 Vasco C. Romão
Vasco C. Romão João Eurico Fonseca
João Eurico Fonseca
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 19 August 2022
Sec. Rheumatology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.689711
This article is part of the Research Topic Risk factors for Rheumatoid Arthritis (RA) and pre-Rheumatoid Arthritis View all 7 articles
In the last decades, the concept of preclinical rheumatoid arthritis (RA) has become established. In fact, the discovery that disease mechanisms start years before the onset of clinical RA has been one of the major recent insights in the understanding of RA pathogenesis. In accordance with the complex nature of the disease, preclinical events extend over several sequential phases. In a genetically predisposed host, environmental factors will further increase susceptibility for incident RA. In the initial steps of preclinical disease, immune disturbance mechanisms take place outside the joint compartment, namely in mucosal surfaces, such as the lung, gums or gut. Herein, the persistent immunologic response to altered antigens will lead to breach of tolerance and trigger autoimmunity. In a second phase, the immune response matures and is amplified at a systemic level, with epitope spreading and widening of the autoantibody repertoire. Finally, the synovial and bone compartment are targeted by specific autoantibodies against modified antigens, initiating a local inflammatory response that will eventually culminate in clinically evident synovitis. In this review, we discuss the elaborate disease mechanisms in place during preclinical RA, providing a broad perspective in the light of current evidence.
The pathophysiology of rheumatoid arthritis (RA) is complex and yet incompletely understood (1, 2). Extensive epidemiological and etiological data emerging over the last decades have made a decisive contribution toward improving the knowledge of the mechanisms involved in the initiation and perpetuation of the disease (3). As demonstrated by the plethora of immune-related host and environmental risk factors for RA, the immune system as a whole plays a central role in the disease pathogenesis (3). It is therefore regarded as a prototypical immune-mediated disease (1, 4).
A comprehensive body of evidence emerging from the last two to three decades has generated many hypotheses, that currently regard RA as a continuum of consecutive phases that unfold longitudinally (Figure 1) (1, 4–7). The first phase corresponds to the general susceptibility to RA, when there are no symptoms or detectable ongoing immune disturbances. Herein, environmental stimuli interact with a genetically predisposed, epigenetically modified host to increase susceptibility to RA [reviewed in detail elsewhere (3)]. The result of these interactions in sites such as the mucosal surfaces (e.g., lung, gums, gut) may lead to the second phase—of preclinical RA—when breach of tolerance occurs and autoimmunity initiates. This seems to be followed by maturation and amplification of the autoimmune response outside the joint (e.g., in secondary lymphoid organs), whereby epitope spreading takes place and blood titers of autoantibodies (e.g., rheumatoid factor [RF], anti-citrullinated protein antibodies [ACPA] and others) increase. In parallel, systemic inflammation expands and levels of circulating cytokines and acute phase reactants rise (4, 6). In a proposed early intermediate step, joint and bone tissue may be targeted by the expanding ACPA repertoire, with minor immune cell infiltration (6, 8). During this phase patients are initially asymptomatic, but soon develop arthralgia and early bone loss (6, 8, 9). Following mostly undisclosed events, which might include a “second hit” (e.g., trauma, infection), elements of the activated adaptive and innate immune systems begin to access synovial tissue as local vascular permeability increases, resulting in overt synovitis that is clinically detectable (1, 6). Replication of this process in several joint sites culminates in the typical polyarticular phenotype of RA, enabling clinical diagnosis. Over time, complex interactions between innate and adaptive immune cells in the inflammatory synovium microenvironment, reinforced by positive feedback loops, synovial neoangiogenesis, insufficient lymphangiogenesis and apoptosis resistance, lead to perpetuation of synovitis (1, 2). A marked stromal tissue response involving fibroblast-like synoviocytes, chondrocytes and osteoclasts, further integrates with the ongoing inflammation to determine tissue reorganization and joint destruction (1, 2, 8). Recently, high-throughput analyses integrating cutting-edge techniques, such as single-cell RNA sequencing and mass cytometry, have shed light on the complexity of RA synovitis by identifying 18 cell populations within the synovial tissue of patients with established disease, including 4 fibroblast subsets, 4 monocyte subsets, 6 T cell subsets and 4 B cell subsets (10). In addition, 4 expanded cell states in the rheumatoid synovia were identified, involving specific subsets of sublining fibroblasts, pro-inflammatory monocytes, autoimmune-associated B cells and T follicular and T peripheral helper cells. These are exciting findings that have been reviewed elsewhere (11) and beyond the scope of the present article. Whether these cell populations and inflammatory cell states are also present in the preclinical or early stages of RA is currently unknown.
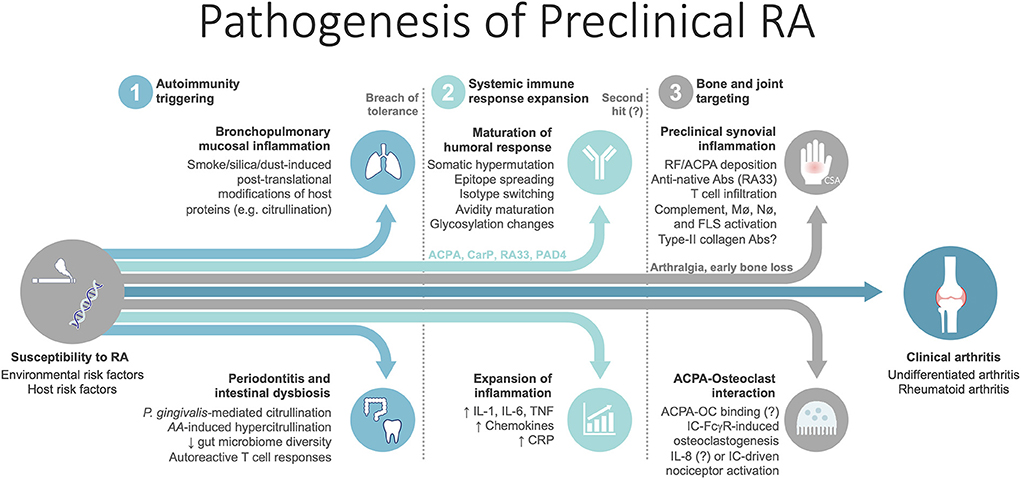
Figure 1. Summary of the pathogenic steps unfolding in the preclinical phase of rheumatoid arthritis. Mechanisms for which evidence is conflicting and significant uncertainty remains are followed by a question mark. AA, Aggregatibacter actinomycetemcomitans; Abs, antibodies; ACPA, anti-citrullinated protein antibodies; CarP, carbamylated protein; CSA, clinically suspect arthralgia; FLS, fibroblast-like synoviocytes; IC, immune complexes; Mø, macrophages; Nø, neutrophils; OC, osteoclast; P. Gingivalis, Porphyromonas gingivalis. PAD4, peptidyl-arginine deiminase 4; RA33, heterogeneous nuclear ribonucleoprotein (hnRNP)-A2, also known as RA33; RF, rheumatoid factor.
It should be highlighted that most of these mechanisms that have been suggested are valid and mostly applied to seropositive (i.e., RF and/or ACPA-positive) RA. There are a few suggestions of comparable phenomena in seronegative disease, but a deep comprehension of the driving forces behind this subtype is still illusive, and far from being a reality (4, 6). Therefore, herein, we will mainly focus on the pathogenesis of seropositive RA, and we will summarize what is currently known regarding seronegative disease.
The first pathophysiological phase, related to the genetic, epigenetic and environmental risk factors for the development of RA, has been addressed elsewhere (3) and is beyond the scope of this review. Herein, we focus on the events that occur during preclinical RA and eventually lead to early RA.
The concept that RA begins years before it is clinically evident has been one of the main advances in the understanding of the disease pathogenesis (4). This was first suggested by seminal work showing that RF (12, 13) and ACPAs (13, 14) often precede the development of clinical RA. Around half of the patients with RA have positive determinations of RF and/or ACPAs for a median of 4.5 years (and up to almost 14 years) before the onset of symptoms (14). Subsequently, ACPA positivity was also demonstrated to antedate and predict future development of arthritis in patients with arthralgia (hazard ratio [HR] 6.0, 95% confidence interval [CI] 1.8–19.8) (15). Similar findings were reported for other RA-related autoantibodies such as anti-carbamylated protein (anti-CarP) antibodies, which may be important in a proportion of ACPA-negative patients (16). Moreover, ACPA status is unlikely to change following the diagnosis, strengthening the hypothesis that these autoantibodies have a direct influence in the development of RA during the preclinical phase (17). Simultaneously, the genetic background of RA started to be dissected and the major contributions of the human leukocyte antigen (HLA) region and of the shared epitope (SE) were revealed, confirming the prominent role of antigen presentation to T cells and T cell-mediated immunity in the pathogenesis of the disease, as already evidenced by the presence of ACPAs (18–20). These studies provided support to a pathogenic model in which breach of tolerance occurs long before clinical disease, due to the interaction of genetic and non-genetic risk factors, and leads to an autoimmune response that is further propelled until RA surfaces. In this process, epigenetic regulation mechanisms are also at play in the interaction between the environment and the genetic background of the host. Indeed, they may contribute to the low concordance rate observed between monozygotic twins (9–15%) (21–23). A recent large epigenome-wide association study found differentially variable methylation signatures in monozygotic twin pairs discordant for RA (24). These altered DNA methylation patterns have been well document in RA patients, suggesting a possible pathogenic role (25–29). Interestingly, specific methylome signatures have even been described across different joints of the same patient, possibly contributing for the characteristic articular pattern of RA (30, 31). Histone modification is another epigenetic process that can influence gene expression. Although less studied, altered acetylation status have been identified in blood and synovial tissue of RA patients (32–35). Further, an interaction of smoking, a major environmental risk factor, with histone deacetylases has been recently described (36, 37). Finally, microRNAs and long non-coding RNAs have also been implicated as regulators of RA susceptibility and severity. microRNA-155 and micro-RNA146a are among the best studied, and have been shown to be increased in blood, synovial tissue/fluid, and specific cell types, with pleotropic effects that promote inflammation and bone erosion pathways (38–41).
Considering the temporal lapse between immune dysregulation and articular disease onset, it arises that early events may be distinct from those in place during the synovitis phase. Indeed, triggering of autoimmunity is thought to take place outside the joint compartment through key interactions between the host and external stimuli, which are mediated by the immune system (6). Genetic and epidemiological studies supplied fundamental evidence implicating mucosal surfaces as relevant sites involved in this process, being the central interfaces between the outer environment and body defenses (1, 6, 42). The lung, gum and gastrointestinal mucosae have all been proposed as possible sites where early pathogenic events take place, summarized in Table 1 (6, 42, 82).

Table 1. Main findings and possible mechanisms supporting a mucosal triggering of autoimmunity in RA.
In this setting, it has been hypothesized that infectious agents, microbiota, cigarette smoke and other stressors interact with resident stromal and immune cells and initiate the chain of events that will eventually culminate in RA-specific autoimmunity. In fact, immunoglobulin (Ig)A-antibodies (ACPAs, RF) and IgA-plasmablasts are enriched in preclinical RA and predict evolution to overt disease, supporting the possible participation of mucosal immunity since the early pathophysiological stages (6, 13, 49).
It should be stressed, however, that the role of mucosal surfaces in initiating the pathogenic process has not been definitely proven and conclusive experimental evidence is lacking. It is likely that different mucosal compartments play different roles in separate patients, with the varying contribution of each surface, according to genetic, epigenetic and environmental factors. It is even possible that in some patients, mucosae play no role in the offset of autoimmunity and breach of tolerance. Thus, much remains to be determined and the content presented here is more appropriately seen as a working theoretical model.
Given its prominent involvement in the pathogenesis of seropositive RA, a brief overview of citrullination is warranted. Citrullination is a physiological process that consists of the post-translational modification of arginine residues into the nonstandard amino acid citrulline. This reaction if catalyzed by calcium-dependent peptidyl-arginine deiminase (PAD) enzymes. Although PADs have been implicated in the pathogenesis of RA, they have multiple physiological roles, from epigenetic regulation to tissue differentiation (e.g., PAD1 in skin keratinization) (95). There are five isoforms of human PAD (PAD1-4 and PAD6), all widely expressed in multiple organs and systems, and each with a specific function [reviewed in detail elsewhere (95–98)]. The most relevant for RA are PAD2 and PAD4, both of which are expressed in immune cells (96). In particular, PAD4 is primarily expressed by neutrophils, eosinophils, and monocytes and is involved in epigenetic regulation through citrullination of histones and transcription factors (97–102). Both PAD2 and PAD4 can generate neoantigens, by catalyzing the citrullination of native widely expressed proteins such as fibrinogen or vimentin (95, 96, 103). Yet, they seem to have partially different targets and patterns of citrullination (95, 96, 103). More specifically, within the articular compartment, PAD4 may be more important for autoantibody generation, whereas PAD2 is more strongly linked to inflammation (96).
In addition to their role in citrullination, PADs have also been proposed to contribute to RA pathogenesis via other mechanisms. Animal studies have suggested both PAD2 and PAD4 enhance the immune response, as their absence leads to reduced antibody production, complement deposition and pro-inflammatory cytokine production, in parallel with skewing of T cell polarization (104–107). This amplifies clinical inflammation and disease severity (96). On the other side, PADs can themselves be targeted by the immune response. Numerous studies have described an increased frequency of anti-PAD antibodies in patients with RA (96, 108–110). Anti-PAD4 antibodies are the most studied, having been described in 22–45% of patients with established RA (96, 108, 109). They are highly specific for RA and associated with the presence of ACPAs, albeit being also detected in 2–19% of patients with seronegative RA (96, 109, 111). In addition, they are associated with disease activity and severity, as well as joint damage and radiographic progression (96, 108–110, 112). Their implication in RA development has been strengthened by the demonstration of their presence in a subset of patients with preclinical disease (113). Anti-PAD2 and anti-PAD3/4 antibodies have also been described in patients with RA (96, 114, 115). Among other mechanisms, it has been hypothesized that anti-PAD antibodies could be generated by exposure of high quantities of extracellular PAD enzymes to autoreactive B/T cells in a setting of intense inflammation, such as within the joint compartment (96, 116).
The implication of citrullination in the pathogenesis of RA has been a matter of debate over recent years (98). Neutrophil extracellular traps (NETs) formation following controlled neutrophil death (also known as NETosis) is an antimicrobial mechanism that has been widely proposed as a source of citrullinated antigens in RA via hypercitrullination (117–119). On the contrary, evidence shows that PAD4 activity actually decreases during NETosis, which is characterized by limited citrullination (e.g., of histone H3) and absence of hypercitrullination (98, 120). Therefore, it is unlikely that NETosis contributes in a significant fashion to the generation of citrullinated autoantigens that characterizes the early stages of RA pathogenesis. Instead, a separate mechanism that may be of relevance in this regard is another form of neutrophil death denominated leukotoxic hypercitrullination (LTH). It is a bacterial-induced immune-evading strategy that features a marked rise in intracellular calcium, resulting in PAD hyperactivation (namely PAD4), cellular protein hypercitrullination, and extrusion of DNA-containing NET-like structures (98, 120–123). The exposed citrullinated residues could then be targeted by the adaptive immune-system, mounting an autoimmune response that eventually leads to the development of RA.
The idea that lungs may be a site in which autoimmunity is generated in some patients is supported by a variety of studies associating smoking and other airborne noxious agents, such as silica and occupational dust, with both RA and ACPA generation (3, 6, 42–46). These exposures were shown to interact with genetic markers (e.g., human HLA-SE) to multiply the risk, thereby suggesting a common pathogenic pathway (42, 47, 48). Moreover, a study reported that 76% of RF/ACPA-positive individuals without arthritis had airway and parenchymal inflammatory abnormalities on high-resolution computed tomography (HRCT), which were similar in type and frequency to those seen in early RA (52). On the other hand, early untreated ACPA-positive RA patients have been shown to have a higher rate of HRCT parenchymal lung changes and of bronchial biopsy microscopic inflammatory infiltrates than those with ACPA-negative RA and healthy controls (53). These data suggest that, in some patients, bronchopulmonary inflammation precedes the onset of synovitis and may possibly play a causal role in triggering RA-inducing autoimmunity.
The mechanisms through which smoking (and other inhaled agents) lead to loss of tolerance is not entirely comprehended. It has been proposed to involve post-translational modifications of common host proteins (1, 6). Citrullination, described above, is the best studied of those changes, but carbamylation (conversion of lysine to homocitrulline), acetylation and others have also been described (4). Smoke, silica or microbes can trigger Toll-like receptors (TLRs; e.g., TLR4) of dendritic cells, B cells and macrophages and increase expression and activation of PAD (6, 54, 55). In fact, smoking has been demonstrated to upregulate expression of PAD2 and citrullinated proteins in the bronchoalveolar lavage (BAL) and bronchial mucosal biopsies of smokers in comparison to non-smokers (42, 54).
However, citrullination per se is not sufficient to spawn autoimmunity as it occurs in the lungs of healthy smokers who do not develop RA and may even serve a physiological role in other tissues (4). Therefore, other factors must be present for loss of tolerance to citrullinated epitopes to occur. Notably, they include HLA molecules, particularly those carrying the SE sequence, which are particularly efficient at presenting citrullinated peptides to T cells (42, 124). Presentation of these self-neoantigens by HLA-SE-positive antigen presenting cells (APCs) leads to the emergence of autoreactive T cells, that interact with and provide help to B cells, resulting in the production of autoantibodies (i.e., ACPAs) and RF (targeting other antibodies) (1, 6). This hypothesis may explain the observation of the multiplicative risk of RA conferred by the coexistence of smoking and the SE (e.g., RR 21.0/1.5 in ever smokers carrying two/no copies of SE genes, respectively) (42).
Simultaneously, B cells attracted to the lungs by smoke substances can undergo local differentiation and recognize modified self-antigens (6). These may then be presented to autoreactive T cells in the context of germinal center-positive ectopic lymphoid structures (ELS) within the induced bronchus-associated lymphoid tissue (6). Whether this latter step is the result of defective thymic regulation of T cell clones recognizing post-translationally modified self-proteins is still unclear.
Several studies have provided circumstantial evidence of a possible involvement of the bronchopulmonary mucosal tissue in the loss of tolerance events that occur in RA. In this regard, a number of relevant observations have been reported: (i) IgA and IgG ACPAs are present in the sputum of early RA patients and enriched in that of at-risk individuals (including seronegative subjects) (50, 51); (ii) in at-risk subjects, ACPAs are associated with markers of local inflammation and NET formation, which can result in further liberation of citrullinated proteins and PAD4 (51); (iii) ACPAs are seen at higher concentrations in the BAL (vs. serum) of patients with early and established disease (53, 56); (iv) infiltration by activated lymphocytes and citrulline-specific plasma cells (e.g., induced bronchus-associated lymphoid tissue with numerous ELS), increased local expression of PAD2, and large areas of citrullinated proteins are commonly found in pulmonary biopsies of patients with early and established ACPA-positive RA (53, 56, 125).
The targets of the pulmonary ACPA response have also been studied. One of the aspects that puzzled investigators at first, but that is currently accepted, is that most ACPAs are directed at citrullinated proteins that are not specific to the synovium, but rather are ubiquitous in many other tissues alike (4–6). These include both intracellular histones and stromal proteins, such as vimentin, fibrinogen, fibronectin, α-enolase, tenascin C or type II collagen (1, 4, 6, 69). The specificities of ACPAs found in the sputum of at-risk individuals, recently reported, frequently included citrullinated forms of fibrinogen, vimentin, fibronectin and apolipoproteins E and A1, as well as non-citrullinated/native forms of some of these proteins (126). Interestingly, the autoantibody pattern of at-risk subjects differed from that of established RA patients, who frequently had ACPAs targeting histones and filaggrin, but also fibronectin and fibrinogen (126). Citrullinated fibrinogen in close relation with plasma cells has also been described in bronchial biopsies of established RA patients (56). These studies confirm that ACPAs generated in the respiratory mucosa match those that have been described in the serum of RA patients and that are commonly detected using commercially available techniques. Recently, five citrullinated peptides of some of these proteins (vimentin [n = 2], annexin A2, histone and actin) were reported to be shared between synovial and bronchial tissue of patients with RA, who also had circulating ACPAs against these targets (127). This is in accordance with previous identification of citrullinated antigens in the rheumatoid joint compartment and strengthens the hypothesis that RA-specific autoimmunity may begin in the lung.
Other parallel or complementary pathogenic mechanisms might possibly be involved since the early phases of immune dysregulation. In fact, it has been suggested that smoke-induced inflammation in the respiratory tree, rather than smoke per se, is responsible for peptide citrullination (128), and that this process is abundant in many tissues and driven by inflammation (129). Moreover, patients with bronchiectasis were shown to have increased rates of serum RF and anti-fibrinogen ACPAs, but the immune response was not citrulline-specific and also targeted native peptides (130). This contrasted with the highly specific ACPA response in RA patients who also had bronchiectasis (130). These findings indicate that chronic bacterial infection associated with bronchiectasis may contribute to induction of autoimmunity that becomes RA-specific following the initial loss of tolerance.
In accordance with this observation, a role for microbial infection and lung mucosal dysbiosis has been proposed. Smoking has been associated with increased bacterial colonization and modification of the lung microbiome, thereby proposing an additional route through which smoke can trigger RA-targeted autoimmunity (131). Moreover, changes in microbiota composition have been demonstrated in sputum samples of ACPA-positive at-risk arthritis-free subjects and in the BAL of early untreated RA patients (132, 133). Distal airway dysbiosis in RA was characterized by a reduction in both diversity and abundance of microbiota, some of which correlated with local/systemic levels of autoantibodies and disease activity (133). Intriguingly, the genera Prevotella and Porphyromonas, which have been linked to RA in other mucosal surfaces, were not increased in the lungs of early RA patients and were even found to be significantly reduced in comparison to healthy controls (133).
It should be noted that although these studies provide a basis to associate the lung with the loss of tolerance mechanisms that unfold in the preclinical stages, they do not establish any direct mechanistic link between smoking and citrullination in the pulmonary mucosal surface. In fact, current evidence on ACPA and PAD lung expression merely suggests, by association, that the lungs may contribute to the early breach of tolerance mechanisms, but is insufficient to establish this mucosal surface as the primary site of RA initiation.
Although there is data supporting lungs as an important site for breach of tolerance to occur, many patients with seropositive disease do not present any pulmonary changes or risk factors. Therefore, within the same framework of mucosal-driven inflammation, other sites have been proposed, most notably the gingival and intestinal mucosae (1, 6, 57, 134). Extensive epidemiological evidence implicates microbiota and external infectious agents with the development of RA and other immune-mediated diseases [reviewed in detail elsewhere (3, 134–138)]. On the other hand, microbiota itself changes throughout the course of disease, raising the question of whether some of the reported disturbances are actually a consequence, rather than a cause, of the disease (138). Nevertheless, there is considerable data implicating specifically intestinal and oral dysbiosis in the etiology of RA. In addition, important mechanistic experimental data is available for these compartments, providing support for the role of oral and intestinal mucosae in preclinical disease.
Periodontitis, results from dysbiosis of the gingival microbiota and is strictly associated with established RA, although the association with incident RA is somewhat inconsistent (57–60). This condition entangles mucosal dysbiosis and various pathogenic agents such as Porphyromonas gingivalis (P. gingivalis), which has the ability to citrullinate peptides through its own PAD (PPAD) (139, 140). Further, PPAD can provide a competitive advantage for P. gingivalis, protecting it from the natural acidification of the oral environment through the ammonia generated in the citrullination process (140, 141). There are, however, several differences between PPAD and the human PADs described above, in that the former can only citrullinate C-terminal arginine residues of autoantigens following its cleavage by arginine gingipains (61, 142). In addition, PPAD has the ability to citrullinate free L-arginine residues and does not require calcium ions for catalysis (140, 143). Importantly, experimental data demonstrate that PPAD can citrullinate several RA-specific autoantigens such as α-enolase, vimentin or fibrinogen (61–63). Of note, antibodies against PPAD-citrullinated vimentin were recently reported in patients with preclinical and early RA (63).
Studies have demonstrated periodontal PAD activity and protein citrullination, as well as local and systemic ACPA responses in patients with periodontitis (64–67). Recently, ACPA-positive first-degree relatives of RA patients were found to have a higher prevalence and severity of periodontitis than ACPA-negative subjects (68). Moreover, signs of molecular mimicry and cross reactivity between ACPAs and anti-P. gingivalis antibodies were shown in all stages of disease, from at-risk individuals to established RA (69–72). Such antibodies had additive interactions with other genetic (HLA-SE) and environmental (smoking) risk factors (70). These aspects fit well with the proposed lung-centered hypothesis explained above, whereby inflammation-induced and P. gingivalis-mediated citrullination occur at the gum and lead to loss of tolerance and ACPA generation.
Nevertheless, several aspects are a topic of controversy, and significant advances have been made in the last years. In particular, PPAD autocitrullination has been proposed as a possible mechanism of RA-specific autoimmunity, with conflicting results (73–76). Quirke et al. reported that a PPAD-mediated autocitrullination could elicit a specific immune response against citrullinated PPAD, which was demonstrated in the serum of a proportion of RA patients (73, 75). Subsequently, Konig et al. demonstrated that PPAD from P. gingivalis was not citrullinated, and was targeted by anti-PPAD antibodies in its native, but not citrullinated, form (74, 76). In addition, anti-PPAD antibodies were not associated with disease activity or ACPA titres in RA patients, and were even decreased in patients with coexisting RA and periodontitis (74). Contrarily, however, a recent study demonstrated that PPAD autocitrullination can occur in vitro, and antibodies against autocitrullinated PPAD can be detected in the serum of patients with preclinical, early or established RA (63). As such, despite the progress, this remains an open area of ongoing discussion.
Besides P. gingivalis, other gum-based pathways have been advanced. Exposure to Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), a Gram-negative periodontitis agent, has been reported at increased levels in different independent cohorts of early and established RA patients, as translated by the presence of antibodies against bacterial proteins or DNA probes (67, 77–79, 142). Of note, in one of such studies, A. actinomycetemcomitans was the only periodontal pathogen that differed significantly between patients with or without RA, regardless of periodontitis status (78). As for preclinical RA, two recent studies failed to show an association between IgG against A. actinomycetemcomitans and presymptomatic stages of the disease (80, 81). Nonetheless, recent exposure to this pathogen, as reflected by IgM anti-leukotoxin A antibodies, may be associated with the development of clinical disease in a proportion of patients (80).
In addition to clinical data, there is also mechanistic evidence supporting the implication of A. actinomycetemcomitans in the pathogenesis of RA. In a recent study, Konig et al. elegantly showed that A. actinomycetemcomitans induces dysregulated activation of neutrophil PADs, through the action of the pore-forming leukotoxin A, mimicking membranolytic mechanisms that occur in the RA joint (67). This results in cellular hypercitrullination (LTH, as described above) that is followed by cytolysis and extracellular release of citrullinated proteins. These autoantigens can then be targeted by ACPAs and, indeed, anti-leukotoxin A antibodies were associated with periodontitis, RA and ACPA and RF-positivity (67). Importantly, the periodontitis citrullinome paralleled that of RA synovial fluid, including several major RA-specific citrullinated neoantigens that are targeted by ACPAs (e.g., vimentin, actin, α-enolase, histones and others), reinforcing the link between the inflamed gingival mucosa and the rheumatoid joint (67). Moreover, the spectrum of citrullinated proteins present and targeted in RA could be generated by A. actinomycetemcomitans, but not P. gingivalis. This interesting study provides a complex mechanistic description of an alternative route through which oral bacteria can trigger RA autoimmunity.
Although Engström et al. have recently suggested that local citrullination and PAD activation induced by chronic gingival inflammation were independent of periodontal pathogens such as P. gingivalis and A. actinomycetemcomitans (66), oral dysbiosis does seem to contribute to the genesis of RA. Differences in the oral microbiome have been described in patients with treatment-naïve RA (area under the curve [AUC] 0.87), that are partially normalized following conventional synthetic disease-modifying antirheumatic drug (csDMARDs) treatment (82). Likewise, subgingival microbiota of patients with new-onset RA differs from that of healthy controls and is characterized by the presence and/or abundance of species such as Anaeroglobus geminatus (associated with ACPA/RF-positivity), Prevotella and Leptotrichia (83). Moreover, an interesting study recently assessed the subgingival microbiome of RA patients without periodontitis and found it to be significantly different from controls, with similar frequencies of P. gingivalis and A. actinomycetemcomitans, but enrichment of proinflammatory gram-negative anaerobes and of citrulline-producing Cryptobacterium curtum (84).
Perhaps some of the most compelling data for a specific role of periodontitis in RA pathogenesis comes from studies evaluating the impact of non-surgical periodontal treatment in RA disease activity. A recent meta-analysis including 9 controlled studies of patients with concomitant RA and periodontitis concluded that non-surgical periodontal treatment resulted in clinical disease activity parameters such as DAS28, swollen and tender joint counts, patient global assessment and C-reactive protein (85). Two other subsequent clinical trials also confirmed these findings (86, 87). On the contrary, an interesting large study including over 400 patients with established RA and low disease activity, 56% of whom with periodontitis, failed to demonstrate a benefit of an oral hygiene intervention in improving disease activity, but did show a reduction of pathogenic bacteria load, including P. gingivalis (144). However, it should be stressed that baseline disease activity was low (mean DAS28 2.7 ± 1.3) and therefore less amenable to improvement, especially in patients with long standing disease (mean disease duration 9.0 ± 0.7 years), in whom remission is often a target difficult to attain. Furthermore, patients with baseline periodontitis seemed to have a greater benefit from oral hygiene measures, reinforcing the importance of periodontal health in the management of RA patients (144).
Finally, the intestinal mucosa has also been put forward as a possible site of early immune dysregulation in RA pathogenesis. The gut has a relevant contribution for the development and maintenance of the immune system, particularly through the interaction with commensal and foreign microbes at the mucosal surface (145). The importance of the intestinal microbiota composition as a determinant of human disease has recently been acknowledged, including in RA (134, 145).
Various studies have shown that the gut microbiome is altered in RA patients, with decreased diversity and expansion of several agents, most notably Prevotella copri (82, 88–92). Recently, Prevotella spp. has been demonstrated to be enriched in stool samples of pre-RA subjects (i.e., ACPA/RF-positive and/or suggestive RA signs/symptoms) in comparison with asymptomatic autoantibody-negative first-degree relatives of RA patients (93). While cross-sectional, these data support a role for intestinal dysbiosis since the early phases of RA development and should be confirmed by longitudinal studies.
Mechanistic studies reinforced this theory by demonstrating that autoreactive RA-specific T cell responses can be triggered by gut dysbiosis and cross-reactivity with intestinal bacterial peptides (91, 92). Furthermore, HLA-DR-presented, RA-specific autoantigens that are highly expressed in the synovial tissue have been shown to possess marked sequence homology with bacterial proteins of Prevotella spp. and other gut bacteria, suggesting a molecular mimicry process (94). T cell responses against the corresponding self and microbial peptides were correlated, providing yet another link between mucosal immunity and joint inflammation (94).
Following loss of tolerance and generation of specific autoantibodies, diverse mechanisms likely carry the pathogenic cascade forward through amplification of systemic autoimmunity (146). Firstly, epitope spreading occurs, during which more antigens are targeted by the immune response (6). While no particular single target has been found to determine the transition from the preclinical to the clinical stage, individuals who develop a wider variety of ACPAs and ACPA-specificities are more likely to progress to clinically apparent RA (13, 147–150). In addition to the broad autoantibody repertoire, ACPA titers (13, 148, 150) and, to a lesser extent, avidity (151) also rise progressively until the onset of disease. Moreover, subjects who go on to develop RA have a more diverse pattern of ACPA isotypes and subclasses (152).
Interestingly, there seems to be an uncoupling of the class-switch recombination and avidity maturation processes of B cell development. Extensive isotype switching is accompanied by only a relatively modest increase in ACPA avidity, which is significantly lower than that observed in other humoral responses against recall antigens (e.g., vaccination) (153). Nonetheless, despite its overall low levels, ACPA avidity maturation does occur during the preclinical phase until disease onset (151). Moreover, once RA becomes evident there does not seem to be a major subsequent variation of the recognition pattern, titer or avidity of ACPAs (149–151). Together, these data suggest that the maturation of the ACPA response occurs before the onset of clinical disease and, most likely, takes place outside the joint, in locations such as secondary lymphoid organs (e.g., lymph nodes), where high-affinity interactions take place between B and T cells (1, 6).
Evidence arising from large twin studies suggests that while environmental (e.g., smoking) and other random events are fundamental for the primary break of tolerance in RA autoimmunity, genetic factors such as HLA-SE are more important in the progression of the immune response described above and in the transition to immune-mediated disease (154). Adaptive immunity, therefore, is at the core of the maturation and amplification of systemic autoimmunity in pre-clinical RA.
Extensive literature directly implicates HLA molecules and their genetic variants as major determinants of the risk of RA (3, 18–20). These observations provide important indirect data for the pathogenesis of the disease, since antigen presentation to T cells is likely to be critical in the process (20). Subsequent studies have detailed the precise mechanisms through which specific amino acids located in the HLA-DR peptide binding groove (positions 11/13, 71, 74) create a positively charged pocket that accommodates neutral citrulline residues – but not positive arginine – of proteins such as vimentin, aggrecan, α-enolase or fibrinogen (6, 124, 155). This explains how post-translational protein modifications may lead to RA in a favorable genetic landscape, by facilitating the presentation of autoantigens by the HLA-DR molecule to T cells, via interaction with the T cell receptor (TCR). Indeed, autoreactive CD4+ T cells specific for citrullinated vimentin and aggrecan were shown to be present in RA patients carrying the risk allele HLA-DRB1*04:01 and its numbers were correlated with disease activity parameters (124). Moreover, the most important genetic risk variant outside the HLA region is the R620W single nucleotide polymorphism (SNP) in the PTPN22 gene, which changes the activation threshold of T cells (and also B cells, to a lesser extent), resulting in the escape from thymic negative selection and in the generation of autoreactive cells (156). These data clearly support a role for T cells since the early stages of the immune response, which react to neoantigens presented by APCs and provide decisive help to B cells in the production of autoantibodies against several targets (e.g., ACPAs).
In this regard, the importance of a novel special subpopulation of T cells in the generation of antibodies and autoantibodies has been revealed in the last two decades. These T cells express an important surface chemokine receptor – CXC-chemokine receptor 5 (CXCR5) – that enables them to access the B cell follicle to provide fundamental help to B cells and were, therefore, named T follicular helper (Tfh) cells (157). More specifically, Tfh cells are instrumental in establishing and driving the germinal center response in secondary lymphoid organs, during which, with their help, B cells undergo clonal expansion, somatic hypermutation and affinity maturation, essential steps in an effective humoral response (157, 158). These functions are balanced by their subsequently described counterparts, T follicular regulatory (Tfr) cells, which possess suppressive capacities (159, 160). Since their discovery, both of these subsets have been implicated in the pathogenesis of RA and other immune-mediated rheumatic diseases characterized by autoantibody production and ELS formation (161). However, studies in RA have, so far, been mostly limited to demonstrating either a role for Tfh cells in the initiation and progression of disease in animal models of autoimmune arthritis (162, 163), or altered frequencies of circulating Tfh and Tfr populations in patients with early or established RA (164–166). Therefore, the disturbances and influence of these cell populations during the preclinical stages of RA, while appealing and of great potential, remain to be confirmed and definitely merit further study.
The implication of B cells in the preclinical stages of RA arises mostly from the wealth of studies (detailed above) demonstrating the complex dynamics of autoantibody production, as a surrogate of autoreactive B cell responses (146). Although earlier studies were unable to directly study B cells, these have been clearly established as major contributors to disease pathogenesis by different mechanisms (167). Besides their role in pathogenic autoantibody production (e.g., RF and ACPA), B cells can serve as potent APCs (presenting self-antigens and activating T cells) (168), can produce proinflammatory cytokines (169), and are critical for ELS formation (167, 170).
Disturbances in circulating B cell subpopulations have been identified since the very early stages (<6 weeks) of RA (171), alongside a cytokine pattern that favors B cell survival and activation (172, 173). Moreover, central and peripheral B cell tolerance checkpoints seem to be impaired, as the frequency of autoreactive B cell clones is increased in early and established RA patients (174). Similarly to T cells, this may be related to SNPs in PTPN22 that affect B cell receptor signaling thresholds and lead to the emergence of mature B cells capable of recognizing citrullinated antigens (146, 156).
Indeed, methodological advances have recently enabled to identify the presence of citrullinated antigen-specific B cells (i.e., ACPA-expressing, IgG or IgA) (175–177), plasmablasts (178–180), and plasma cells (181) at relatively high levels in the peripheral blood of RA patients. These ACPAs are notable for their broad multi-reactivity, targeting an expanded set of citrullinated antigens, including those located at the joints (e.g., α-enolase or fibrinogen) (176–181). Additionally, they were found to directly promote the development of arthritis in mice (176) and to highly stimulate the production of TNF by macrophages (180). Importantly, these ACPAs presented highly mutated variable regions, which is suggestive of T cell help, with positive antigen selection through successive passages across germinal centers in secondary lymphoid organs, mucosae or, potentially, synovial ELS (176–178, 180–182). In fact, plasma cells expressing highly somatically mutated and multireactive ACPAs have also been recently identified in the synovial fluid of RA patients (181). These antibodies targeted distinct amino acid motifs and partially overlapping protein targets, supporting the hypothesis that multiple antigen encounters and selection processes occur sequentially, at different sites, such as the mucosae or the joints (181).
Further evidence directly implicating B cells in the initiation of RA was recently provided by a study demonstrating that the presence of dominant B cell receptor clones in peripheral blood of at-risk subjects is associated with a 6-fold increase in the risk of arthritis development (183). Moreover, as clinical disease ensued, these clones migrated to the synovial compartment, strongly implicating them as pathogenic (183).
Another important aspect in the maturation of the RA immune response that has recently been unraveled, relates to the glycosylation profile of both the fragment crystallizable (Fc) and variable region of the ACPA molecule. It has been demonstrated that in established RA, up to more than 90% of IgG ACPAs are glycosylated in their variable domain, a much higher figure in comparison to polyclonal human IgG of healthy donors (15–25%) (146, 182, 184). Of note, the N-linked glycosylation site has been confirmed to be introduced during the process of somatic hypermutation (182, 185), which is in line with the studies demonstrating extensive mutation of ACPA IgG variable regions (176–178, 180–182), and with the absence of such changes in IgM ACPA (186).
Importantly, the extensive variable region N-glycosylation seems not to result from random accumulation of mutations, but rather reflects a competitive advantage for survival and differentiation of ACPA-positive B cell clones, allowing them to escape tolerance checkpoints and generate RA-specific autoimmunity (146, 182, 184, 185). The relevance of these findings for the development of RA has been most recently confirmed in two independent longitudinal studies, which have found that variable domain glycans are present in IgG ACPA up to 15 years before RA onset and their frequency increases progressively up until overt clinical disease (187, 188). Glycosylation of IgG ACPA variable region was higher in at-risk individuals who later developed RA (HR 6.07, 95% CI 1.46–25.2) (187). Further, it was associated with the presence of HLA-SE alleles, thereby suggesting a novel link with this genetic risk marker (188).
In contrast, the Fc tail glycosylation of serum IgG is reduced in patients with RA, as a result of the loss of terminal galactose and sialic acid residues (189–191). This hypoglycosylation profile is associated with a pro-inflammatory phenotype, as the IgG molecule more easily binds Fc receptors (FcRs) and activates complement, being correlated with disease activity and severity (146, 190, 192). Moreover, the Fc galactosylation and sialylation is lower in ACPA-IgG, when compared to total IgG, and in synovial fluid vs. serum ACPA-IgG (193). Notably, a decrease in galactosylated IgG and ACPA-IgG precedes the onset of RA by 3.5 years and 3 months, respectively (190, 191). This is paralleled by a rise in systemic inflammation, as assessed by ESR (191). Data coming from mice studies have implicated the interleukin (IL)-23-T helper (Th)17 axis (via IL-21 and IL-22) in the reduction of IgG Fc sialylation that will lead to arthritis (146). Whether a similar mechanism can be present in humans is an attractive, though unproven, hypothesis that would directly link Fc glycosylation with the evolution of autoimmunity to clinical disease.
Finally, the amplification of the immune response that predates RA is also characterized by expanding systemic inflammation. Circulating cytokines (e.g., IL-1, IL-6, IL-12, tumor necrosis factor [TNF], interferon-γ, granulocyte–macrophage colony-stimulating factor), chemokines (e.g., monocyte chemotactic protein-1, macrophage inflammatory protein-1) and common markers of inflammation such as C-reactive protein have all been shown to increase progressively during the preclinical stages of RA, paralleling or even preceding the appearance and maturation of pathogenic autoantibodies (194–196). Such changes reflect the true systemic nature of the inflammatory mechanisms that will eventually culminate in overt clinical disease.
Despite remarkable progress in the comprehension of the disease pathogenesis, the mechanisms behind the transition from RA systemic autoimmunity, initiated at distant compartments like the mucosal surfaces, to localized and specific joint inflammation remain incompletely understood. The wealth of epidemiological data discussed above, showing a robust association of RF and ACPA with RA development, activity and severity, suggest that these autoantibodies are likely to be directly involved in the initiation of the arthritis that is characteristic of the disease. However, notwithstanding some major advances in recent years, this is still an area of ongoing controversy and a clear-cut pathogenic effect of RF/ACPA remains to be convincingly demonstrated and replicated (197).
A few important observations regarding the putative role of ACPAs and RF in joint targeting have been made. First, maternal transference of ACPAs and RF to the fetal circulation fails to provoke disease in the newborn (6). Second, ACPAs are multi-reactive (see above), can bind a number of citrullinated forms of proteins, which in their native form can be found in several tissues, and the relevant antigen target and tissue in vivo remains unknow (4–6, 197, 198). Third, ACPAs are, by themselves, insufficient to provoke synovitis, but can exacerbate and perpetuate an ongoing arthritic process (5, 6, 199). Under this model, an additional, perhaps preceding event, that causes an acute or subacute form of arthritis – a second hit, such as trauma, a viral infection or other unclear microvascular or neuroimmune factors – would be required to, in the presence of circulating ACPAs and/or RF, set in motion the pathogenic chain of events that will ultimately result in an autonomous, persistent, chronic synovitis (1, 6).
Nevertheless, it should be stressed that the pathogenicity of ACPAs has, to date, not been demonstrated in animal models. They have, in fact, been shown to be absent from common experimental arthritis models in mice and rats, such as collagen-induced arthritis (CIA) (200, 201), collagen antibody-induced arthritis (CAIA) (201), human TNF-transgenic mice (201, 202), adjuvant induced arthritis (201), and pristane-induced arthritis (PIA) (201, 203). Instead, what has been identified in such models, is the presence of measurable anti-CarP antibodies in mice with CIA and rats with CIA, PIA and AIA (200, 201). These arthritis models share the common feature of requiring the direct involvement of the adaptive immune system. This suggests that the tolerance to carbamylated antigens is breached early in the pathogenic process, unlike what is seen for citrullinated peptides, again questioning the pathogenicity of ACPAs. Another key aspect that is observed in these animal models, is the targeting of uncitrullinated forms of autoantigens widely recognized in RA, such as vimentin, histones, filaggrin or heterogeneous nuclear ribonucleoprotein (hnRNP)-A2 (also known as RA33) (107, 202, 203).
Autoantibodies against native (i.e., unmodified, uncitrullinated) proteins have been reported to be common in seropositive and seronegative RA. Among the most frequently described are FcV receptor (targeted by RF), PADs (most notably, PAD4), hnRNP-A2 (RA33) and (112, 114, 115, 204–207). The former two have been addressed above, while the latter has experienced a renewed interest in recent years. As previously mentioned, anti-RA33 antibodies have been identified in animal models of arthritis (202, 203). This antigen is overexpressed in the joints of human TNF-transgenic mice and PIA rats, and a specific humoral response targeting RA33, but not ACPAs, takes place in both preclinical and clinical phases of the disease. An important study by Konig et al. recently demonstrated a dual antibody response against both native and citrullinated forms of RA33 in the synovial compartment of patients with RA (205). The authors proposed an antigen-centered model, wherein an initial immunologic response would target RA33 in its unmodified form. According to this model, citrullination would only occur at a later stage, amplifying, but not starting, the immune response. This is demonstrated by the association of antibodies targeting citrullinated RA33 (i.e., ACPAs) with disease duration and erosive disease, in contrast with anti-RA33 antibodies, which were limited to early, mild, non-erosive RA. This transition from an anti-native to an ACPA response would depend on genetic (e.g., HLA-DR risk alleles) and environmental factors (e.g., smoking, infections, LTH). This is supported by studies demonstrating that the immune response seen in chronic bacterial infections, such as periodontitis or bronchiectasis, mainly targets non-citrullinated antigens in its earlier stages (130, 208). Future studies investigating this hypothesis are expected to provide further insight in the specific mechanisms involved in the targeting of rheumatoid joints.
Prior to the emergence of clinically detectable arthritis, however, an earlier intermediate step is postulated to take place. This is when the first symptoms begin to appear, usually in the form of arthralgia (7). In this stage, most patients have evidence of ongoing autoimmunity (e.g., ACPA positivity) and start to experience what has been defined as clinically suspicious arthralgia (9). The reasons for these symptoms have been a matter of debate. In fact, evident imaging and histological synovitis were shown to be absent in subjects at high-risk of progressing to RA and could not discriminate which patients would develop the disease, suggesting that mechanisms other than synovial inflammation could explain patient symptoms (209, 210).
Nonetheless, there are indications that at least some degree of joint inflammation, not detectable by clinical examination, could be present before the onset of full-blown arthritis. In one of the studies addressing at-risk subjects, synovial infiltration by T cells was marginally associated with later development of arthritis (HR 2.8, 95% CI 0.9–9.1), particularly when circulating ACPAs were also positive (HR 3.8, 95% CI 1.3–11.3) (210). Moreover, the presence of CD8+ T cells in the synovium was strongly associated with ACPA positivity (OR 16.0, 95% CI 1.7–151.1), suggesting that specific joint citrullinated antigens may be targeted by the RA immune response (210). Synovial macrophage and T cell infiltration have also been demonstrated prior to the onset of arthritis in immunized monkeys and in clinically uninvolved joints of RA patients (211). It is thus possible that at least part of the symptoms experienced by patients during this phase, already reflects some degree of synovial inflammation, not clinically identifiable, that may be related to the presence of ACPAs and RF, closely preceding the installation of flagrant arthritis. This concept is supported by more recent studies reporting increased rates of subclinical synovial and bone inflammation defined by magnetic resonance imaging (212–214) and ultrasound (215, 216) in patients with clinically suspicious arthralgia. Of note, these changes were able to predict progression to RA in both ACPA-positive (215, 216) and ACPA-negative disease (214).
It seems apparent that, at least in the final stages of preclinical phase, targeting of the synovial compartment starts to occur. In this regard, ACPAs and RF have been proposed to play a pathogenic role (197). Recognition and binding of joint citrullinated antigens by ACPAs can lead to immune complex formation and activation of the complement cascade, that will result in recruitment of immune cells into the synovial compartment (217, 218). Incoming and resident cells, such as macrophages, mast cells, neutrophils and synovial fibroblasts, can then be activated by these immune complexes via interaction with surface FcγRs (and also TLRs) (197, 218–222). Cellular activation leads to the release of cytokines (e.g., TNF, IL-6) and other inflammatory and vasoactive mediators, enhancing and propagating the synovitis process (197, 218–222). Importantly, RF also seems to contribute significantly to this process, as it binds to synovial ACPA immune complexes and boosts their pro-inflammatory actions of complement activation and cytokine production (223–225). Moreover, the discussed ACPA Fc hypoglycosylation observed in RA and pre-RA patients further contributes to greatly increase the immune complex-induced activation of FcγR and complement, reinforcing the hypothesis that ACPAs are closely linked to synovitis initiation (146, 190, 191, 193). Overall, these studies support the working model in which the developing humoral response culminates, possibly following a predisposing insult, in the targeting of synovial citrullinated antigens by ACPAs and RF. These have, together and individually, enhanced ability to spark the inflammatory response typical of synovitis, which, albeit not clinically recognizable at first, becomes evident as the process advances.
Even considering that pre-arthritis symptoms may be due to undetectable degrees of synovitis, the possibility still remains that other alternative pathways are in place. Accordingly, a new theory that directly implicates ACPAs – in an osteoclast-centered, synovitis-independent process – has been proposed in recent years, generating great controversy in the field (6, 8, 197). In this model, not only arthralgia but also early cortical bone loss, reported to be significantly increased in ACPA-positive pre-RA patients (226), could be the result of the interaction of ACPAs with key bone constituents. Osteoclast precursors have been shown to express citrullinating enzymes (i.e., PAD) and surface citrullinated proteins (e.g., vimentin, actin) that can be directly targeted by ACPAs (227–229). This induces the release of TNF and CXCL8 (i.e., IL-8), which promote osteoclast differentiation and activation in an autocrine fashion, leading to osteopenia and bone resorption in vivo (181, 227, 229). Notably, these processes were blocked by inhibition of PAD (229), CXCL8 (229), or TNF (227). These findings were initially replicated in a separate study (228), but the citrulline-binding specificity of the supposed ACPAs was subsequently not confirmed (230), suggesting that other mechanisms or technical reasons (e.g., antibody aggregation) might be involved (197, 231).
An alternative mechanism linking ACPAs and bone targeting was later proposed, as the interaction of immune complexes with FcγRs of pre-osteoclasts was demonstrated to promote osteoclastogenesis (232). This effect was dependent on IgG sialylation status, with desialylated (but not sialylated) immune complexes (and ACPAs) greatly stimulating osteoclast differentiation in vitro and in vivo (232). These findings are in accordance with the discussed increased pathogenicity of Fc hypoglycosylated ACPAs and total IgG reported in pre-RA and RA (190, 191), as well as with the observation that RA patients with lower IgG and ACPA sialylation had more bone loss (232). Although these studies lack unequivocal replication, the interaction of ACPAs – either in Fc-dependent or independent fashion – with developing osteoclasts represents an appealing explanation for both the targeting of bone and joints by the autoimmune response started in other distant compartments and the early bone loss observed in pre-RA patients, prior to the dawn of inflammation (6, 226).
In the second complementary part of this theory, ACPAs have also been proposed to have a direct link with joint pain in pre-RA, independently from synovitis (6, 233). The hypothesis proposed that osteoclast-bound ACPAs would induce the release of CXCL8, which could then bind and activate its receptors (CXCR1 and CXCR2) on nociceptive nerve terminals (233). In mice, this resulted in pain-like behavior that could be abolished by CXCR1/2 blockade (233). However, following the discovery that the antibodies used in this study were not ACPAs (230), this hypothesis has been questioned and, to date, could not be replicated (197). An alternative explanation could stem from CAIA models, in which antibodies against type II collagen are injected and bind to cartilage surface, occasioning immune complex deposition and florid synovial inflammation (234). In this model, mice experience pain-like behavior before the onset of arthritis and independently of complement activation, due to local immune complexes deposition which activate FcγRs expressed on sensory neurons (235). Interestingly, this mechanism could partially explain the results observed in the unconfirmed study (228), through cross-reactivity of polyclonal ACPAs that would recognize and bind citrullinated epitopes of type II collagen (235).
Finally, a few other pathways have been suggested to explain joint targeting in RA, mainly based on data arising from animal models and lacking substantial epidemiological support (1, 5, 6). These include the immune response to unmodified forms of ubiquitous enzymes such as glucose-6-phosphate isomerase (236) or joint-specific cartilage constituents (e.g., type II collagen) (237). While the former has been found not to be common or specific for RA (238), the latter has been one of the most studied and has recently gained renewed attention (237, 239). Anti-type II collagen antibodies have been found to be present in a subgroup of RA patients (3–27%), particularly during early disease, and may be associated with a particular phenotype of marked acute inflammation in a similar fashion to what is observed in CAIA and CIA experimental models (237, 240). These antibodies can lead to arthritis via immune-complex deposition and are also believed to have direct damaging effects in the cartilage (237). More recently, the relevance of type II collagen citrullination was proposed, as citrullinated forms of type II collagen were identified in the synovial fluid of RA patients (239) and were targeted by cross-reacting circulating ACPAs in 17–27% of patients (241). Additionally, ACPAs highly specific to citrullinated type II collagen have been demonstrated to be pathogenic in mice (239) and to be present in about a third of RA patients, linking ACPA and cartilage-targeting pathways (242). Whether, as proposed above, these ACPAs against citrullinated type II collagen are the result of the evolving initial immune response targeting native cartilage constituents, is a possibility that deserves further exploration.
The events described above are applicable to seropositive disease, that is, where ACPA (and usually also RF) are present in circulation. This is the major RA subtype, which accounts for 70–80% of patients, both in the context of research and daily practice. Substantially less is known about the mechanisms involved in the pathogenesis of the seronegative subset of disease, that is, that without either ACPA or RF (243). It is important to address a few aspects concerning the definition and characteristics of this patient population. Although seronegative RA has long been recognized as a distinct clinical entity, there is still debate as to whether this really is the case. While some argue that these patients have a phenotype compatible with RA, but present different etiological and pathogenic pathways, others regard this classification as the result of unmeasured/unknown circulating autoantibodies (both ACPAs and non-ACPAs) in “true” RA patients. Others, still, consider this an heterogeneous population, possibly including patients who were misclassified or will evolve into other diagnoses over time (e.g., spondyloarthritis) (244). Although the majority of the driving forces behind seronegative RA are undetermined, some progress has been made over the last decade. Evidence that can inform in this regard is mostly indirect, but a few points can be made (summarized in Table 2).
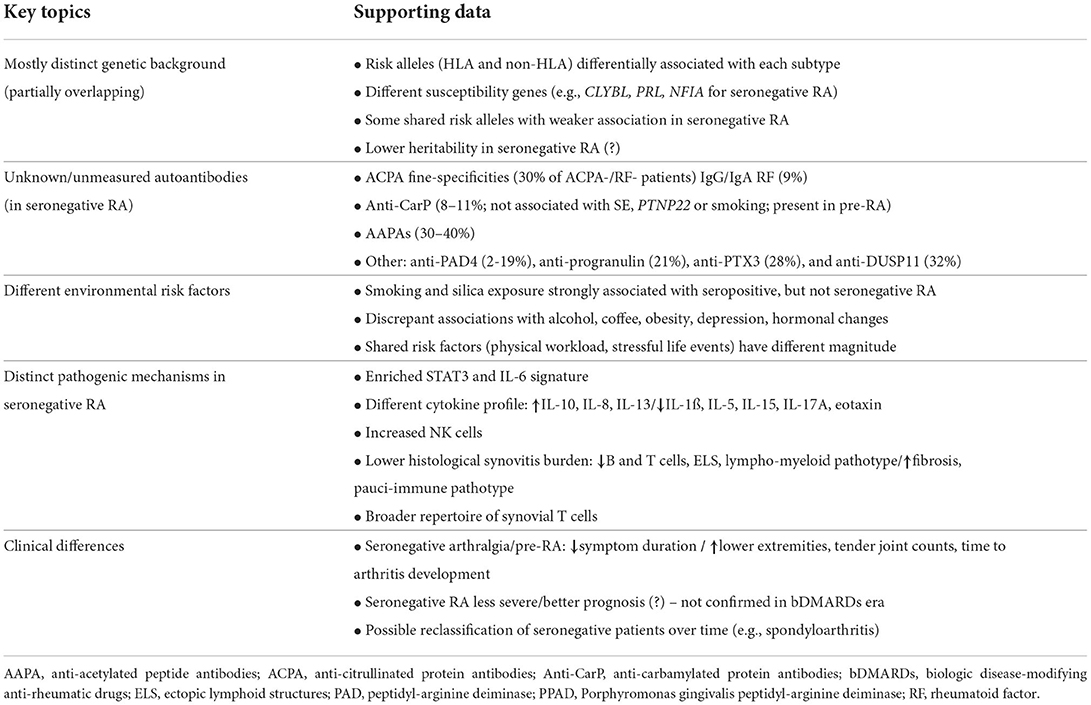
Table 2. Essential aspects differentiating seropositive and seronegative RA.
First, there is evidence that ACPA-positive and ACPA-negative RA have mostly distinct genetic backgrounds (243). This has been suggested by several studies, with risk alleles differentially associated with each subtype being identified both within and outside the HLA region (245–252). The latter included some genes that are also associated with seropositive RA (e.g., TNFAIP3, PTPN22), as well as a few novel susceptibility genes (e.g., CLYBL, PRL, NFIA) (245, 250). Also, the strength of the associations seen for shared risk alleles is different between both subsets in a number of studies, with lower effect sizes in ACPA-negative RA (245–250). Likewise, differences were even noted in specific amino acid residues of the HLA molecule for both disease subsets (246). These findings are in accordance with the recently reported lower heritability in seronegative RA (~20%), in comparison with seropositive disease (50%) (253). However, other studies have suggested that genetic risk of both subsets greatly overlaps (254–256). Therefore, the current view is that ACPA-positive and ACPA-negative RA do share some genetic risk markers, but there are substantial differences between these two subsets, with several susceptibility genes more strongly associated with one or another subtype.
Second, another possibility is that patients classified as seronegative have other unknown or unmeasured circulating autoantibodies. A recent study showed that many classically-defined seronegative patients had ACPA fine-specificities (30%), IgA/IgG RF (9.4%) or anti-CarP antibodies (16%), thus underlining that a considerable proportion of “seronegative” patents are wrongly classified as such (257). Anti-CarP antibodies had been originally identified also in 16% of ACPA-negative patients, who had a more severe disease course than those negative for both anti-CarP and ACPA (258). The presence of these autoantibodies in ACPA-negative patients was replicated in other cohorts (8–14%), and was not associated with HLA-DRB1 SE alleles, PTPN22 or smoking, suggesting distinct biological mechanisms in comparison with ACPA-positive RA (259). Anti-CarP antibodies have now been shown to be present years before the onset of clinical disease, in around 10–11% of ACPA/RF-negative pre-RA patients, and predict incident RA independently of ACPA (16, 260, 261). These findings suggest that anti-CarP is involved in the pathogenesis of RA, including of ACPA/RF-negative disease. Anti-acetylated peptide antibodies (AAPA) are another important class of non-ACPA autoantibodies that was recently described in 30–40% of patients with early or established seronegative RA (262, 263). Further, diagnostic accuracy was greatly increased when positivity against more than one acetylated peptide was observed, with AAPAs against three acetylated peptides being 100% specific for RA (262). Together, ACPAs, anti-CarP and AAPAs may be classified as anti-modified-protein antibodies (AMPAs) (264). The various AMPAs may present cross-reactivity between each other to different extents, i.e., to one, two or all three of them (264–267). In patients with new-onset RA, triple reactivity has even been shown to be associated with higher rates of radiographic progression at 12 months than single reactivity (265). Finally, novel antibodies were recently identified in a substantial proportion of seronegative patients, such as anti-PAD4 (2–19%) (96, 109, 111), anti-progranulin (21%), anti-PTX3 (28%), and anti-DUSP11 (32%) antibodies, further expanding the comprehension of ACPA-negative disease (268, 269).
Third, an additional argument in favor of a distinct pathogenesis between ACPA-positive and ACPA-negative RA is supported by the differences observed in environmental risk factors for both subsets (3). Some of the major risk factors for RA, such as smoking (270) or silica (46) exposure, are strongly associated with seropositive, but not seronegative RA (6). Similarly discrepant findings have been reported for variables as different as alcohol consumption (271), coffee intake (272), obesity (273, 274), depression (275) or hormonal changes (276). Of note, as for genetic background, some environmental risk factors, such as physical workload (277) or stressful life events (278), associate with both subtypes, but on a different magnitude. Again, these data are indicative that the mechanisms occurring in the preclinical phases of seropositive and seronegative RA are at least partially distinct.
Fourth, although significantly less is known about the mechanisms leading to, and driving, seronegative RA, some evidence has emerged over the last decade. Circulating CD4+ T cells of untreated undifferentiated arthritis patients have been shown to have a transcriptional signature enriched in STAT3-inducible genes, which predicts the evolution to RA, especially to seronegative disease (279). This gene expression pattern was correlated with IL-6 levels, which were higher in patients with ACPA-negative RA (279). IL-6 signals mainly via STAT3, and an IL-6-driven STAT3 signaling pathway in CD4+ T cells has been identified in the early phases of seronegative RA (280). These pairs well with the strong genetic association of ACPA-negative RA with ANKRD5, a gene greatly expressed by CD4+ T cells, which lies close to the locus encoding glycoprotein 130 (gp130), a signal transducing component of the functional receptor complexes for IL-6 (249, 252). In addition to a putative prominent role of IL-6 in seronegative RA, another study demonstrated a distinct cytokine profile of patients with early ACPA/RF-negative disease, with increased IL-10 and decreased IL-1ß, IL-15 and eotaxin in levels in comparison with patients with seropositive RA (281). Further, IL-5 and IL-17A were not altered in seronegative arthralgia patients (contrary to RF-positive disease), whereas an increase in IL-8 and IL-13 could be seen in this population (194). The former could directly induce pain by binding to nociceptors, while the latter promotes synovial hyperplasia and is secreted by CD56bright natural killer (NK) cells, which are increased in seronegative RA and also produce IL-10 (282, 283). Thus, however slim and lacking replication, these data suggest that distinctive mechanisms may underlie the preclinical phases of seronegative RA.
Additional evidence stems from the comparison of synovial tissue features of ACPA-positive and ACPA-negative patients. Patients with established seronegative RA were reported to have a lower synovitis burden, translated by a lower infiltration of B and T cells (especially CD8+), less lymphoid aggregates (and corresponding lympho-myeloid pathotype), more fibrosis, and a higher representation of a pauci-immune pathotype (284–286). In addition, synovial T cells have a more restricted repertoire in ACPA-positive, in comparison with ACPA-negative patients, suggesting a more antigen-driven pathway in seropositive disease (287). Interestingly, CD68+ sublining macrophages have similar distributions in both subsets (284–286).
Fifth, a few words are warranted for the clinical aspects of seronegative RA. A recent study has demonstrated that the preclinical phases leading to RA are different in ACPA-positive and ACPA-negative patients (288). Patients with seronegative arthralgia had more often involvement of the lower extremities, higher tender joint counts, shorter symptom duration, and more difficulty making a fist. However, after presenting with arthralgia, ACPA-negative patients took longer to develop arthritis, thus suggesting that seropositive and seronegative disease develop differently. In terms of disease course, RF/ACPA-negative RA has traditionally been regarded as less severe, and indeed, seropositivity is a recognized poor prognostic factor (289–291). However, studies of patients treated in the novel biologic treatment era have demonstrated that patients with seronegative RA have higher baseline disease activity, disability, patient reported outcomes and erosive burden (292, 293). Further, although these patients responded well to DMARD therapy, the treatment intensity and requirement was similar to RF/ACPA-positive patients, and serological status was not associated with remission (293). Likewise, functional, social and occupational impairment were similar, contradicting the view of seronegative RA as a less impacting disease (292). Finally, it should be acknowledged that the group of patients classified as seronegative RA may include other diseases that present in a RA-like fashion and develop manifestations of other conditions later in the disease course. In fact, recent studies demonstrated that a significant proportion of seronegative patients are reclassified as having another diagnosis over long-term follow-up, most notably spondyloarthritis (244, 294).
In summary, the pathogenesis of seronegative RA is still mostly unknown. The limited available data suggest that the mechanisms leading up to the onset of clinically evident disease are at least partially different from those described for ACPA-positive RA. In addition, novel autoantibodies are being described, which could lead to a significant reduction in the pool of real ‘seronegative’ patients with RA. Much more so, when considering that many of them will eventually be reclassified as having another disease in the future. This is an expanding field, where our understanding of the true nature and driving forces behind seronegative RA is likely to increase substantially over the next years.
Tremendous progress has been made over the last decades in the understanding of early pathological events that occur before the onset of RA. One of the major insights that has since arisen is the concept that the initial immune disturbance mechanisms take place outside the joint compartment. Indeed, albeit being mostly localized to the joints, RA is truly a systemic disease even since its primordial stages. In this regard, mucosal tissue plays with a high probability a crucial role, as the interface between self and environment where the initial immunologic response to altered antigens commence. The lungs are the foremost site driving the process that will culminate in breach of tolerance and triggering of autoimmunity, closely followed by the gingival and intestinal mucosae.
What is unique in this theoretical model is that it clearly explains the close connection between environment (e.g., smoking) and host (e.g., HLA-SE) in determining the development and onset of disease. It provides a basis for the strong epidemiological observations that pinpoint specific risk factors for incident RA. Further, it strengthens the recommendations made for at-risk subjects, such as quitting smoking, maintaining good oral and periodontal health, and following a balanced diet, maintaining a low body mass index.
Attesting to the systemic nature of RA, the adaptive immune system will likely drive the amplification of the immune response that follows mucosal loss of tolerance, expanding it to secondary lymphoid organs. Herein, APCs, T cells, B cells, and plasma cells, closely interact, maturing the immune response over time, and resulting in a broader and more specific antibody repertoire. This is paralleled by modifications in the glycosylation status of immunoglobulins, including ACPA and RF, that increases their pathogenic and inflammatory potential, as well as a steady overall rise in systemic inflammatory markers.
Finally, the inflammatory response reaches the joint, probably prior to the advent of clinical symptoms. Circulating ACPAs recognize and bind synovial citrullinated antigens, activating an immune complex-driven local inflammatory response that is further amplified by RF. Other attractive mechanisms including ACPA-induced activation and differentiation of osteoclasts have been proposed and deserve further study. Once started, the synovitis process evolves into clinically recognizable arthritis.
The better comprehension of these preclinical pathophysiological events allows for an overall comprehension of RA as a complex systemic disease. It facilitates the improvement of prediction models and biomarkers, particularly useful for at-risk subjects (295); it contributes to the identification of key mediators of disease, which can be targeted by novel therapeutic agents (296); and it can ultimately revolutionize the paradigm of RA care, by directly interfering in the early preclinical stages of the disease with lifestyle modifications and specific therapies that can halt, or at least delay, the ongoing pathogenic immune response (297). These ideas have recently been tested in several observational studies and clinical trials (Table 3). In fact, the PRAIRI study provided proof-of-concept that such a strategy could be useful, by demonstrating that a single infusion of B cell-depleting rituximab delayed the onset of arthritis in at-risk individuals (302). The recently reported results of ARIAA study were also in line with this, wherein targeting T cell co-stimulation with abatacept for 6 months significantly decreased subclinical joint inflammation and the incidence of new-onset arthritis in ACPA+ subjects with arthralgia and imaging evidence of joint inflammation (306). Other studies are investigating a similar effect with treatments such as methotrexate or hydroxychloroquine (Table 3) (304, 305, 308).
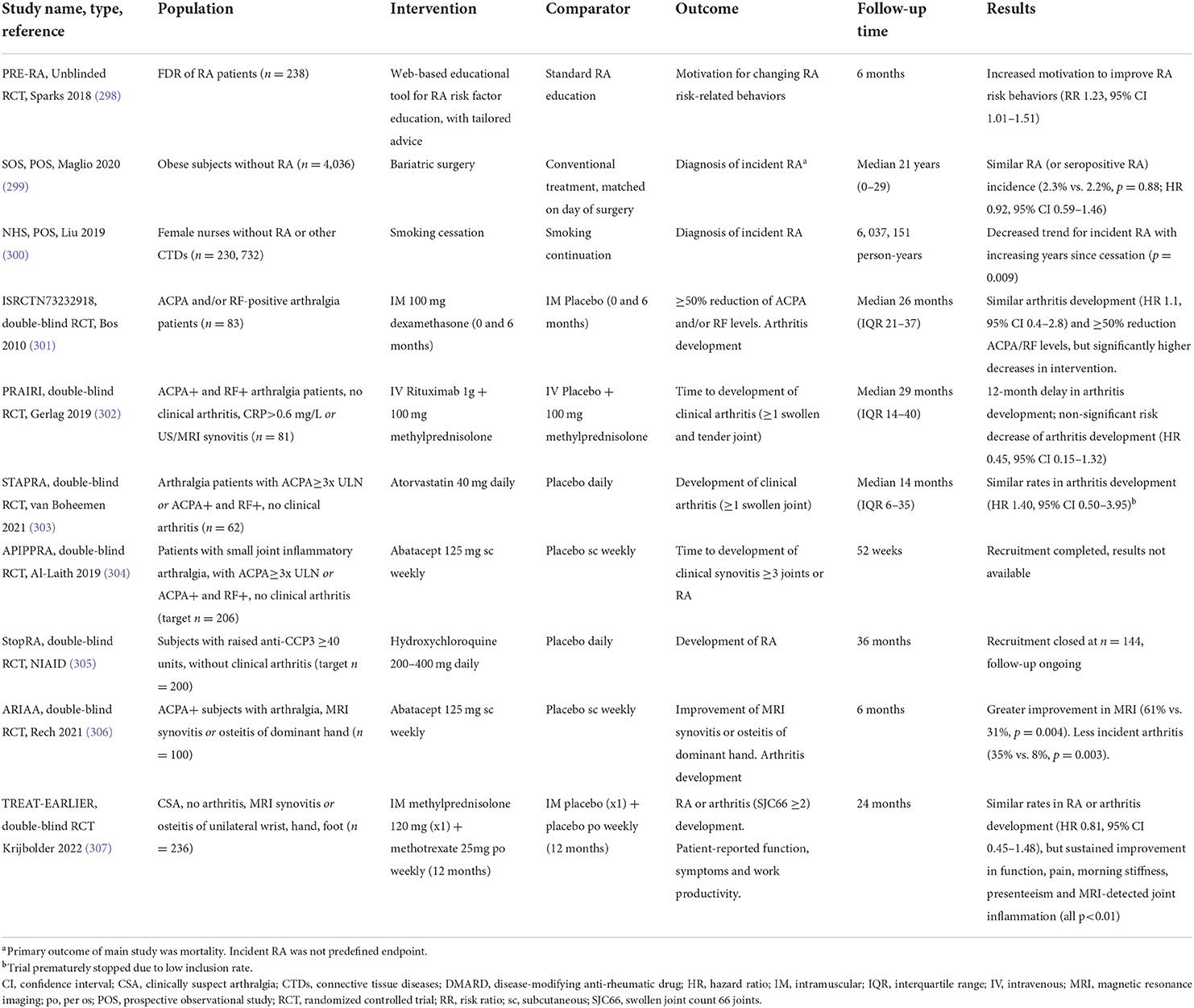
Table 3. Summary of studies investigating lifestyle and pharmacological interventions in preclinical RA.
In this regard, it should be stressed that the designation of at-risk individuals has multiple definitions across studies. Such examples include first-degree relatives of RA patients, ACPA+ asymptomatic subjects, seronegative patients with CSA, patients with subclinical imaging synovitis, to name only a few. These subject groups may or may not be analyzed together or in different combinations, potentially diluting or scattering important signals arising from studies, translating independent mechanistic pathways. Thus, terminology standardization according to published guidelines is key in order to allow robust comparison between studies (7). In addition, types and timing of therapeutic or preventive interventions also vary, as do outcomes of interest. To tackle these issues, a data-driven consensus statement was recently published, providing additional guidance for research studies focusing on this population (309).
In conclusion, the pathogenesis of RA initiates well before the onset of clinical symptoms. In line with the complex nature of the disease, and the multitude of etiological risk factors, intricate pathogenic mechanisms take place both outside and within the joints, evolving over time until clinical disease becomes apparent. Future developments in the field are likely to provide new insightful data, potentially leading to a novel era of ever-earlier treatment institution, arresting disease progression and aiming at prevention of RA.
VCR and JEF have contributed to study conception and design, collected and analyzed the data, and drafted the manuscript. All authors have critically reviewed the manuscript for important intellectual content, have read, and approved its final version.
VCR work was funded by Fundação para a Ciência e Tecnologia (Interno Doutorando Bursary reference SFRH/SINTD/95030/2013); European League Against Rheumatism (EULAR Scientific Training Bursary 2014); and Sociedade Portuguesa de Reumatologia (Fundo de Apoio à Investigação da SPR 2015 & 2016 to VCR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
2. Smolen JS, Aletaha D, Mcinnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
3. Romão VC, Fonseca JE. Etiology and risk factors for rheumatoid arthritis: a state-of-the-art review. Front Med. (2021) 8:e689698. doi: 10.3389/fmed.2021.689698
4. Hochberg MC, Gravallese EM, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH. Rheumatology. 7th ed. Philadelphia, PA: Elsevier Inc. (2019).
5. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Prim. (2018) 4:18001. doi: 10.1038/nrdp.2018.1
6. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat Rev Immunol. (2017) 17:60–75. doi: 10.1038/nri.2016.124
7. Gerlag DM, Raza K, Van Baarsen LGM, Brouwer E, Buckley CD, Burmester GR, et al. EULAR recommendations for terminology and research in individuals at risk of rheumatoid arthritis: Report from the Study Group for Risk Factors for Rheumatoid Arthritis. Ann Rheum Dis. (2012) 71:638–41. doi: 10.1136/annrheumdis-2011-200990
8. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. (2012) 8:656–64. doi: 10.1038/nrrheum.2012.153
9. van Steenbergen HW, Aletaha D, Beaart-van de Voorde LJJ, Brouwer E, Codreanu C, Combe B, et al. EULAR definition of arthralgia suspicious for progression to rheumatoid arthritis. Ann Rheum Dis. (2017) 76:491–6. doi: 10.1136/annrheumdis-2016-209846
10. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. (2019) 20:928–42. doi: 10.1038/s41590-019-0378-1
11. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol. (2021) 22:10–8. doi: 10.1038/s41590-020-00816-x
12. Puente A Del, Knowler WC, Pettitt DJ, Bennett PH. The incidence of rheumatoid arthritis is predicted by rheumatoid factor titer in a longitudinal population study. Arthritis Rheum. (1988) 31:1239–44. doi: 10.1002/art.1780311004
13. Rantapää-Dahlqvist S, De Jong BAW, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and iga rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. (2003) 48:2741–9. doi: 10.1002/art.11223
14. Nielen MMJ, Van Schaardenburg D, Reesink HW, Van De Stadt RJ, Van Der Horst-Bruinsma IE, De Koning MHMT, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50:380–6. doi: 10.1002/art.20018
15. Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, De Vries N, Van Der Horst-Bruinsma IE, et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: A prospective cohort study. Ann Rheum Dis. (2010) 69:490–4. doi: 10.1136/ard.2008.105759
16. Shi J, Van De Stadt LA, Levarht EWN, Huizinga TWJ, Toes REM, Trouw LA, et al. Anti-carbamylated protein antibodies are present in arthralgia patients and predict the development of rheumatoid arthritis. Arthritis Rheum. (2013) 65:911–5. doi: 10.1002/art.37830
17. Rönnelid J, Wick MC, Lampa J, Lindblad S, Nordmark B, Klareskog L, et al. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: Anti-CP status predicts worse disease activity and greater radiological progression. Ann Rheum Dis. (2005) 64:1744–9. doi: 10.1136/ard.2004.033571
18. Deighton CM, Walker DJ, Griffiths ID, Roberts DF. The contribution of HLA to rheumatoid arthritis. Clin Genet. (1989) 36:178–82. doi: 10.1111/j.1399-0004.1989.tb03185.x
19. Zanelli E, Breedveld F, de Vries RR. Hla class II association with rheumatoid arthritis. Hum Immunol. (2000) 61:1254–61. doi: 10.1016/S0198-8859(00)00185-3
20. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. Arthritis Rheum. (1987) 30:1205–12. doi: 10.1002/art.1780301102
21. Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. (1993) 32:903–7. doi: 10.1093/rheumatology/32.10.903
22. Aho K, Koskenvuo M, Tuominen J, Kaprio J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J Rheumatol. (1986) 13:899–902.
23. Svendsen AJ, Kyvik KO, Houen G, Junker P, Christensen K, Christiansen L, et al. On the origin of rheumatoid arthritis: the impact of environment and genes—a population based twin study. Ahuja SK, editor. PLoS ONE. (2013) 8:e57304. doi: 10.1371/journal.pone.0057304
24. Webster AP, Plant D, Ecker S, Zufferey F, Bell JT, Feber A, et al. Increased DNA methylation variability in rheumatoid arthritis-discordant monozygotic twins. Genome Med. (2018) 10:64. doi: 10.1186/s13073-018-0575-9
25. Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. (2009) 60:3613–22. doi: 10.1002/art.25018
26. Glossop JR, Emes RD, Nixon NB, Packham JC, Fryer AA, Mattey DL, et al. Genome-wide profiling in treatment-naive early rheumatoid arthritis reveals DNA methylome changes in T and B lymphocytes. Epigenomics. (2016) 8:209–24. doi: 10.2217/epi.15.103
27. de Andres MC, Perez-Pampin E, Calaza M, Santaclara FJ, Ortea I, Gomez-Reino JJ, et al. Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther. (2015) 17:233. doi: 10.1186/s13075-015-0748-5
28. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. (2013) 31:142–7. doi: 10.1038/nbt.2487
29. Zhu H, Wu LF, Mo XB, Lu X, Tang H, Zhu XW, et al. Rheumatoid arthritis-associated DNA methylation sites in peripheral blood mononuclear cells. Ann Rheum Dis. (2019) 78:36–42. doi: 10.1136/annrheumdis-2018-213970
30. Frank-Bertoncelj M, Trenkmann M, Klein K, Karouzakis E, Rehrauer H, Bratus A, et al. Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun. (2017) 8:14852. doi: 10.1038/ncomms14852
31. Ai R, Hammaker D, Boyle DL, Morgan R, Walsh AM, Fan S, et al. Joint-specific DNA methylation and transcriptome signatures in rheumatoid arthritis identify distinct pathogenic processes. Nat Commun. (2016) 7:11849. doi: 10.1038/ncomms11849
32. Huber LC, Brock M, Hemmatazad H, Giger OT, Moritz F, Trenkmann M, et al. Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum. (2007) 56:1087–93. doi: 10.1002/art.22512
33. Kawabata T, Nishida K, Takasugi K, Ogawa H, Sada K, Kadota Y, et al. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res Ther. (2010) 12:R133. doi: 10.1186/ar3071
34. Horiuchi M, Morinobu A, Chin T, Sakai Y, Kurosaka M, Kumagai S. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J Rheumatol. (2009) 36:1580–9. doi: 10.3899/jrheum.081115
35. Niederer F, Ospelt C, Brentano F, Hottiger MO, Gay RE, Gay S, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis. (2011) 70:1866–73. doi: 10.1136/ard.2010.148957
36. Engler A, Niederer F, Klein K, Gay RE, Kyburz D, Camici GG, et al. SIRT6 regulates the cigarette smoke-induced signalling in rheumatoid arthritis synovial fibroblasts. J Mol Med. (2014) 92:757–67. doi: 10.1007/s00109-014-1139-0
37. Engler A, Tange C, Frank-Bertoncelj M, Gay RE, Gay S, Ospelt C. Regulation and function of SIRT1 in rheumatoid arthritis synovial fibroblasts. J Mol Med. (2016) 94:173–82. doi: 10.1007/s00109-015-1332-9
38. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci USA. (2011) 108:11193–8. doi: 10.1073/pnas.1019536108
39. Chan EKL, Ceribelli A, Satoh M. MicroRNA-146a in autoimmunity and innate immune responses. Ann Rheum Dis. (2013) 72(suppl 2):ii90–5. doi: 10.1136/annrheumdis-2012-202203
40. Vicente R, Noël D, Pers YM, Apparailly F, Jorgensen C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat Rev Rheumatol. (2016) 12:211–20. doi: 10.1038/nrrheum.2015.162
41. Klein K, Karouzakis E, Gay S. Rheumatoid Arthritis and Epigenetics. In: The Epigenetics of Autoimmunity. Elsevier. (2018). doi: 10.1016/B978-0-12-809912-4.00007-6
42. Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. (2006) 54:38–46. doi: 10.1002/art.21575
43. Sugiyama D, Nishimura K, Tamaki K, Tsuji G, Nakazawa T, Morinobu A, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: A meta-analysis of observational studies. Ann Rheum Dis. (2010) 69:70–81. doi: 10.1136/ard.2008.096487
44. Anderson R, Meyer PWA, Ally MMTM, Tikly M. Smoking and air pollution as pro-inflammatory triggers for the development of rheumatoid arthritis. Nicotine Tob Res. (2016) 18:1556–65. doi: 10.1093/ntr/ntw030
45. Too CL, Muhamad NA, Ilar A, Padyukov L, Alfredsson L, Klareskog L, et al. Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case–control study. Ann Rheum Dis. (2016) 75:997–1002. doi: 10.1136/annrheumdis-2015-208278
46. Stolt P, Källberg H, Lundberg I, Sjögren B, Klareskog L, Alfredsson L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: Results from the Swedish EIRA study. Ann Rheum Dis. (2005) 64:582–6. doi: 10.1136/ard.2004.022053
47. Källberg H, Ding B, Padyukov L, Bengtsson C, Rönnelid J, Klareskog L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: Estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. (2011) 70:508–11. doi: 10.1136/ard.2009.120899
48. Criswell LA, Saag KG, Mikuls TR, Cerhan JR, Merlino LA, Lum RF, et al. Smoking interacts with genetic risk factors in the development of rheumatoid arthritis among older Caucasian women. Ann Rheum Dis. (2006) 65:1163–7. doi: 10.1136/ard.2005.049676
49. Kinslow JD, Blum LK, Deane KD, Demoruelle MK, Okamoto Y, Parish MC, et al. Elevated IgA plasmablast levels in subjects at risk of developing rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:2372–83. doi: 10.1002/art.39771
50. Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, et al. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. (2013) 65:2545–54. doi: 10.1002/art.38066
51. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti–citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol. (2017) 69:1165–75. doi: 10.1002/art.40066
52. Demoruelle MK, Weisman MH, Simonian PL, Lynch DA, Sachs PB, Pedraza IF, et al. Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arthritis Rheum. (2012) 64:1756–61. doi: 10.1002/art.34344
53. Reynisdottir G, Karimi R, Joshua V, Olsen H, Hensvold AH, Harju A, et al. Structural changes and antibody enrichment in the lungs are early features of anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:31–9. doi: 10.1002/art.38201
54. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJW, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
55. Stolt P, Yahya A, Bengtsson C, Källberg H, Rönnelid J, Lundberg I, et al. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann Rheum Dis. (2010) 69:1072–6. doi: 10.1136/ard.2009.114694
56. Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. (2006) 116:3183–94. doi: 10.1172/JCI28756
57. Potempa J, Mydel P, Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:606–20. doi: 10.1038/nrrheum.2017.132
58. Fuggle NR, Smith TO, Kaul A, Sofat N. Hand to mouth: a systematic review and meta-analysis of the association between rheumatoid arthritis and periodontitis. Front Immunol. (2016) 7:80. doi: 10.3389/fimmu.2016.00080
59. Arkema E V, Karlson EW, Costenbader KH. A prospective study of periodontal disease and risk of rheumatoid arthritis. J Rheumatol. (2010) 37:1800–4. doi: 10.3899/jrheum.091398
60. Demmer RT, Molitor JA, Jacobs DR, Michalowicz BS. Periodontal disease, tooth loss and incident rheumatoid arthritis: results from the First National Health and Nutrition Examination Survey and its epidemiological follow-up study. J Clin Periodontol. (2011) 38:998–1006. doi: 10.1111/j.1600-051X.2011.01776.x
61. Wegner N, Wait R, Sroka A, Eick S, Nguyen K, Lundberg K, et al. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. (2010) 62:2662–72. doi: 10.1002/art.27552
62. Montgomery AB, Kopec J, Shrestha L, Thezenas ML, Burgess-Brown NA, Fischer R, et al. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: Implications for autoimmunity in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1255–61. doi: 10.1136/annrheumdis-2015-207656
63. Jenning M, Marklein B, Ytterberg J, Zubarev RA, Joshua V, Van Schaardenburg D, et al. Bacterial citrullinated epitopes generated by Porphyromonas gingivalis infection-a missing link for ACPA production. Ann Rheum Dis. (2020) 79:1194–202. doi: 10.1136/annrheumdis-2019-216919
64. Harvey GP, Fitzsimmons TR, Dhamarpatni AASSK, Marchant C, Haynes DR, Bartold PM. Expression of peptidylarginine deiminase-2 and−4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J Periodontal Res. (2013) 48:252–61. doi: 10.1111/jre.12002
65. Terao C, Asai K, Hashimoto M, Yamazaki T, Ohmura K, Yamaguchi A, et al. Significant association of periodontal disease with anti-citrullinated peptide antibody in a Japanese healthy population – The Nagahama study. J Autoimmun. (2015) 59:85–90. doi: 10.1016/j.jaut.2015.03.002
66. Engström M, Eriksson K, Lee L, Hermansson M, Johansson A, Nicholas AP, et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J Transl Med. (2018) 16:214. doi: 10.1186/s12967-018-1588-2
67. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
68. Loutan L, Alpizar-Rodriguez D, Courvoisier DS, Finckh A, Mombelli A, Giannopoulou C. Periodontal status correlates with anti-citrullinated protein antibodies in first-degree relatives of individuals with rheumatoid arthritis. J Clin Periodontol. (2019) 46:690–8. doi: 10.1111/jcpe.13117
69. Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. (2008) 58:3009–19. doi: 10.1002/art.23936
70. Kharlamova N, Jiang X, Sherina N, Potempa B, Israelsson L, Quirke AM, et al. Antibodies to Porphyromonas gingivalis indicate interaction between oral infection, smoking, and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol. (2016) 68:604–13. doi: 10.1002/art.39491
71. Johansson L, Sherina N, Kharlamova N, Potempa B, Larsson B, Israelsson L, et al. Concentration of antibodies against Porphyromonas gingivalis is increased before the onset of symptoms of rheumatoid arthritis. Arthritis Res Ther. (2016) 18:201. doi: 10.1186/s13075-016-1100-4
72. Mikuls TR, Thiele GM, Deane KD, Payne JB, O'Dell JR, Yu F, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheum. (2012) 64:3522–30. doi: 10.1002/art.34595
73. Quirke AM, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis. (2014) 73:263–9. doi: 10.1136/annrheumdis-2012-202726
74. Konig MF, Paracha AS, Moni M, Bingham CO, Andrade F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann Rheum Dis. (2015) 74:2054–61. doi: 10.1136/annrheumdis-2014-205385
75. Quirke AM, Lundberg K, Potempa J, Mikuls TR, Venables PJ. PPAD remains a credible candidate for inducing autoimmunity in rheumatoid arthritis: Comment on the article by Konig et al. Ann Rheum Dis. (2015) 74:e7. doi: 10.1136/annrheumdis-2014-206665
76. Konig MF, Bingham CO, Andrade F. PPAD is not targeted as a citrullinated protein in rheumatoid arthritis, but remains a candidate for inducing autoimmunity. Ann Rheum Dis. (2015) 74:100–2. doi: 10.1136/annrheumdis-2014-206681
77. Yoshida A, Nakano Y, Yamashita Y, Oho I, Ito H, Kondo M, et al. Immunodominant region of Actinobacillus actinomycetemcomitans 40-kilodalton heat shock protein in patients with rheumatoid arthritis. J Dent Res. (2001) 80:346–50. doi: 10.1177/00220345010800010901
78. Laugisch O, Wong A, Sroka A, Kantyka T, Koziel J, Neuhaus K, et al. Citrullination in the periodontium—a possible link between periodontitis and rheumatoid arthritis. Clin Oral Investig. (2016) 20:675–83. doi: 10.1007/s00784-015-1556-7
79. Volkov M, Dekkers J, Loos BG, Bizzarro S, Huizinga TWJ, Praetorius HA, et al. Comment on “Aggregatibacter actinomycetemcomitans –induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis.” Sci Transl Med. (2018) 10:33–6. doi: 10.1126/scitranslmed.aan8349
80. Gomez-Bañuelos E, Johansson L, Konig MF, Lundquist A, Paz M, Buhlin K, et al. Exposure to aggregatibacter actinomycetemcomitans before symptom onset and the risk of evolving to rheumatoid arthritis. J Clin Med. (2020) 9:1–11. doi: 10.3390/jcm9061906
81. Manoil D, Courvoisier DS, Gilbert B, Möller B, Walker UA, Von Muehlenen I, et al. Associations between serum antibodies to periodontal pathogens and preclinical phases of rheumatoid arthritis. Rheumatol. (2021) 60:4755–64. doi: 10.1093/rheumatology/keab097
82. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. (2015) 21:895–905. doi: 10.1038/nm.3914
83. Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. (2012) 64:3083–94. doi: 10.1002/art.34539
84. Lopez-Oliva I, Paropkari AD, Saraswat S, Serban S, Yonel Z, Sharma P, et al. Dysbiotic subgingival microbial communities in periodontally healthy patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:1008–13. doi: 10.1002/art.40485
85. Sun J, Zheng Y, Bian X, Ge H, Wang J, Zhang Z. Non-surgical periodontal treatment improves rheumatoid arthritis disease activity: a meta-analysis. Clin Oral Investig. (2021) 25:4975–85. doi: 10.1007/s00784-021-03807-w
86. Nguyen VB, Nguyen TT, Huynh NCN, Nguyen KD, Le TA, Hoang HT. Effects of non-surgical periodontal treatment in rheumatoid arthritis patients: A randomized clinical trial. Dent Med Probl. (2021) 58:97–105. doi: 10.17219/dmp/131266
87. Moura MF, Cota LOM, Silva TA, Cortelli SC, Ferreira GA, López MM, et al. Clinical and microbiological effects of non-surgical periodontal treatment in individuals with rheumatoid arthritis: a controlled clinical trial. Odontology. (2021) 109:484–93. doi: 10.1007/s10266-020-00566-0
88. Horta-Baas G, Romero-Figueroa M del S, Montiel-Jarquín AJ, Pizano-Zárate ML, García-Mena J, Ramírez-Durán N. Intestinal dysbiosis and rheumatoid arthritis: a link between gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res. (2017) 2017:4835189. doi: 10.1155/2017/4835189
89. Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. (2013) 2:e01202. doi: 10.7554/eLife.01202
90. Liu X, Zou Q, Zeng B, Fang Y, Wei H. Analysis of fecal lactobacillus community structure in patients with early rheumatoid arthritis. Curr Microbiol. (2013) 67:170–6. doi: 10.1007/s00284-013-0338-1
91. Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, et al. Dysbiosis contributes to arthritis development via activation of autoreactive t cells in the intestine. Arthritis Rheumatol. (2016) 68:2646–61. doi: 10.1002/art.39783
92. Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE, et al. Evidence of the immune relevance of prevotella copri, a gut microbe, in patients with rheumatoid arthritis. Arthritis Rheumatol. (2017) 69:964–75. doi: 10.1002/art.40003
93. Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, et al. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. (2019) 78:590–3. doi: 10.1136/annrheumdis-2018-214514
94. Pianta A, Arvikar SL, Strle K, Drouin EE, Wang Q, Costello CE, et al. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J Clin Invest. (2017) 127:2946–56. doi: 10.1172/JCI93450
95. Vossenaar ER, Zendman AJW, Van Venrooij WJ, Pruijn GJM. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays. (2003) 25:1106–18. doi: 10.1002/bies.10357
96. Curran AM, Naik P, Giles JT, Darrah E. PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol. (2020) 16:301–15. doi: 10.1038/s41584-020-0409-1
97. Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y. Citrullination and Autoimmunity. In: Mosaic of Autoimmunity. Elsevier. (2019). doi: 10.1016/B978-0-12-814307-0.00014-1
98. Konig MF, Andrade F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front Immunol. (2016) 7:e00461. doi: 10.3389/fimmu.2016.00461
99. Sharma P, Azebi S, England P, Christensen T, Møller-Larsen A, Petersen T, et al. Citrullination of Histone H3 Interferes with HP1-Mediated Transcriptional Repression. PLoS Genet. (2012) 8:1–15. doi: 10.1371/journal.pgen.1002934
100. Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature. (2014) 507:104–8. doi: 10.1038/nature12942
101. Tanikawa C, Espinosa M, Suzuki A, Masuda K, Yamamoto K, Tsuchiya E, et al. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun. (2012) 3:e016716. doi: 10.1038/ncomms1676
102. Sun B, Dwivedi N, Bechtel TJ, Paulsen JL, Muth A, Bawadekar M, et al. Citrullination of NF-B p65 promotes its nuclear localization and TLR-induced expression of IL-1 and TNF. Sci Immunol. (2017) 2:1–13. doi: 10.1126/sciimmunol.aal3062
103. Assohou-Luty C, Raijmakers R, Benckhuijsen WE, Stammen-Vogelzangs J, De Ru A, Van Veelen PA, et al. The human peptidylarginine deiminases type 2 and type 4 have distinct substrate specificities. Biochim Biophys Acta - Proteins Proteomics. (2014) 1844:829–36. doi: 10.1016/j.bbapap.2014.02.019
104. Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K, et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep. (2015) 5:1–7. doi: 10.1038/srep13041
105. Suzuki A, Kochi Y, Shoda H, Seri Y, Fujio K, Sawada T, et al. Decreased severity of experimental autoimmune arthritis in peptidylarginine deiminase type 4 knockout mice. BMC Musculoskelet Disord. (2016) 17:1–15. doi: 10.1186/s12891-016-1055-2
106. Bawadekar M, Shim D, Johnson CJ, Warner TF, Rebernick R, Damgaard D, et al. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J Autoimmun. (2017) 80:39–47. doi: 10.1016/j.jaut.2017.01.006
107. Shelef MA, Sokolove J, Lahey LJ, Wagner CA, Sackmann EK, Warner TF, et al. Peptidylarginine deiminase 4 contributes to tumor necrosis factor α-induced inflammatory arthritis. Arthritis Rheumatol. (2014) 66:1482–91. doi: 10.1002/art.38393
108. Nissinen R, Paimela L, Julkunen H, Tienari PJ, Leirisalo-Repo M, Palosuo T, et al. Peptidylarginine deiminase, the arginine to citrulline converting enzyme, is frequently recognized by sera of patients with rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren syndrome. Scand J Rheumatol. (2003) 32:337–42. doi: 10.1080/03009740410004990
109. Zhao J, Zhao Y, He J, Jia R, Li Z. Prevalence and significance of anti-peptidylarginine deiminase 4 antibodies in rheumatoid arthritis. J Rheumatol. (2008) 35:969–74.
110. Halvorsen EH, Pollmann S, Gilboe IM, Van Der Heijde D, Landewé R, Ødegård S, et al. Serum IgG antibodies to peptidylarginine deiminase 4 in rheumatoid arthritis and associations with disease severity. Ann Rheum Dis. (2008) 67:414–7. doi: 10.1136/ard.2007.080267
111. Reyes-Castillo Z, Palafox-Sánchez CA, Parra-Rojas I, Martínez-Bonilla GE, del Toro-Arreola S, Ramírez-Dueñas MG, et al. Comparative analysis of autoantibodies targeting peptidylarginine deiminase type 4, mutated citrullinated vimentin and cyclic citrullinated peptides in rheumatoid arthritis: Associations with cytokine profiles, clinical and genetic features. Clin Exp Immunol. (2015) 182:119–31. doi: 10.1111/cei.12677
112. Harris ML, Darrah E, Lam GK, Bartlett SJ, Giles JT, Grant A V, et al. Association of autoimmunity to peptidyl arginine deiminase type 4 with genotype and disease severity in rheumatoid arthritis. Arthritis Rheum. (2008) 58:1958–67. doi: 10.1002/art.23596
113. Kolfenbach JR, Deane KD, Derber LA, O'Donnell CI, Gilliland WR, Edison JD, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum. (2010) 62:2633–9. doi: 10.1002/art.27570
114. Darrah E, Giles JT, Ols ML, Bull HG, Andrade F, Rosen A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci Transl Med. (2013) 5:1–19. doi: 10.1126/scitranslmed.3005370
115. Darrah E, Giles JT, Davis RL, Naik P, Wang H, Konig MF, et al. Autoantibodies to peptidylarginine deiminase 2 are associated with less severe disease in rheumatoid arthritis. Front Immunol. (2018) 9:1–9. doi: 10.3389/fimmu.2018.02696
116. Spengler J, Lugonja B, Jimmy Ytterberg A, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. (2015) 67:3135–45. doi: 10.1002/art.39313
117. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
118. Thieblemont N, Wright HL, Edwards SW, Witko-Sarsat V. Human neutrophils in auto-immunity. Semin Immunol. (2016) 28:159–73. doi: 10.1016/j.smim.2016.03.004
119. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
120. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med. (2013) 5:209. doi: 10.1126/scitranslmed.3006869
121. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
122. Darrah E, Rosen A, Giles JT, Andrade F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: Novel insights into autoantigen selection in rheumatoid arthritis. Ann Rheum Dis. (2012) 71:92–8. doi: 10.1136/ard.2011.151712
123. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. (2012) 3:1–11. doi: 10.3389/fimmu.2012.00307
124. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
125. Reynisdottir G, Olsen H, Joshua V, Engström M, Forsslund H, Karimi R, et al. Signs of immune activation and local inflammation are present in the bronchial tissue of patients with untreated early rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1722–7. doi: 10.1136/annrheumdis-2015-208216
126. Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, et al. Antibody responses to citrullinated and noncitrullinated antigens in the sputum of subjects with rheumatoid arthritis and subjects at risk for development of rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:516–27. doi: 10.1002/art.40401
127. Ytterberg AJ, Joshua V, Reynisdottir G, Tarasova NK, Rutishauser D, Ossipova E, et al. Shared immunological targets in the lungs and joints of patients with rheumatoid arthritis: Identification and validation. Ann Rheum Dis. (2015) 74:1772–7. doi: 10.1136/annrheumdis-2013-204912
128. Lugli EB, Correia R, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther. (2015) 17:9. doi: 10.1186/s13075-015-0520-x
129. Makrygiannakis D, Af Klint E, Lundberg IE, Löfberg R, Ulfgren AK, Klareskog L, et al. Citrullination is an inflammation-dependent process. Ann Rheum Dis. (2006) 65:1219–22. doi: 10.1136/ard.2005.049403
130. Quirke AM, Perry E, Cartwright A, Kelly C, De Soyza A, Eggleton P, et al. Bronchiectasis is a model for chronic bacterial infection inducing autoimmunity in rheumatoid arthritis. Arthritis Rheumatol. (2015) 67:2335–42. doi: 10.1002/art.39226
131. Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. (2013) 187:1067–75. doi: 10.1164/rccm.201210-1913OC
132. Demoruelle MK, Norris JM, Holers VM, Harris JK, Deane KD. The lung microbiome differs in asymptomatic subjects at elevated risk of future rheumatoid arthritis compared with healthy control subjects. Ann Am Thorac Soc. (2014) 11(Supplement 1):S74. doi: 10.1513/AnnalsATS.201306-185MG
133. Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Öckinger J, Kullberg S, et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome. (2016) 4:60. doi: 10.1186/s40168-016-0206-x
134. Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ. (2018) 360:j5145. doi: 10.1136/bmj.j5145
135. Scher JU, Littman DR, Abramson SB. Review: Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol. (2016) 68:35–45. doi: 10.1002/art.39259
136. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. (2016) 535:75–84. doi: 10.1038/nature18848
137. Van De Wiele T, Van Praet JT, Marzorati M, Drennan MB, Elewaut D. How the microbiota shapes rheumatic diseases. Nat Rev Rheumatol. (2016) 12:398–411. doi: 10.1038/nrrheum.2016.85
138. Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. (2017) 17:219–32. doi: 10.1038/nri.2017.7
139. Rosenstein ED, Greenwald RA, Kushner LJ, Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. (2004) 28:311–8. doi: 10.1007/s10753-004-6641-z
140. McGraw WT, Potempa J, Farley D, Travis J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect Immun. (1999) 67:3248–56. doi: 10.1128/IAI.67.7.3248-3256.1999
141. Marquis RE, Bender GR, Murray DR, Wong A. Arginine deiminase system and bacterial adaptation to acid environments. Appl Environ Microbiol. (1987) 53:198–200. doi: 10.1128/aem.53.1.198-200.1987
142. Gómez-Bañuelos E, Mukherjee A, Darrah E, Andrade F. Rheumatoid arthritis-associated mechanisms of porphyromonas gingivalis and aggregatibacter actinomycetemcomitans. J Clin Med. (2019) 8:e91309. doi: 10.3390/jcm8091309
143. Abdullah S-N, Farmer E-A, Spargo L, Logan R, Gully N. Porphyromonas gingivalis peptidylarginine deiminase substrate specificity. Anaerobe. (2013) 23:102–8. doi: 10.1016/j.anaerobe.2013.07.001
144. Mariette X, Perrodeau E, Verner C, Struillou X, Picard N, Schaeverbeke T, et al. Role of good oral hygiene on clinical evolution of rheumatoid arthritis: A randomized study nested in the ESPOIR cohort. Rheumatol. (2020) 59:988–96. doi: 10.1093/rheumatology/kez368
145. Guerreiro CS, Calado Â, Sousa J, Fonseca JE. Diet, microbiota, and gut permeability—the unknown triad in rheumatoid arthritis. Front Med. (2018) 5:349. doi: 10.3389/fmed.2018.00349
146. Scherer HU, Huizinga TWJ, Krönke G, Schett G, Toes REM. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol. (2018) 14:157–69. doi: 10.1038/nrrheum.2018.10
147. Van De Stadt LA, Van Der Horst AR, De Koning MHMT, Bos WH, Jan Wolbink G, Van De Stadt RJ, et al. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann Rheum Dis. (2011) 70:128–33. doi: 10.1136/ard.2010.132662
148. Van De Stadt LA, De Koning MHMT, Van De Stadt RJ, Wolbink G, Dijkmans BAC, Hamann D, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. (2011) 63:3226–33. doi: 10.1002/art.30537
149. Van Der Woude D, Rantapää-Dahlqvist S, Ioan-Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, et al. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis. (2010) 69:1554–61. doi: 10.1136/ard.2009.124537
150. Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. Matloubian M, editor. PLoS ONE. (2012) 7:e35296. doi: 10.1371/journal.pone.0035296
151. Suwannalai P, Van De Stadt LA, Radner H, Steiner G, El-Gabalawy HS, Zijde CMJ Van Der, et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. (2012) 64:1323–8. doi: 10.1002/art.33489
152. Verpoort KN, Jol-Van Der Zijde CM, Papendrecht-Van Der Voort EAM, Ioan-Facsinay A, Drijfhout JW, Van Tol MJD, et al. Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum. (2006) 54:3799–808. doi: 10.1002/art.22279
153. Suwannalai P, Scherer HU, Van Der Woude D, Ioan-Facsinay A, Jol-van Der Zijde CM, Van Tol MJD, et al. Anti-citrullinated protein antibodies have a low avidity compared with antibodies against recall antigens. Ann Rheum Dis. (2011) 70:373–9. doi: 10.1136/ard.2010.135509
154. Hensvold AH, Magnusson PKE, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: An epidemiological investigation in twins. Ann Rheum Dis. (2015) 74:375–80. doi: 10.1136/annrheumdis-2013-203947
155. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. (2012) 44:291–6. doi: 10.1038/ng.1076
156. Stanford SM, Bottini N. PTPN22: The archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. (2014) 10:602–11. doi: 10.1038/nrrheum.2014.109
157. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
158. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
159. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
160. Fonseca VR, Agua-Doce A, Maceiras AR, Pierson W, Ribeiro F, Romão VC, et al. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci Immunol. (2017) 2:eaan1487. doi: 10.1126/sciimmunol.aan1487
161. Deng J, Wei Y, Fonseca VR, Graca L, Yu D. T follicular helper cells and T follicular regulatory cells in rheumatic diseases. Nat Rev Rheumatol. (2019) 15:475–90. doi: 10.1038/s41584-019-0254-2
162. Moschovakis GL, Bubke A, Friedrichsen M, Falk CS, Feederle R, Förster R. T cell specific Cxcr5 deficiency prevents rheumatoid arthritis. Sci Rep. (2017) 7:8933. doi: 10.1038/s41598-017-08935-6
163. Chevalier N, Macia L, Tan JK, Mason LJ, Robert R, Thorburn AN, et al. The role of follicular helper T cell molecules and environmental influences in autoantibody production and progression to inflammatory arthritis in mice. Arthritis Rheumatol. (2016) 68:1026–38. doi: 10.1002/art.39481
164. Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol. (2012) 2012:827480. doi: 10.1155/2012/827480
165. Liu C, Wang D, Lu S, Xu Q, Zhao L, Zhao J, et al. Increased circulating follicular treg cells are associated with lower levels of autoantibodies in patients with rheumatoid arthritis in stable remission. Arthritis Rheumatol. (2018) 70:711–21. doi: 10.1002/art.40430
166. Romão VC, Fonseca JE, Agua-Doce A, Graca L. T follicular regulatory cells are decreased in patients with established treated rheumatoid arthritis with active disease: comment on the article by Liu et al. Arthritis Rheumatol. (2018) 70:1893–5. doi: 10.1002/art.40586
167. Moura RA, Graca L, Fonseca JE. To B or Not to B the conductor of rheumatoid arthritis orchestra. Clin Rev Allergy Immunol. (2012) 43:281–91. doi: 10.1007/s12016-012-8318-y
168. Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen-antibody complexes by rheumatoid factor B cells. J Exp Med. (1991) 173:487–9. doi: 10.1084/jem.173.2.487
169. Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, et al. Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. (1988) 18:1797–802. doi: 10.1002/eji.1830181122
170. Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. (2001) 167:4710–8. doi: 10.4049/jimmunol.167.8.4710
171. Moura RA, Weinmann P, Pereira PA, Caetano-Lopes J, Canhao H, Sousa E, et al. Alterations on peripheral blood B-cell subpopulations in very early arthritis patients. Rheumatology. (2010) 49:1082–92. doi: 10.1093/rheumatology/keq029
172. Moura RA, Cascao R, Perpetuo I, Canhao H, Vieira-Sousa E, Mourao AF, et al. Cytokine pattern in very early rheumatoid arthritis favours B-cell activation and survival. Rheumatology. (2011) 50:278–82. doi: 10.1093/rheumatology/keq338
173. Moura R a, Canhão H, Polido-Pereira J, Rodrigues AM, Navalho M, Mourão AF, et al. BAFF and TACI gene expression are increased in patients with untreated very early rheumatoid arthritis. J Rheumatol. (2013) 40:1293–302. doi: 10.3899/jrheum.121110
174. Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. (2005) 201:1659–67. doi: 10.1084/jem.20042321
175. Kerkman PF, Fabre E, Van Der Voort EIH, Zaldumbide A, Rombouts Y, Rispens T, et al. Identification and characterisation of citrullinated antigen-specific B cells in peripheral blood of patients with rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1170–6. doi: 10.1136/annrheumdis-2014-207182
176. Titcombe PJ, Wigerblad G, Sippl N, Zhang N, Shmagel AK, Sahlström P, et al. Pathogenic citrulline-multispecific B cell receptor clades in rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:1933–45. doi: 10.1002/art.40590
177. van de Stadt LA, van Schouwenburg PA, Bryde S, Kruithof S, van Schaardenburg D, Hamann D, et al. Monoclonal anti-citrullinated protein antibodies selected on citrullinated fibrinogen have distinct targets with different cross-reactivity patterns. Rheumatology. (2013) 52:631–5. doi: 10.1093/rheumatology/kes371
178. Li S, Yu Y, Yue Y, Liao H, Xie W, Thai J, et al. Autoantibodies from single circulating plasmablasts react with citrullinated antigens and porphyromonas gingivalis in rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:614–26. doi: 10.1002/art.39455
179. Tan YC, Kongpachith S, Blum LK, Ju CH, Lahey LJ, Lu DR, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:2706–15. doi: 10.1002/art.38754
180. Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity maturation drives epitope spreading and generation of proinflammatory anti–citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:1946–58. doi: 10.1002/art.40587
181. Steen J, Forsström B, Sahlström P, Odowd V, Israelsson L, Krishnamurthy A, et al. Recognition of amino acid motifs, rather than specific proteins, by human plasma cell–derived monoclonal antibodies to posttranslationally modified proteins in rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:196–209. doi: 10.1002/art.40699
182. Vergroesen RD, Slot LM, Hafkenscheid L, Koning MT, Van Der Voort EIH, Grooff CA, et al. B-cell receptor sequencing of anti-citrullinated protein antibody (ACPA) IgG-expressing B cells indicates a selective advantage for the introduction of N-glycosylation sites during somatic hypermutation. Ann Rheum Dis. (2018) 77:955–7. doi: 10.1136/annrheumdis-2017-212052
183. Tak PP, Doorenspleet ME, De Hair MJH, Klarenbeek PL, Van Beers-Tas MH, Van Kampen AHC, et al. Dominant B cell receptor clones in peripheral blood predict onset of arthritis in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. (2017) 76:1924–30. doi: 10.1136/annrheumdis-2017-211351
184. Rombouts Y, Willemze A, Van Beers JJBC, Shi J, Kerkman PF, Van Toorn L, et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:578–85. doi: 10.1136/annrheumdis-2014-206598
185. Vergroesen RD, Slot LM, van Schaik BDC, Koning MT, Rispens T, van Kampen AHC, et al. N-glycosylation site analysis of citrullinated antigen-specific B-cell receptors indicates alternative selection pathways during autoreactive B-cell development. Front Immunol. (2019) 10:2092. doi: 10.3389/fimmu.2019.02092
186. Kempers AC, Hafkenscheid L, Dorjée AL, Moutousidou E, Van De Bovenkamp FS, Rispens T, et al. The extensive glycosylation of the ACPA variable domain observed for ACPA-IgG is absent from ACPA-IgM. Ann Rheum Dis. (2018) 77:1087–8. doi: 10.1136/annrheumdis-2017-211533
187. Hafkenscheid L, Moel E, Smolik I, Tanner S, Meng X, Jansen BC, et al. N -linked glycans in the variable domain of igg anti–citrullinated protein antibodies predict the development of rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:1626–33. doi: 10.1002/art.40920
188. Kissel T, van Schie KA, Hafkenscheid L, Lundquist A, Kokkonen H, Wuhrer M, et al. On the presence of HLA-SE alleles and ACPA-IgG variable domain glycosylation in the phase preceding the development of rheumatoid arthritis. Ann Rheum Dis. (2019) 78:1616–20. doi: 10.1136/annrheumdis-2019-215698
189. Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature. (1985) 316:452–7. doi: 10.1038/316452a0
190. Ercan A, Cui J, Chatterton DEW, Deane KD, Hazen MM, Brintnell W, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. (2010) 62:2239–48. doi: 10.1002/art.27533
191. Rombouts Y, Ewing E, Van De Stadt LA, Selman MHJ, Trouw LA, Deelder AM, et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. (2015) 74:234–41. doi: 10.1136/annrheumdis-2013-203565
192. van Zeben D, Rook GA, Hazes JM, Zwinderman AH, Zhang Y, Ghelani S, et al. Early agalactosylation of IgG is associated with a more progressive disease course in patients with rheumatoid arthritis: results of a follow-up study. Br J Rheumatol. (1994) 33:36–43. doi: 10.1093/rheumatology/33.1.36
193. Scherer HU, Van Der Woude D, Ioan-Facsinay A, El Bannoudi H, Trouw LA, Wang J, et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. (2010) 62:1620–9. doi: 10.1002/art.27414
194. Kokkonen H, Söderström I, Rocklöv J, Hallmans G, Lejon K, Dahlqvist SR. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. (2010) 62:383–91. doi: 10.1002/art.27186
195. Deane KD, O'Donnell CI, Hueber W, Majka DS, Lazar AA, Derber LA, et al. The number of elevated cytokines and chemokines in preclinical seropositive rheumatoid arthritis predicts time to diagnosis in an age-dependent manner. Arthritis Rheum. (2010) 62:3161–72. doi: 10.1002/art.27638
196. Nielen MMJ, Van Schaardenburg D, Reesink HW, Twisk JWR, Van De Stadt RJ, Van Der Horst-Bruinsma IE, et al. Simultaneous development of acute phase response and autoantibodies in preclinical rheumatoid arthritis. Ann Rheum Dis. (2006) 65:535–7.
197. Toes R, Pisetsky DS. Pathogenic effector functions of ACPA: Where do we stand? Ann Rheum Dis. (2019) 78:716–21. doi: 10.1136/annrheumdis-2019-215337
198. Vossenaar ER, Smeets TJM, Kraan MC, Raats JM, Van Venrooij WJ, Tak PP. The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum. (2004) 50:3485–94. doi: 10.1002/art.20584
199. Kuhn KA, Robinson WH, Holers VM, Kuhn KA, Kulik L, Tomooka B, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis Find the latest version : Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. (2006) 116:961–73. doi: 10.1172/JCI25422
200. Stoop JN, Liu BS, Shi J, Jansen DTSL, Hegen M, Huizinga TWJ, et al. Antibodies specific for carbamylated proteins precede the onset of clinical symptoms in mice with collagen induced arthritis. PLoS ONE. (2014) 9:1–9. doi: 10.1371/journal.pone.0102163
201. Stoop JN, Fischer A, Hayer S, Hegen M, Huizinga TWJ, Steiner G, et al. Anticarbamylated protein antibodies can be detected in animal models of arthritis that require active involvement of the adaptive immune system. Ann Rheum Dis. (2015) 74:949–50. doi: 10.1136/annrheumdis-2014-206797
202. Hayer S, Tohidast-Akrad M, Haralambous S, Jahn-Schmid B, Skriner K, Trembleau S, et al. Aberrant expression of the autoantigen heterogeneous nuclear ribonucleoprotein-A2 (RA33) and spontaneous formation of rheumatoid arthritis-associated anti-RA33 autoantibodies in TNF-α transgenic mice. J Immunol. (2005) 175:8327–36. doi: 10.4049/jimmunol.175.12.8327
203. Hoffmann MH, Tuncel J, Skriner K, Tohidast-Akrad M, Türk B, Pinol-Roma S, et al. The rheumatoid arthritis-associated autoantigen hnRNP-A2 (RA33) is a major stimulator of autoimmunity in rats with pristane-induced arthritis. J Immunol. (2007) 179:7568–76. doi: 10.4049/jimmunol.179.11.7568
204. Nell-Duxneuner V, Machold K, Stamm T, Eberl G, Heinzl H, Hoefler E, et al. Autoantibody profiling in patients with very early rheumatoid arthritis: A follow-up study. Ann Rheum Dis. (2010) 69:169–74. doi: 10.1136/ard.2008.100677
205. Konig MF, Giles JT, Nigrovic PA, Andrade F. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:2022–8. doi: 10.1136/annrheumdis-2015-208529
206. Steiner G, Hartmuth K, Skriner K, Maurer-Fogy I, Sinski A, Thalmann E, et al. Purification and partial sequencing of the nuclear autoantigen RA33 shows that it is indistinguishable from the A2 protein of the heterogeneous nuclear ribonucleoprotein complex. J Clin Invest. (1992) 90:1061–6. doi: 10.1172/JCI115921
207. Hassfeld W, Steiner G, Hartmuth K, Kolarz G, Scherak O, Graninger W, et al. Demonstration of a new antinuclear antibody (ANTI-RA33) that is highly specific for rheumatoid arthritis. Arthritis Rheum. (1989) 32:1515–20. doi: 10.1002/anr.1780321204
208. De Pablo P, Dietrich T, Chapple ILC, Milward M, Chowdhury M, Charles PJ, et al. The autoantibody repertoire in periodontitis: A role in the induction of autoimmunity to citrullinated proteins in rheumatoid arthritis? Ann Rheum Dis. (2014) 73:580–6. doi: 10.1136/annrheumdis-2012-202701
209. van de Sande MGH, de Hair MJH, van der Leij C, Klarenbeek PL, Bos WH, Smith MD, et al. Different stages of rheumatoid arthritis: features of the synovium in the preclinical phase. Ann Rheum Dis. (2011) 70:772–7. doi: 10.1136/ard.2010.139527
210. de Hair MJH, van de Sande MGH, Ramwadhdoebe TH, Hansson M, Landewé R, van der Leij C, et al. Features of the synovium of individuals at risk of developing rheumatoid arthritis: implications for understanding preclinical rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:513–22. doi: 10.1002/art.38273
211. Kraan MC, Versendaal H, Jonker M, Bresnihan B, Post WJ, 'T Hart BA, et al. A symptomatic synovitis precedes clinically manifest arthritis. Arthritis Rheum. (1998) 41:1481–8. doi: 10.1002/1529-0131(199808)41:8<1481::AID-ART19>3.0.CO;2-O
212. Krabben A, Stomp W, Van Der Heijde DMFM, Van Nies JAB, Bloem JL, Huizinga TWJ, et al. MRI of hand and foot joints of patients with anticitrullinated peptide antibody positive arthralgia without clinical arthritis. Ann Rheum Dis. (2013) 72:1540–4. doi: 10.1136/annrheumdis-2012-202628
213. Van Steenbergen HW, Van Nies JAB, Huizinga TWJ, Bloem JL, Reijnierse M, Van Der Helm-Van Mil AHM, et al. Characterising arthralgia in the preclinical phase of rheumatoid arthritis using MRI. Ann Rheum Dis. (2015) 74:1225–32. doi: 10.1136/annrheumdis-2014-205522
214. van Steenbergen HW, van Nies JAB, Huizinga TWJ, Reijnierse M, van der Helm-van Mil AHM. Subclinical inflammation on MRI of hand and foot of anticitrullinated peptide antibody–negative arthralgia patients at risk for rheumatoid arthritis. Arthritis Res Ther. (2014) 16:R92. doi: 10.1186/ar4536
215. Nam JL, Hensor EMA, Hunt L, Conaghan PG, Wakefield RJ, Emery P. Ultrasound findings predict progression to inflammatory arthritis in anti-CCP antibody-positive patients without clinical synovitis. Ann Rheum Dis. (2016) 75:2060–7. doi: 10.1136/annrheumdis-2015-208235
216. van Beers-Tas MH, Blanken AB, Nielen MMJ, Turkstra F, van der Laken CJ, Meursinge Reynders M, et al. The value of joint ultrasonography in predicting arthritis in seropositive patients with arthralgia: a prospective cohort study. Arthritis Res Ther. (2018) 20:279. doi: 10.1186/s13075-018-1767-9
217. Trouw LA, Haisma EM, Levarht EWN, Van Der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. (2009) 60:1923–31. doi: 10.1002/art.24622
218. Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FMA, Boackle SA, et al. Arthritis critically dependent on innate immune system players. Immunity. (2002) 16:157–68. doi: 10.1016/S1074-7613(02)00275-3
219. Clavel C, Nogueira L, Laurent L, Iobagiu C, Vincent C, Sebbag M, et al. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. (2008) 58:678–88. doi: 10.1002/art.23284
220. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. (2011) 63:53–62. doi: 10.1002/art.30081
221. Suurmond J, Rivellese F, Dorjée AL, Bakker AM, Rombouts YJPC, Rispens T, et al. Toll-like receptor triggering augments activation of human mast cells by anti-citrullinated protein antibodies. Ann Rheum Dis. (2015) 74:1915–23. doi: 10.1136/annrheumdis-2014-205562
222. Sanchez-Pernaute O, Filkova M, Gabucio A, Klein M, MacIejewska-Rodrigues H, Ospelt C, et al. Citrullination enhances the pro-inflammatory response to fibrin in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. (2013) 72:1400–6. doi: 10.1136/annrheumdis-2012-201906
223. Sokolove J, Johnson DS, Lahey LJ, Wagner CA, Cheng D, Thiele GM, et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:813–21. doi: 10.1002/art.38307
224. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor– and complement-dependent effector functions of the disease-specific anti–citrullinated protein autoantibodies. J Immunol. (2015) 194:3664–74. doi: 10.4049/jimmunol.1402334
225. Laurent L, Anquetil F, Clavel C, Ndongo-Thiam N, Offer G, Miossec P, et al. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann Rheum Dis. (2015) 74:1425–31. doi: 10.1136/annrheumdis-2013-204543
226. Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, et al. Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis. (2014) 73:854–60. doi: 10.1136/annrheumdis-2012-202958
227. Harre U, Bozec A, Axmann R, Baum W, Schett G, Georgess D, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. (2012) 122:1791–802. doi: 10.1172/JCI60975
228. Krishnamurthy A, Joshua V, Hensvold AH, Jin T, Sun M, Vivar N, et al. Identification of a novel chemokine-dependent molecular mechanism underlying Rheumatoid arthritisassociated autoantibody-mediated bone loss. Ann Rheum Dis. (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
229. Krishnamurthy A, Ytterberg AJ, Sun M, Sakuraba K, Steen J, Joshua V, et al. Citrullination controls dendritic cell transdifferentiation into osteoclasts. J Immunol. (2019) 202:3143–50. doi: 10.4049/jimmunol.1800534
230. Ge C, Xu B, Liang B, Lönnblom E, Lundström SL, Zubarev RA, et al. Structural basis of cross-reactivity of anti–citrullinated protein antibodies. Arthritis Rheumatol. (2019) 71:210–21. doi: 10.1002/art.40698
231. Krishnamurthy A, Joshua V, Haj Hensvold A. Correction: Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. (2019) 78:866. doi: 10.1136/annrheumdis-2015-208093corr1
232. Harre U, Lang SC, Pfeifle R, Rombouts Y, Frühbeißer S, Amara K, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. (2015) 6:6651. doi: 10.1038/ncomms7651
233. Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to citrullinated proteins may induce joint pain independent of inflammation. Ann Rheum Dis. (2016) 75:730–8. doi: 10.1136/annrheumdis-2015-208094
234. Nandakumar KS, Collin M, Happonen KE, Lundström SL, Croxford AM, Xu B, et al. Streptococcal Endo-β-N-acetylglucosaminidase suppresses antibody-mediated inflammation in vivo. Front Immunol. (2018) 9:1623. doi: 10.3389/fimmu.2018.01623
235. Bersellini Farinotti A, Wigerblad G, Nascimento D, Bas DB, Morado Urbina C, Nandakumar KS, et al. Cartilage-binding antibodies induce pain through immune complex–mediated activation of neurons. J Exp Med. (2019) 216:1904–24. doi: 10.1084/jem.20181657
236. Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Mathis D, et al. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. (2002) 3:360–5. doi: 10.1038/ni772
237. Rowley MJ, Nandakumar KS, Holmdahl R. The role of collagen antibodies in mediating arthritis. Mod Rheumatol. (2008) 18:429–41. doi: 10.1007/s10165-008-0080-x
238. Matsumoto I, Lee DM, Goldbach-Mansky R, Sumida T, Hitchon CA, Schur PH, et al. Low prevalence of antibodies to glucose-6-phosphate isomerase in patients with rheumatoid arthritis and a spectrum of other chronic autoimmune disorders. Arthritis Rheum. (2003) 48:944–54. doi: 10.1002/art.10898
239. Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E, Engström Å, et al. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med. (2009) 206:449–62. doi: 10.1084/jem.20081862
240. Mullazehi M, Mathsson L, Lampa J, Rönnelid J. High anti-collagen type-II antibody levels and induction of proinflammatory cytokines by anti-collagen antibody-containing immune complexes in vitro characterise a distinct rheumatoid arthritis phenotype associated with acute inflammation at the time of d. Ann Rheum Dis. (2007) 66:537–41. doi: 10.1136/ard.2006.064782
241. Haag S, Schneider N, Mason DE, Tuncel J, Andersson IE, Peters EC, et al. Identification of new citrulline-specific autoantibodies, which bind to human arthritic cartilage, by mass spectrometric analysis of citrullinated type II collagen. Arthritis Rheumatol. (2014) 66:1440–9. doi: 10.1002/art.38383
242. Ge C, Tong D, Liang B, Lönnblom E, Schneider N, Hagert C, et al. Anti-citrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight. (2017) 2:e93688. doi: 10.1172/jci.insight.93688
243. Pratt AG, Isaacs JD. Seronegative rheumatoid arthritis: Pathogenetic and therapeutic aspects. Best Pract Res Clin Rheumatol. (2014) 28:651–9. doi: 10.1016/j.berh.2014.10.016
244. Paalanen K, Rannio K, Rannio T, Asikainen J, Hannonen P, Sokka T. Does early seronegative arthritis develop into rheumatoid arthritis? A 10-year observational study. Clin Exp Rheumatol. (2019) 37:37–43.
245. Bossini-Castillo L, de Kovel C, Kallberg H, van ‘t Slot R, Italiaander A, Coenen M, et al. A genome-wide association study of rheumatoid arthritis without antibodies against citrullinated peptides. Ann Rheum Dis. (2015) 74:e15. doi: 10.1136/annrheumdis-2013-204591
246. Han B, Diogo D, Eyre S, Kallberg H, Zhernakova A, Bowes J, et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am J Hum Genet. (2014) 94:522–32. doi: 10.1016/j.ajhg.2014.02.013
247. Padyukov L, Seielstad M, Ong RTH, Ding B, Rönnelid J, Seddighzadeh M, et al. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann Rheum Dis. (2011) 70:259–65. doi: 10.1136/ard.2009.126821
248. Terao C, Ohmura K, Kochi Y, Ikari K, Maruya E, Katayama M, et al. A large-scale association study identified multiple HLA-DRB1 alleles associated with ACPA-negative rheumatoid arthritis in Japanese subjects. Ann Rheum Dis. (2011) 70:2134–9. doi: 10.1136/annrheumdis-2011-200353
249. Viatte S, Plant D, Bowes J, Lunt M, Eyre S, Barton A, et al. Genetic markers of rheumatoid arthritis susceptibility in anti-citrullinated peptide antibody negative patients. Ann Rheum Dis. (2012) 71:1984–90. doi: 10.1136/annrheumdis-2011-201225
250. Viatte S, Massey J, Bowes J, Duffus K, Eyre S, Barton A, et al. Replication of associations of genetic loci outside the HLA region with susceptibility to anti–cyclic citrullinated peptide–negative rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:1603–13. doi: 10.1002/art.39619
251. Ohmura K, Terao C, Maruya E, Katayama M, Matoba K, Shimada K, et al. Anti-citrullinated peptide antibody-negative RA is a genetically distinct subset: A definitive study using only bone-erosive ACPA-negative rheumatoid arthritis. Rheumatology. (2010) 49:2298–304. doi: 10.1093/rheumatology/keq273
252. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. (2012) 44:1336–40. doi: 10.1038/ng.2462
253. Frisell T, Holmqvist M, Källberg H, Klareskog L, Alfredsson L, Askling J. Familial risks and heritability of rheumatoid arthritis: Role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum. (2013) 65:2773–82. doi: 10.1002/art.38097
254. Frisell T, Hellgren K, Alfredsson L, Raychaudhuri S, Klareskog L, Askling J. Familial aggregation of arthritis-related diseases in seropositive and seronegative rheumatoid arthritis: A register-based case-control study in Sweden. Ann Rheum Dis. (2016) 75:183–9. doi: 10.1136/annrheumdis-2014-206133
255. Terao C, Ohmura K, Kochi Y, Ikari K, Okada Y, Shimizu M, et al. Anti-citrullinated peptide/protein antibody (ACPA)-negative RA shares a large proportion of susceptibility loci with ACPA-positive RA: A meta-analysis of genome-wide association study in a Japanese population. Arthritis Res Ther. (2015) 17:1–11. doi: 10.1186/s13075-015-0623-4
256. Van Der Woude D, Houwing-Duistermaat JJ, Toes REM, Huizinga TWJ, Thomson W, Worthington J, et al. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. (2009) 60:916–23. doi: 10.1002/art.24385
257. Reed E, Hedström AK, Hansson M, Mathsson-Alm L, Brynedal B, Saevarsdottir S, et al. Presence of autoantibodies in “seronegative” rheumatoid arthritis associates with classical risk factors and high disease activity. Arthritis Res Ther. (2020) 22:1–11. doi: 10.1186/s13075-020-02191-2
258. Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GMC, van Veelen PA, et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA. (2011) 108:17372–7. doi: 10.1073/pnas.1114465108
259. Jiang X, Trouw LA, Van Wesemael TJ, Shi J, Bengtsson C, Källberg H, et al. Anti-CarP antibodies in two large cohorts of patients with rheumatoid arthritis and their relationship to genetic risk factors, cigarette smoking and other autoantibodies. Ann Rheum Dis. (2014) 73:1761–8. doi: 10.1136/annrheumdis-2013-205109
260. Gan RW, Trouw LA, Shi J, Toes REM, Huizinga TWJ, Demoruelle MK, et al. Anti-carbamylated protein antibodies are present prior to rheumatoid arthritis and are associated with its future diagnosis. J Rheumatol. (2015) 42:572–9. doi: 10.3899/jrheum.140767
261. Shi J, Van De Stadt LA, Levarht EWN, Huizinga TWJ, Hamann D, Van Schaardenburg D, et al. Anti-carbamylated protein (anti-CarP) antibodies precede the onset of rheumatoid arthritis. Ann Rheum Dis. (2014) 73:780–3. doi: 10.1136/annrheumdis-2013-204154
262. Studenic P, Alunno A, Sieghart D, Bang H, Aletaha D, Blüml S, et al. Presence of anti-acetylated peptide antibodies (AAPA) in inflammatory arthritis and other rheumatic diseases suggests discriminative diagnostic capacity towards early rheumatoid arthritis. Ther Adv Musculoskelet Dis. (2021) 13:1759720X2110225. doi: 10.1177/1759720X211022533
263. Juarez M, Bang H, Hammar F, Reimer U, Dyke B, Sahbudin I, et al. Identification of novel antiacetylated vimentin antibodies in patients with early inflammatory arthritis. Ann Rheum Dis. (2016) 75:1099–107. doi: 10.1136/annrheumdis-2014-206785
264. Scherer HU, van der Woude D, Toes REM. From risk to chronicity: evolution of autoreactive B cell and antibody responses in rheumatoid arthritis. Nat Rev Rheumatol. (2022) 18:371–83. doi: 10.1038/s41584-022-00786-4
265. Nijjar JS, Morton FR, Bang H, Buckley CD, van der Heijde D, Gilmour A, et al. The impact of autoantibodies against citrullinated, carbamylated, and acetylated peptides on radiographic progression in patients with new-onset rheumatoid arthritis: an observational cohort study. Lancet Rheumatol. (2021) 3:e284–93. doi: 10.1016/S2665-9913(20)30381-7
266. Kissel T, Reijm S, Slot LM, Cavallari M, Wortel CM, Vergroesen RD, et al. Antibodies and B cells recognising citrullinated proteins display a broad cross-reactivity towards other post-translational modifications. Ann Rheum Dis. (2020) 79:472–80. doi: 10.1136/annrheumdis-2019-216499
267. Sahlström P, Hansson M, Steen J, Amara K, Titcombe PJ, Forsström B, et al. Different hierarchies of anti–modified protein autoantibody reactivities in rheumatoid arthritis. Arthritis Rheumatol. (2020) 72:1643–57. doi: 10.1002/art.41385
268. Li K, Mo W, Wu L, Wu X, Luo C, Xiao X, et al. Novel autoantibodies identified in ACPA-negative rheumatoid arthritis. Ann Rheum Dis. (2021) 80:739–47. doi: 10.1136/annrheumdis-2020-218460
269. Assmann G, Zinke S, Gerling M, Bittenbring JT, Preuss KD, Thurner L. Progranulin-autoantibodies in sera of rheumatoid arthritis patients negative for rheumatoid factor and anti-citrullinated peptide antibodies. Clin Exp Rheumatol. (2020) 38:94–8.
270. Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. (2003) 62:835–41. doi: 10.1136/ard.62.9.835
271. Scott IC, Tan R, Stahl D, Steer S, Lewis CM, Cope AP. The protective effect of alcohol on developing rheumatoid arthritis: A systematic review and meta-analysis. Rheumatol (United Kingdom). (2013) 52:856–67. doi: 10.1093/rheumatology/kes376
272. Lee YH, Bae SC, Song GG. Coffee or tea consumption and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol. (2014) 33:1575–83. doi: 10.1007/s10067-014-2631-1
273. Feng J, Chen Q, Yu F, Wang Z, Chen S, Jin Z, et al. Body mass index and risk of rheumatoid arthritis. Medicine (Baltimore). (2016) 95:e2859. doi: 10.1097/MD.0000000000002859
274. Qin B, Yang M, Fu H, Ma N, Wei T, Tang Q, et al. Body mass index and the risk of rheumatoid arthritis: a systematic review and dose-response meta-analysis. Arthritis Res Ther. (2015) 17:86. doi: 10.1186/s13075-015-0601-x
275. Sparks JA, Malspeis S, Hahn J, Wang J, Roberts AL, Kubzansky LD, et al. Depression and subsequent risk for incident rheumatoid arthritis among women. Arthritis Care Res. (2021) 73:78–89. doi: 10.1002/acr.24441
276. Bengtsson C, Malspeis S, Orellana C, Sparks JA, Costenbader KH, Karlson EW. Association between menopausal factors and the risk of seronegative and seropositive rheumatoid arthritis: results from the nurses' health studies. Arthritis Care Res. (2017) 69:1676–84. doi: 10.1002/acr.23194
277. Zeng P, Klareskog L, Alfredsson L, Bengtsson C. Physical workload is associated with increased risk of rheumatoid arthritis: results from a Swedish population-based case–control study. RMD Open. (2017) 3:e000324. doi: 10.1136/rmdopen-2016-000324
278. Wesley A, Bengtsson C, Skillgate E, Saevarsdottir S, Theorell T, Holmqvist M, et al. Association between life events and rheumatoid arthritis: Results from a population-based case-control study. Arthritis Care Res. (2014) 66:844–51. doi: 10.1002/acr.22230
279. Pratt AG, Swan DC, Richardson S, Wilson G, Hilkens CMU, Young DA, et al. A CD4 T cell gene signature for early rheumatoid arthritis implicates interleukin 6-mediated STAT3 signalling, particularly in anti-citrullinated peptide antibody-negative disease. Ann Rheum Dis. (2012) 71:1374–81. doi: 10.1136/annrheumdis-2011-200968
280. Anderson AE, Pratt AG, Sedhom MAK, Doran JP, Routledge C, Hargreaves B, et al. IL-6-driven STAT signalling in circulating CD4+ lymphocytes is a marker for early anticitrullinated peptide antibody-negative rheumatoid arthritis. Ann Rheum Dis. (2016) 75:466–73. doi: 10.1136/annrheumdis-2014-205850
281. Chalan P, Bijzet J, Van Den Berg A, Kluiver J, Kroesen BJ, Boots AMH, et al. Analysis of serum immune markers in seropositive and seronegative rheumatoid arthritis and in high-risk seropositive arthralgia patients. Sci Rep. (2016) 6:1–9. doi: 10.1038/srep26021
282. Chalan P, Bijzet J, Kroesen BJ, Boots AMH, Brouwer E. Altered natural killer cell subsets in seropositive arthralgia and early rheumatoid arthritis are associated with autoantibody status. J Rheumatol. (2016) 43:1008–16. doi: 10.3899/jrheum.150644
283. Molendijk M, Hazes JMW, Lubberts E. From patients with arthralgia, pre-RA and recently diagnosed RA: What is the current status of understanding RA pathogenesis? RMD Open. (2018) 4:1–11. doi: 10.1136/rmdopen-2016-000256
284. Van Oosterhout M, Bajema I, Levarht EWN, Toes REM, Huizinga TWJ, Van Laar JM. Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum. (2008) 58:53–60. doi: 10.1002/art.23148
285. Orr C, Najm A, Biniecka M, McGarry T, Ng CT, Young F, et al. Synovial immunophenotype and anti–citrullinated peptide antibodies in rheumatoid arthritis patients: relationship to treatment response and radiologic prognosis. Arthritis Rheumatol. (2017) 69:2114–23. doi: 10.1002/art.40218
286. Lliso-Ribera G, Humby F, Lewis M, Nerviani A, Mauro D, Rivellese F, et al. Synovial tissue signatures enhance clinical classification and prognostic/treatment response algorithms in early inflammatory arthritis and predict requirement for subsequent biological therapy: results from the pathobiology of early arthritis cohort. Ann Rheum Dis. (2019) 78:1642–52. doi: 10.1136/annrheumdis-2019-215751
287. Cantaert T, Brouard S, Thurlings RM, Pallier A, Salinas GF, Braud C, et al. Alterations of the synovial T cell repertoire in anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheum. (2009) 60:1944–56. doi: 10.1002/art.24635
288. Burgers LE, van Steenbergen HW, ten Brinck RM, Huizinga TWJ, van der Helm-van Mil AHM. Differences in the symptomatic phase preceding ACPA-positive and ACPA-negative RA: a longitudinal study in arthralgia during progression to clinical arthritis. Ann Rheum Dis. (2017) 76:1751–4). doi: 10.1136/annrheumdis-2017-211325
289. Dadoun S, Zeboulon-Ktorza N, Combescure C, Elhai M, Rozenberg S, Gossec L, et al. Mortality in rheumatoid arthritis over the last fifty years: Systematic review and meta-analysis. Jt Bone Spine. (2013) 80:29–33. doi: 10.1016/j.jbspin.2012.02.005
290. Ajeganova S, Humphreys JH, Verheul MK, Van Steenbergen HW, Van Nies JAB, Svensson B, et al. Factor are associated with increased mortality but with different causes of death in patients with rheumatoid arthritis: A longitudinal study in three European cohorts. Ann Rheum Dis. (2016) 75:1924–32. doi: 10.1136/annrheumdis-2015-208579
291. Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 2020:685–99.
292. Boer AC, Boonen A, van der Helm van Mil AHM. Is anti–citrullinated protein antibody–positive rheumatoid arthritis still a more severe disease than anti–citrullinated protein antibody–negative rheumatoid arthritis? A longitudinal cohort study in rheumatoid arthritis patients diagnosed from 2000 Onwar. Arthritis Care Res. (2018) 70:987–96. doi: 10.1002/acr.23497
293. Barra L, Pope JE, Orav JE, Boire G, Haraoui B, Hitchon C, et al. Prognosis of seronegative patients in a large prospective cohort of patients with early inflammatory arthritis. J Rheumatol. (2014) 41:2361–9. doi: 10.3899/jrheum.140082
294. Paalanen K, Puolakka K, Nikiphorou E, Hannonen P, Sokka T. Is seronegative rheumatoid arthritis true rheumatoid arthritis? A nationwide cohort study. Rheumatol (United Kingdom). (2021) 60:2391–5. doi: 10.1093/rheumatology/keaa623
295. van Boheemen L, van Schaardenburg D. Predicting Rheumatoid Arthritis in At-Risk Individuals. Clin Ther. (2019) 2019:1–13. doi: 10.1016/j.clinthera.2019.04.017
296. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
297. Deane KD, Holers VM. Rheumatoid arthritis pathogenesis, prediction, and prevention: an emerging paradigm shift. Arthritis Rheumatol. (2021) 73:181–93. doi: 10.1002/art.41417
298. Sparks JA, Iversen MD, Yu Z, Triedman NA, Prado MG, Miller Kroouze R, et al. Disclosure of personalized rheumatoid arthritis risk using genetics, biomarkers, and lifestyle factors to motivate health behavior improvements: a randomized controlled trial. Arthritis Care Res. (2018) 70:823–33. doi: 10.1002/acr.23411
299. Maglio C, Zhang Y, Peltonen M, Andersson-Assarsson J, Svensson PA, Herder C, et al. Bariatric surgery and the incidence of rheumatoid arthritis - a Swedish Obese Subjects study. Rheumatol (United Kingdom). (2020) 59:303–9. doi: 10.1093/rheumatology/kez275
300. Liu X, Tedeschi SK, Barbhaiya M, Leatherwood CL, Speyer CB, Lu B, et al. Impact and timing of smoking cessation on reducing risk of rheumatoid arthritis among women in the nurses' health studies. Arthritis Care Res (Hoboken). (2019) 71:914–24. doi: 10.1002/acr.23837
301. Bos WH, Dijkmans BAC, Boers M, Van De Stadt RJ, Van Schaardenburg D. Effect of dexamethasone on autoantibody levels and arthritis development in patients with arthralgia: A randomised trial. Ann Rheum Dis. (2010) 69:571–4. doi: 10.1136/ard.2008.105767
302. Gerlag DM, Safy M, Maijer KI, Tang MW, Tas SW, Starmans-Kool MJF, et al. Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: The PRAIRI study. Ann Rheum Dis. (2019) 78:179–85. doi: 10.1136/annrheumdis-2017-212763
303. Van Boheemen L, Turk S, Beers-Tas M Van, Bos W, Marsman D, Griep EN, et al. Atorvastatin is unlikely to prevent rheumatoid arthritis in high risk individuals: Results from the prematurely stopped STAtins to Prevent Rheumatoid Arthritis (STAPRA) trial. RMD Open. (2021) 7:e001591. doi: 10.1136/rmdopen-2021-001591
304. Al-Laith M, Jasenecova M, Abraham S, Bosworth A, Bruce IN, Buckley CD, et al. Arthritis prevention in the pre-clinical phase of RA with abatacept (the APIPPRA study): A multi-centre, randomised, double-blind, parallel-group, placebo-controlled clinical trial protocol. Trials. (2019) 20:1–15. doi: 10.1186/s13063-019-3403-7
305. National Institute of Allergy Infectious Diseases S. Strategy to Prevent the Onset of Clinically-Apparent Rheumatoid Arthritis (StopRA).
306. Rech J, Ostergaard M, Tascilar K, Hagen M, Valor Mendez L, Kleyer A, et al. Abatacept reverses subclinical arthritis in patients with high-risk to develop rheumatoid arthritis -results from the randomized, placebo-controlled ariaa study in RA-at risk patients. Arthritis Rheumatol. (2021) 73(Suppl 10):41–4. Available online at: https://acrabstracts.org/abstract/abatacept-reverses-subclinical-arthritis-in-patients-with-high-risk-to-develop-rheumatoid-arthritis-results-from-the-randomized-placebo-controlled-ariaa-study-in-ra-at-risk-patients/
307. Krijbolder DI, Verstappen M, van Dijk BT, Dakkak YJ, Burgers LE, Boer AC, et al. Intervention with methotrexate in patients with arthralgia at risk of rheumatoid arthritis to reduce the development of persistent arthritis and its disease burden (TREAT EARLIER): a randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet. (2022) 400:283–94. doi: 10.1016/S0140-6736(22)01193-X
308. Niemantsverdriet E, Dakkak YJ, Burgers LE, Bonte-Mineur F, Steup-Beekman GM, van der Kooij SM, et al. TREAT Early Arthralgia to Reverse or Limit Impending Exacerbation to Rheumatoid arthritis (TREAT EARLIER): a randomized, double-blind, placebo-controlled clinical trial protocol. Trials. (2020) 21:1–18. doi: 10.1186/s13063-020-04731-2
Keywords: rheumatoid arthritis, Pre-RA, preclinical rheumatoid arthritis, pathogenesis, etiology
Citation: Romão VC and Fonseca JE (2022) Disease mechanisms in preclinical rheumatoid arthritis: A narrative review. Front. Med. 9:689711. doi: 10.3389/fmed.2022.689711
Received: 01 April 2021; Accepted: 04 August 2022;
Published: 19 August 2022.
Edited by:
Mitsuhiro Takeno, Nippon Medical School Musashi Kosugi Hospital, JapanReviewed by:
Ryusuke Yoshimi, Yokohama City University, JapanCopyright © 2022 Romão and Fonseca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: João Eurico Fonseca, amVjZm9uc2VjYUBnbWFpbC5jb20=
†ORCID: João Eurico Fonseca orcid.org/0000-0003-1432-3671
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.