causes of TMA will be reviewed here. Moreover, the different clinical trials evaluating the use of complement-inhibitors for the treatment of patients suffering from different TMA-associated disorders are summarized, as a clear example of the entry into a new era of personalized medicine in its management.
Thrombotic microangiopathies (TMA) constitute a group of disorders characterized by micoangiopathic hemolysis, platelet consumption and systemic organ damage. The identification of the underlying etiology among TMA-associated disorders is a major challenge due to the variability of clinical presentation and the absence of pathognomonic histological findings. Despite these factors, its assessment is crucial for directed therapy (1).
The vascular endothelium plays a pivotal role in the regulation of the hemostatic balance. In this regard, endothelial cells (EC) provide a non-thrombogenic inner layer of the vascular wall which maintains the blood fluidity, prevent thrombosis through different anticoagulant, and antiplatelet mechanisms, and regulate clot formation, limiting it to those areas without vascular integrity (2). Endothelial injury is the common underlying mechanism among different TMA, leading to the microvasculature thrombosis observed in these conditions (3) (Figure 1).
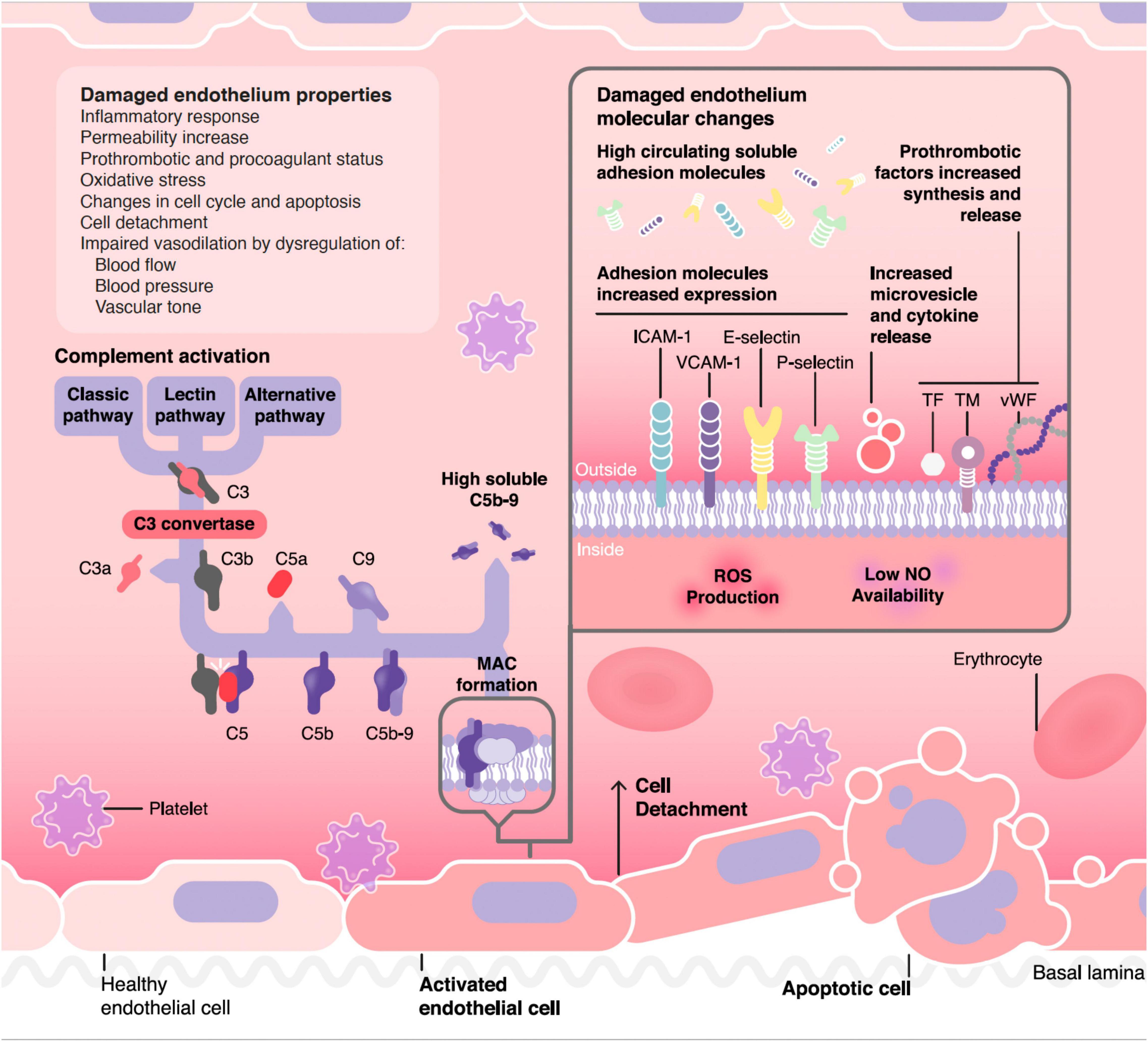
Figure 1. Pathophysiology of complement mediated endothelial damage in TMA. ICAM-1, Intercellular Adhesion Molecule 1; VCAM-1, Vascular cell adhesion protein 1; TF, tissue factor; TM, thrombomodulin; vWF, von Willebrand factor; MAC, membrane attack complex.
The complement system (CS) is a central component of innate immunity and bridges the innate to the adaptive immune response. Activation of complement pathways under physiological conditions facilitates the clearance of microbes, damaged cells and immune complexes from an organism, promotes inflammation, and attacks pathogen’s cell membrane. To prevent undesirable activation and tissue damage, complement activation is strictly controlled by numerous regulators under normal circumstances (4).
During the last decades, the contribution of dysregulated complement activation to endothelial damage has been widely demonstrated in some TMA forms. Among them, the prototype is atypical hemolytic syndrome (aHUS), in which complement dysregulation occurs as the primary event (genetic and/or adquired alternative complement pathway dysregulation). Complement involvement has also been demonstrated in thrombotic thrombocytopenic purpura (TTP) and in Shiga toxin-producing Escherichia coli hemolytic uremic syndrome (STEC-HUS), but, in these cases, it occurs as a secondary event, triggered either by ADAMTS13-deficiency or toxin-mediated injury, respectively (5). Finally, the role of complement has also been suggested to be involved in other TMA-associated disorders, although it has not been completely elucidated (6).
Distinguishing among the different causes of TMA may be challenging, leading to a delay in etiologic diagnosis and, therefore, in the initiation of the most appropriate treatment. The development of diagnostic tools based on functional and genetic studies to assess the involvement of complement in the pathogenesis of the different clinical entities is crucial (1). In this regard, new management approaches of TMA are being evaluated, and complement-targeted therapies are gaining importance since they selectively block this inflammation pathway, thus avoiding the side effects of some traditional therapies (7).
This review addresses the relationship between endothelial damage and complement dysregulation among the main causes of TMA. We also summarize the clinical trials carried out in TMA-associated disorders treated with complement inhibitors (Table 1) and the need to consolidate and develop different biomarkers that may allow an individualized treatment in the near future.
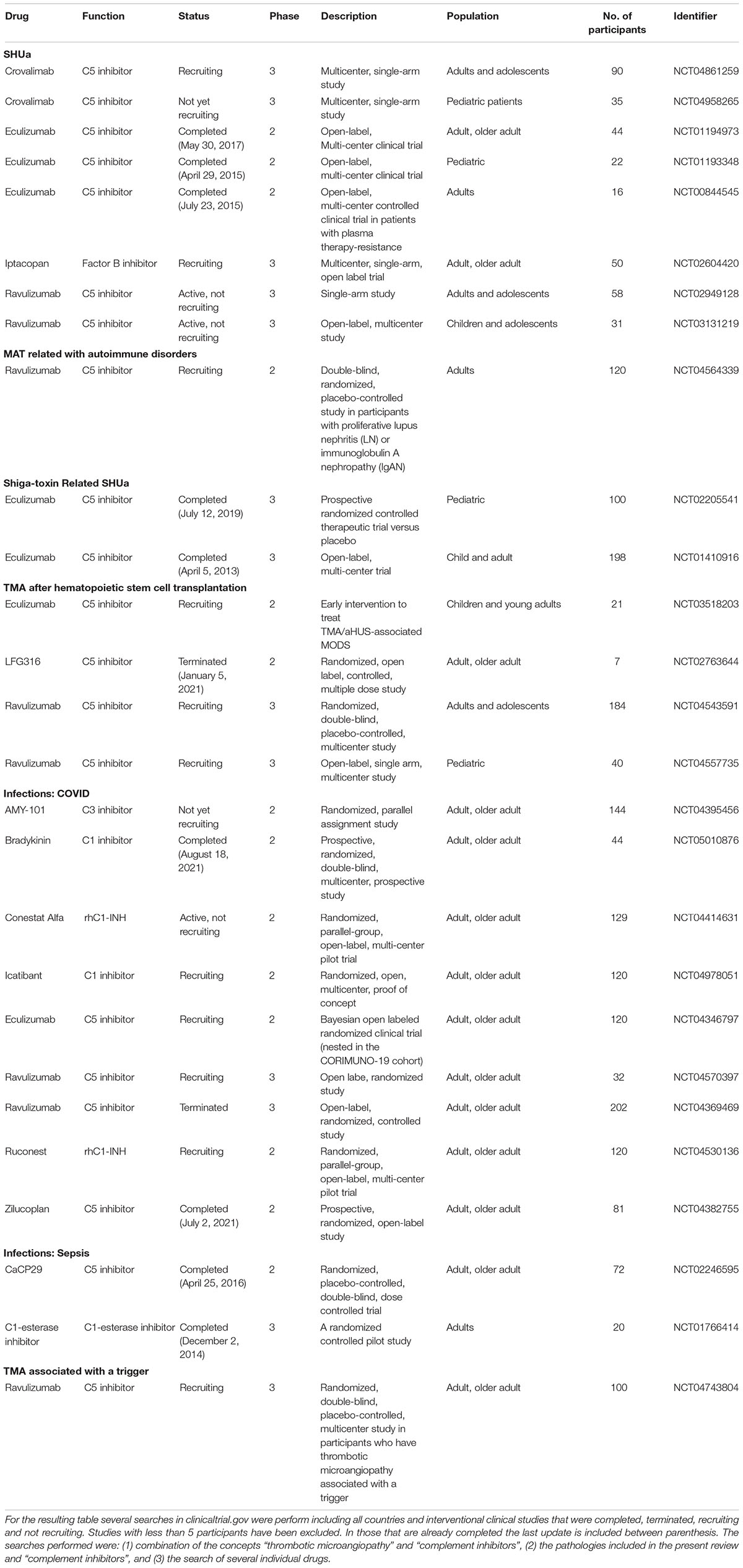
Table 1. Complement inhibitors clinical trials.
Dysregulation of the alternative complement pathway over EC surfaces is the main cause of TMA development in atypical hemolytic uremic syndrome (aHUS) (8, 9). Genetic variants in complement components (gain or loss function mutations) and/or acquired conditions (antibodies against complement factor H) have been identified in about 40–60% of patients (10–12). Coexistence of these predisposing factors with environmental triggers or complement amplifying conditions (i.e., infections, drugs, surgery, pregnancy) may lead to disease onset (13, 14). aHUS is an ultra-rare disease, that affects both pediatric and adult populations, of a systemic nature and with highly morbidity and mortality rates (14–16). These characteristics, together with the need for a clinical diagnosis (exclusion of all potential TMA causes), could delay an early etiological treatment with terminal complement blockers (17–22), decreasing organ function recovery outcomes (especially kidney function).
Endothelial activation and injury in aHUS occur mainly due to immune innate system dysregulation and the loss of protection at cell surfaces. Disease phenotype and severity will depend on pro- and anti-inflammatory cytokines balance and regulation of individual complement components (23, 24). Once the complement cascade is amplified, the terminal phase will play a key role, especially, the cleavage of C5 molecule into C5a and the subsequent generation of membrane attack complex (MAC), also called C5b-9 (16). C5a is a potent anaphylatoxin, promoting the recruitment of platelets and leukocytes to the endothelial surface. In addition, it causes endothelial retraction with the consequent exposure of the underlying basement membrane and overexpression of procoagulant tissue factor (25). MAC forms pores in pathogens or targeted cell membranes leading to osmolysis (cytolytic effector of innate and adaptative immunity). It can also induce cell activation and intracellular signaling (26), promoting a switch from an anti-coagulant and anti-inflammatory endothelial phenotype to a highly active, pro-coagulant phenotype. Although it may not be identified in all cases, aHUS patients present a defect of AP regulation over EC surfaces, allowing an abnormal formation of terminal complement phase (C5a and C5b-9) on the endothelium, which lead to cell apoptosis and TMA development, with a special predilection for the kidney vasculature (27).
ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) deficiency is the key event in the pathophysiology of TTP (28). When functional, ADAMTS-13 cleaves the ultra-Large von Willebrand factor (ULVWF) multimers, which are highly reactive to platelets, before VWF is released to plasma. ADAMTS-13 deficiency, which can be congenital or acquired, leads to the accumulation of ULVWF secreted by the endothelium, causing platelet aggregation, formation of microthrombi and ultimately endothelial damage (29). However, an important factor in the pathogenesis of this disease is the different susceptibility of the vascular beds. The selective injury to dermal, renal, and cerebral microvasculature does not occur in EC from lung and liver (30). Notably, this different susceptibility is not appreciated in cells with a macrovascular origin. This phenomenon has not been described only in vitro experiments (31) but also in animal models (32), and offers the potential explanation for tissue distribution damage in TTP.
Although the role of complement dysregulation in TTP is not as well-defined as it is in aHUS etiology, it has been demonstrated through several approaches. Increased levels of complement anaphylatoxins and soluble C5b-9 (sC5b-9), have been detected in patients with acute TTP when compared with remission and also with healthy controls (33, 34). Furthermore, significant decrease of C3a and sC5b9 has been observed during plasma exchange in TTP patients (34). Higher complement activity has been also reported in patients dying during an acute TTP episode compared to patients responding to treatment (35). From the three different pathways through which the CS can be activated, the classical is the one triggered by immunocomplexes. The majority of TTP cases are idiopathic and mediated by immunoglobulin G antibodies to ADAMTS-13 (29). Therefore, antigen-antibody complexes formed by ADAMTS-13 and anti-ADAMTS-13 IgG directly activate the classical complement pathway, leading to downstream activation of C3 and formation of C3a (34). Moreover, the increased thrombi formation in acute TTP could also accelerate complement activation (36) through its activity as C5 convertase by forming positive feedback loops (37). In keeping with this, it has been documented that TTP could trigger aHUS in susceptible individuals presenting mutations in CFH or other complement-related proteins (38). Finally, there is evidence showing that anti-C5 therapy is effective in refractory TTP unresponsive to plasma (39).
In addition, the role of the lectin complement pathway (LP) has recently been postuated in TTP through the finding of mannose-binding lectin associated serine protease (MASP2) elevated levels in the sera from acute phase TTP patients. Moreover, in vitro experiments using plasma from PTT patients suggest a role of this pathway in microvascular endothelial cell injury through an specific caspase 8 activation that can be blocked by the anti-MASP2 human monoclonal antibody narsoplimab (40).
Shiga toxin (Stx)-producing Escherichia coli (STEC) O157:H7 is the main cause of hemorrhagic colitis, occurring mainly in childhood. In 5–15% of cases it progresses to hemolytic uremic syndrome (HUS), and constitutes the leading cause of HUS worldwide (41).
Endothelial damage plays a central role in the underlying pathogenesis of STEC-HUS, and Stx (principally Stx subtype 2) is thought to be the key in this microangiopathic process through several mechanisms. In vitro studies have demonstrated that Stx upregulates the generation of adhesive molecules (E-selectin, ICAM-1, and VCAM-1) and chemokines (MCP-1, IL-8, fractalkine), promoting the adhesion of leukocytes to cultured human EC under flow conditions (42, 43). Stx also induces rapid release of ULVWF multimers from EC and inhibits the multimer cleavage by ADAMTS13, thus favoring platelet adhesion and clot formation in the microvasculature (41, 44). Moreover, Stx promote changes in gene expression, stimulating mRNA and protein production of chemokines and cytokines which may exacerbate endothelial damage (45). All these events generate changes in the endothelial phenotype, resulting in a prothrombogenic intravascular environment.
Another key element in STEC-HUS pathogenesis is dysregulation of the CS, and, likewise, it is mainly driven by Stx. Stx activates complement in the fluid phase, with reduced plasma levels of C3 and augmented levels of C3a, Bb and sC5b-9 during the active phase of the disease (46, 47). This evidence, along with the demonstration in murine models of disease that factor B knockout mices were protected from Stx induced renal damage (48), indicates that complement activation is produced via alternative pathway (AP) in STEC-HUS patients. Moreover, deposits of C3 and C9 have been detected on platelet-leukocyte heterotypic aggregates and microvesicles obtained from these patients (49, 50), thus indicating that complement AP might contribute to endothelial damage and thrombosis in STEC-HUS.
Two double-blinded placebo controlled trials with eculizumab (C5 inhibitor) has been performed to evaluate its efficacy for STEC-HUS treatment: ECULISHU (NCT02205541) in France looking at renal outcome in pediatric population, completed in 2019, and ECUSTEC (NCT01410916) in the United Kingdom looking at overall disease severity, completed in 2015 (Table 1). Until results become available from these prospective, randomized control trials, practitioners will continue to rely on small case series, single-center studies, and case reports to guide the eculizumab off-label indication for STEC-HUS treatment.
Transplantation associated thrombotic microangiopathy (TA-TMA) is a severe complication associated with allogeneic hematopoietic cell transplantation (HCT) that occurs when endothelial dysfunction triggers the CS inducing the formation of platelet enriched thrombi in the microvasculature and a consequent hemolytic anemia (51). This complication can constitute a mild self-limiting disease or be associated with multiorgan disorder leading to death (52). The incidence and mortality of TA-TMA varies widely among studies and hospitals due to heterogeneous diagnostic criteria and under-recognition (53).
The endothelial damage in HCT is multifactorial and cumulative (54, 55) due to the effect of the conditioning regimen, calcineurin inhibitors, infections, graft vs. host disease and processes inherent to HCT such as the engraftment syndrome, among others (56–59). This endothelial damage leads to a release of proinflammatory cytokines and procoagulant factors together with nitric oxide depletion and an increase in the expression of adhesion molecules at cell surface. All these phenomena promote further endothelial injury and leads to platelet aggregation and the initiation and propagation of the complement cascade (60, 61). There is evidence showing that neutrophil extracellular traps (NETs) could constitute the mechanistical link between endothelial damage and complement activation (62). Several approaches have identified increased levels of C3b and sC5b-9 (63, 64), being the last included in TMA diagnostic algorithm proposed by Jodele et al. (63). In this algorithm the authors assessed that TA-TMA should be suspected in HCT recipients with an acute elevation of LDH, proteinuria >30 mg/dL, and hypertension more severe than expected with calcineurin or steroid therapy, and that clinical interventions should be considered for patients with both proteinuria >30 mg/dL and elevated sC5b-9. All these phenomena have been included in the “Three-Hit Hypothesis” postulated by Dvorak et al. (65). This hypothesis includes the different risk factors for the development of TA-TMA in three potential consecutive events that culminate in a cycle of activation of endothelial cells: Hit 1, an underlying predisposition to complement activation (genetical susceptibility due to complement gene variants) or pre-existing endothelial injury; Hit 2, the direct effect on endothelium of toxic conditioning regimen; and Hit 3, the additional insults triggered by medications, alloreactivity, infections, and/or antibodies that occur post-transplant. Furthermore, the anti-C5 monoclonal antibody eculizumab has proven to be an effective therapeutic strategy for TA-TMA, both in pediatric (66) and adult (67) patients, supporting the involvement of CS in the pathogenesis of TA-TMA.
TMA associated with kidney transplantation may occur as a recurrence of aHUS or without previous history of thrombotic microangiopathy (de novo TMA). Kidney transplantation recipients are exposed to many triggers that can damage the endothelium and produce TMA, which can have a significant impact on allograft survival (68). The incidence is concentrated in the first months after transplantation and the main causes are ischemia-reperfusion damage, acute humoral rejection, medications, and opportunistic infections (69). Unlike TMA that occurs in native kidneys, graft biopsy is very useful since it can offer an etiological diagnosis in most cases.
aHUS recurrence after transplantation depends on genetic predisposition and endothelial damage is mediated by a dysregulation of AC pathway (70, 71). Endothelium could also be injured by important number of factors in transplanted patients, triggering de novo TMA. Calcineurin inhibitors (CNI) can induce TMA through arteriolar vasoconstriction (increased synthesis of endothelin and thromboxane A2, and reduced expression of prostacyclin and prostaglandin E2) (72–74), platelet activation, anti-fibrinolytic and pro-coagulant effects (75) and, by activation of AC pathway (microparticles released by injured EC) (76). m-TOR inhibitors block cell cycle progression and proliferation, induce endothelial progenitor cells death, increase procoagulant status, reduce fibrinolytic state and decrease renal expression of endothelial growth factor (77–80). All these processes can contribute to the endothelial damage that triggers de novo TMA onset, described with the use of CNI, m-TOR, or a combination of both (possibly related to elevated serum levels) (81, 82). Acute humoral rejection is an important cause of endothelial damage in kidney grafts, frequently associated with TMA development. Donor specific antibodies bind to major histocompatibility complexes on EC, fixing the CS and activating them through the over-expression of pro-inflammatory genes. Terminal complement phase (C5b-9) has been also related to allograft vasculopathy development in humoral response, since it can activate EC, favoring T cell recruitment and secretion of cytokines and interferon γ. Finally, ischemia-reperfusion injury is also associated with endothelial damage and TMA onset. The changes induced on EC produce complement activation that in turn accentuates and perpetuates endothelial damage at the expense of MAC (83). Animal models have shown a clear benefit of blocking the terminal phase of complement by different therapeutic strategies (84–86).
Antiphospholipid syndrome (APS) and systemic erythematous lupus (SLE) are the autoimmune disorders most frequently associated with TMA, which can result in a life-threating complication in these contexts.
APS is a prothrombotic disorder characterized by thrombosis and/or pregnancy morbidity, which occurs in the presence of antiphospholipid antibodies (aPL). Among them, anti-β2-glycoprotein I (B2GPI) antibodies are one of the main drivers of the endothelial damage, since they upregulate the endothelial expression of prothrombotic factors and adhesion molecules. B2GPI also interacts with von Willebrand factor (vWF), leading to platelet activation (87). Moreover, murine models have demonstrated that increased complement activation plays a role in APS pathogenesis, and the interaction of C5a with its receptor C5aR leads to inflammation, placental insufficiency, and thrombosis (88, 89). APS can occur in isolation or in association with other autoimmune diseases, such as SLE.
SLE is a multisystem autoimmune disorder associated with the presence of autoantibodies against double-stranded DNA (ds-DNA), among others, which can produce endothelial damage, either directly binding to EC or forming circulating immunocomplexes that deposit on vessels. In this regard, it has been demonstrated that several indicators of endothelial dysfunction (Pentraxin 3, E-selectin, VCAM-1) are higher in patients with SLE compared to healthy controls (90). CS is also involved in SLE pathogenesis, since these immunocomplexes activate the classical complement pathway and cause tissue damage, as can be observed in kidney biopsies of patients with lupus nephritis, in which C1q, C3 and C4d deposition is common (90, 91). Moreover, there is increasing evidence that CS is linked with thrombotic events occurring is SLE. An example is the relationship that has been observed between the deposition of complement activation products (C1q, C3d, C4d) on platelets surfaces with an increased risk for venous thrombosis in SLE patients (92).
Besides the above-mentioned STEC-HUS, TMA has been associated with a large number of infectious diseases, specially viral (6). Mechanisms of TMA-associated with infectious diseases are complex, and differ depending on the pathogens causing them.
Cytomegalovirus (CMV) can directly damage EC and cause platelet adhesion by inducing the expression of adhesion molecules and release of vWF (93). Moreover, CMV activates the complement classical pathway by binding of C1q to CMV infected cells (94).
TMA is also a known complication of HIV infection, with different forms of presentation. Cases of both classic TTP and aHUS have been described in HIV-infected patients (95, 96), although the exact mechanisms involved remain unclear. Most cases present without a severe reduction in ADAMTS-13 activity levels, which support the hypothesis that the underlying mechanism may be different from classic TTP. It has been suggested that endothelial cells can be infected by HIV (96).
Moreover, TMA induced by influenza A virus (H1N1) infection was reported during the 2009 pandemic. The neuraminidase produced by this virus, as well as by the bacteria Streptococcus pneumoniae, causes erythrocyte fusion and hemolysis, activation of platelets and generation of thrombin, leading to TMA in both cases. Furthermore, low levels of ADAMTS-13 and elevated sC5b-9 have been detected in patients with H1N1 infection (97).
More recently, the severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) has emerged as another clear example of viral infection in which endothelial damage occurs in parallel to an overactivation of complement cascade. Patients with moderate and severe COVID-19 disease exhibit elevated C5a and sC5b-9 plasma levels compared with healthy controls, the later correlating with vWF plasma levels and disease severity (98). Moreover, COVID-19 patients were also found to have C5b-9 and C4d skin deposits, as well as mannose-binding lectin (MBL) deposits in lungs, suggesting the overactivation of alternative and lectin-complement pathways (99–101). These observations may reflect the close relationship between endothelial stress and complement dysregulation in this condition. In this regard, isolated experiences using complement inhibitors under a compassionate-use program, such as narsoplimab (102), or AMY-101, a compstatin-based C3 inhibitor (103) has been publicated. The former (narsoplimab) demonstrated rapid and sustained reduction of circulating endothelial cells, as well as with decreased circulating inflammatory cytokines, while the second (AMY-101) was associated with a favorable clinical evolution in a patient with COVID-19 severe pneumonia. All together, these results suggest that CS plays a central role in the pathophysiology of COVID-19-related lung injury.
Moreover, several clinical trials targeting the complement system are currently ongoing in patients with COVID-19 (Table 1). Among them, two have been completed: AntagoBrad-Cov (NCT05010876), a prospective, randomized, double-blind, multicenter study of three parallel groups of patients comparing the efficacy of human C1 inhibitor, administered alone or in combination with icatibant (a specific bradykinin B2 receptor antagonist) on the pulmonary manifestations of COVID-19 infections; and ZILU-COV (NCT04382755), a prospective, randomized, open-label, study to investigate the efficacy of a complement C5 inhibitor (Zilucoplan®) in improving oxygenation and short-and long-term outcome of COVID-19 patients with acute hypoxic respiratory failure. The results of the latter haven’t been published yet.
Interestingly, samples from septic shock (SS) patients induced higher C5b-9 deposition on EC than those from COVID-19 patients, whereas there were no differences regarding sC5b-9 levels between both groups. Thus, COVID-19 endotheliopathy may differs from SS, in which endothelial damage and complement may also play an important pathogenic role (98). In this regard, two clinical trials have been performed (Table 1). The first one, completed in 2014, is VECTORII Study (NCT01766414), a randomized, controlled, pilot study to evaluate the role of a C1-esterasa inhibitor in the modulation of innate immune response in a human endotoxemia model. The inhibition of C1-esterase exerted anti-inflammatory effects in the absence of classic complement activation (104). The second one, completed in 2016, is SCIENS Study (NCT02246595), a phase II clinical trial conducted to study safety, tolerability, pharmacokinetics, and pharmacodynamics of Vilobelimab (IFX-1; CaCP 29), a recombinant monoclonal antibody against C5a, in patients with severe sepsis or septic shock. Vilobelimab demonstrated to neutralize selectively C5a in a dose-dependent manner without blocking formation of the membrane attack complex, and without resulting in detected safety issues (105).
Clinical TMA management (diagnostic-therapeutic process) is a great challenge due to its systemic nature (variable signs and symptoms) and high associated morbidity and mortality. Enhancing the etiological knowledge of the different clinical entities involved in TMA will allow the development of targeted therapies that may improve their poor prognosis. The cornerstone for thrombi development in the microvasculature is the endothelial cell damage, common to all TMA-associated disorders. However, these pathological processes exhibit differential patterns and mechanisms of endothelial injury, although occasionally there may be also a pathophysiological overlap.
CS, a key element of the innate immune system, requires precise regulation. Defects in the elements involved result in dysregulation and over-activation. In the case of TMA, an acquired or congenital dysregulation of the alternative complement pathway is primarily responsible for the endothelial damage that occurs in aHUS patients. Therefore, the development of monoclonal antibodies that block the terminal complement phase (C5) has led to a great advance in aHUS management, especially with early therapy introduction. The excellent response to these treatments has led to the research of the potential pathological role of the CS in other TMA forms with higher prevalence and without known etiological treatment.
Figure 1 summarizes the pathophysiological process through which complement could damage the endothelium in different TMA forms, as well as the resulting functional and molecular endothelial changes. One of the difficulties faced in making a diagnosis of complement mediated TMA is the lack of reliable markers of CS hyperactivation. For instance, in the case of the paradigmatic complement mediated TMA, aHUS, relatively few patients have consumption of complement factors, complement mutations are heterozygous-and the corresponding protein concentrations in blood are not consistently abnormal- and genetic screenings are slow and even uninformative in up to 50% of cases (106). In general, the quantification of levels of individual complement components in serum is not a straightforward approach in TMA diagnosis, as soluble levels are not reliable biomarkers of complement activation in any form of TMA (107), although the quantification of sC5b-9 has shown some usefulness in the management of patients with TA-TMA (64) and is available clinically (commercial kits). For this reason, new diagnostic tools are needed for TMAs management. In this regard, functional studies may help to demonstrate the role of complement activation not only at diagnosis, but also to monitor treatment response. Among them, the analysis of C5b-9 deposition on endothelial cells culture could be an attractive option, since it has shown a great correlation in different clinical stages of aHUS patients, as well as in certain secondary TMA forms by different research groups (108–112). However, there is still an open discussion about their utility, especially because it is not a quick procedure, requires specialized and trained personnel, and is not commercially available (1).
In section 2, we have reviewed the evidence generated regarding the participation of the CS in various TMA forms. Since the approval of eculizumab for the treatment of aHUS in 2011, the development of therapies that directly target CS at different levels has enabled a large number of clinical trials focused on complement blockers in patients with different TMA-associated disorders (Table 1). Most of them have evaluated the role of C5-inhibitors, mainly eculizumab and ravulizumab (a long-acting C5-inhibitor), in the treatment of various conditions. Among them, those performed with eculizumab in patients with aHUS and STEC-HUS have been already completed. In recent decades, a large number of potential complement-related targets have progressively emerged, such as C3, factor B and C1q. Probably, this fact may be related with the large amount of scientific evidence generated regarding the role of complement in the pathogenesis of the different conditions reviewed in this work. As shown in Table 1, several clinical trials evaluating these factors are ongoing, and their results may probably help to extend the therapeutic options for TMA-associated conditions in the near future.
In conclusion, endothelial damage in TMA can have different origins depending on the responsible pathological process. However, the potential role of complement system in many of them represents a real and current treatment opportunity. Identifying the exact factors involved in each disease will allow an individualized patient management. This is a challenge that will require a great effort involving clinicians, immunologists, geneticists, and basic researchers.
MB reviewed the bibliography and wrote aHUS, kidney transplantation associated TMA and discussion sections. MP reviewed and wrote the TTP and TA-TMA sections, and designed Table 1 and Figure 1 with the support of MB and EG-O. EG-O reviewed and wrote introduction, STEC-HUS, TMA related to autoimmune disorders, and TMA associated with infections sections. MD-R contributed to the manuscript with her critical review and comments. All authors contributed to the article and approved the submitted version.
This work was supported by Fundació Miarnau (Spain), Fundació La Marató de TV3 (202026-10), Instituto de Salud Carlos III from Spanish Government (PI19/00888), Shook Studio for figure design, and Ayuda a la Investigación de la Fundación Senefro 2020.
MB reports advisory boards and symposium speaker honoraria from Alexion Pharmaceuticals. MD-R has received research funding and speaker fees from Jazz Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Blasco M, Guillén E, Quintana LF, Garcia-Herrera A, Piñeiro G, Poch E, et al. Thrombotic microangiopathies assessment: mind the complement. Clin Kidney J. (2021) 14:1055–66. doi: 10.1093/ckj/sfaa195
2. Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, et al. The vascular endothelium and human diseases. Int J Biol Sci. (2013) 9:1057–69. doi: 10.7150/ijbs.7502
3. Mathew RO, Nayer A, Asif A. The endothelium as the common denominator in malignant hypertension and thrombotic microangiopathy. J Am Soc Hypertens. (2016) 10:352–9. doi: 10.1016/j.jash.2015.12.007
4. Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol. (2019) 19:503–16. doi: 10.1038/s41577-019-0168-x
5. Gavriilaki E, Anagnostopoulos A, Mastellos DC. Complement in thrombotic microangiopathies: unraveling Ariadne’s thread into the labyrinth of complement therapeutics. Front Immunol. (2019) 10:337. doi: 10.3389/fimmu.2019.00337
6. Palma LMP, Sridharan M, Sethi S. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. (2021) 6:11–23. doi: 10.1016/j.ekir.2020.10.009
7. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov. (2019) 18:707–29. doi: 10.1038/s41573-019-0031-6
8. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. (2016) 31:15–39. doi: 10.1007/s00467-015-3076-8
9. Goodship THJ, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “kidney disease: improving global outcomes” (KDIGO) controversies conference. Kidney Int. (2017) 91:539–51. doi: 10.1016/j.kint.2016.10.005
10. Rodríguez De Córdoba S, Hidalgo MS, Pinto S, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. (2014) 40:422–30. doi: 10.1055/s-0034-1375296
11. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, De Cordoba SR, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. (2013) 24:475–86. doi: 10.1681/ASN.2012090884
12. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. (2010) 5:1844–59. doi: 10.2215/CJN.02210310
13. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship THJ, et al. Atypical aHUS: state of the art. Mol Immunol. (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
14. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 361:1676–87. doi: 10.1056/NEJMra0902814
15. Constantinescu AR, Bitzan M, Weiss LS, Christen E, Kaplan BS, Cnaan A, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. (2004) 43:976–82. doi: 10.1053/j.ajkd.2004.02.010
16. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. (2011) 6:60. doi: 10.1186/1750-1172-6-60
17. Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis. (2016) 68:84–93. doi: 10.1053/j.ajkd.2015.12.034
18. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor Eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med. (2013) 368:2169–81. doi: 10.1056/NEJMoa1208981
19. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. (2015) 87:1061–73. doi: 10.1038/ki.2014.423
20. Menne J, Delmas Y, Fakhouri F, Licht C, Lommelé Å, Minetti EE, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. (2019) 20:125. doi: 10.1186/s12882-019-1314-1
21. Barbour T, Scully M, Ariceta G, Cataland S, Garlo K, Heyne N, et al. Long-term efficacy and safety of the long-acting complement C5 inhibitor Ravulizumab for the treatment of atypical hemolytic uremic syndrome in adults. Kidney Int Rep. (2021) 6:1603–13. doi: 10.1016/j.ekir.2021.03.884
22. Tanaka K, Adams B, Aris AM, Fujita N, Ogawa M, Ortiz S, et al. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr Nephrol. (2021) 36:889–98. doi: 10.1007/s00467-020-04774-2
23. Kerr H, Richards A. Complement-mediated injury and protection of endothelium: lessons from atypical haemolytic uraemic syndrome. Immunobiology. (2012) 217:195–203. doi: 10.1016/j.imbio.2011.07.028
24. Esparza-Gordillo J, Jorge EG, Garrido CA, Carreras L, López-Trascasa M, Sánchez-Corral P, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. (2006) 43:1769–75. doi: 10.1016/j.molimm.2005.11.008
25. Manthey HD, Woodruff TM, Taylor SM, Monk PN. Complement component 5a (C5a). Int J Biochem Cell Biol. (2009) 41:2114–7. doi: 10.1016/j.biocel.2009.04.005
26. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. (2016) 7:10587. doi: 10.1038/ncomms10587
27. Taylor CM, Chua C, Howie AJ, Risdon RA. Clinico-pathological findings in diarrhoea-negative haemolytic uraemic syndrome. Pediatr Nephrol. (2004) 19:419–25. doi: 10.1007/s00467-003-1385-9
28. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. (2017) 1:590–600. doi: 10.1182/bloodadvances.2017005124
29. Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, et al. von Willebrand factor–cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic–uremic syndrome. N Engl J Med. (1998) 339:1578–84. doi: 10.1056/NEJM199811263392202
30. Mitra D, Jaffe EA, Weksler B, Hajjar KA, Soderland C, Laurence J. Thrombotic thrombocytopenic purpura and sporadic hemolytic-uremic syndrome plasmas induce apoptosis in restricted lineages of human microvascular endothelial cells. Blood. (1997) 89:1224–34. doi: 10.1182/blood.v89.4.1224
31. Stefanescu R, Bassett D, Modarresi R, Santiago F, Fakruddin M, Laurence J. Synergistic interactions between interferon-γ and TRAIL modulate c-FLIP in endothelial cells, mediating their lineage-specific sensitivity to thrombotic thrombocytopenic purpura plasma–associated apoptosis. Blood. (2008) 112:340–9. doi: 10.1182/blood-2007-10-119552
32. Le Besnerais M, Favre J, Denis CV, Lenormand B, Mulder P, Nicol L, et al. Assessment of endothelial damage and cardiac injury in a mouse model mimicking thrombotic thrombocytopenic purpura. Blood. (2013) 122:447. doi: 10.1111/jth.13439
33. Westwood J-P, Langley K, Heelas E, Machin SJ, Scully M. Complement and cytokine response in acute thrombotic thrombocytopenic purpura. Br J Haematol. (2014) 164:858–66. doi: 10.1111/bjh.12707
34. Réti M, Farkas P, Csuka D, Rázsó K, Schlammadinger Á, Udvardy ML, et al. Complement activation in thrombotic thrombocytopenic purpura. J Thromb Haemost. (2012) 10:791–8. doi: 10.1111/j.1538-7836.2012.04674.x
35. Wu TC, Yang S, Haven S, Holers VM, Lundberg AS, Wu H, et al. Complement activation and mortality during an acute episode of thrombotic thrombocytopenic purpura. J Thromb Haemost. (2013) 11:1925–7. doi: 10.1111/jth.12369
36. Markiewski MM, Nilsson B, Nilsson Ekdahl K, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
37. Kurosawa S, Stearns-Kurosawa DJ. Complement, thrombotic microangiopathy and disseminated intravascular coagulation. J Intensive Care. (2014) 2:61. doi: 10.1186/s40560-014-0061-4
38. Noris M, Bucchioni S, Galbusera M, Donadelli R, Bresin E, Castelletti F, et al. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol. (2005) 16:1177–83. doi: 10.1681/ASN.2005010086
39. Chapin J, Weksler B, Magro C, Laurence J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol. (2012) 157:772–4. doi: 10.1111/j.1365-2141.2012.09084.x
40. Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 levels are elevated in thrombotic microangiopathies: association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin Exp Immunol. (2020) 203:96–104. doi: 10.1111/cei.13497
41. Petruzziello-Pellegrini TN, Moslemi-Naeini M, Marsden PA. New insights into Shiga toxin-mediated endothelial dysfunction in hemolytic uremic syndrome. Virulence. (2013) 4:556–63. doi: 10.4161/viru.26143
42. Morigi M, Galbusera M, Binda E, Imberti B, Gastoldi S, Remuzzi A, et al. Verotoxin-1–induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood. (2001) 98:1828–35. doi: 10.1182/blood.v98.6.1828
43. Zoja C, Buelli S, Morigi M. Shiga toxin triggers endothelial and podocyte injury: the role of complement activation. Pediatr Nephrol. (2019) 34:379–88. doi: 10.1007/s00467-017-3850-x
44. Zoja C, Angioletti S, Donadelli R, Zanchi C, Tomasoni S, Binda E, et al. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-κB dependent up-regulation of IL-8 and MCP-11. Kidney Int. (2002) 62:846–56. doi: 10.1046/j.1523-1755.2002.00503.x
45. Nolasco LH, Turner NA, Bernardo A, Tao Z, Cleary TG, Dong J, et al. Hemolytic uremic syndrome–associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. (2005) 106:4199–209. doi: 10.1182/blood-2005-05-2111
46. Matussek A, Lauber J, Bergau A, Hansen W, Rohde M, Dittmar KEJ, et al. Molecular and functional analysis of Shiga toxin–induced response patterns in human vascular endothelial cells. Blood. (2003) 102:1323–32. doi: 10.1182/blood-2002-10-3301
47. Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. (2009) 4:1920–4. doi: 10.2215/CJN.02730409
48. Keir LS, Langman CB. Complement and the kidney in the setting of Shiga-toxin hemolytic uremic syndrome, organ transplantation, and C3 glomerulonephritis. Transfus Apher Sci. (2016) 54:203–11. doi: 10.1016/j.transci.2016.04.010
49. Ståhl A, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli–induced hemolytic uremic syndrome. Blood. (2011) 117:5503–13. doi: 10.1182/blood-2010-09-309161
50. Conway EM. HUS and the case for complement. Blood. (2015) 126:2085–90. doi: 10.1182/blood-2015-03-569277
51. Young JA, Pallas CR, Knovich MA. Transplant-associated thrombotic microangiopathy: theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transplant. (2021) 56:1805–17. doi: 10.1038/s41409-021-01283-0
52. Dandoy CE, Rotz S, Alonso PB, Klunk A, Desmond C, Huber J, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv. (2021) 5:1–11. doi: 10.1182/bloodadvances.2020003455
53. Epperla N, Li A, Logan B, Fretham C, Chhabra S, Aljurf M, et al. Incidence, risk factors for and outcomes of transplant-associated thrombotic microangiopathy. Br J Haematol. (2020) 189:1171–81. doi: 10.1111/bjh.16457
54. Palomo M, Diaz-Ricart M, Carreras E. endothelial dysfunction in hematopoietic cell transplantation. Clin Hematol Int. (2019) 1:45. doi: 10.2991/chi.d.190317.001
55. Carreras E. Vascular endothelial syndromes after HCT: 2020 update. Bone Marrow Transplant. (2020) 55:1885–7. doi: 10.1038/s41409-020-0852-2
56. Carmona A, Díaz-Ricart M, Palomo M, Molina P, Pino M, Rovira M, et al. Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus–sirolimus on endothelial cells: protective effect of defibrotide. Biol Blood Marrow Transplant. (2013) 19:1439–45. doi: 10.1016/j.bbmt.2013.07.001
57. Mir E, Palomo M, Rovira M, Pereira A, Escolar G, Penack O, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. (2017) 52:1317–25. doi: 10.1038/bmt.2017.121
58. Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Escolar G, et al. The release of soluble factors contributing to endothelial activation and damage after hematopoietic stem cell transplantation is not limited to the allogeneic setting and involves several pathogenic mechanisms. Biol Blood Marrow Transplant. (2009) 15:537–46. doi: 10.1016/j.bbmt.2009.01.013
59. Palomo M, Diaz-Ricart M, Carbo C, Rovira M, Fernandez-Aviles F, Martine C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. (2010) 16:985–93. doi: 10.1016/j.bbmt.2010.02.008
60. Goldberg RJ, Nakagawa T, Johnson RJ, Thurman JM. The role of endothelial cell injury in thrombotic microangiopathy. Am J Kidney Dis. (2010) 56:1168–74. doi: 10.1053/j.ajkd.2010.06.006
61. Cohen H, Bull HA, Seddon A, Enayat MS, Hill FGH, Woolf N, et al. Vascular endothelial cell function and ultrastructure in thrombotic microangiopathy following allogeneic bone marrow transplantation. Eur J Haematol. (2009) 43:207–14. doi: 10.1111/j.1600-0609.1989.tb00284.x
62. Gloude NJ, Khandelwal P, Luebbering N, Lounder DT, Jodele S, Alder MN, et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood. (2017) 130:1259–66. doi: 10.1182/blood-2017-05-782870
63. Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. (2014) 124:645–53. doi: 10.1182/blood-2014-03-564997
64. Qi J, Wang J, Chen J, Su J, Tang Y, Wu X, et al. Plasma levels of complement activation fragments C3b and sC5b-9 significantly increased in patients with thrombotic microangiopathy after allogeneic stem cell transplantation. Ann Hematol. (2017) 96:1849–55. doi: 10.1007/s00277-017-3092-9
65. Dvorak CC, Higham C, Shimano KA. Transplant–associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Front Pediatr. (2019) 7:133. doi: 10.3389/fped.2019.00133
66. Jodele S, Dandoy CE, Lane A, Laskin BL, Teusink-Cross A, Myers KC, et al. Complement blockade for TA-TMA: lessons learned from large pediatric cohort treated with eculizumab. Blood. (2020) 135:1049–57. doi: 10.1182/blood.2019004218
67. Chapin J, Shore T, Forsberg P, Desman G, Van Besien K, Laurence J. Hematopoietic transplant-associated thrombotic microangiopathy: case report and review of diagnosis and treatments. Clin Adv Hematol Oncol. (2014) 12:565–73.
68. Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. (2010) 10:1517–23. doi: 10.1111/j.1600-6143.2010.03156.x
69. Radha S, Tameem A, Sridhar G, Rajaram K, Aiyangar A, Prasad R, et al. Thrombotic microangiopathy in renal allografts. Indian J Nephrol. (2014) 24:24. doi: 10.4103/0971-4065.125052
70. Artz MA, Steenbergen EJ, Hoitsma AJ, Monnens LAH, Wetzels JFM. Renal transplantation in patients with hemolytic uremic syndrome: high rate of recurrence and increased incidence of acute rejections1. Transplantation. (2003) 76:821–6. doi: 10.1097/01.tp.0000085083.74065.1b
71. Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. (2011) 7:23–35. doi: 10.1038/nrneph.2010.155
72. Remuzzi G, Bertani T. Renal vascular and thrombotic effects of cyclosporine. Am J Kidney Dis. (1989) 13:261–72. doi: 10.1016/s0272-6386(89)80032-0
73. Ramírez C, Olmo A, O’Valle F, Masseroli M, Aguilar M, Gómez-Morales M, et al. Role of intrarenal endothelin 1, endothelin 3, and angiotensin II expression in chronic cyclosporin A nephrotoxicity in rats. Nephron Exp Nephrol. (2000) 8:161–72. doi: 10.1159/000020664
74. Sahin G, Akay OM, Bal C, Yalcin AU, Gulbas Z. The effect of calcineurin inhibitors on endothelial and platelet function in renal transplant patients. Clin Nephrol. (2011) 76:218–25.
75. Oyen O, Strom EH, Midtvedt K, Bentdal O, Hartmann A, Bergan S, et al. Calcineurin inhibitor-free immunosuppression in renal allograft recipients with thrombotic microangiopathy/hemolytic uremic syndrome. Am J Transplant. (2006) 6:412–8. doi: 10.1111/j.1600-6143.2005.01184.x
76. Renner B, Klawitter J, Goldberg R, McCullough JW, Ferreira VP, Cooper JE, et al. Cyclosporine induces endothelial cell release of complement-activating microparticles. J Am Soc Nephrol. (2013) 24:1849–62. doi: 10.1681/ASN.2012111064
77. Baas MC, Gerdes VEA, ten Berge IJM, Heutinck KM, Florquin S, Meijers JCM, et al. Treatment with everolimus is associated with a procoagulant state. Thromb Res. (2013) 132:307–11. doi: 10.1016/j.thromres.2013.07.004
78. Keller K, Daniel C, Schöcklmann H, Endlich K-H, Kerjaschki D, Johnson RJ, et al. Everolimus inhibits glomerular endothelial cell proliferation and VEGF, but not long-term recovery in experimental thrombotic microangiopathy. Nephrol Dial Transplant. (2006) 21:2724–35. doi: 10.1093/ndt/gfl340
79. Miriuka SG, Rao V, Peterson M, Tumiati L, Delgado DH, Mohan R, et al. mTOR inhibition induces endothelial progenitor cell death. Am J Transplant. (2006) 6:2069–79. doi: 10.1111/j.1600-6143.2006.01433.x
80. Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, et al. Sirolimus–induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant. (2005) 5:2441–7. doi: 10.1111/j.1600-6143.2005.01047.x
81. Nava F, Cappelli G, Mori G, Granito M, Magnoni G, Botta C, et al. Everolimus, cyclosporine, and thrombotic microangiopathy: clinical role and preventive tools in renal transplantation. Transplant Proc. (2014) 46:2263–8. doi: 10.1016/j.transproceed.2014.07.062
82. Fortin M-C, Raymond M-A, Madore F, Fugere J-A, Paquet M, St-Louis G, et al. Increased risk of thrombotic microangiopathy in patients receiving a cyclosporin-sirolimus combination. Am J Transplant. (2004) 4:946–52. doi: 10.1111/j.1600-6143.2004.00428.x
83. de Vries DK, van der Pol P, van Anken GE, van Gijlswijk DJ, Damman J, Lindeman JH, et al. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation. (2013) 95:816–20. doi: 10.1097/TP.0b013e31827e31c9
84. Xie CB, Qin L, Li G, Fang C, Kirkiles-Smith NC, Tellides G, et al. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-γ–primed human endothelium. Circ Res. (2019) 124:1747–59. doi: 10.1161/circresaha.119.314845
85. Marshall KM, He S, Zhong Z, Atkinson C, Tomlinson S. Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. J Exp Med. (2014) 211:1793–805. doi: 10.1084/jem.20131902
86. Hu C, Li L, Ding P, Li L, Ge X, Zheng L, et al. Complement inhibitor CRIg/FH ameliorates renal ischemia reperfusion injury via activation of PI3K/AKT signaling. J Immunol. (2018) 201:3717–30. doi: 10.4049/jimmunol.1800987
87. Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. (2013) 368:1033–44. doi: 10.1056/NEJMra1112830
88. Romay-Penabad Z, Liu XX, Montiel-Manzano G, De Martínez EP, Pierangeli SS. C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci. (2007) 1108:554–66. doi: 10.1196/annals.1422.058
89. Pierangeli SS, Girardi G, Vega-Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody-mediated thrombophilia. Arthritis Rheum. (2005) 52:2120–4. doi: 10.1002/art.21157
90. Atehortúa L, Rojas M, Vásquez GM, Castaño D. Endothelial alterations in systemic lupus erythematosus and rheumatoid arthritis: potential effect of monocyte interaction. Mediators Inflamm. (2017) 2017:9680729. doi: 10.1155/2017/9680729
91. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis. (2014) 73:1601–6. doi: 10.1136/annrheumdis-2014-205287
92. Petri MA, Conklin J, O’Malley T, Dervieux T. Platelet-bound C4d, low C3 and lupus anticoagulant associate with thrombosis in SLE. Lupus Sci Med. (2019) 6:e000318. doi: 10.1136/lupus-2019-000318
93. Rahbar A, Söderberg-Nauclér C. Human Cytomegalovirus infection of endothelial cells triggers platelet adhesion and aggregation. J Virol. (2005) 79:2211–20. doi: 10.1128/JVI.79.4.2211-2220.2005
94. Loenen WAM, Bruggeman CA, Wiertz EJHJ. Immune evasion by human cytomegalovirus: lessons in immunology and cell biology. Semin Immunol. (2001) 13:41–9. doi: 10.1006/smim.2001.0294
95. Saab KR, Elhadad S, Copertino D, Laurence J. Thrombotic microangiopathy in the setting of HIV infection: a case report and review of the differential diagnosis and therapy. AIDS Patient Care STDS. (2016) 30:359–64. doi: 10.1089/apc.2016.0124
96. Park YA, Hay SN, Brecher ME. ADAMTS13 activity levels in patients with human immunodeficiency virus-associated thrombotic microangiopathy and profound CD4 deficiency. J Clin Apher. (2009) 24:32–6. doi: 10.1002/jca.20189
97. Lopes da Silva RL. Viral-associated thrombotic microangiopathies. Hematol Oncol Stem Cell Ther. (2011) 4:51–9. doi: 10.5144/1658-3876.2011.51
98. Fernández S, Moreno-Castaño AB, Palomo M, Martinez-Sanchez J, Torramadé-Moix S, Téllez A, et al. Distinctive biomarker features in the endotheliopathy of COVID-19 and septic syndromes. Shock. (2021). [Epub ahead of print]. doi: 10.1097/SHK.0000000000001823
99. Castro P, Palomo M, Moreno-Castaño AB, Fernández S, Torramadé-Moix S, Pascual G, et al. Is the endothelium the missing link in the pathophysiology and treatment of COVID-19 complications? Cardiovasc Drugs Ther. (2021). [Epub ahead of print]. doi: 10.1007/s10557-021-07207-w
100. Wang X, Sahu KK, Cerny J. Coagulopathy, endothelial dysfunction, thrombotic microangiopathy and complement activation: potential role of complement system inhibition in COVID-19. J Thromb Thrombolysis. (2021) 51:657–62. doi: 10.1007/s11239-020-02297-z
101. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
102. Rambaldi A, Gritti G, Micò MC, Frigeni M, Borleri G, Salvi A, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology. (2020) 225:152001. doi: 10.1016/j.imbio.2020.152001
103. Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. (2020) 215:108450. doi: 10.1016/j.clim.2020.108450
104. Dorresteijn MJ, Visser T, Cox LA, Bouw MP, Pillay J, Koenderman AH, et al. C1-esterase inhibitor attenuates the inflammatory response during human endotoxemia. Crit Care Med. (2011) 39:243. doi: 10.1097/CCM.0b013e3181f17be4
105. Bauer M, Weyland A, Marx G, Bloos F, Weber S, Weiler N, et al. Efficacy and safety of vilobelimab (IFX-1), a novel monoclonal anti-C5a antibody, in patients with early severe sepsis or septic shock-A randomized, placebo-controlled, double-blind, multicenter, phase IIa trial (SCIENS study). Crit Care Explor. (2021) 3:e0577. doi: 10.1097/CCE.0000000000000577
106. Gavriilaki E, Yuan X, Ye Z, Ambinder AJ, Shanbhag SP, Streiff MB, et al. Modified ham test for atypical hemolytic uremic syndrome. Blood. (2015) 125:3637–46. doi: 10.1182/blood-2015-02-629683
107. Bu F, Meyer NC, Zhang Y, Borsa NG, Thomas C, Nester C, et al. Soluble C5b-9 as a biomarker for complement activation in atypical hemolytic uremic syndrome. Am J Kidney Dis. (2015) 65:968–9. doi: 10.1053/j.ajkd.2015.02.326
108. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. (2014) 124:1715–26. doi: 10.1182/blood-2014-02-558296
109. Timmermans SAMEG, Abdul-Hamid MA, Potjewijd J, Theunissen ROMFIH, Damoiseaux JGMC, Reutelingsperger CP, et al. C5b9 formation on endothelial cells reflects complement defects among patients with renal thrombotic microangiopathy and severe hypertension. J Am Soc Nephrol. (2018) 29:2234–43. doi: 10.1681/ASN.2018020184
110. Galbusera M, Noris M, Gastoldi S, Bresin E, Mele C, Breno M, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis. (2019) 74:56–72. doi: 10.1053/j.ajkd.2018.11.012
111. Palomo M, Blasco M, Molina P, Lozano M, Praga M, Torramade-Moix S, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. (2019) 14:1719–32. doi: 10.2215/CJN.05830519
Keywords: C5b-9 deposition, complement system activation, complement blockade, endothelia cells, membrane attack complex, thrombotic microangiopathies
Citation: Blasco M, Guillén-Olmos E, Diaz-Ricart M and Palomo M (2022) Complement Mediated Endothelial Damage in Thrombotic Microangiopathies. Front. Med. 9:811504. doi: 10.3389/fmed.2022.811504
Received: 08 November 2021; Accepted: 14 March 2022;
Published: 25 April 2022.
Edited by:
Eleni Gavriilaki, G. Papanikolaou General Hospital, GreeceReviewed by:
Peter Hughes, Royal Melbourne Hospital, AustraliaCopyright © 2022 Blasco, Guillén-Olmos, Diaz-Ricart and Palomo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Palomo, bXBhbG9tb0BjYXJyZXJhc3Jlc2VhcmNoLm9yZw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.