 Ye Cheng1†Yanna Chen1†Guodong Wang1Pei Liu1Guiling Xie1Huan Jing1Hongtao Chen2Youlin Fan3Min Wang4
Ye Cheng1†Yanna Chen1†Guodong Wang1Pei Liu1Guiling Xie1Huan Jing1Hongtao Chen2Youlin Fan3Min Wang4 Jun Zhou1*
Jun Zhou1*
- 1Department of Anesthesiology, The Third Affiliated Hospital of Southern Medical University, Guangzhou, China
- 2Department of Anesthesiology, The Eighth People’s Hospital of Guangzhou, Guangzhou, China
- 3Department of Anesthesiology, Guangzhou Panyu Central Hospital of Panyu District, Guangzhou, China
- 4Department of Anesthesiology, The Gaoming People’s Hospital, Foshan, China
Chronic kidney disease (CKD) is defined by persistent urine aberrations, structural abnormalities, or impaired excretory renal function. Diabetes is the leading cause of CKD. Their common pathological manifestation is renal fibrosis. Approximately half of all patients with type 2 diabetes and one-third with type 1 diabetes will develop CKD. However, renal fibrosis mechanisms are still poorly understood, especially post-transcriptional and epigenetic regulation. And an unmet need remains for innovative treatment strategies for preventing, arresting, treating, and reversing diabetic kidney disease (DKD). People believe that protein methylation, including histone and non-histone, is an essential type of post-translational modification (PTM). However, prevalent reviews mainly focus on the causes such as DNA methylation. This review will take insights into the protein part. Furthermore, by emphasizing the close relationship between protein methylation and DKD, we will summarize the clinical research status and foresee the application prospect of protein methyltransferase (PMT) inhibitors in DKD treatment. In a nutshell, our review will contribute to a more profound understanding of DKD’s molecular mechanism and inspire people to dig into this field.
Introduction
Chronic kidney disease (CKD) is a fatal cause of mortality worldwide. The prevalence of CKD has increased steadily over the past decade. Besides the increased mortality, CKD also significantly lessens patients’ quality of life and imposes a financial burden on the economy (1–4). CKD is diagnosed when there is a chronic reduction in kidney function and chronic damage in the structure. Both events are the final common pathological manifestations of renal fibrosis. Renal fibrosis represents unsuccessful wound healing of the kidney tissue. This fibrosis is characterized by glomerulosclerosis, tubular atrophy, and interstitial fibrosis. As fibrosis evolves, an increasing number of nephrons lose their function (5, 6).
Approximately half of all patients with type 2 diabetes and one-third with type 1 diabetes will develop CKD, which is clinically defined by the presence of impaired renal function or elevated urinary albumin excretion or both (7, 8). DKD has been traditionally viewed as a microvascular disorder, clustered along with retinopathy and neuropathy, and separate from the macrovascular disease that contributes to coronary heart disease (CHD), peripheral vascular disease, and cerebrovascular disease. However, each disorder can be considered tissue-specific manifestations of the same pathogenetic process. DKD is the renal manifestation of the same glucose-driven process at susceptible sites elsewhere in the body. Although all cells are chronically exposed to high plasma glucose levels in diabetic patients, only some manifest progressive dysfunction. The endothelial cells lining the vasculature are a prime example. Specifically, the inability of endothelial cells to down-regulate their glucose transport in response to high glucose leads to an overwhelming flux of intracellular glucose, which triggers the generation of pathogenetic mediators that contribute to the development of diabetic complications, including DKD (9, 10).
Despite genetic risks, environmental influence may be strongly associated with susceptibility to DKD. Epigenetic modifications refer to gene transcription changes that manifest in the phenotype, which can be inherited through multiple cell divisions. As the most pervasive epigenetic modifications, DNA methylation and histone modifications cause stable gene expression via chromatin remodeling. These mechanisms play a role in determining the developmental cell fate and human physiological and pathological processes. As the most intensively investigated PTM, protein methylation has become a prominent research topic of interest. Protein methylation is part of many critical biological functions that play a role in healthy physiological development and diseases, such as obesity and type 2 diabetes (11).
In this review, to present existing knowledge on the underlying mechanism of protein methylation, we consider the biological function of protein methylation, followed by the relationship between protein methylation and DKD. The evidence presented here supports the expectation that drugs targeting protein methylation will prove value for DKD treatment.
Biological Functions of Protein Methylation
Protein methylation, including histone methylation and non-histone protein methylation, is an essential type of PTM (12). Protein methylation mainly occurs on lysine and arginine. The methylation is performed by protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs), including members of the PRMT family, SET gene family, and non-SET gene family (13–15). Methylation occurs at different amino acid sites and in various forms. For example, lysine can be monomethylated (me1), dimethylated (me2), or trimethylated (me3), while arginine can be monomethylated (me1), symmetrically dimethylated (me2s), or asymmetrically dimethylated (me2a) on its guanidine group (16). These results indicate that protein methylation is flexible and diverse (17). Methylation affects protein activity, protein-protein interactions, and interaction with other PTMs (17).
Histones are widely studied owing to their abundance and relatively easy detection. Protein methylation is often associated with histones and their roles in gene regulation (18). Histone methylation is associated with gene repression or activation, depending on which residue is modified (19). Methylation of histone H3 at lysines 4 or 36 positively correlates with the transcription rate of RNA polymerase (pol) II. Histone methyltransferases (HMTs) for these methyl groups interact with elongated RNA pol II to form histone methylations in transcribed regions. In contrast, transcription repression factors recruit repressive HMTs for H3K9 or H3K27 methylation (20–24). Lysine and arginine methylation have been found in hundreds of non-histone proteins, and the terms, PKMT and PRMT, are now more frequently used than the original term, HMT. In addition to histone-centric roles, the crucial role of non-histone proteins should not be ignored. The latter have significant implications for human health and the treatment of human diseases (25, 26) (Figure 1).
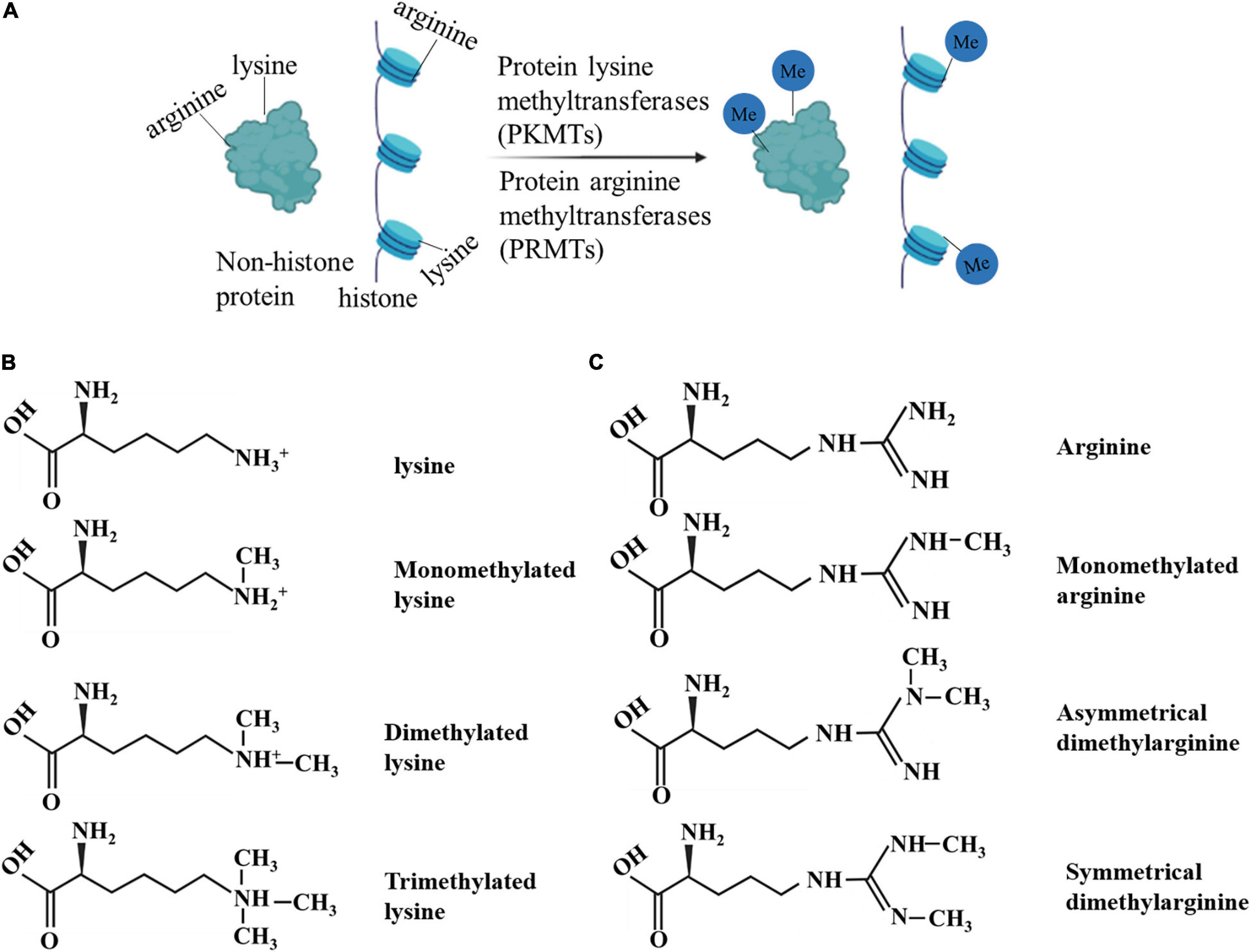
Figure 1. (A–C) Diverse protein methylation.
Protein Methylation in Diabetic Kidney Disease
The associations between the dysregulation of PMT and the progression of many human diseases have been described (16, 27–29). SET9-mediated methylation of inhibitory Smad7, a crucial regulator of transforming growth factor-beta (TGF-β)/bone morphogenetic protein signaling by negative feedback loops, might lead to its degradation, thereby affecting TGF-β-dependent activation of extracellular matrix (ECM) genes. Critical components of mitogen-activated protein kinase, such as redundant acronym syndrome and retinoblastoma protein, are subject to lysine and arginine methylation, often in a complex manner (30, 31). Numerous studies have already revealed that protein methylation is associated with DKD and that the final common pathway in the transition from CKD to ESRD is fibrosis (6, 32). It is still in its infancy to understand protein methylation in DKD, including its occurence, progression, and treatment. However, research on the connection between histone methylation and non-histone methylation has flourished recently (33). Below, we summarize studies on the relationship between DKD and protein methylation, including histone methylation and non-histone methylation.
Histone Methylation in Diabetic Kidney Disease
Methylation is the most widely studied modification of histone proteins. Methylation of histone proteins changes the transcription machinery, creating an active or repressive chromatin structure and transcriptional marks (34). In fact, histone methylation is usually considered a prevalent modification among core histone tails and is one of the most stable PTMs. Histone methylation mainly includes the methylation of lysine and arginine residues. Among these, lysine is the most well-studied. Histone methylation is dynamic and reversible. Histone methylation has a mixed gene expression effect, unlike histone acetylation linked to an “open” chromatin state and gene expression activation. Therefore, they can be marks of the active and repressive states of chromatin, thereby producing different developmental effects. Researchers have found that histone methylation plays an essential role in DKD progression (35, 36).
Histone Lysine Methylation in Diabetic Kidney Disease
Different modified-residue of histone lysine corresponds to gene repression or activation. Methylation at H3K4, H3K36, and H3K79 correlates with gene transcriptional activation, whereas methylation at H3K9, H3K27, and H4K20 is related to transcriptional repression (37). The burgeoning research in this field has included investigations of the biological and pathological functions of histone methylation in some common diseases, including diabetic nephropathy (DN), one of the most common CKDs. Below, we focus on the role of histone lysine methylation in the development and progression of DN based on active and repressive chromatin marks (38).
Active Marks of Histone Lysine Methylation in Diabetic Kidney Disease
H3K4me1/2/3, H3K36me2/3, and H3K79me2 are associated with transcriptionally active regions. In addition, increasing evidence suggests that histone methylation regulates ECM and inflammatory genes in almost all renal cell types related to the pathogenesis of DKD (29, 39).
Transforming growth factor-beta 1-induced expression of ECM genes plays an important part in the development of chronic renal diseases, such as DN. Although many key transcription factors are studied clearly, it remains unclear how the modulation of nuclear chromatin influences the expression of ECM gene. Epigenetic chromatin marks, such as H3Kme, might play a role in TGF-β1-induced gene expression in rat mesangial cells (RMC) under normal and high-glucose (HG) conditions. When treated with HG and TGF-β1, the levels of H3K4me marks (H3K4me1/2/3) at their respective promoters are increased (40, 41). The gene expression levels of collagen 1alpha1 (Col1α1), plasminogen activator inhibitor 1 (PAI-1), and connective tissue growth factor (CTGF) in RMC are also upregulated. Metabolic memory existing in vascular dysfunction has been associated with H3K4me modification (42). One critical study reported the significance of alterations in histone methylation (specifically in the lysine residues) in acute kidney injury (AKI). H3K4me3 at pro-inflammatory genes (monocyte chemotactic protein 1 [MCP-1] and tumor necrosis factor-alpha [TNF-α]) and profibrotic genes (TGF-β1 and collagen III) were increased in renal ischemia reperfusion injury (IRI) animal models (43, 44). The TNF-α and MCP-1 genes characteristically showed H3K4m3 methylation in mouse models of AKI induced by IRI, endotoxin, unilateral ureteral obstruction, and maleate (43, 45, 46). In another study on patients with AKI, the levels of H3K4m3 at exon 1 of the hydroxy methylglutaryl-CoA (HMG-CoA) reductase genes were also upregulated (47). The upregulation of histone H3K4 me3 may be involved in podocyte dysfunction, which is the most common cause of primary nephrotic syndrome in the middle-aged and elderly. Collectively, the results indicate that H3K4 methylation is essential in the progression of DKD in diverse aspects, including both inflammatory and oxidant stress, all of which will result in renal fibrosis.
H3K36me3 is a chromatin marker associated with transcriptional elongation (24, 48). The levels of H3K36me3 were found to be higher at the monocyte chemoattractant protein 1 (MCP-1) and RAGE loci, and in the plasminogen activator inhibitor-1 (PAI-1) gene in db/db mice treated with water. After treatment with losartan for 10 weeks, the H3K36me3 levels decreased at the RAGE and PAI-1 loci (49). These changes imply the role of H3K36me in DN progression. Unlike most methylated sites located in the histone H3 tail, the H3K79 methylated site is situated in the histone globular domain. Methylation of H3K79 catalyzed by a disruptor of telomeric silencing-1H3K79me plays an essential role in cell cycle regulation, embryonic development, DNA damage response, hematopoiesis, cardiac function, and development of leukemia (37). One report described the involvement of the dynamic regulation of H3K79me in fluid reabsorption, which is essential for blood pressure control and electrolyte homeostasis in kidney collecting ducts. The downregulation of H3K79me at the epithelial sodium channel promoter can lead to increased gene expression in response to aldosterone signaling (50–52), and decreased H3K79me2 may contribute to the changes in DN patients and mouse cortical collecting duct M1 cell models (53). These results suggest that H3K36me and H3K79me may play a crucial part in fluid reabsorption and chronic changes of kidney structure, which might severely affect the normal function of the kidney.
Repressive Marks of Histone Methylation in Diabetic Kidney Disease
H3K9me2/3, H3K27me3, and H4K20me3 are generally associated with gene silencing or repression. Histone methylation may be responsible for the “metabolic memory” phenomenon that leads to long-term changes in diabetic complications, including DN, and plays essential role in the progression of KD in fibrotic, inflammatory, and oxidative stress pathways.
Based on multiple evidence, H3K9me plays a crucial role in the development of DN. HG can stimulate decreased H3K9me3 levels at the promoters of critical inflammatory genes, such as interleukin (IL)-6, macrophage colony-stimulating factor, and MCP-1. These events increase the expression of these inflammatory genes in normal human vascular smooth muscle cells (VSMCs). Similar chromatin lysine methylation changes were demonstrated in the VSMCs of db/db mice compared to non-diabetic control db/ + mice (54). TNF-α induction can also lead to sustained decreases in H3K9me3 at promoters following increased inflammatory gene expression in the VSMC of db/db mice (51, 55). Of note, a similar change was noted in RMC models following treatment with TGF-β and HG. The reduced levels of the repressive marks, H3K9me2 (di-methylation at the 9th lysine residue of the histone H3 protein) and H3K9me3, at their respective promoters (40, 49) can induce the upregulation of the Col1α1, PAI-1, and CTGF genes.
H3K27me is associated with gene repression (56, 57). H3K27me3 levels at PAI-1 promoters were reportedly decreased in an animal model of type 2 diabetes. In another study, cyclo-oxygenase-2 (COX2) and MCP-1 were upregulated in the kidneys of OVE26 mice. The OVE26 mouse is characterized by transgenic overexpression of calmodulin in pancreatic β cells, deficient insulin production, and type I diabetes. This phenomenon has been associated with decreased H3K27me3 levels and H3K27me3 demethylase KDM6A/UTX) (58). Altogether, these findings regarding H3K27me3 further imply the role of histone methylation in DKD (Figure 2).
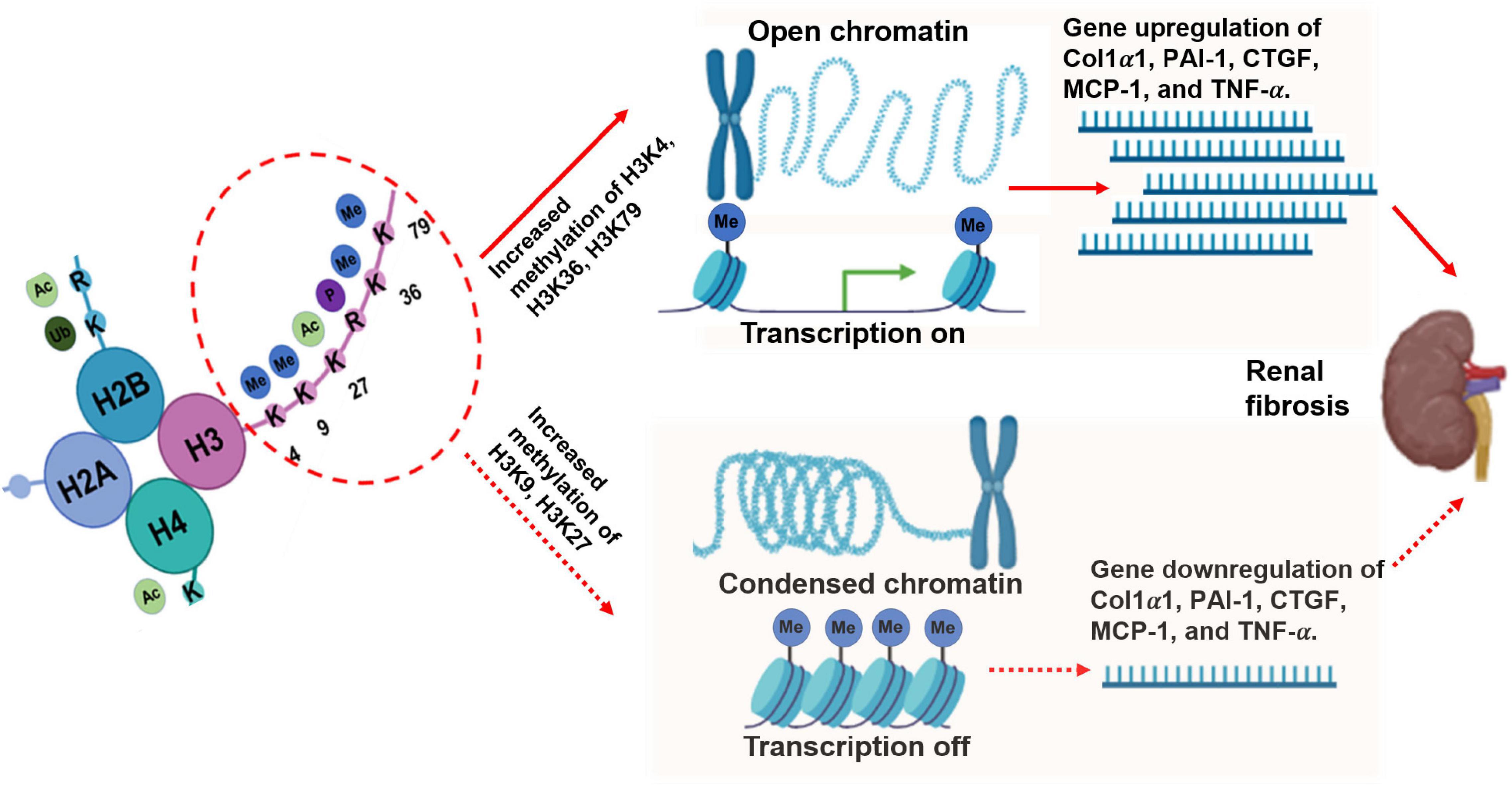
Figure 2. Histone lysine methylation in DKD.
Histone Arginine Methylation in Diabetic Kidney Disease
The methylation of arginine residues by PRMTs is involved in regulating basic cellular processes, including DNA damage response, RNA transcription and processing, signal transduction cascades, and liquid-liquid phase separation. Recent evidence has considerably advanced the identification of clinically relevant PRMT inhibitors (59) based on the defined physiological roles of PRMTs. The inhibitors have been linked to cancer, metabolic diseases, neurodegenerative disorders, and some kidney diseases (15, 60), thereby enabling the development of experimental tools.
Asymmetric dimethylarginine (ADMA) is a naturally occurring dimethylated analog of the amino acid. ADMA inhibits nitric oxide synthase and is an established cardiovascular risk factor in many diseases, including CKD (61–63). Atherosclerotic cardiovascular disease is the leading cause of death in patients with CKD. However, the underlying vascular disease mechanisms are not fully elucidated. Altered arginine methylation is associated with the degree of atherosclerosis in a CKD mouse model, suggesting the therapeutic potential of interrupting this pathway in CKD-related atherosclerosis (64). Indeed, elevated plasma levels of ADMA have been observed in patients with hypertension and CKD (61, 65–67). Furthermore, Andrade et al. observed that patients who received a renal transplant displayed only moderately decreased renal function but presented disturbances in the methylation cycle and arginine-creatine pathway that lead to increased plasma values of homocysteine (Hcys), S-adenosylhomocysteine, and ADMA. Defective methylation also contributes to endothelial dysfunction due to the impaired production of NO, which has been observed in patients that received a renal transplants (68). Further investigation of the possible benefits of appropriate therapeutic measures is warranted (69).
Non-Histone Protein Methylation in Diabetic Kidney Disease
Although some methylating enzymes are histone-specific, many have now been found to modify both histone and non-histone substrates (70–72). Non-histone protein methylation has recently emerged as a PTM with wide-ranging cellular functions (73–76). The methylation of non-histones plays a significant part in the pathogenesis of DKD. SMYD2 is a SET and MYND domain-containing HMT that methylates histone and non-histone proteins. For example, the tumor suppressors RB, p53, and the molecular chaperone heat shock protein 90 have been identified in the pathogenesis of autosomal dominant polycystic kidney disease (ADPKD) (77–81). The transcriptional regulators, STAT3 and p65, are SMYD2 non-histone substrates. Upregulation of SMYD2 leads to methylation of STAT3 at lysine 685, methylation of p65 at lysine 310, and partial methylation at lysine 221, followed by their phosphorylation and activation to regulate proliferation and apoptosis of renal epithelial cells (82). SMYD2 is overexpressed in cystic renal epithelial cells, it might serve as a promising target for novel ADPKD therapies (83). The enormous potential of drugs that target protein methylation will likely only be realized if their effects on non-histone protein methylation are better understood.
Protein Methylation as New Directions for the Diagnosis and Treatment of Diabetic Kidney Disease
All PMTs share a common catalytic mechanism. The SAM donor and peptide methyl-acceptor bind to different but connected surfaces within the active site to form a functional ternary complex. Following the assembly of the complex, direct transfer of the methyl group from SAM to the substrate proceeds via a classical bimolecular nucleophilic substitution (SN2) reaction. Chemical inhibition targets either cofactor or substrate-binding sites, as well as by allosteric means (Figure 3).
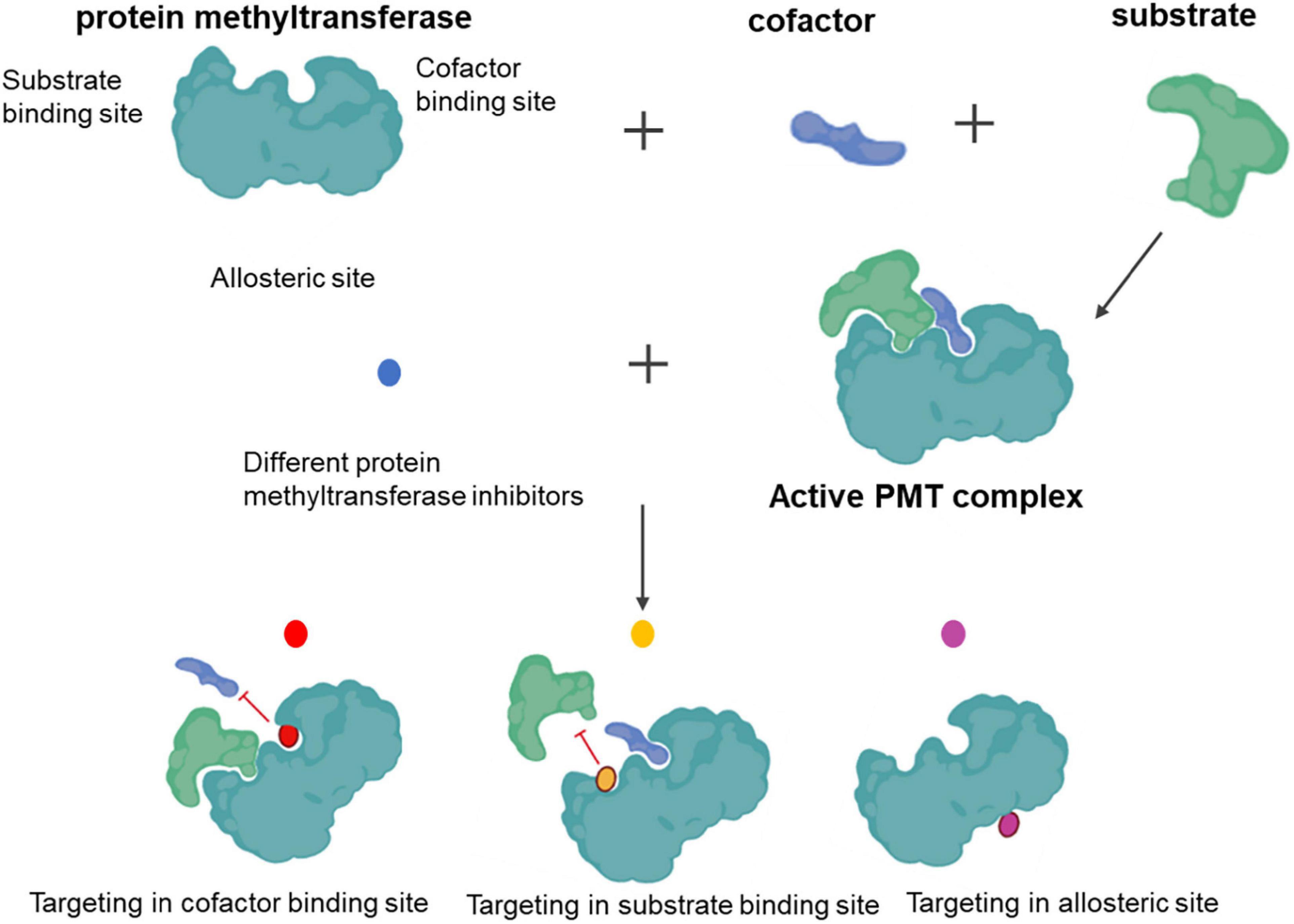
Figure 3. Functional mechanism of protein methyltransferase inhibitors.
Knowledge of the pharmacological modulation of proteins that write, read, and erase methyl marks is needed (84, 85). We have witnessed advances in PMT inhibitors from tool compounds to precision medicines. Indeed, several histone methyltransferase inhibitors have already reached clinical application (86), demonstrating the availability of PMTs as a target class. In addition to histone-centric roles, PMTs are also vital for regulating non-histone proteins, with significant implications for human health and disease treatment (25, 26). Well-defined sets and selective chemical probes have provided valuable tools for investigating biological mechanisms. Panels of well-validated protein methyltransferase probes with demonstrated utility in interrogating complex biological systems have also been described (87). Selective, potent, and cell-active inhibitors of both lysine and arginine methyltransferases have been developed by exploiting the cofactor-binding site, substrate peptide-binding site, and, less commonly, distal allosteric pockets. In 2007, the first selective small-molecule inhibitor of PKMT, BIX-01294, was reported (88). Since its discovery, this inhibitor has been used to probe G9a involvement in cellular reprogramming (89, 90). Unfortunately, the poor separation between cytotoxic and functional effects has limited the broader utility and adoption of this compound. The discovery of UNC0638 primarily overcame this issue. UNC0638 can reduce the abundance of H3K9me2 and can reactivate G9a-silenced genes and a retroviral reporter in mouse embryonic stem cells, demonstrating its usefulness in studying the biology of G9a/glucagon-like peptides (91). Although numerous studies have linked G9a to disease (92, 93), no G9a inhibitors are currently used in the clinic. The five SMYD lysine methyltransferase members methylate both histone and non-histone proteins (94). Although limited SMYD2 inhibitors have been reported, we believe that its application prospects are vast.
Renal fibrosis is the common pathological hallmark of chronic kidney disease, and the SET domain-containing lysine methyltransferase 7 (SETD7) promotes renal fibrosis considerably. Ice treated with PFI-2, an inhibitor of SETD7, presented less bone marrow-derived myofibroblasts, fewer CD206 + /α-smooth muscle actin + cells, and developed less renal fibrosis. Furthermore, SETD7 inhibition reduced the infiltration of inflammatory cells and decreased the production of pro-inflammatory cytokines and chemokines in the kidneys after folic acid treatment (95). Some in vivo and in vitro studies demonstrate that emodin reduced extracellular collagen deposition and inhibited Smad3 and CTGF pro-fibrotic signaling pathways, which were correlated with the down-regulation of EZH2 and reduced trimethylation of histone H3 on lysine 27 (H3k27me3) in NRK-49F fibrotic cells and UUO kidneys (96). Disruptor of telomeric silencing-1 like (DOT1L) protein specifically catalyzes the methylation of histone H3 on Lys79 (H3K79) and is implicated in tumors. DOT1L inhibition increased expression of phosphatase and tensin homolog, a protein associated with dephosphorylation of tyrosine kinase receptors, and prevented the decline in levels of Klotho and Smad7, two renoprotective factors. Targeting DOT1L attenuates renal fibrosis by inhibiting renal fibroblasts and EMT by suppressing the activation of multiple profibrotic signaling pathways while retaining the expression of renoprotective factors (97).
From a therapeutic point of view, reader antagonism may provide alternative routes to modulate methyl signaling pathways in DKD, which may be far-reaching when resistance has developed to existing clinical candidates (98, 99). Recent advances in the development of methyl-lysine reader antagonists present opportunities to selectively intervene in the downstream signaling of the methyl mark or as alternative sites to target PMTs themselves (100). In addition, individual proteins often contain several distinct reader modules with different binding capabilities (101). Potent, selective, and cell-active antagonists of reader function are, therefore, valuable tools to decipher the individual contributions of distinct reader domains in addition to uncovering potential therapeutic value. For example, a-366 was recently shown to antagonize recognition of H3K4me3 by the Tudor domain of Spindin1, a methyl-lysine reader (IC50 = 182.6 ± 9.1 nM). Although significant progress has been made, much remains to be learned about the pharmacology and biology of most PMTs (99, 102). Furthermore, such knowledge is expected to considerably advance CKD treatment.
Conclusion and Future Perspectives
Both histone methylation and non-histone methylation are associated with various renal diseases. Moreover, they participate in regulating ECM and inflammatory genes in almost all renal cell types (19, 103). The search for the link between protein methylation and DKD mainly focuses on histones, especially histone lysine methylations (104, 105). Notably, histones can be marks of the active, “poised,” and repressive chromatin states. An increasing number of PMTs could serve as new epigenetic therapy agents for multiple diseases, including DKD, in the future (106, 107). In addition to studying protein methylation, the cross-interaction among DNA methylation, protein methylation, and protein acetylation is an essential area of study regarding the pathogenesis, development, and progression of DKD. Further efforts are urgently needed and should not be limited to a single layer.
Author Contributions
YeC and GW: writing – original draft preparation. GX, PL, and HJ: writing – review and editing. YaC and MW: visualization. HC, YF, and JZ: funding acquisition.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (2019A1515011087 to JZ and 2021A1515012453 to HC), Natural Science Foundation of Xinjiang Uygur Autonomous Region (2018D01C016 to YF), and Guangzhou Science and Technology Innovation Commission (202002030038 to HC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. JAMA. (2007) 298:2038–47.
2. Hsu C-Y, Vittinghoff E, Lin F, Shlipak MG. The incidence of end-stage renal disease is increasing faster than the prevalence of chronic renal insufficiency. Ann Intern Med. (2004) 141:95–101. doi: 10.7326/0003-4819-141-2-200407200-00007
3. Plantinga LC, Boulware LE, Coresh J, Stevens LA, Miller ER, Saran R, et al. Patient awareness of chronic kidney disease: trends and predictors. Arch Intern Med. (2008) 168:2268–75. doi: 10.1001/archinte.168.20.2268
4. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. (2013) 382:260–72. doi: 10.1016/S0140-6736(13)60687-X
5. Andrassy KM. Comments on ‘KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease’. Kidney Int. (2013) 84:622–3. doi: 10.1038/ki.2013.243
6. Zoccali C, Vanholder R, Massy ZA, Ortiz A, Sarafidis P, Dekker FW, et al. The systemic nature of CKD. Nat Rev Nephrol. (2017) 13:344–58. doi: 10.1038/nrneph.2017.52
7. Dwyer JP, Parving HH, Hunsicker LG, Ravid M, Remuzzi G, Lewis JB. Renal dysfunction in the presence of normoalbuminuria in type 2 diabetes: results from the DEMAND study. Cardiorenal Med. (2012) 2:1–10. doi: 10.1159/000333249
8. Thomas MC, Weekes AJ, Broadley OJ, Cooper ME, Mathew TH. The burden of chronic kidney disease in Australian patients with type 2 diabetes (the NEFRON study). Med J Aust. (2006) 185:140–4. doi: 10.5694/j.1326-5377.2006.tb00499.x
9. Lundbaek K. Diabetic angiopathy: a specific vascular disease. Lancet. (1954) 266:377–9. doi: 10.1016/s0140-6736(54)90924-1
10. Root HF, Pote WH Jr, Frehner H. Triopathy of diabetes; sequence of neuropathy, retinopathy, and nephropathy in one hundred fifty-five patients. AMA Arch Intern Med. (1954) 94:931–41. doi: 10.1001/archinte.1954.00250060065006
11. Liu L, Liu F, Guan Y, Zou J, Zhang C, Xiong C, et al. Critical roles of SMYD2 lysine methyltransferase in mediating renal fibroblast activation and kidney fibrosis. FASEB J. (2021) 35:e21715. doi: 10.1096/fj.202000554RRR
12. Ambler RP, Rees MW. Epsilon-N-methyl-lysine in bacterial flagellar protein. Nature. (1959) 184:56–7. doi: 10.1038/184056b0
13. Trievel RC, Beach BM, Dirk LM, Houtz RL, Hurley JH. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell. (2002) 111:91–103. doi: 10.1016/s0092-8674(02)01000-0
14. Lee S, Oh S, Jeong K, Jo H, Choi Y, Seo HD, et al. Dot1 regulates nucleosome dynamics by its inherent histone chaperone activity in yeast. Nat Commun. (2018) 9:240. doi: 10.1038/s41467-017-02759-8
15. Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. (2017) 65:8–24. doi: 10.1016/j.molcel.2016.11.003
16. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. (2012) 13:343–57. doi: 10.1038/nrg3173
17. Wu Z, Connolly J, Biggar KK. Beyond histones – the expanding roles of protein lysine methylation. FEBS J. (2017) 284:2732–44. doi: 10.1111/febs.14056
18. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
19. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
20. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. (2007) 128:707–19. doi: 10.1016/j.cell.2007.01.015
21. Buratowski S, Kim T. The role of cotranscriptional histone methylations. Cold Spring Harb Symp Quant Biol. (2010) 75:95–102. doi: 10.1101/sqb.2010.75.036
22. Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. (2005) 122:517–27. doi: 10.1016/j.cell.2005.06.026
23. Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, et al. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. (2005) 3:e328. doi: 10.1371/journal.pbio.0030328
24. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
25. Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer. (2015) 15:110–24. doi: 10.1038/nrc3884
26. Buuh ZY, Lyu Z, Wang RE. Interrogating the roles of post-translational modifications of non-histone proteins. J Med Chem. (2018) 61:3239–52. doi: 10.1021/acs.jmedchem.6b01817
27. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
28. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. (2010) 28:1057–68. doi: 10.1038/nbt.1685
29. Li X, Li C, Li X, Cui P, Li Q, Guo Q, et al. Involvement of histone lysine methylation in p21 gene expression in rat kidney in vivo and rat mesangial cells in vitro under diabetic conditions. J Diabetes Res. (2016) 2016:3853242. doi: 10.1155/2016/3853242
30. Murn J, Shi Y. The winding path of protein methylation research: milestones and new frontiers. Nat Rev Mol Cell Biol. (2017) 18:517–27. doi: 10.1038/nrm.2017.35
31. Clarke SG. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem Sci. (2013) 38:243–52. doi: 10.1016/j.tibs.2013.02.004
32. Lu L, Zhong Z, Gu J, Nan K, Zhu M, Miao C. ets1 associates with KMT5A to participate in high glucose-mediated EndMT via upregulation of PFN2 expression in diabetic nephropathy. Mol Med. (2021) 27:74. doi: 10.1186/s10020-021-00339-7
33. Sharma N, Sankrityayan H, Kale A, Gaikwad AB. Role of SET7/9 in the progression of ischemic renal injury in diabetic and non-diabetic rats. Biochem Biophys Res Commun. (2020) 528:14–20. doi: 10.1016/j.bbrc.2020.05.075
34. Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta. (2014) 1839:1362–72. doi: 10.1016/j.bbagrm.2014.02.007
35. Huang T, Li X, Wang F, Lu L, Hou W, Zhu M, et al. The CREB/KMT5A complex regulates PTP1B to modulate high glucose-induced endothelial inflammatory factor levels in diabetic nephropathy. Cell Death Dis. (2021) 12:333. doi: 10.1038/s41419-021-03629-4
36. Soofi A, Kutschat AP, Azam M, Laszczyk AM, Dressler GR. Regeneration after acute kidney injury requires PTIP-mediated epigenetic modifications. JCI Insight. (2020) 5:e130204. doi: 10.1172/jci.insight.130204
37. Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. (2011) 25:1345–58. doi: 10.1101/gad.2057811
38. Jia Y, Reddy MA, Das S, Oh HJ, Abdollahi M, Yuan H, et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-beta1-induced gene expression in mesangial cells and diabetic kidney. J Biol Chem. (2019) 294:12695–707. doi: 10.1074/jbc.RA119.007575
39. Majumder S, Thieme K, Batchu SN, Alghamdi TA, Bowskill BB, Kabir MG, et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J Clin Invest. (2018) 128:483–99. doi: 10.1172/JCI95946
40. Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. (2010) 21:2069–80. doi: 10.1681/ASN.2010060633
41. Shuttleworth VG, Gaughan L, Nawafa L, Mooney CA, Cobb SL, Sheerin NS, et al. The methyltransferase SET9 regulates TGFB1 activation of renal fibroblasts via interaction with SMAD3. J Cell Sci. (2018) 131:jcs207761. doi: 10.1242/jcs.207761
42. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. (2008) 205:2409–17. doi: 10.1084/jem.20081188
43. Naito M, Zager RA, Bomsztyk K. BRG1 increases transcription of proinflammatory genes in renal ischemia. J Am Soc Nephrol. (2009) 20:1787–96. doi: 10.1681/ASN.2009010118
44. Zager RA, Johnson ACM. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol. (2009) 296:F1032–41. doi: 10.1152/ajprenal.00061.2009
45. Naito M, Bomsztyk K, Zager RA. Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J Am Soc Nephrol. (2008) 19:1321–30. doi: 10.1681/ASN.2007121368
46. Naito M, Bomsztyk K, Zager RA. Renal ischemia-induced cholesterol loading: transcription factor recruitment and chromatin remodeling along the HMG CoA reductase gene. Am J Pathol. (2009) 174:54–62. doi: 10.2353/ajpath.2009.080602
47. Johnson AC, Ware LB, Himmelfarb J, Zager RA. HMG-CoA reductase activation and urinary pellet cholesterol elevations in acute kidney injury. Clin J Am Soc Nephrol. (2011) 6:2108–13. doi: 10.2215/CJN.02440311
48. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. (2008) 40:897–903. doi: 10.1038/ng.154
49. Reddy MA, Sumanth P, Lanting L, Yuan H, Wang M, Mar D, et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. (2014) 85:362–73. doi: 10.1038/ki.2013.387
50. Zhang D, Yu ZY, Cruz P, Kong Q, Li S, Kone BC. Epigenetics and the control of epithelial sodium channel expression in collecting duct. Kidney Int. (2009) 75:260–7. doi: 10.1038/ki.2008.475
51. Villeneuve LM, Reddy MA, Natarajan R. Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol. (2011) 38:451–9. doi: 10.1111/j.1440-1681.2011.05497.x
52. Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. (2006) 281:18059–68. doi: 10.1074/jbc.M601903200
53. Wu H, Chen L, Zhang X, Zhou Q, Li JM, Berger S, et al. Aqp5 is a new transcriptional target of Dot1a and a regulator of Aqp2. PLoS One. (2013) 8:e53342. doi: 10.1371/journal.pone.0053342
54. Liebisch M, Bondeva T, Franke S, Hause S, Wolf G. Growth arrest specific 2-like protein 1 expression is upregulated in podocytes through advanced glycation end-products. Nephrol Dial Transplant. (2017) 32:641–53. doi: 10.1093/ndt/gfw313
55. Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA. (2008) 105:9047–52. doi: 10.1073/pnas.0803623105
56. Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. (2006) 441:349–53. doi: 10.1038/nature04733
57. Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci USA. (2006) 103:15782–7. doi: 10.1073/pnas.0607617103
58. Komers R, Mar D, Denisenko O, Xu B, Oyama TT, Bomsztyk K. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab Invest. (2013) 93:543–52. doi: 10.1038/labinvest.2013.47
59. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. (2019) 20:642–57. doi: 10.1038/s41580-019-0155-x
60. Zhu Y, Yu C, Zhuang S. Protein arginine methyltransferase 1 mediates renal fibroblast activation and fibrogenesis through activation of Smad3 signaling. Am J Physiol Renal Physiol. (2020) 318:F375–87. doi: 10.1152/ajprenal.00487.2019
61. Böger RH, Maas R, Schulze F, Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality–an update on patient populations with a wide range of cardiovascular risk. Pharmacol Res. (2009) 60:481–7. doi: 10.1016/j.phrs.2009.07.001
62. Tain Y-L, Huang L-T. Asymmetric dimethylarginine: clinical applications in pediatric medicine. J Formos Med Assoc. (2011) 110:70–7. doi: 10.1016/S0929-6646(11)60012-0
63. Teerlink T, Luo Z, Palm F, Wilcox CS. Cellular ADMA: regulation and action. Pharmacol Res. (2009) 60:448–60. doi: 10.1016/j.phrs.2009.08.002
64. Mathew AV, Zeng L, Byun J, Pennathur S. Metabolomic profiling of arginine metabolome links altered methylation to chronic kidney disease accelerated atherosclerosis. J Proteomics Bioinform. (2015). doi: 10.4172/jpb.S14-001
65. Fliser D, Kronenberg F, Kielstein JT, Morath C, Bode-Böger SM, Haller H, et al. Asymmetric dimethylarginine and progression of chronic kidney disease: the mild to moderate kidney disease study. J Am Soc Nephrol. (2005) 16:2456–61. doi: 10.1681/ASN.2005020179
66. Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. (2007) 293:H3227–45. doi: 10.1152/ajpheart.00998.2007
67. Kuo HC, Hsu CN, Huang CF, Lo MH, Chien SJ, Tain YL. Urinary arginine methylation index associated with ambulatory blood pressure abnormalities in children with chronic kidney disease. J Am Soc Hypertens. (2012) 6:385–92. doi: 10.1016/j.jash.2012.09.003
68. Chen YY, Peng XF, Liu GY, Liu JS, Sun L, Liu H, et al. Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2563–75. doi: 10.1016/j.bbadis.2019.06.001
69. Andrade F, Rodriguez-Soriano J, Prieto JA, Aguirre M, Ariceta G, Lage S, et al. Methylation cycle, arginine-creatine pathway and asymmetric dimethylarginine in paediatric renal transplant. Nephrol Dial Transplant. (2011) 26:328–36. doi: 10.1093/ndt/gfq404
70. Liu H, Galka M, Mori E, Liu X, Lin YF, Wei R, et al. A method for systematic mapping of protein lysine methylation identifies functions for HP1beta in DNA damage response. Mol Cell. (2013) 50:723–35. doi: 10.1016/j.molcel.2013.04.025
71. Carlson SM, Moore KE, Green EM, Martin GM, Gozani O. Proteome-wide enrichment of proteins modified by lysine methylation. Nat Protoc. (2014) 9:37–50. doi: 10.1038/nprot.2013.164
72. Lee TY, Chang CW, Lu CT, Cheng TH, Chang TH. Identification and characterization of lysine-methylated sites on histones and non-histone proteins. Comput Biol Chem. (2014) 50:11–8. doi: 10.1016/j.compbiolchem.2014.01.009
73. Xie Q, Bai Y, Wu J, Sun Y, Wang Y, Zhang Y, et al. Methylation-mediated regulation of E2F1 in DNA damage-induced cell death. J Recept Signal Transduct Res. (2011) 31:139–46. doi: 10.3109/10799893.2011.552914
74. Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, et al. Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kappaB signaling. Nat Immunol. (2011) 12:29–36. doi: 10.1038/ni.1968
75. Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. (2006) 127:1361–73. doi: 10.1016/j.cell.2006.10.043
76. Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. (2014) 510:283–7. doi: 10.1038/nature13320
77. Hamamoto R, Toyokawa G, Nakakido M, Ueda K, Nakamura Y. SMYD2-dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Lett. (2014) 351:126–33. doi: 10.1016/j.canlet.2014.05.014
78. Zhou X, Fan LX, Sweeney WE Jr, Denu JM, Avner ED, Li X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J Clin Invest. (2013) 123:3084–98. doi: 10.1172/JCI64401
79. Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. (2006) 444:629–32. doi: 10.1038/nature05287
80. Saddic LA, West LE, Aslanian A, Yates JR III, Rubin SM, Gozani O, et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. (2010) 285:37733–40. doi: 10.1074/jbc.M110.137612
81. Seeger-Nukpezah T, Proia DA, Egleston BL, Nikonova AS, Kent T, Cai KQ, et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci USA. (2013) 110:12786–91. doi: 10.1073/pnas.1301904110
82. Li LX, Fan LX, Zhou JX, Grantham JJ, Calvet JP, Sage J, et al. Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J Clin Invest. (2017) 127:2751–64. doi: 10.1172/JCI90921
83. Cho HS, Hayami S, Toyokawa G, Maejima K, Yamane Y, Suzuki T, et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia. (2012) 14:476–86. doi: 10.1593/neo.12656
84. Jones PA, Issa J-PJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. (2016) 17:630–41. doi: 10.1038/nrg.2016.93
85. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. (2012) 11:384–400. doi: 10.1038/nrd3674
86. Copeland RA. Protein methyltransferase inhibitors as precision cancer therapeutics: a decade of discovery. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170080. doi: 10.1098/rstb.2017.0080
87. Scheer S, Ackloo S, Medina TS, Schapira M, Li F, Ward JA, et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat Commun. (2019) 10:19. doi: 10.1038/s41467-018-07905-4
88. Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. (2007) 25:473–81. doi: 10.1016/j.molcel.2007.01.017
89. Shi Y, Desponts C, Do JT, Hahm HS, Schöler HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. (2008) 3:568–74. doi: 10.1016/j.stem.2008.10.004
90. Shi Y, Do JT, Desponts C, Hahm HS, Schöler HR, Ding S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. (2008) 2:525–8. doi: 10.1016/j.stem.2008.05.011
91. Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. (2011) 7:566–74. doi: 10.1038/nchembio.599
92. Casciello F, Windloch K, Gannon F, Lee JS. Functional role of G9a histone methyltransferase in cancer. Front Immunol. (2015) 6:487. doi: 10.3389/fimmu.2015.00487
93. Irifuku T, Doi S, Sasaki K, Doi T, Nakashima A, Ueno T, et al. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. (2016) 89:147–57. doi: 10.1038/ki.2015.291
94. Zhang X, Huang Y, Shi X. Emerging roles of lysine methylation on non-histone proteins. Cell Mol Life Sci. (2015) 72:4257–72. doi: 10.1007/s00018-015-2001-4
95. Liu B, Nie J, Liang H, Liang Z, Huang J, Yu W, et al. Pharmacological inhibition of SETD7 by PFI-2 attenuates renal fibrosis following folic acid and obstruction injury. Eur J Pharmacol. (2021) 901:174097. doi: 10.1016/j.ejphar.2021.174097
96. Xu L, Gao J, Huang D, Lin P, Yao D, Yang F, et al. Emodin ameliorates tubulointerstitial fibrosis in obstructed kidneys by inhibiting EZH2. Biochem Biophys Res Commun. (2021) 534:279–85. doi: 10.1016/j.bbrc.2020.11.094
97. Liu L, Zou J, Guan Y, Zhang Y, Zhang W, Zhou X, et al. Blocking the histone lysine 79 methyltransferase DOT1L alleviates renal fibrosis through inhibition of renal fibroblast activation and epithelial-mesenchymal transition. FASEB J. (2019) 33:11941–58. doi: 10.1096/fj.201801861R
98. Yang C, Chen Z, Yu H, Liu X. Inhibition of disruptor of telomeric silencing 1-like alleviated renal ischemia and reperfusion injury-induced fibrosis by blocking PI3K/AKT-mediated oxidative stress. Drug Des Devel Ther. (2019) 13:4375–87. doi: 10.2147/DDDT.S224909
99. Shi Y, Xu L, Tao M, Fang L, Lu J, Gu H, et al. Blockade of enhancer of zeste homolog 2 alleviates renal injury associated with hyperuricemia. Am J Physiol Renal Physiol. (2019) 316:F488–505. doi: 10.1152/ajprenal.00234.2018
100. Guo Q, Li X, Han H, Li C, Liu S, Gao W, et al. Histone lysine methylation in TGF-beta1 mediated p21 gene expression in rat mesangial cells. Biomed Res Int. (2016) 2016:6927234. doi: 10.1155/2016/6927234
101. Sasaki K, Doi S, Nakashima A, Irifuku T, Yamada K, Kokoroishi K, et al. Inhibition of SET domain-containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J Am Soc Nephrol. (2016) 27:203–15. doi: 10.1681/ASN.2014090850
102. Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R, et al. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol. (2016) 27:2092–108. doi: 10.1681/ASN.2015040457
103. Reddy MA, Tak Park J, Natarajan R. Epigenetic modifications in the pathogenesis of diabetic nephropathy. Semin Nephrol. (2013) 33:341–53. doi: 10.1016/j.semnephrol.2013.05.006
105. Susztak K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J Am Soc Nephrol. (2014) 25:10–7. doi: 10.1681/ASN.2013050461
106. Tan J-Z, Yan Y, Wang X-X, Jiang Y, Xu HE. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. (2014) 35:161–74. doi: 10.1038/aps.2013.161
Keywords: chronic kidney disease, renal fibrosis, protein methylation, histone methylation, nonhistone methylation
Citation: Cheng Y, Chen Y, Wang G, Liu P, Xie G, Jing H, Chen H, Fan Y, Wang M and Zhou J (2022) Protein Methylation in Diabetic Kidney Disease. Front. Med. 9:736006. doi: 10.3389/fmed.2022.736006
Received: 04 July 2021; Accepted: 07 March 2022;
Published: 12 May 2022.
Edited by:
Björn Tampe, University Medical Center Göttingen, GermanyReviewed by:
Shougang Zhuang, Brown University, United StatesMurugavel Ponnusamy, Qingdao University, China
Copyright © 2022 Cheng, Chen, Wang, Liu, Xie, Jing, Chen, Fan, Wang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Zhou, emhvdWp1bjc4NDNAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship