 Mohammad Reza Khazdair
Mohammad Reza Khazdair Mohammad Hossein Boskabady
Mohammad Hossein Boskabady- 1Cardiovascular Diseases Research Center, Birjand University of Medical Sciences, Birjand, Iran
- 2Applied Biomedical Research Center, Mashhad University of Medical Sciences, Mashhad, Iran
- 3Department of Physiology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
Sulfur mustard (SM) is one of the major potent chemical warfare that caused the death of victims in World War I and the Iraq-Iran conflict (1980–1988). The respiratory system is the main target of SM exposure and there are no definitive therapeutic modalities for SM-induced lung injury. The effects of the new pharmaceutical drugs on lung injury induced by SM exposure were summarized in this review. Literature review on PubMed, ScienceDirect, and Google Scholar databases was performed to find papers that reported new treatment approach on SM-exposure-induced injury in the respiratory system until October 2019. The search was restricted to sulfur mustard AND induced injury (in vitro studies, animal experiments, and clinical trials) AND respiratory system OR lung, AND treatment in all fields. Two hundred and eighty-three relevant articles were identified that 97 retrieved articles were eligible and were included in the review. Some new pharmaceutical drugs have shown therapeutic potential in controlling various characteristics of lung injury due to SM exposure. Recent studies showed therapeutic effects of mucolytic drugs, non-steroidal drugs, and antibiotics on reducing lung inflammation, oxidative stress responses, and modulating of the immune system as well as improving of respiratory symptoms and pulmonary function tests. Studies on the therapeutic effects of new agents with amelioration or treatment of SM-induced lung injury were reviewed and discussed.
Introduction
Sulfur mustard (SM) is a toxic vesicant that was first used in 1917 as a chemical weapons (CW) agent and was repeatedly used during Iraq–Iran conflict (1983–1988) (1). For a period (5 years) from August 1983 to July 1988, Iran was attacked several times with CW by the Iraqi army. Iraq used large amounts of CW against Iranian military and civilian people. Sardasht (the Kurdish cities, North West of Iran) was attacked several times in July 1987 and June 1988. The last Iraq chemical attack was in Feb 1988 in the town of Oshnaviyeh (North West of Iran) which injured thousands of civilians. The use of CW against civilian population in eight locations in Oshnaviyeh (Sheikh Othman District) has been confirmed by United Nation’s experts (2).
Sulfur mustard is a cell poison agent which can alter DNA and other nuclear components (3), and cause short and long term injury to heart, lung, nervous, and digestive systems (4, 5) depending on the dose and duration of the exposure (6). The lungs are major targets of SM which led to destruction of bronchial tissues (7), obstruction of airways (8), and oxidative stress (9). Edema and erythema of the pharynx and bronchial tree have occurred minutes to several hours after exposure to SM (10), hemorrhagic pulmonary edema, secondary pneumonia, and respiratory failure 24–48 h after severe exposure (11) and bronchopneumonia 36–48 h after SM exposure (12) in the veterans. Various pathological changes of the lungs (13) in SM exposed individuals can lead to asthma and/or chronic obstructive pulmonary disease (COPD) like symptoms (14).
Releasing of several inflammatory mediators by phagocytic leukocytes (15, 16), increased bronchoalveolar lavage fluid (BALF) cells (17, 18) which were shown in animals exposed to SM that indicates the role of phagocytic leukocytes and their inflammatory mediators in the pathogenic response to inhaled SM (19). Pulmonary and systemic inflammation such as changes in serum levels of cytokines (20), increased C-reactive protein, (21) and the serum levels of the pro-apoptotic protein, soluble Fas l (22) were observed in individuals long time after exposing to SM. Increased capillary leakage (23) and permeability in the cultured vascular endothelial cell monolayers (24) were also reported due to SM exposure. Hematological changes such as increased WBC count, increased respiratory symptoms, and reduced pulmonary function test (PFT) were reported in veterans long term after SM exposure (25). The possible mechanisms of SM-induced lung disorders were shown in Figure 1.
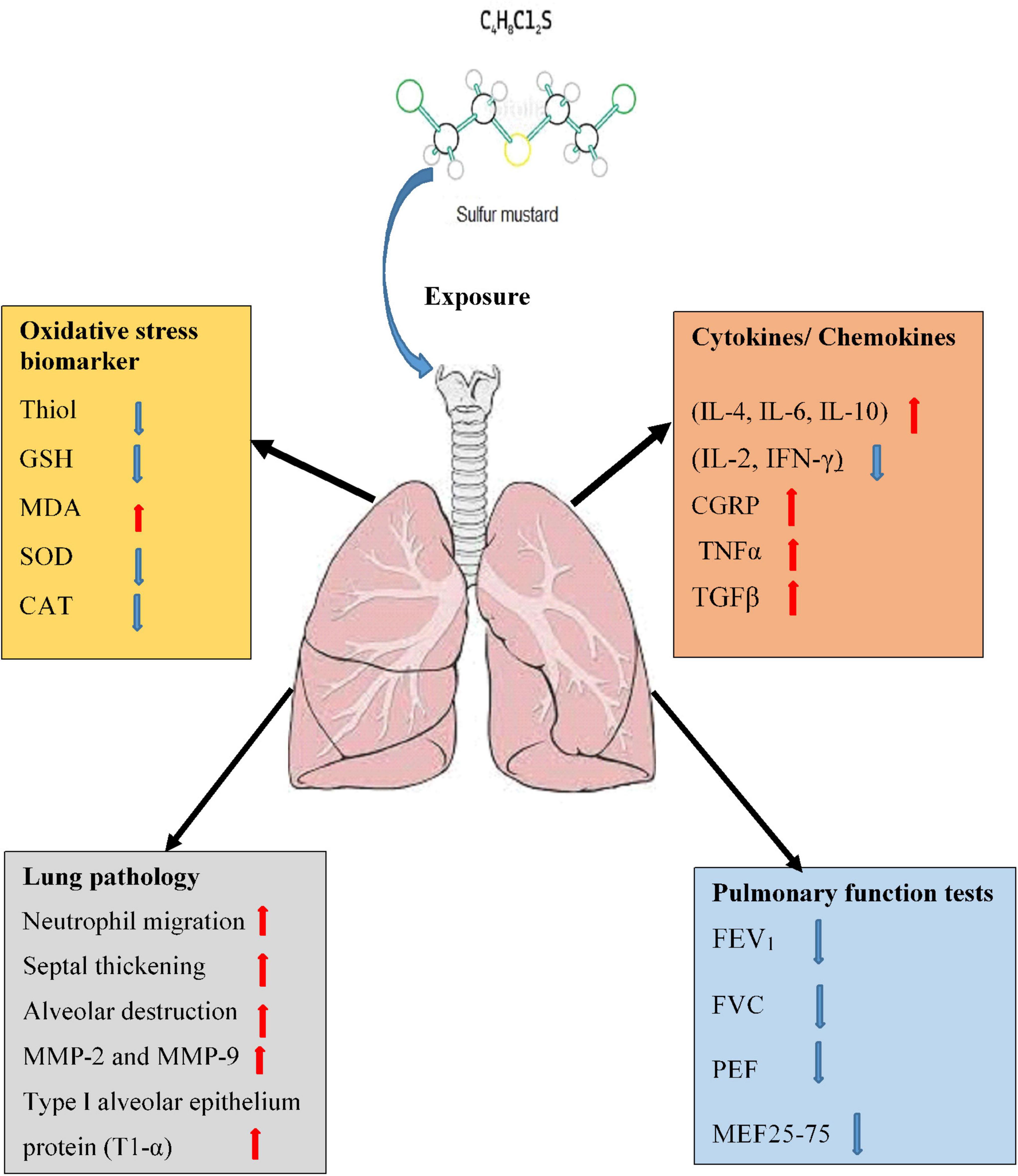
Figure 1. Possible mechanisms of lung injury induced by SM exposure. MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; CAT, catalase; TGF-β1, transforming growth factor-beta1; IL, interleukin; IFNγ, interferon gamma; TNFα, tumor necrosis factor-α; MMPs, matrix metalloproteinase; FVC, forced volume capacity; FEV1, volume in one second; PEF, peak expiratory flow; MEF, maximal expiratory flow.
Presently, there is no approved medication for the treatment of SM exposure-induced lung injury (26), but a combination of vitamin E and corticosteroids was used for protection against acute phases of SM-induced lung injury (27). Moreover, L-nitroarginine methyl ester (L-NAME) and L-thiocitrullline (L-TC) are two protective drugs against the acute toxicity of SM in the in vitro and in vivo studies (28, 29). The results of these studies showed that different treatments still have no optimal effectiveness and also have known adverse effects in this stage of disease (30). After the acute phase, respiratory problems such as chronic bronchitis (59%), tracheobronchial stenosis (24%), asthma (11%), or bronchiectasis (9%) are the leading causes of long-term disability among patients with exposure to SM (31, 32). Inhaled corticosteroids are extensively used to resolve exacerbation of respiratory symptoms for the treatment of long-term toxic effects of SM (33). In addition, bronchodilators can be applied for treatment of increased airway hyper-reactivity in SM exposed patients (34). The combination of β-agonist and anticholinergic with corticosteroids has been found to be more effective than any of the other bronchodilators used alone in SM exposed patients (35). The possible therapeutic effects of natural products on SM induced complication were also reported (36).
Exposure to SM could induced lung injury including long-term effects like COPD and fibrosis, even decades after exposure. It is essential to identify efficacious treatments for chronic diseases induced by SM. Therefore, this review aimed to present available experimental and clinical publications on the efficacy of the synthesized drugs in the prevention and/or treatment of lung disorders due to SM exposure.
Possible Treatment Approaches for SM Induced Lung Disorders
Anti-inflammatory Agents
Interferon-Gamma
Interferon-gamma (IFN-γ) and transforming growth factor-beta1 (TGF-β1) showed inhibitory effects on several aspects of the inflammatory process. TGF-β1 induced stimulation of collagen transcription in fibroblasts independent of Stat1–promoter interactions but IFN-γ inhibited these effects (37, 38). IFN-γ and TGFβ may also provide opposing signals to macrophages (39).
Experimental Studies
Interferon-gamma modified mesenchymal stem cells (MSCs) induced apoptosis in lung tumor cells through caspase-3 activation. The percentage of activated-caspase-3-positive tumor cells in IFN-γ modified cultured MSCs was significantly higher than in control cultured MSCs. The results of this study provide a new strategy for tumor therapy that utilizes IFN-γ modified MSCs (40).
Anti-inflammatory effect of IFN-γ via down-regulation of TGF-β1 and pro-collagen I and III gene expression in a mouse model of lung fibrosis was demonstrated (41). Interactions of IFN-γ and IL-13, on the mouse model of airway inflammation, showed a negative correlation between increased IFN-γ (number of Th1 cells) with airway hyperreactivity. Also, IFN-γ is able to modulate the effects of IL-13-induced airway hyperreactivity and goblet cell hyperplasia. Intranasal administration of IFN-γ inhibits IL-13-induced goblet cell hyperplasia and airway eosinophilia. Co-administration of IFN-γ and IL-13 showed synergic effects on increased IL-6 level as well as numbers of natural killer (NK) cells and CD11c-positive cells in the airways of mice (42).
Clinical Studies
Interferon-gamma has been shown to be effective in the treatment of patients with idiopathic pulmonary fibrosis (IPF) (43). Decreased IFN-γ levels in leukocyte cultures from SM exposed patients were also reported (44). In a clinical study, 36 SM exposed patients with SM bronchiolitis were divided into case and control groups. The case group was treated with a combination of 200 mg IFN-γ and 7.5 mg prednisolone for 6 months. Pulmonary function tests (PFT) including FEV1 and forced vital capacity (FVC) were not significantly different at the baseline between the two groups. However, FEV1 and FVC were significantly increased in the case group during the subsequent months (45). In another similar study, the effect of interferon IFN-γ on respiratory symptoms, quality-of-life, and oxidative parameters in SM exposed patients was investigated. IFN-γ (100 μg) was administered every other day for 6 months. Severity and frequency of respiratory symptoms, quality-of-life, serum levels of different cytokines, and oxidative stress parameters were assessed at the baseline and at the end of the study. The results showed that IFN-γ therapy not only elevated FEV1, but also reduced the severity of cough, dyspnea, and frequency of sputum occurrence. IFN-γ therapy also is associated with improvements in quality-of-life. Serum levels of IL-4, IL-6, IL-10, calcitonin gene-related peptide (CGRP), MMP-9, tumor necrosis factor-alpha (TNFα), TGFβ, and malondialdehyde (MDA) as oxidative stress marker were also decreased while the level of IL-2, IFN-γ, and glutathione (GSH) were increased at the end of study (46).
The above studies suggest the therapeutic potential of IFN-γ in SM-induced lung injury by reduction of inflammatory mediators and oxidative stress as well as respiratory symptoms but increased PFT values in SM exposed patients with lung disorders.
Theophylline
Theophylline in high-dose has bronchodilatory effects and in low concentration may have immunomodulatory and anti-inflammatory properties (47). Theophylline suppressed TGF-β-induced type I collagen mRNA expression in lung fibroblasts and also inhibited fetal bovine serum (FBS)-stimulated fibroblast proliferation and TGF-β-induced α-smooth muscle actin protein (48).
Experimental Studies
The administration of theophylline (10 or 20 μg/ml) 2 h before human TGF-β1 stimulation suppressed TGF-β-induced type I collagen (COL1) mRNA expression in lung fibroblasts and also inhibited fibroblast proliferation (48). It was reported that theophylline (1–50 mg/kg–1, i.p.) significantly inhibited the inflammation at the early (4 h) and late (48 h) phases of inflammatory reaction induced by carrageenin (1%) in a murine model of pleurisy (49). Theophylline reduced airway inflammation and pathologic changes of lung tissue induced by cigarette smoke in a COPD model of rats (50). Theophylline (3 and 30 mg/kg) reduced the influx of inflammatory cells into the BALF of Guinea pigs exposed to LPS (30 μg/mL). Furthermore, theophylline improved histological changes induced by LPS, including accumulation of inflammatory cells in the lung parenchyma, swelling of the alveolar walls, and goblet cell hyperplasia in the airways (51).
Clinical Studies
Treatment of children with moderate to severe cystic fibrosis (CF) by theophylline for 10 days showed protective effects on arterial oxygen desaturation during sleep, increased wakefulness and decreased sleep efficiency (52). Histone deacetylase complex (HDAC) is the main enzyme responsible for regulating inflammatory gene expression activity induced by oxidative stress. Theophylline can restore reduction of this enzyme. Moreover, theophylline may be able to reverse steroid resistance in COPD and other inflammatory lung diseases (53). Treatment of asthmatic smokers with theophylline (400 mg) in combination with inhaled beclometasone (200 mg) per day after four weeks significantly improved peak expiratory flow (PEF) and FVC but a borderline increase in pre-bronchodilator FEV1 (54).
In a clinical study, SM-exposed patients were treated with oral slow releasing (SR) theophylline (250 mg), omeprazole (20mg), NAC (600 mg), salmetrol, and fluxitide (2 puffs) twice a day, during 8 weeks of therapy. The results showed that the low dose of theophylline and other mentioned drugs was partially able to decelerate the reductions in PFT values of these patients (55).
The decline of the reductions in PFT values in SM-exposed patients and its effect on CF and improvement of PFT values in asthmatic smokers by theophylline suggest its therapeutic effect on SM-induced lung disorders.
Protease Inhibitor Agents
Doxycycline
Doxycycline (DOX) is an antibiotic used to treat bacterial infections, pneumonia, and other respiratory tract infections that has been reported to exhibit non-specific matrix metalloproteinase (MMPs) inhibitory activity (56). The important role of MMPs in the toxicity of SM in different tissues including, lungs, skin, and eyes were indicated (57).
Experimental Studies
It was reported that doxycycline (2 mg/kg, p.o.) reduced lung pathology (neutrophil migration, septal thickening, and alveolar destruction), intra-alveolar inflammatory cells (mainly neutrophils), bacterial number, and MMP-2 and MMP-9 lipopolysaccharide-induced lung injury in C57Bl/6 mice (58). The effects of doxycycline (20 mg/kg or 60 mg/kg, orally), on virulent influenza A/Aichi/2/68 (H3N2) virus-induced lung injury in mice showed a significant decrease in inflammation and protein leakage in the lungs. Treatment with doxycycline also reduced the levels of MMP-2 and MMP-9 activity, T1-α (type I alveolar epithelium protein), and thrombomodulin (endothelial protein) compared to the non-treated group. These results demonstrated that doxycycline treatment is able to reduce lung damage but it did not affect virus titers and body weights (59).
Administration of doxycycline (2 mg/kg) in the drinking water in mice challenged with intratracheal LPS, significantly decreased the number of neutrophils and shed syndecan-1 in BALF. In addition, doxycycline had no significant effect on total BALF protein and the whole lung caspase-3 activity (60). The protective effect of doxycycline on acute lung injury induced by cardiopulmonary bypass (CPB) in 30 healthy mongrel dogs showed that administration of doxycycline (30 mg/kg) in feeding food significantly decreased WBC count, alveolar-arterial oxygen difference, respiratory index, total protein, and myeloperoxidase (MPO) activity in the BALF compared to the control group. Also, doxycycline (60 mg/kg) significantly decreased the concentration of MMP-9 compared to the control group (61).
Administration of doxycycline (2 mg/kg) in drinking water prior to intratracheally administration of bleomycin which induced pulmonary fibrosis, significantly decreased the number of neutrophils, although the total number of cells in the BALF remained unchanged. Dxycycline also attenuated gelatinase activities and reduced production of gelatinase B in BALF (62). Pretreatment of SM exposed guinea pigs with doxycycline resulted in the reduction of gelatinases activity (MMP-2 and MMP-9), decreased lung inflammation (cellularity and protein levels in BAL), and decreased lung pathological changes (epithelial lesions) (63). These results indicated that doxycycline has potent therapeutic effects on LPS and bleomycin-induced pulmonary fibrosis and inflammation in animal models with a similar action to SM-induced lung injury.
Clinical Studies
In a clinical study, pharyngeal Chlamydia trachomatis patients were treated with azithromycin (n = 78) and doxycycline (n = 64). Treatment failure in patients treated with azithromycin and doxycycline was 8.78 (10%) and 1.64 (2%), respectively. Treatment with doxycycline (100 mg) twice a day for 7 days was associated with less treatment failure of oropharyngeal chlamydia compared with azithromycin (1 g), (64). The effect of doxycycline on cystic fibrosis exacerbation in a randomized, double-blind, placebo-controlled study was studied and bio-specimens were collected at the start and the end of the study. Treatment with doxycycline (100 mg) orally twice daily in participants (n = 20) was given over an 8-day period during hospitalization, significantly reduced the levels of MMP-9 in sputum, which was also associated with the reduction in active MMP-9 levels (56.5%), and increased TIMP-1 in sputum. Doxycycline also improved forced expiratory volume in the first second (FEV1) and increased the time to next exacerbation compared to the placebo participants (n = 19). Furthermore, treatment with doxycycline reduced total hospital days compared with the placebo-treated group (65).
Using nuclear magnetic resonance (NMR)-based metabolomics to obtain serum metabolic profiles of doxycycline-treated (n = 60) and standard therapy of COPD patients (n = 40), doxycycline (100 mg) significantly increased the values of FEV1/forced vital capacity (FVC) and improved the COPD assessment test (CAT) scores after 3 months’ treatment. In addition, doxycycline significantly down-regulated serum levels of lactate and fatty acid, while, up-regulated the levels of formate, citrate, imidazole, and L-arginine compared to the pre-treatment level. The post doxycycline treatment significantly reduced serum level of the folate compared to COPD patients (66).
The results of experimental and clinical studies indicated that doxycycline could attenuate lung injury and pulmonary edema through anti-inflammatory effects such as degradation of the cell membrane, pulmonary neutrophil infiltration, and PFT test. These results suggest its therapeutic value in COPD and lung injury induced by SM.
Tissue Plasminogen Activator
Tissue plasminogen activator (tPA) is a potent fibrinolytic agent that currently used as first-line therapy in clot-associated diseases, such as stroke (67).
Experimental Studies
Sulfur mustard analog exposure led to the formation of fibrin-rich in the airways which caused airway obstruction (68). The intra-tracheal administration of tPA (0.15–0.7 mg/kg, 5.5 and 6.5 h) in adult rats exposed to SM analog completely eliminated mortality at 48 h (0%), and improved morbidity (90–100%). Treatment with tPA also normalized plastic bronchitis and hypercarbia. It also improved respiratory distress, pulmonary gas exchange, and oxygen utilization while reduced airway fibrin casts (69).
Intra-tracheal tPA treatment reduced mortality at 48 h (0%) and significantly improved lung injury after lethal SM inhalation (100% death in controls). In addition, tPA improved respiratory distress and normalized hypoxemia, hypercarbia, and lactic acidosis induced by SM. Moreover, tPA was given via airway 6h after SM exposure prevented death from lethal SM inhalation, and normalized oxygenation and ventilation defects (70).
Clinical Studies
The effect of intravenous recombinant human tPA (rt-PA) was compared to urokinase in 45 patients with pulmonary embolism (PE). The results showed improvement in lung scan reperfusion in the two treatment groups after 24 h. The reduction in fibrinogen did not differ significantly between the two treated groups. These results also indicated that rt-PA acts more rapidly and is safer than urokinase in the treatment of acute PE (71).
Therefore, tPA reduced mortality and respiratory distress as well as normalized hypoxemia and hypercarbia induced by lethal SM inhalation which suggest the therapeutic effect of tPA on SM-induced lung disorders.
Tissue Factor Pathway Inhibitor
Tissue factor pathway inhibitor (TFPI) is a 276 amino acid glycoprotein with three distinct structural domains and an acidic N terminus which is an important physiologic inhibitor of the extrinsic pathway of the coagulation system (72). It was reported that TFPI-2 inhibits tumor invasion and angiogenesis in vitro and in vivo, and also suggested a potentially important therapeutic role for recombinant TFPI-2 in the treatment of malignant esophageal carcinomas (73).
Experimental Studies
Pre and post-exposure treatment of rats with recombinant TFPI (rTFPI) significantly inhibited LPS-induced pulmonary vascular injury and coagulation abnormalities. Treatment with rTFPI also significantly inhibited increases of TNF-α, cytokine-induced neutrophil chemo-attractant, and myeloperoxidase in lung tissue. The administration of rTFPI significantly reduced the expression of TNF-α messenger RNA (mRNA) and inhibited TNF-α production in the lungs after stimulation by LPS. The results of this study suggested the effect of rTFPI on pulmonary vascular injury by inhibiting leukocyte activation in LPS-administered rats (74). Intravenous injection of rTFPI immediately before the introduction of tumor cells also reduced metastasis by 83% in mice (75).
Intra-tracheal administration of TFPI decreased fibrin-containing formation and limited severe hypoxemia in SM analog-induced lung injury in animals. TFPI limited thrombin activation in airways by reduction of prothrombin consumption and decreased thrombin- antithrombin complex (TAT) in BALF which led to a reduced airway obstruction and an improved gas exchange in rats (76). Therefore, the therapeutic effect of TFPI on LPS and SM analog-induced lung injury was documented.
Antioxidant Agents
N-Acetyl Cysteine
N-acetyl cysteine (NAC) is a thiol compound and categorized as a mucolytic drug-containing sulfhydryl groups. It is a reactive oxygen species (ROS) scavenger and also reduces GSH and therefore can regulate the oxidation status in cells. In addition, it interferes with several signaling pathways including regulating apoptosis, cell growth and arrest, angiogenesis, redox-regulated gene expression, and inflammatory response (77).
Experimental Studies
The involvement of IL-8 in the pathogenesis of Bleomycin (BLM)-induced lung injury has been suggested and its level was elevated in a bronchial epithelial cell line (BEAS-2B cells). However, BLM-induced expression of IL-8 protein and mRNA in BEAS-2B cells was inhibited by NAC (78). Pre-incubation with NAC (5 × 10–5M, 24 h) also significantly reduced peroxynitrite (ONOO–) and O2–production in lung macrophages obtained from systemic sclerosis (SSc) patients (79).
Administration of NAC in lead-exposed animals, reduced or reversed lead-induced oxidative stress. Lead-exposed rats resulted in signs of anemia such as anisocytosis, poikilocytosis, and alterations in hemoglobin and hematocrit. It also causes an alteration in lipid peroxidation such as, increased MDA content and a decrease in GSH which are reversed by NAC (80). It has been reported that pretreated animals with NAC or liposomally-entrapped N-acetylcysteine (L- NAC) (25 mg/kg, iv), and challenged with lipopolysaccharide (LPS) (Escherichia coli, LPS 0111:B4) prevents increased lung weights, decreased lung angiotensin-converting enzyme (ACE) (the injury marker for pulmonary endothelial cells), and significantly reduced the LPS-induced increase in plasma TNF-α levels. In addition, NAC significantly decreased myeloperoxidase (MPO) and chloramine concentrations in the lungs and the extent of lipid peroxidation in liver tissues (81).
The enhancement of endothelial survival via GSH against SM injury of the endothelium was also reported. In this study, exposure to 500 μM SM resulted in increased activation of the nuclear transcription factor (NFkB) binding to its consensus sequence 5 h after exposure. Pretreatment with NAC suppressed SM-induced NFkB activation which is an important transcription factor for a number of cytokine genes (e.g., TNF) and is activated following stress in endothelial cells (82). GSH-induced enhancement of cell viability at 2.5 and 5 μM was shown after SM exposure. Pretreatment with hioninesulfoximine (BSO) an inhibitor of GSH synthesis, alone did not show toxicity effect but potentiated the toxicity of SM (82). Similarly, SM exposure led to a dose and time-dependent decrease in GSH content in human skin fibroblast cell line (HF2FF). NAC increased intracellular GSH level and protected the cells against SM-induced reactive oxygen species formation and lactate dehydrogenase leakage. In contrast, BSO pretreatment reduced cellular GSH and enhanced the cytotoxic effects of SM on HF2FF cells (83).
Treatment with intra-tracheal administration of NAC on pulmonary edema formation induced by phosgene exposure in rabbit lungs lowered pulmonary artery pressure, lung weight gain, the concentration of peptide leukotrienes LTC4, D4, and E4, and lipid peroxidation which were increased following exposure to phosgene (84).
Animal exposure to SM leads to inflammatory cell accumulation in the airways and lung as well as structural and functional alterations in the respiratory tract (18, 85). The role of oxygen species and free radicals in the pathophysiology of inflammation-induced pulmonary lesions was described previously (86). However, treatment with NAC reduced the number of neutrophils in mice that were exposed to SM and developed lung injuries (87). Following animal exposure to SM vapor (100 μg/kg) for 10 min, arterial blood oxygen saturation levels (SaO2), arterial blood pH and bicarbonate (HCO) were significantly decreased. Also, arterial blood carbon dioxide (PaCO2) and shunt fraction were significantly increased. After treatment with inhaled doses of NAC (1 ml of 200 mg) arterial blood oxygen saturation, HCO level, and shunt fraction were significantly improved compared to those of the SM exposure. In addition, infiltration of neutrophils and concentrations of protein in BALF were significantly decreased compared to the SM group (88).
Clinical Studies
In clinical studies, treatment with NAC in patients with COPD, asthma, and acute bronchitis improved respiratory symptom, lung function, and quality of life. Treatment with NAC also scavenged reactive oxygen species and inhibited mediator release (89, 90). It was reported that the administration of NAC, 600 mg daily for 12 months in patients with COPD, reduced oxidative stress (91, 92) and also the oxidative burst of poly morphonuclear (PMN) cells and showed protective effects on peripheral granulocytes in COPD patients (93, 94).
Intravenous administration of NAC (190 mg/kg/day) in adult patients (n = 42) with respiratory distress syndrome (Pao2/Fio2 ≤ 200 mm Hg) significantly decreased the lung injury score between days 1 and 3 compared to the placebo group (95).
Treatment with NAC (40 mg/kg/day) intravenously for 3 days in 61 adult patients with mild-to-moderate acute lung injury (NAC, n = 32 and the placebo groups, n = 29 patients) improved ventilation compared to the placebo group. The oxygenation index (PaO2/FIo2) significantly was improved from day 0 to day 3 only in the NAC treated group and the lung injury score was improved in the NAC treated group during the first 10 days of treatment but, no change was observed in the placebo group (96).
Treatment of SM-induced bronchiolitis obliterans patients (n = 144) with NAC (1,200 mg daily), improved dyspnea, wake-up dyspnea, and cough after 4 months of treatment compared to the control group. Additionally, NAC reduced sputum from (76.9%) of cases before the study to (9.6%) of cases at the end of trial. The values of FEV1, FVC, and FEV1/FVC were also significantly improved in NAC treated patients compared to the placebo group (97).
In another clinical study, treatment with NAC (1,800 mg daily) in 144 bronchiolitis obliterans patients 18 years after SM exposure improved clinical signs and symptoms of patients including; dyspnea, cough, sputum, wake-up dyspnea, and also increased PFT values (98). It has been reported that administration of NAC (oral, IV, IP or IT) is effective in the management of SM induced acute lung injury due to inhibition of oxidative stress, inflammatory responses and apoptosis in a time-dependent manner. Furthermore, oral NAC alone at high doses for 4 months or in combination with clarithromycin (500 mg/day) at a dose 600 mg/day for 6 months improved clinical and paraclinical pulmonary parameters of patients with bronchiolitis obliterans induced by SM exposure (99). The efficacy of NAC in reducing SM toxicity in different studies (in vitro and in vivo) model of lung injury as well as the safe and efficacy of NAC in treating patients suffering the long-term and chronic pulmonary effects of SM exposure in veterans was reported (100).
Both experimental and clinical studies suggest the possible therapeutic effect of NAC on SM-induced lung disorders and other respiratory diseases via possible anti-inflammatory and anti-oxidant mechanisms.
Vitamin E
Experimental Studies
The effects of vitamin C, vitamin E, and rINN, alone or in combination in placental villi culture after exposure to nicotine-induced endothelial dysfunction reduced placental cell proliferation until cell death (101). Vitamin E reduced phospholipidosis in cultured human skin fibroblasts chronically exposed to amiodarone and desethylamiodarone (DEA) and inhibited cumulative uptake of the drugs in a dose-dependent manner (102). Vitamin E pretreatment on human fibroblast cells significantly protected against ciprofloxacin (CPFX)-induced cytotoxicity. It also significantly increased the total GSH content and reduced the level of lipid peroxidation (103).
The effects of liposome-encapsuled vitamin E on a mouse model for airway inflammation induced by inhalation exposure to the alkylating nitrogen mustard melphalan showed that vitamin E (50 mg/kg) reduced inflammatory cell influx, and inhibited collagen formation in lung tissue (27). The administration of vitamin E (600 mg/kg), dexamethasone (5 mg/kg) or their combination in guinea pigs exposed to SM caused improvement in the pathological changes in the livers and kidneys (104).
The effect of vitamin E on tracheal responsiveness (TR) and lung inflammation in SM exposed animals also showed that TR to methacholine, total and differential WBC count in lung lavage, and serum levels of IL-4 were significantly decreased in treated animals with vitamin E compared to the untreated SM-exposed group (105). In addition, vitamin E treatment reduced pathological changes in guinea pigs exposed to SM. The antioxidant effect of tocopherol acetate (vitamin E) was also reported (106, 107) which supports the above results.
Tocopherol acetate (200 mg/kg, i.p.) increased GSH levels and decreased MDA level in systemic toxicity due to percutaneous administration of SM in animals. Furthermore, total antioxidant status was increased but red blood corpuscles and hemoglobin content were significantly decreased in the tocopherol acetate treated group (108).
Liposomes containing tocopherol (α, γ, δ) showed protective effects on the accumulation of RBC in the bronchi, alveolar space, arterioles and veins, and fibrin and collagen deposition in the alveolar space on 2-chloroethyl ethyl sulfide (CEES) (a mono-functional analog of sulfur mustard)-induced lung injury in a guinea pig. Moreover, tocopherol decreased lipid peroxidation level and hydroxyproline in the lungs (109). The treatment with liposome of tocopherol (α, γ, δ) significantly blocked the CEES-induced increase in the protein levels of the cell cycle protein (cyclin D1), and a cell differentiation marker (PCNA). Additionally, tocopherol protected the lungs against CEES-induced inflammation and alveolar infiltration of neutrophils and eosinophils (110).
The effect of vitamin E on CEES and alkylating nitrogen mustard-induced lung inflammation, as well as its effect on lung oxidative stress, lung inflammation, lung pathology, and tracheal responsiveness due to similar mechanisms in SM-exposed subjects indicated its therapeutic value in SM and other chemical-induced lung injuries.
Clinical Studies
Dietary intakes of vitamin E in 178 men and women aged (70–96 year) indicated that intake of vitamin E may influence lung function by increasing FVC, FEV1 and showed protective effects on wheeze in elderly in three European countries including Finland (n = 1,248), Italy (n = 1,386), and the Netherlands (111, 112). Treatment of 33 mild atopic asthmatics subjects for 16 weeks, with high doses of -α-tocopheryl acetate significantly decreased F2-isoprostanes, and reduced allergen-provoked concentrations of IL-3 and IL-4, and increased levels of IL-12 in BALF. In addition, natural source of vitamin E improved airway responsiveness to methacholine (113).
In a placebo-controlled randomized clinical trial the effects of natural vitamin E (500 mg) or matched placebo in 72 participants aged (18–60 years) for 6 weeks, had no significant impact on FEV1, asthma symptom scores, or serum immunoglobulin levels (114). Investigation of the influence of maternal antioxidant intake in pregnancy on the development of asthma and eczema in children showed that maternal vitamin E intake during pregnancy is negatively associated with wheeze in the absence of “cold” and childhood eczema in the children’s with 24 months of age (115).
The therapeutic effects of pharmaceutical drugs on SM-induced lung disorders in experimental and clinical studies were summarized in Tables 1, 2, respectively. The possible mechanisms of the therapeutic effects of pharmaceutical drugs on SM-induced lung disorders were also summarized in Figure 2.
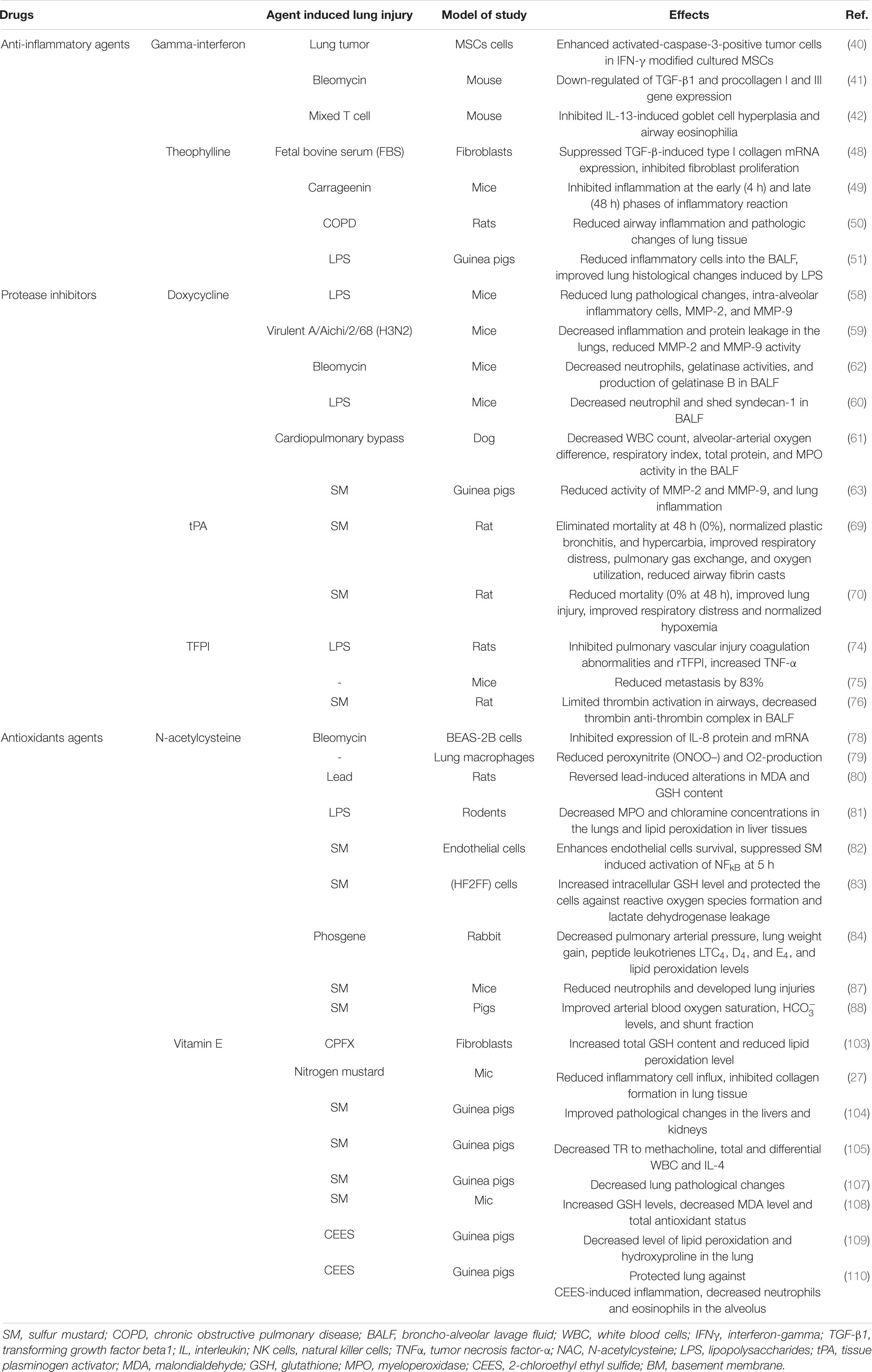
Table 1. Therapeutic effects of pharmaceutical drugs on SM-induced lung injuries, experimental evidence.
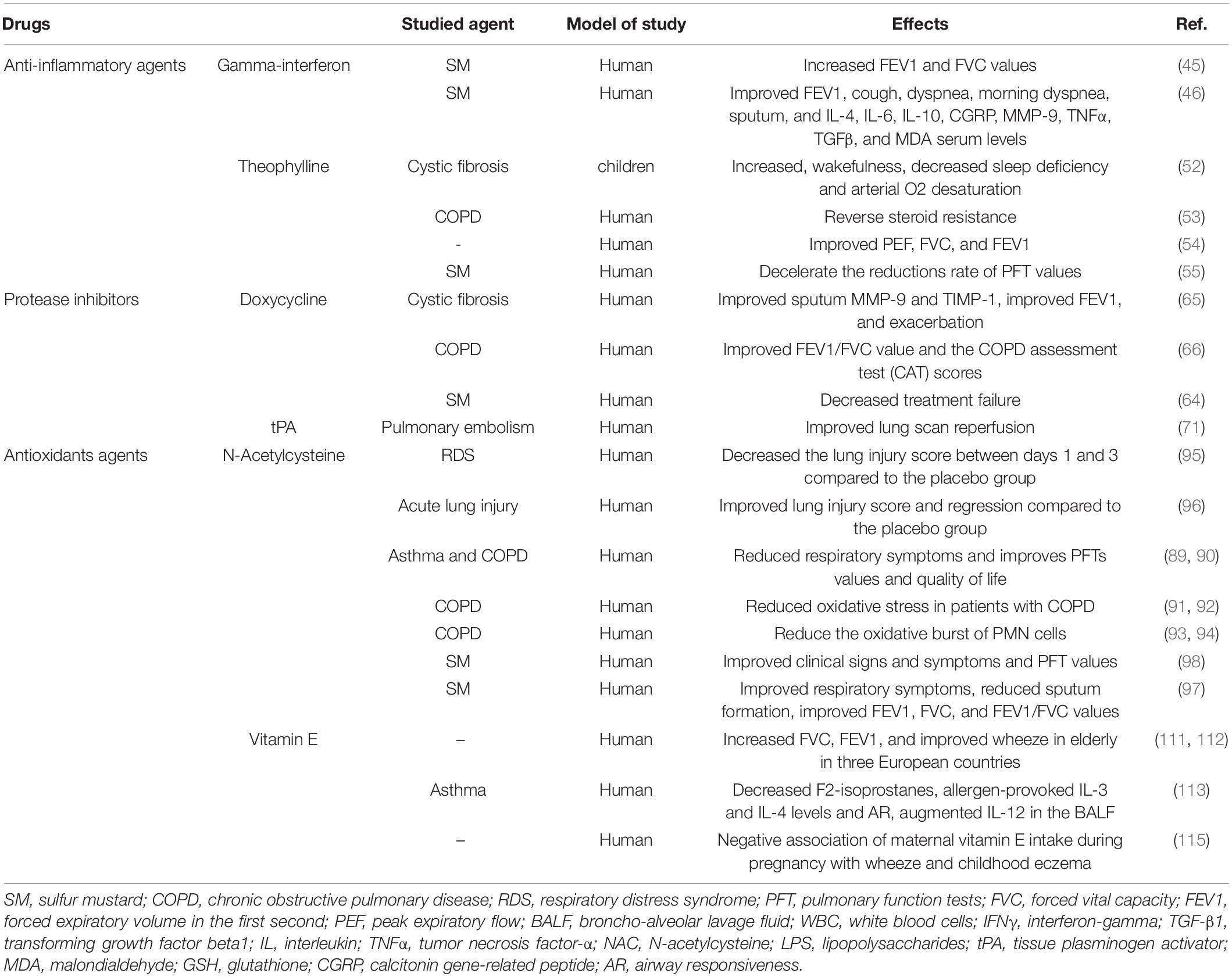
Table 2. Therapeutic effects of pharmaceutical drugs on SM-induced lung injuries, clinical studies.
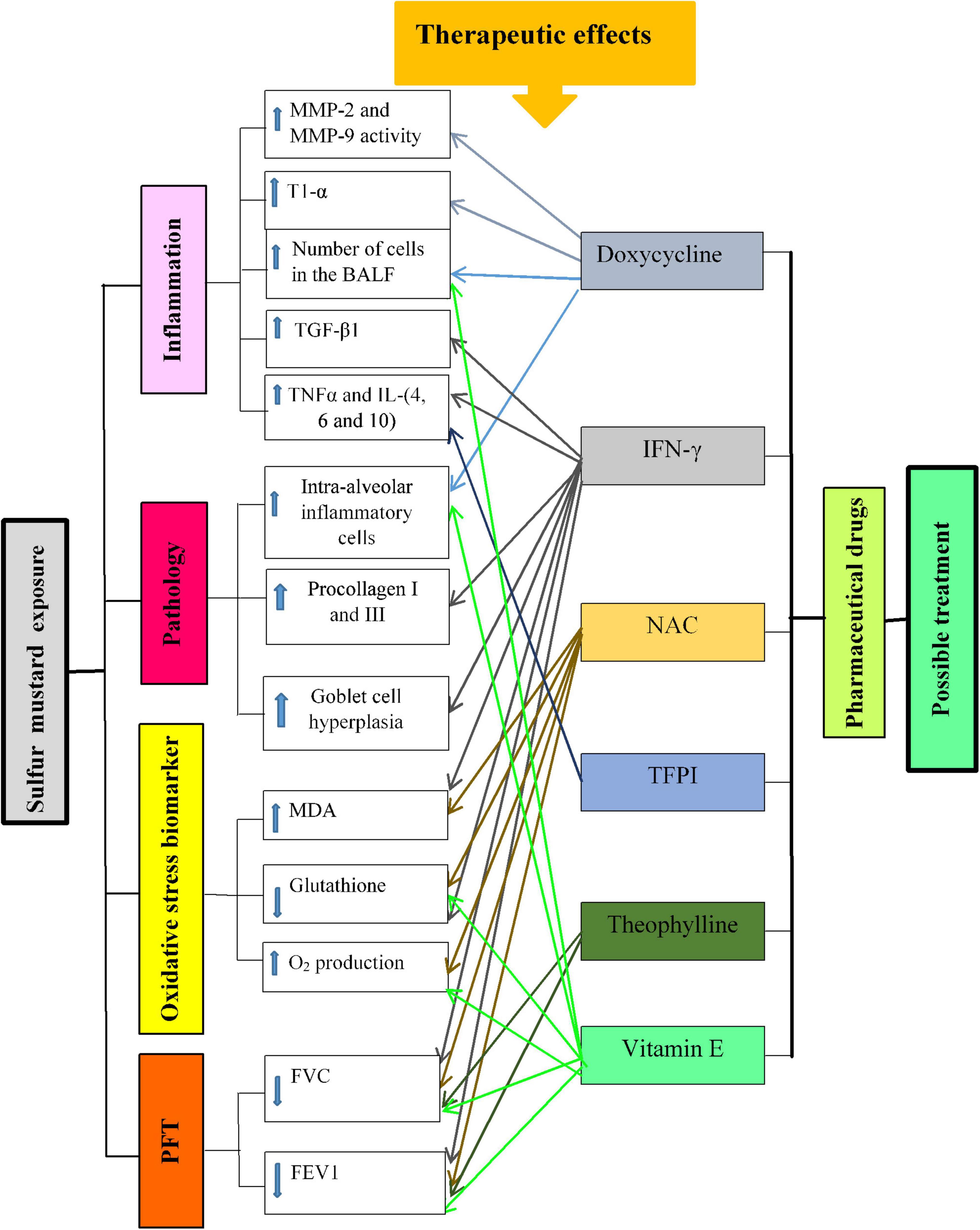
Figure 2. Possible mechanisms of lung injury induced by SM exposure and the effect of pharmaceutical drugs in these changes. T1-α, type I alveolar epithelium protein; MMPs, matrix metalloproteinase; BALF, bronchoalveolar lavage fluid; TGF-β1, transforming growth factor-beta1; IL, interleukin; IFNγ, interferon gamma; TNFα, tumor necrosis factor-α; NO, nitric oxide; MDA, malondialdehyde; FVC, forced volume capacity; FEV1, volume in one second.
Conclusion
Sulfur mustard, CEES, and nitrogen mustard exposure due to similar actions induces various acute and chronic diseases such as lung disorders. Unfortunately, there is still no definitive medication for SM-induced diseases in clinics. However, several studies suggested the therapeutic effects of individual or a combination of pharmaceutical drug products on SM and its analogs-induced lung disorders in animals as well as in some clinical studies. In this review, published papers regarding the new approach for the treatment of SM-induced lung disorders by pharmaceutical drugs were discussed.
The results of the reviewed papers showed that pharmaceutical drugs improved inflammatory cells, inflammatory mediators, oxidative stress biomarkers, and lung pathological changes in lung injury induced by various agents including SM exposure. The pharmaceutical agents also showed a reduction of clinical symptoms, improvement of PFT values, and health-related quality of life as well as systemic and lung inflammation and oxidative stress in patients suffering from various chronic lung diseases including SM-induced pulmonary complications. Therefore the present review present various potential treatment strategy for preventing and treatment of lung injury patients who are exposed to SM in mean of inflammation and oxidative stress. However, more studies especially clinical trials are needed to examine the exact mechanisms of action of pharmaceutical drugs and their long-term outcome as well as exploring new drugs for the treatment of the serious condition of lung disorders induced by SM exposure.
Author Contributions
MRK searched the literature and wrote the first draft of the manuscript. MHB designed the study and critically edited and revised the manuscript. Both authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Ach, acetylcholine; BALF, broncho-alveolar lavage fluid; CGRP, calcitonin gene-related peptide; COPD, chronic obstructive pulmonary disease; DOX, doxycycline; FDA, Food and Drug Administration; FEV1, forced expiratory volume in the first second; FVC, forced vital capacity; GSH, glutathione; HDAC, histone deacetylase complex; hPBMCs, human peripheral blood mononuclear cells; HRQoL, health-related quality of life; hs-CRP, high-sensitivity C-reactive protein; IFNγ, interferon-gamma; IL, interleukin; iNOS, induced nitric oxide synthase; IPF, idiopathic pulmonary fibrosis; LPS, lipopolysaccharides; MCHC, mean corpuscular hemoglobin concentration; MCP-1, monocyte chemotactic protein-1; MCV, mean corpuscular volume; MDA, malondialdehyde; MMEF, maximum mid-expiratory flow; MMPs, matrix metalloproteinase; NAC, N-Acetylcysteine; NFkB, nuclear transcription factor; NK cells, natural killer cells; OVA, ovalbumin; PaCO2, arterial blood carbon dioxide; PE, pulmonary embolism; PEF, peak expiratory flow; PFT, pulmonary function tests; ROS, reactive oxygen species; SaO2, arterial blood oxygenation saturation; SIL-BS, silibinin-bis-succinat; SM, sulfur mustard; TAT, thrombin anti-thrombin; TFPI, tissue factor pathway inhibitor; TGF-β1, transforming growth factor beta1; TNFα, tumor necrosis factor-α; tPA, tissue plasminogen activator; TR, tracheal responsiveness; TSM, tracheal smooth muscle; WBC, white blood cells.
References
1. WHO. Report of the Mission Dispatched by the Secretary-General to Investigate Allegations of the Use of Chemical Weapons in the Conflict Between Iran and Iraq. New York, NY: United Nations (1986). p. 12.
2. Seyed MR, Mahdiyeh SR, Mohsen P, Payman S. Iraq-Lran chemical war: calendar, mortality and morbidity. Chin J Traumatol. (2014) 17:165–9.
3. Lodhi IJ, Sweeney JF, Clift RE, Hinshaw DB. Nuclear dependence of sulfur mustard-mediated cell death. Toxicol Appl Pharmacol. (2001) 170:69–77. doi: 10.1006/taap.2000.9083
4. Somani S, Babu S. Toxicodynamics of sulfur mustard. Int J Clin Pharmacol Ther Toxicol. (1989) 27:419–35.
5. Evison D, Hinsley D, Rice P. Regular review: chemical weapons. BMJ Br Med J. (2002) 324:332. doi: 10.1136/bmj.324.7333.332
6. Kehe K, Szinicz L. Medical aspects of sulphur mustard poisoning. Toxicology. (2005) 214:198–209. doi: 10.1016/j.tox.2005.06.014
7. Bahadori M, Pishva N, Masjedi M. History of pulmonary lesions in victims of chemical warfare. Iran J Med Sci. (1989) 14:999–1005.
8. Emad A, Rezaian G, Hosseini K, Ghayyoomi S. Chronic pulmonary sequelae of sulfur mustard gas exposure in man: a report of 36 cases. Iran J Med Sci. (1995) 20:1–4.
9. Laskin JD, Black AT, Jan YH, Sinko PJ, Heindel ND, Sunil V, et al. Oxidants and antioxidants in sulfur mustard–induced injury. Ann N Y Acad Sci. (2010) 1203:92–100. doi: 10.1111/j.1749-6632.2010.05605.x
10. Chan A, Allen R. Bronchiolitis obliterans: an update. Curr Opin Pulm Med. (2004) 10:133–41. doi: 10.1097/00063198-200403000-00008
11. Ghanei M, Khalili ARH, Arab MJ, Mojtahedzadeh M, Aslani J, Lessan-Pezeshki M, et al. Diagnostic and therapeutic value of short-term corticosteroid therapy in exacerbation of mustard gas-induced chronic bronchitis. Basic Clin Pharmacol Toxicol. (2005) 97:302–5. doi: 10.1111/j.1742-7843.2005.pto_187.x
12. Hosseini K, Moradi A, Mansouri A, Vessal K. Pulmonary manifestations of mustard gas injury: a review of 61 cases. Iran J Med Sci. (1989) 14:20–6.
13. Emad A, Rezaian GR. The diversity of the effects of sulfur mustard gas inhalation on respiratory system 10 years after a single, heavy exposure analysis of 197 cases. Chest J. (1997) 112:734–8. doi: 10.1378/chest.112.3.734
14. Khazdair MR, Boskabady MH, Ghorani V. Respiratory effects of sulfur mustard exposure, similarities and differences with asthma and COPD. Inhalz Toxicol. (2015) 27:731–44. doi: 10.3109/08958378.2015.1114056
15. McClintock SD, Till GO, Smith MG, Ward PA. Protection from half-mustard-gas-induced acute lung injury in the rat. J Appl Toxicol. (2002) 22:257–62. doi: 10.1002/jat.856
16. Gao X, Ray R, Xiao Y, Ishida K, Ray P. Macrolide antibiotics improve chemotactic and phagocytic capacity as well as reduce inflammation in sulfur mustard-exposed monocytes. Pulm Pharmacol Ther. (2010) 23:97–106. doi: 10.1016/j.pupt.2009.10.010
17. Anderson DR, Yourick JJ, Moeller RB, Petrali JP, Young GD, Byers SL. Pathologic changes in rat lungs following acute sulfur mustard inhalation. Inhal Toxicol. (1996) 8:285–97. doi: 10.3109/08958379609005436
18. Malaviya R, Sunil VR, Cervelli J, Anderson DR, Holmes WW, Conti ML, et al. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol Appl Pharmacol. (2010) 248:89–99. doi: 10.1016/j.taap.2010.07.018
19. Amir A, Chapman S, Kadar T, Gozes Y, Sahar R, Allon N. Sulfur mustard toxicity in macrophages: effect of dexamethasone. J Appl Toxicol. (2000) 20:S51–8. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat689>3.0.co;2-5
20. Pourfarzam S, Ghazanfari T, Yaraee R, Ghasemi H, Hassan ZM, Faghihzadeh S, et al. Serum levels of IL-8 and IL-6 in the long term pulmonary complications induced by sulfur mustard: Sardasht-Iran cohort study. Int Immunopharmacol. (2009) 9:1482–8. doi: 10.1016/j.intimp.2009.09.002
21. Attaran D, Lari SM, Khajehdaluee M, Ayatollahi H, Towhidi M, Asnaashari A, et al. Highly sensitive C-reactive protein levels in Iranian patients with pulmonary complication of sulfur mustard poisoning and its correlation with severity of airway diseases. Hum Exp Toxicol. (2009) 28:739–45. doi: 10.1177/0960327109354311
22. Ghazanfari T, Sharifnia Z, Yaraee R, Pourfarzam S, Kariminia A, Mahlojirad M, et al. Serum soluble fas ligand and nitric oxide in long-term pulmonary complications induced by sulfur mustard: Sardasht-Iran Cohort Study. Int Immunopharmacol. (2009) 9:1489–93. doi: 10.1016/j.intimp.2009.08.019
23. Feister AJ. Medical Defense Against Mustard Gas: Toxic Mechanisms and Pharmacological Implications. Boca Raton, FL: CRC Press (1991). p. 43–78.
24. Hinshaw DB, Lodhi IJ, Hurley LL, Atkins KB, Dabrowska MI. Activation of poly [ADP-ribose] polymerase in endothelial cells and keratinocytes: role in anin vitromodel of sulfur mustard-mediated vesication. Toxicol Appl Pharmacol. (1999) 156:17–29. doi: 10.1006/taap.1999.8634
25. Khazdair MR, Boskabady MH. Long term effect of sulfur mustard exposure on hematologic and respiratory status, a case control study. Drug Chem Toxicol. (2019) 42:295–9. doi: 10.1080/01480545.2018.1463239
26. Razavi SM, Karbakhsh M, Salamati P. Preventive measures against the mustard gas: a review. Med J Islam Repub Iran. (2013) 27:83.
27. Wigenstam E, Rocksén D, Ekstrand-Hammarström B, Bucht A. Treatment with dexamethasone or liposome-encapsuled vitamin E provides beneficial effects after chemical-induced lung injury. Inhal Toxicol. (2009) 21:958–64. doi: 10.1080/08958370802596298
28. Sawyer TW. Characterization of the protective effects of L-nitroarginine methyl ester (L-NAME) against the toxicity of sulphur mustard in vitro. Toxicology. (1998) 131:21–32. doi: 10.1016/s0300-483x(98)00120-6
29. Callaway S, Pearce K. Protection against systemic poisoning by mustard gas, di (2-chloroethyl) sulphide, by sodium thio-sulphate and thiocit in the albino rat. Br J Pharmacol Chemother. (1958) 13:395–8. doi: 10.1111/j.1476-5381.1958.tb00227.x
30. Poursaleh Z, Harandi AA, Vahedi E, Ghanei M. Treatment for sulfur mustard lung injuries; new therapeutic approaches from acute to chronic phase. DARU J Pharm Sci. (2012) 20:27. doi: 10.1186/2008-2231-20-27
31. Aghanouri R, Ghanei M, Aslani J, Keivani-Amine H, Rastegar F, Karkhane A. Fibrogenic cytokine levels in bronchoalveolar lavage aspirates 15 years after exposure to sulfur mustard. Am J Physiol Lung Cell Mol Physiol. (2004) 287:L1160–4. doi: 10.1152/ajplung.00169.2003
32. Hosseini-Khalili A, Haines DD, Modirian E, Soroush M, Khateri S, Joshi R, et al. Mustard gas exposure and carcinogenesis of lung. Mutat Res. (2009) 678:1–6. doi: 10.1016/j.mrgentox.2009.05.022
33. Culpitt SV, Maziak W, Loukidis S, Nightingale JA, Matthews JL, Barnes PJ. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Resp Crit Care Med. (1999) 160:1635–9. doi: 10.1164/ajrccm.160.5.9811058
34. Aslani J. Late respiratory complications of sulfur mustard. In: Cheraghali AM, editor. Prevention and Treatment of Complications of Chemical Warfare Agents. Tehran: Chemical Warfare Research Center (2000). p. 76.
35. Ghanei M, Shohrati M, Harandi AA, Eshraghi M, Aslani J, Alaeddini F, et al. Inhaled corticosteroids and long-acting β2-agonists in treatment of patients with chronic bronchiolitis following exposure to sulfur mustard. Inhal Toxicol. (2007) 19:889–94. doi: 10.1080/08958370701432132
36. Khazdair MR, Boskabady MH. Possible treatment with medicinal herbs and their ingredients of lung disorders induced by sulfur mustard exposures: a review. Environ Sci Poll Res. (2021) 28:54191–208. doi: 10.1007/s11356-021-15697-2
37. Varga J, Olsen A, Herhal J, Constantine G, Rosenbloom J, Jimenez S. Interferon−γ reverses the stimulation of collagen but not fibronectin gene expression by transforming growth factor−β in normal human fibroblasts. Eur J Clin Invest. (1990) 20:487–93. doi: 10.1111/j.1365-2362.1990.tb01890.x
38. Yufit T, Vining V, Wang L, Brown RR, Varga J. Inhibition of type I collagen mRNA expression independent of tryptophan depletion in interferon-γ-treated human dermal fibroblasts. J Invest Dermatol. (1995) 105:388–93. doi: 10.1111/1523-1747.ep12320990
39. Hausmann E, Hao S-Y, Pace JL, Parmely MJ. Transforming growth factor beta 1 and gamma interferon provide opposing signals to lipopolysaccharide-activated mouse macrophages. Infect Immunity. (1994) 62:3625–32. doi: 10.1128/iai.62.9.3625-3632.1994
40. Yang X, Du J, Xu X, Xu C, Song W. IFN-γ-secreting-mesenchymal stem cells exert an antitumor effect in vivo via the TRAIL pathway. J Immunol Res. (2014) 2014:318098. doi: 10.1155/2014/318098
41. Gurujeyalakshmi G, Giri S. Molecular mechanisms of antifibrotic effect of interferon gamma in bleomycin-mouse model of lung fibrosis: downregulation of TGF-β and procollagen I and III gene expression. Exp Lung Res. (1995) 21:791–808. doi: 10.3109/01902149509050842
42. Ford JG, Rennick D, Donaldson DD, Venkayya R, Mcarthur C, Hansell E, et al. IL-13 and IFN-γ: interactions in lung inflammation. J Immunol. (2001) 167:1769–77. doi: 10.4049/jimmunol.167.3.1769
43. Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block L-H. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Eng J Med. (1999) 341:1264–9. doi: 10.1056/nejm199910213411703
44. Hassan ZM, Ebtekar M. Immunological consequence of sulfur mustard exposure. Immunol Lett. (2002) 83:151–2. doi: 10.1016/s0165-2478(02)00076-7
45. Ghanei M, Panahi Y, Mojtahedzadeh M, Aslani J. Effect of gamma interferon on lung function of mustard gas exposed patients, after 15 years. Pulm Pharmacol Ther. (2006) 19:148–53. doi: 10.1016/j.pupt.2005.07.003
46. Panahi Y, Ghanei M, Vahedi E, Ghazvini A, Parvin S, Madanchi N, et al. Effect of recombinant human IFNγ in the treatment of chronic pulmonary complications due to sulfur mustard intoxication. J Immunotoxicol. (2013) 11:72–7. doi: 10.3109/1547691X.2013.797525
47. Kidney J, Dominguez M, Taylor PM, Rose M, Chung KF, Barnes PJ. Immunomodulation by theophylline in asthma. Demonstration by withdrawal of therapy. Am J Resp Crit Care Med. (1995) 151:1907–14. doi: 10.1164/ajrccm.151.6.7767539
48. Yano Y, Yoshida M, Hoshino S, Inoue K, Kida H, Yanagita M, et al. Anti-fibrotic effects of theophylline on lung fibroblasts. Biochem Biophys Res Commun. (2006) 341:684–90. doi: 10.1016/j.bbrc.2006.01.018
49. Saleh TSF, Calixto JB, Medeiros YS. Anti-inflammatory effects of theophylline, cromolyn and salbutamol in a murine model of pleurisy. Br J Pharmacol. (1996) 118:811–9. doi: 10.1111/j.1476-5381.1996.tb15472.x
50. Sun X, Li Q, Gong Y, Ren L, Wan H, Deng W. Low-dose theophylline restores corticosteroid responsiveness in rats with smoke-induced airway inflammation. Can J Physiol Pharmacol. (2012) 90:895–902. doi: 10.1139/y2012-079
51. Kaneko Y, Takashima K, Suzuki N, Yamana K. Effects of theophylline on chronic inflammatory lung injury induced by LPS exposure in guinea pigs. Allergol Int. (2007) 56:445–56. doi: 10.2332/allergolint.O-07-490
52. Avital A, Sanchez I, Holbrow J, Kryger M, Chernick V. Effect of theophylline on lung function tests, sleep quality, and nighttime 8a02 in children with cystic fibrosis1-3. Am Rev Respir Dis. (1991) 144:1245–9. doi: 10.1164/ajrccm/144.6.1245
53. Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. COPD. (2005) 2:445–55. doi: 10.1080/15412550500346683
54. Spears M, Donnelly I, Jolly L, Brannigan M, Ito K, Mcsharry C, et al. Effect of theophylline plus beclometasone on lung function in smokers with asthma-a pilot study. Eur Resp J. (2009) 33:1010–7. doi: 10.1183/09031936.00158208
55. Panahi Y, Poursaleh Z, Amini-Harandi A, Saburi A, Shohrati M, Ghanei M. Study on effectiveness of low dose theophylline as add-on to inhaled corticosteroid for patients with sulfur mustard induced bronchiolitis. Asia Pac J Med Toxicol. (2013) 2:126–30.
56. Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg. (1996) 23:336–46. doi: 10.1016/s0741-5214(96)70279-3
57. Shakarjian MP, Heck DE, Gray JP, Sinko PJ, Gordon MK, Casillas RP, et al. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol Sci. (2010) 114:5–19. doi: 10.1093/toxsci/kfp253
58. Fujita M, Harada E, Ikegame S, Ye Q, Ouchi H, Inoshima I, et al. Doxycycline attenuated lung injury by its biological effect apart from its antimicrobial function. Pulm Pharmacol Ther. (2007) 20:669–75. doi: 10.1016/j.pupt.2006.08.006
59. Ng H, Narasaraju T, Phoon M, Sim M, Seet J, Chow VT. Doxycycline treatment attenuates acute lung injury in mice infected with virulent influenza H3N2 virus: involvement of matrix metalloproteinases. Exp Mol Pathol. (2012) 92:287–95. doi: 10.1016/j.yexmp.2012.03.003
60. Moon A, Gil S, Gill SE, Chen P, Matute-Bello G. Doxycycline impairs neutrophil migration to the airspaces of the lung in mice exposed to intratracheal lipopolysaccharide. J Inflamm. (2012) 9:31. doi: 10.1186/1476-9255-9-31
61. Zhang C, Gong W, Liu H, Guo Z, Ge S. Inhibition of matrix metalloproteinase-9 with low-dose doxycycline reduces acute lung injury induced by cardiopulmonary bypass. Int J Clin Exp Med. (2014) 7:4975.
62. Fujita M, Ye Q, Ouchi H, Harada E, Inoshima I, Kuwano K, et al. Doxycycline attenuated pulmonary fibrosis induced by bleomycin in mice. Antimicrob Agents Chemother. (2006) 50:739–43. doi: 10.1128/AAC.50.2.739-743.2006
63. Guignabert C, Taysse L, Calvet J-H, Planus E, Delamanche S, Galiacy S, et al. Effect of doxycycline on sulfur mustard-induced respiratory lesions in guinea pigs. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L67–74. doi: 10.1152/ajplung.00475.2004
64. Manavi K, Hettiarachchi N, Hodson J. Comparison of doxycycline with azithromycin in treatment of pharyngeal chlamydia infection. Int J STD AIDS. (2016) 27:1303–8. doi: 10.1177/0956462415614723
65. Xu X, Abdalla T, Bratcher PE, Jackson PL, Sabbatini G, Wells JM, et al. Doxycycline improves clinical outcomes during cystic fibrosis exacerbations. Eur Respir J. (2017) 49:1601102. doi: 10.1183/13993003.01102-2016
66. Singh B, Jana SK, Ghosh N, Das SK, Joshi M, Bhattacharyya P, et al. Metabolomic profiling of doxycycline treatment in chronic obstructive pulmonary disease. J Pharm Biomed Anal. (2017) 132:103–8. doi: 10.1016/j.jpba.2016.09.034
67. Barreto AD, Alexandrov AV, Lyden P, Lee J, Martin-Schild S, Shen L, et al. The argatroban and tissue-type plasminogen activator stroke study final results of a pilot safety study. Stroke. (2012) 43:770–5. doi: 10.1161/strokeaha.111.625574
68. Veress LA, O’neill HC, Hendry-Hofer TB, Loader JE, Rancourt RC, White CW. Airway obstruction due to bronchial vascular injury after sulfur mustard analog inhalation. Am J Resp Crit Care Med. (2010) 182:1352–61. doi: 10.1164/rccm.200910-1618OC
69. Veress LA, Hendry-Hofer TB, Loader JE, Rioux JS, Garlick RB, White CW. Tissue plasminogen activator prevents mortality from sulfur mustard analog–induced airway obstruction. Am J Resp Cell Mol Biol. (2013) 48:439–47. doi: 10.1165/rcmb.2012-0177oc
70. Veress LA, Anderson DR, Hendry-Hofer TB, Houin PR, Rioux JS, Garlick RB, et al. Airway tissue plasminogen activator prevents acute mortality due to lethal sulfur mustard inhalation. Toxicol Sci. (2015) 143:178–84. doi: 10.1093/toxsci/kfu225
71. Goldhaber S, Heit J, Sharma G, Nagel JS, Kim D, Parker JA, et al. Randomised controlled trial of recombinant tissue plasminogen activator versus urokinase in the treatment of acute pulmonary embolism. Lancet. (1988) 332:293–8. doi: 10.1016/s0140-6736(88)92354-9
72. Lwaleed BA, Bass PS. Tissue factor pathway inhibitor: structure, biology and involvement in disease. J Pathol. (2006) 208:327–39. doi: 10.1002/path.1871
73. Ran Y, Pan J, Hu H, Zhou Z, Sun L, Peng L, et al. A novel role for tissue factor pathway inhibitor-2 in the therapy of human esophageal carcinoma. Hum Gene Ther. (2009) 20:41–9. doi: 10.1089/hum.2008.129
74. Enkhbaatar P, Okajima K, Murakami K, Uchiba M, Okabe H, Okabe K, et al. Recombinant tissue factor pathway inhibitor reduces lipopolysaccharide-induced pulmonary vascular injury by inhibiting leukocyte activation. Am J Resp Crit Care Med. (2000) 162:1752–9. doi: 10.1164/ajrccm.162.5.9911018
75. Amirkhosravi A, Meyer T, Chang J-Y, Amaya M, Siddiqui F, Desai H, et al. Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb Haemostas. (2002) 88:930–6. doi: 10.1055/s-0037-1613114
76. Rancourt RC, Veress LA, Ahmad A, Hendry-Hofer TB, Rioux JS, Garlick RB, et al. Tissue factor pathway inhibitor prevents airway obstruction, respiratory failure and death due to sulfur mustard analog inhalation. Toxicol Appl Pharmacol. (2013) 272:86–95. doi: 10.1016/j.taap.2013.05.020
78. Gon Y, Hashimoto S, Nakayama T, Matsumoto K, Koura T, Takeshita I, et al. N-acetyl-L-cysteine inhibits bleomycin-induced interleukin-8 secretion by bronchial epithelial cells. Respirology. (2000) 5:309–13. doi: 10.1046/j.1440-1843.2000.00268.x
79. Failli P, Palmieri L, D’alfonso C, Giovannelli L, Generini S, Del Rosso A, et al. Effect of N-acetyl-L-cysteine on peroxynitrite and superoxide anion production of lung alveolar macrophages in systemic sclerosis. Nitric Oxide. (2002) 7:277–82. doi: 10.1016/s1089-8603(02)00120-9
80. Gürer H, Özgünes H, Neal R, Spitz DR, Erçal N. Antioxidant effects of N-acetylcysteine and succimer in red blood cells from lead-exposed rats. Toxicology. (1998) 128:181–9. doi: 10.1016/s0300-483x(98)00074-2
81. Mitsopoulos P, Omri A, Alipour M, Vermeulen N, Smith MG, Suntres ZE. Effectiveness of liposomal-N-acetylcysteine against LPS-induced lung injuries in rodents. Int J Pharm. (2008) 363:106–11. doi: 10.1016/j.ijpharm.2008.07.015
82. Atkins KB, Lodhi IJ, Hurley LL, Hinshaw DB. N-acetylcysteine and endothelial cell injury by sulfur mustard. J Appl Toxicol. (2000) 20:S125–8. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat671>3.0.co;2-u
83. Zaree Mahmoudabad A, Saberi M, Pirzad J. Critical role of GSH in sulfur mustard-induced oxidative stress and cytotoxicity in human skin fibroblast cell line. Iran J Pharm Res. (2010) 7:35–41. doi: 10.1016/j.toxlet.2015.09.002
84. Sciuto AM, Strickland PT, Kennedy TP, Gurtner GH. Protective effects of N-acetylcysteine treatment after phosgene exposure in rabbits. Am J Resp Crit Care Med. (1995) 151:768–72. doi: 10.1164/ajrccm.151.3.7881668
85. Calvet J-H, Jarreau P-H, Levame M, D’ortho M, Lorino H, Harf A, et al. Acute and chronic respiratory effects of sulfur mustard intoxication in guinea pig. J Appl Physiol. (1994) 76:681–8. doi: 10.1152/jappl.1994.76.2.681
86. Čulić O, Eraković V, Parnham MJ. Anti-inflammatory effects of macrolide antibiotics. Eur J Pharmacol. (2001) 429:209–29. doi: 10.1016/s0014-2999(01)01321-8
87. Anderson DR, Byers SL, Vesely KR. Treatment of sulfur mustard (HD)-induced lung injury†. J Appl Toxicol. (2000) 20:S129–32.
88. Jugg B, Fairhall S, Smith A, Rutter S, Mann T, Perrott R, et al. N-acetyl-L-cysteine protects against inhaled sulfur mustard poisoning in the large swine. Clin Toxicol. (2013) 51:216–24. doi: 10.3109/15563650.2013.780208
89. Kupczyk M, Kuna P. [Mucolytics in acute and chronic respiratory tract disorders. II. Uses for treatment and antioxidant properties]. Polski Merkuriusz Lekarski. (2002) 12:248–52.
90. Kupczyk M, Kuna P. [Mucolytics in acute and chronic respiratory tract disorders. I. Pathophysiology and mechanisms of action]. Polski Merkuriusz Lekarski. (2002) 12:245–7.
91. Grassi C, Morandini G. A controlled trial of intermittent oral acetylcysteine in the long-term treatment of chronic bronchitis. Eur J Clin Pharmacol. (1976) 9:393–6. doi: 10.1007/BF00606554
92. Völkl K, Schneider B. [Therapy of respiratory tract diseases with n-acetylcysteine. An open therapeutic observation study of 2,512 patients]. Fortschritte der Medizin. (1992) 110:346–50.
93. Allegra L, Dal Sasso M, Bovio C, Massoni C, Fonti E, Braga PC. Human neutrophil oxidative bursts and their in vitro modulation by different N-acetylcysteine concentrations. Arzneimittel Forschung. (2001) 52:669–76. doi: 10.1055/s-0031-1299949
94. Jankowska R, Passowicz-Muszyńska E, Medrala W, Banaś T, Marcinkowska A. [The influence of N-acetylcysteine on chemiluminescence of granulocytes in peripheral blood of patients with chronic bronchitis]. Pneumonol Alergol Polska. (1992) 61:586–91.
95. Domenighetti G, Suter PM, Schaller M-D, Ritz R, Perret C. Treatment with N-acetylcysteine during acute respiratory distress syndrome: a randomized, double-blind, placebo-controlled clinical study. J Crit Care. (1997) 12:177–82. doi: 10.1016/s0883-9441(97)90029-0
96. Suter PM, Domenighetti G, Schaller M-D, Laverrière M-C, Ritz R, Perret C. N-acetylcysteine enhances recovery from acute lung injury in man: a randomized, double-blind, placebo-controlled clinical study. Chest. (1994) 105:190–4. doi: 10.1378/chest.105.1.190
97. Ghanei M, Shohrati M, Jafari M, Ghaderi S, Alaeddini F, Aslani J. N-acetylcysteine improves the clinical conditions of mustard gas-exposed patients with normal pulmonary function test. Basic Clin Pharmacol Toxicol. (2008) 103:428–32. doi: 10.1111/j.1742-7843.2008.00318.x
98. Shohrati M, Aslani J, Eshraghi M, Alaedini F, Ghanei M. Therapeutics effect of N-acetyl cysteine on mustard gas exposed patients: evaluating clinical aspect in patients with impaired pulmonary function test. Resp Med. (2008) 102:443–8. doi: 10.1016/j.rmed.2007.10.004
99. Shohrati M, Karimzadeh I, Saburi A, Khalili H, Ghanei M. The role of N-acetylcysteine in the management of acute and chronic pulmonary complications of sulfur mustard: a literature review. Inhal Toxicol. (2014) 26:507–23. doi: 10.3109/08958378.2014.920439
100. Sawyer TW. N-acetylcysteine as a treatment for sulphur mustard poisoning. Free Radic Biol Med. (2020) 161:305–20. doi: 10.1016/j.freeradbiomed.2020.09.020
101. Gallo C, Renzi P, Loizzo S, Loizzo A, Piacente S, Festa M, et al. Potential therapeutic effects of vitamin E and C on placental oxidative stress induced by nicotine: an in vitro evidence. Open Biochem J. (2010) 4:77. doi: 10.2174/1874091X01004010077
102. Honegger UE, Scuntaro I, Wiesmann UN. Vitamin E reduces accumulation of amiodarone and desethylamiodarone and inhibits phospholipidosis in cultured human cells. Biochem Pharmacol. (1995) 49:1741–5. doi: 10.1016/0006-2952(95)00100-e
103. Gürbay A, Garrel C, Osman M, Richard M, Favier A, Hincal F. Cytotoxicity in ciprofloxacin-treated human fibroblast cells and protection by vitamin E. Human Exp Toxicol. (2002) 21:635–41. doi: 10.1191/0960327102ht305oa
104. Boskabady MH, Tabatabayee A, Amiri S, Vahedi N. The effect of vitamin E on pathological changes in kidney and liver of sulphur mustard-exposed guinea pigs. Toxicol Indust Health. (2012) 28:216–21. doi: 10.1177/0748233711410908
105. Boskabady MH, Amery S, Vahedi N, Khakzad MR. The effect of vitamin E on tracheal responsiveness and lung inflammation in sulfur mustard exposed guinea pigs. Inhal Toxicol. (2011) 23:157–65. doi: 10.3109/08958378.2011.558934
106. Ozdil S, Bolkent Ş, Yanardag R, Arda-Pirincci P. Protective effects of ascorbic acid, dl-α-tocopherol acetate, and sodium selenate on ethanol-induced liver damage of rats. Biol Trace Element Res. (2004) 97:149–61. doi: 10.1385/BTER:97:2:149
107. Gholamnezhad Z, Boskabady MH, Amery S, Vahedi N, Tabatabaei A, Boskabady M, et al. The effect of vitamin E on lung pathology in sulfur mustard-exposed guinea pigs. Toxicol Indust Health. (2016) 32:1971–7. doi: 10.1177/0748233715600986
108. Vijayaraghavan R, Gautam A, Sharma M, Satish H, Pant S, Ganesan K. Comparative evaluation of some flavonoids and tocopherol acetate against the systemic toxicity induced by sulphur mustard. India J Pharmacol. (2008) 40:114. doi: 10.4103/0253-7613.42304
109. Mukherjee S, Stone WL, Yang H, Smith MG, Das SK. Protection of half sulfur mustard gas–induced lung injury in guinea pigs by antioxidant liposomes. J Biochem Mol Toxicol. (2009) 23:143–53. doi: 10.1002/jbt.20279
110. Mukhopadhyay S, Mukherjee S, Stone WL, Smith M, Das SK. Role of MAPK/AP-1 signaling pathway in the protection of CEES-induced lung injury by antioxidant liposome. Toxicology. (2009) 261:143–51. doi: 10.1016/j.tox.2009.05.010
111. Dow L, Tracey M, Villar A, Coggon D, Margetts BM, Campbell MJ, et al. Does dietary intake of vitamins C and E influence lung function in older people? Am J Resp Crit Care Med. (1996) 154:1401–4. doi: 10.1164/ajrccm.154.5.8912755
112. Tabak C, Smit HA, Räsänen L, Fidanza F, Menotti A, Nissinen A, et al. Dietary factors and pulmonary function: a cross sectional study in middle aged men from three European countries. Thorax. (1999) 54:1021–6. doi: 10.1136/thx.54.11.1021
113. Hoskins A, Roberts JL, Milne G, Choi L, Dworski R. Natural-source D-α-tocopheryl acetate inhibits oxidant stress and modulates atopic asthma in humans in vivo. Allergy. (2012) 67:676–82. doi: 10.1111/j.1398-9995.2012.02810.x
114. Pearson P, Lewis S, Britton J, Fogarty A. Vitamin E supplements in asthma: a parallel group randomised placebo controlled trial. Thorax. (2004) 59:652–6. doi: 10.1136/thx.2004.022616
Keywords: sulfur mustard (SM), treatment, pharmaceutical drugs, chemical agents, lung injury
Citation: Khazdair MR and Boskabady MH (2022) Possible Treatment Approaches of Sulfur Mustard-Induced Lung Disorders, Experimental and Clinical Evidence, an Updated Review. Front. Med. 9:791914. doi: 10.3389/fmed.2022.791914
Received: 07 December 2021; Accepted: 12 April 2022;
Published: 29 April 2022.
Edited by:
Nahal Mansouri, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Sermet Sezigen, University of Health Sciences, TurkeyPai-Chien Chou, Taipei Medical University Hospital, Taiwan
Copyright © 2022 Khazdair and Boskabady. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Hossein Boskabady, Ym9za2FiYWR5bWhAbXVtcy5hYy5pcg==; Ym9za2FiYWR5bWgyQGdtYWlsLmNvbQ==