 Jiawei Xu
Jiawei Xu Yiyi Xiong
Yiyi Xiong Zhou Xu
Zhou Xu Hongquan Xing1,2
Hongquan Xing1,2 Xinyi Zhang
Xinyi Zhang- 1Department of Respiratory Diseases, The Second Affiliated Hospital of Nanchang University, Nanchang, China
- 2The Second Clinical Medical College of Nanchang University, Nanchang, China
- 3International Education College, Jiangxi University of Chinese Medicine, Nanchang, China
Lung cancer (LC) is one of the most incident malignancies and a leading cause of cancer mortality worldwide. Common tumorigenic drivers of LC mainly include genetic alterations of EGFR, ALK, KRAS, BRAF, ROS1, and MET. Small inhibitory molecules and antibodies selectively targeting these alterations or/and their downstream signaling pathways have been approved for treatment of LC. Unfortunately, following initial positive responses to these targeted therapies, a large number of patients show dismal prognosis due to the occurrence of resistance mechanisms, such as novel mutations of these genes and activation of alternative signaling pathways. Over the past decade, it has become clear that there is no possible cure for LC unless potent antitumor immune responses are induced by therapeutic intervention. Immunogenic cell death (ICD) is a newly emerged concept, a form of regulated cell death that is sufficient to activate adaptive immune responses against tumor cells. It transforms dying cancer cells into a therapeutic vaccine and stimulates long-lasting protective antitumor immunity. In this review, we discuss the key targetable genetic aberrations and the underlying mechanism of ICD in LC. Various agents inducing ICD are summarized and the possibility of harnessing ICD in LC immunotherapy is further explored.
1 Introduction
Over decades, lung cancer (LC) has remained one of the most frequently diagnosed cancers and ranks as the leading cause of cancer-related death in human globally (1). The 5-year survival rate is disappointing: only 19% of patients have survived overall, and most of them have suffered from a high risk of cancer relapse (2, 3). In the past decade, significant progress has been made in the treatment of LC. Researchers found several driver gene mutations of LC and deeply investigated molecular targeted therapy, which obviously improves the landscape of non-small cell lung carcinoma (NSCLC) treatment (4). Genetic alterations, such as mutations of epidermal growth factor receptor (EGFR), KRAS, MET, and rearrangements of ALK and ROS1, are the main contributors to LC tumorigenesis and progression (5), which dysregulate proliferation, apoptosis, migration, and invasion of cancer cells through various downstream signaling pathways. Targeting these abnormal genes or/and their downstream signaling has been used for treatment of LC. However, owing to the high heterogeneity of LC and acquired resistance to treatment, therapeutic efficacy of the targeted therapy is not guaranteed (6, 7).
Immunogenic cell death (ICD) is a novel form of regulated cell death which can evoke an adaptive immune response against cancer cells (8). Dying cancer cells can secrete damage-associated molecular patterns (DAMPs), mainly including high mobility group box 1 (HMGB1), calreticulin (CRT), adenosine triphosphate (ATP) and Type I interferon (Type I IFN). Recognized by the pattern recognition receptors (PRRs), the DAMPs enhance function of the antigen-presenting cells (APCs), activate T cells, increase the immunogenicity of tumor cells and ultimately trigger ICD (9). ICD can be triggered by many kinds of anti-cancer therapies, including chemotherapy, radiation, targeted drugs, photodynamic therapy (PDT) and immune checkpoints inhibitors (ICIs) (10–12). A shared characteristic of these various ICD is their ability to provoke endoplasmic reticulum (ER) stress and reactive oxygen species (ROS) generation. By restoring the immunogenicity of poor ICD triggers and stimulating DAMPs secretion, (ER) stress and ROS are believed to be indispensable for ICD (13, 14). Therefore, harnessing ICD to maintain the efficacy of anti-tumor therapies is crucial and challenging for LC treatment (15).
2 Genetic alterations affecting signaling pathways in lung cancer
Mutations of EGFR, KRAS, BRAF, and MET, and rearrangements of ALK and ROS1 have aroused great interest in recent years. Plenty of studies have pointed out the significance of targeting tumor driver genes in cancer therapies nowadays due to their key roles in promoting cancer survival, proliferation and cell-cycle progression through modulating downstream signaling pathways in lung cancer (Figure 1).
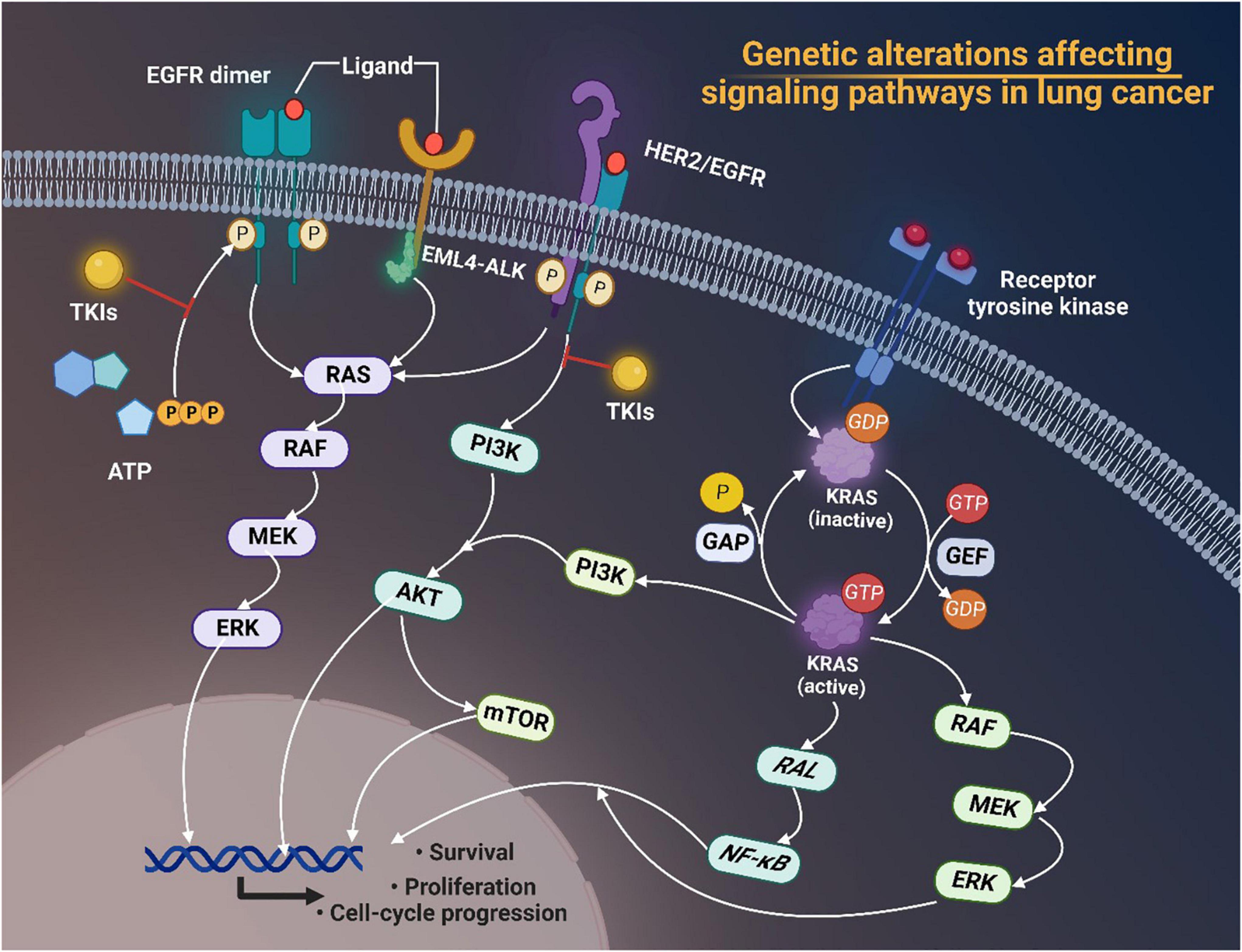
Figure 1. Genetic alterations of EGFR, ALK, and HER2 affecting signaling pathways in lung cancer. Tyrosine kinase inhibitors (TKIs) target receptor tyrosine kinases and prevent phosphorylation of the TK domain receptor of EGFR, thus inhibiting the activation of downstream signaling pathways such as the RAS/RAF/MEK/ERK pathway and the phosphatidylinositol 3-kinase (PI3K)-AKT pathway, thereby interfering with cell proliferation, differentiation, migration and survival. Activated KRAS proteins principally activate the downstream PI3K-AKT-mTOR signaling pathway that regulates cell proliferation and the RAS-RAF-MEK-ERK signaling pathway that regulates cell growth. Tumorigenic alterations contribute to EGFR overexpression, which ultimately increases the risk of cancer development by regulating related signaling pathways. EGFR, epidermal growth factor receptor; TKI, tyrosine kinase inhibitors; EML4-ALK, echinoderm microtubule associated protein like 4-activin-like kinase; HER2, human epidermal growth factor receptor 2; ATP, adenosine triphosphate; MEK, MAP kinase-ERK kinase; ERK, extracellular regulated protein kinases; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; GAP, growth-associated protein; GDP, guanosine diphosphate; GTP, guanosine triphosphate; GEF, Granule, Effervescent; RAL, Ras-like; NF-κB, nuclear factor-kappa B; RAF, rheumatoid arthritis factor; ERK, extracellular regulated protein kinases.
2.1 EGFR mutations
For Asian patients with lung cancer, EGFR mutant NSCLC is the most prevalent subtype (16). Over the past decades, more and more studies have showed that EGFR mutation is a common driver of tumorigenesis, and lung cancer is no exception (17, 18). EGFR mutations include in-frame mutations or point mutations and insertions, which typically occurs in exon18-21, encoding a portion of the EGFR kinase domain. In-frame deletions in exon 19 and the L858R point mutation in exon 21 account for nearly 90% of EGFR mutations and confer high sensitivity to clinical target therapies (19). These mutations confer higher sensitivity to clinical target therapies due to increased affinity of TKIs to the ATP-binding pocket of mutant EGFR compared to its wild-type. However, insertion mutation and T790M point mutation in exon 20 are often resistant to TKI (20). EGFR mutations are responsible for activation of constitutive ligand-independent receptor and regulation of downstream signaling pathways, promoting cancer proliferation and cell survival (21). Regulated downstream signaling pathways include activation of RAS/RAF/MEK/ERK, phospholipase C (PLCγ) and phosphoinositide 3-kinase (PI3K)-AKT, but inhibition the p38/MAPK and JNK/STAT pathway (22) (Figure 2). Oncogenic alterations may result in EGFR overexpression as well, which eventually increases the cancer incidence risk via regulating related signaling pathway (23).
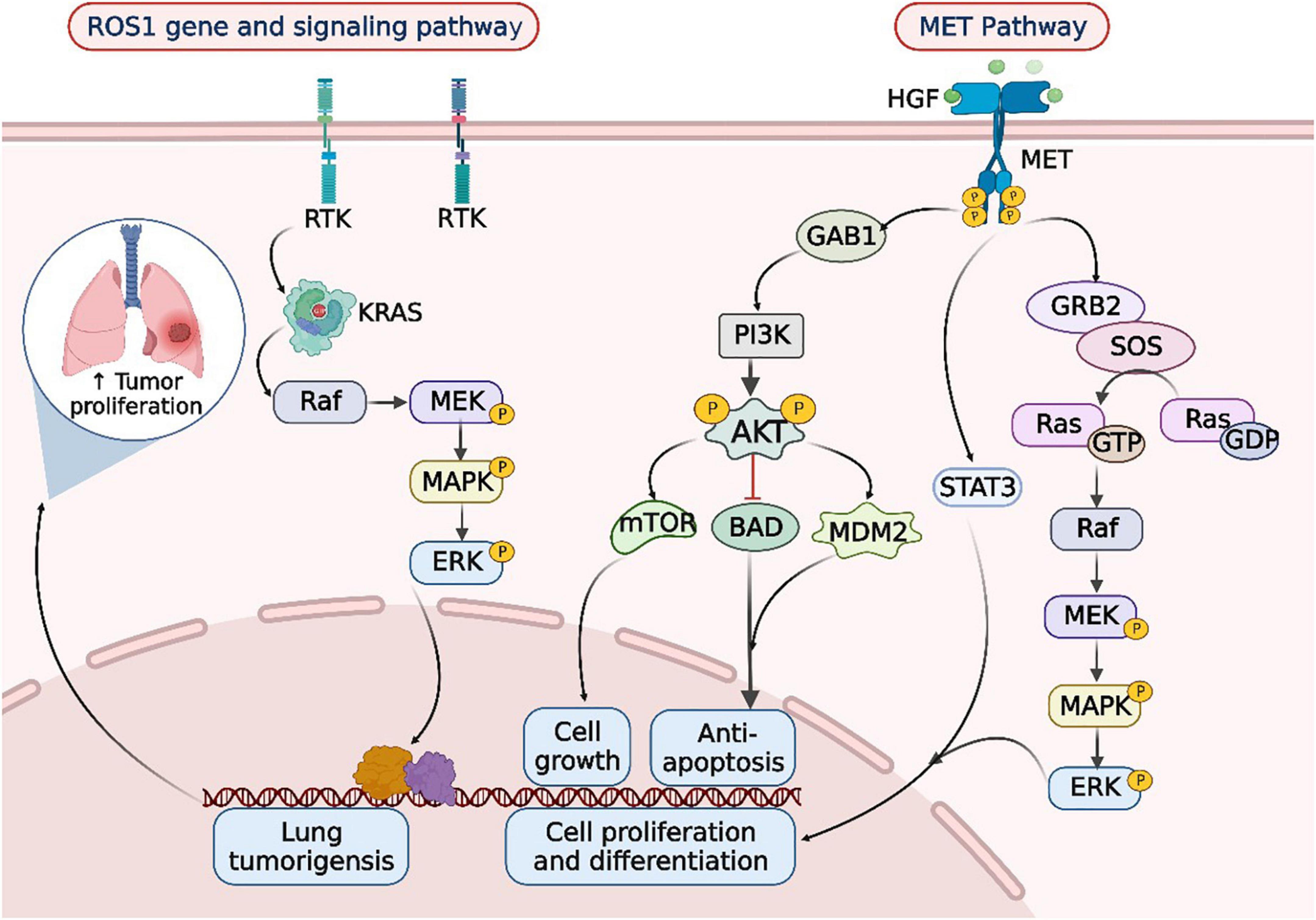
Figure 2. ROS1 rearrangements and MET mutations in the occurrence and development of lung cancer. When lung cancer is developed, the tyrosine kinase receptor (RTK) encoded by the ROS1 gene synergizes with oncogenic drivers such as KRAS to enhance MAPK-ERK signaling. Upon MET activation, PI3K associates with GAB1 and activates AKT/protein kinase. AKT inactivates the pro-apoptotic protein BCL-2 cell death antagonist (BAD) and triggers the E3 ubiquitin-protein ligase MDM2, thereby inhibiting apoptosis and promoting cell survival. In addition, AKT activates the mammalian target of rapamycin (mTOR) protein, promoting protein synthesis and cell growth, MET activation signals through the RAS-MAPK pathway as well. The nucleotide exchange protein Son of Sevenless (SOS) activates RAS upon binding to GRB2, which leads to activation of the v-Raf murine sarcoma viral oncogene homolog B1 (RAF) kinase, followed by stimulation of MAPK effector kinase (MEK) and resulting in MAPK activation. MAPK phosphorylates ERK, the ultimate effector of the cascade. The RAS-MAPK pathway is responsible for cell proliferation, cell motility, and cell cycle progression. Moreover, MET could relay signals to the activator of the transcription 3 (STAT3) pathway. STAT3 directly binds to MET, enabling STAT3 phosphorylation, which regulates cell transformation and invasion. RTK, receptor tyrosine kinases; Raf, rheumatoid arthritis factor; MEK, MAP kinase-ERK kinase; ERK, extracellular regulated protein kinases; HGF, hepatic growth factor; MET, mesenchymal to epithelial transition factor; GAB1, Grb2-associated binders 1; GAB2, Grb2-associated binders 2; PI3K, phosphoinositide-3 kinase; AKT, protein kinase B; mTOR, mammalian target of rapamycin; BAD, Bcl-xL/Bcl-2 associated death promoter; MDM2, murine double minute 2; SOS, son of sevenless; GTP, guanosine triphosphate; GDP, guanosine diphosphate; STAT3, signal transducer and activator of transcription 3.
Mutant EGFR could inhibit apoptosis via inhibiting BH3 -domain proteins, such as pro-apoptotic BIM and BMF (24). Trever et al. proposed that the FAS/NF-κB pathway, which promotes tumor growth, could rescue EGFR mutant lung cancer cells from EGFR inhibition (25). Karachaliou et al. found that knockdown of Orphan receptor 1 (ROR1) could inhibit the growth of NCI-H1975 cells [harboring EGFR L858R and T790M mutations (24)] via the ROR1/MEK/ERK signaling pathway, indicating the potential of ROR1 as therapeutic target for EGFR positive lung adenocarcinoma (LUAD) (26). The study of Kinehara et al. proposed that the GPI-anchored protein semaphorin 7A (SEMA7A) is overexpressed via induction of mTOR signaling in LUAD. Mutant EGFR lung cancer could upregulate SEMA7A/ITBG1 axis, which normally activates ERK signaling and leads to apoptosis resistance (27). Zhang et al. reported that phosphohistidine phosphatase 1 (PHPT1), often overexpressed and caused poor survival in lung cancer patients, could activate ERK/MAPK pathway targeting F-box protein 32 (FBXO32) as E3 ubiquitin ligase in EGFR mutant lung cancer (28). Previous studies have revealed that betacellulin (BTC) could binds to members of the ErbB family and mediating cancer development. Chava et al. firstly proposed that BTC could suppress apoptosis and promote cancer growth in EGFR-mutant LUAD in a MAP kinase-dependent way (29). The F-box protein FBXL2, a potential therapeutic target for EGFR mutant LC, could suppress EGFR-driven NSCLC cell growth. Niu et al. showed that glucose-regulated protein 94 (Grp94) protects the stability of EGFR via blockage of FBXL2, thereby promoting EGFR mutant cell proliferation and anti-apoptosis (30).
Epidermal growth factor receptor-positive lung cancer could confer potent invasive ability. Tsai et al.’s study showed that EGFR-L858R mutant LUAD could activate CXCL12-CXCR4 axis to enhance metastasis (31). Feng et al. reached a similar conclusion in their study that EGFR 19 exon deletion can promote the expression of MMP-2 and MMP-9 by enhancing the CXCR4/CXCL12 signaling pathway, leading to higher proliferation, migration, and invasion abilities (32). Li et al.’s study found that the EGFR-mutant NSCLC related brain metastasis is associated with downregulation of WNT5A by E2F1 via ERK1/2 pathway (33). A recent study elucidated that Y-box Binding protein (YB-1), an important drug sensitivity modulator, could activate AKT signaling and epithelial-to-mesenchymal transition (EMT) via targeting major vault protein (MVP), especially in EGFR mutant LAUD (34).
Moreover, the crosstalk of EGFR with the tumor microenvironment (TME) could affect the immunity to cancer. High level of IL-6 is a biomarker in lung cancer patients, Cao et al. revealed that mutant EGFR could upregulate IL-6 via gp130/JAK signaling pathway targeting signal transducer and activator of transcription (STAT) 3, a known oncogenic protein (35). Patients with EGFR activation showed upregulated CD73/adenosine via EGFR-ERK signaling pathway, which contributes to the immune-inert environment for EGFR-mutant NSCLC (36–39). Chen et al. reported that activated EGFR NSCLC cells enhance ILT4 expression, which suppresses T cell proliferation and immunity and thereby leads to immune escape (40).
2.2 KRAS mutations
RAS is the most frequent mutant oncogene in cancer (41, 42). Among all kinds of isoforms, KRAS, which belongs to GFPase superfamily, accounts for 86% RAS-mutants (43). Research demonstrated that 20–40% of LUAD and 30% NSCLC have been observed with KRAS mutation, which is more prevalent in smokers (44). KRAS mutation could destruct the activation of GTPase, which would lead to the accumulation of KRAS under GTP binding condition and thus cause the activation of basic downstream pathways related with cellular life events, including MAPK, PI3K, and Ras-like (Ral) 2 guanine nucleotide exchange factor (RalGEF) and so on (45, 46).
KRAS mutations act as strong drivers for tumorigenesis by modulating multiple signaling pathways. It has been demonstrated that mutant RAS gene, could activate Raf–MEK–ERK phosphorylation cascade to enhance tumorigenesis (47). SIRT1, an oncogene or tumor suppressor, was found decreased by KRAS in a PI3K and MEK dependent way, which contributes to lung carcinogenesis (48). RASSF1A is believed to be a weak suppressor of human tumors. Transgenic mice with RASSF1A-defective background demonstrated that the loss of RASSF1A apparently enhances the RAS-driven lung cancer (49). EGFR palmitoylation has been shown to inhibit EGFR activity and alter downstream signaling in the KRAS mutant lung cancer. Blocking EGFR palmitoylation decreased PI3K signaling, negatively regulating lung carcinogenesis (50, 51). The ERBB/EGFR signaling pathway is also dysregulated in lung cancer. The activation of KRAS induces the phosphorylation of iRhom2, which induces excessive shedding of ERBB ligand and tumorigenesis (52). Escaping from oncogene surveillance is a vital part in tumorigenesis. RUNX3, which serves as a mediator of multiple tumor suppressor pathways, is inactivated in KRAS mutant lung cancer. KRAS-activated cells could develop into ADCs when Runx3-mediated tumor suppress signaling pathways are abrogated (53).
Accumulating evidence suggests that inflammation is an essential factor for tumor promotion (54). The JAK-STAT pathway is considered as a central player for inflammation mediated tumorigenesis and targeting this pathway in KRAS-driven LC has been proposed. KRAS mutant LC secrete pro-inflammatory cytokines which activate JAK1 and JAK2, thereby improving cell survival. Besides, deletion of STAT3 could enhance KRAS-driven lung cancer development (55). Previous studies have found that the abnormal activation of STAT3 in the development of KRAS mutant lung cancer, which will be attenuated under anti-IL-6 therapy, suggesting a tight association between IL-6/STAT3 signaling and inflammation in KRAS-activated tumorigenesis (56, 57). A study established a murine model and confirmed that this route is gender-specific, deletion of epithelial STAT3 in KRAS mutant LC female mice will decrease tumorigenesis, while the outcome is completely opposite in male mice (58). Later, their team showed that NF-κB is activated in KRAS-driven mouse model of LUAD (59). Bassères et al.’s work indicated that NF-κB is significant in KRAS-driven tumorigenesis, as the deficiency of p65/RelA profoundly impairs KRAS-driven lung tumorigenesis. Besides, inhibition of IKKβ expression suppressed NF-κB expression in KRAS-driven lung cells (60).
More and more studies have explained the underlying mechanism from various perspectives including proliferation, invasion and EMT. By upregulating DUSP6, a negative regulator of p-ERK, KRAS mutant lung cancer retrained the ERK1/2 mediated toxicity and promote cell proliferation (61). Wang et al. demonstrated that mutant KRAS could enhance the Cathepsin L/CUX1 axis, thereby promoting lung cancer invasion and migration (62). Hsu et al. firstly reported Yes-associated protein (YAP) in LUAD, their study stressed that YAP promote the brain metastasis of NSCLC cell lines H2030-BrM3 (KRASG12C mutation), especially by, targeting the downstream genes CTGF and CYR61 (63).
2.3 BRAF mutations
V-raf murine sarcoma viral oncogene homolog B (BRAF), which plays a vital part in cell proliferation, differentiation, and growth through mediating the MAPK pathway, belongs to the RAF family of serine/threonine protein kinase (64). Over 40 missense mutations have been discovered in human, while the most common BRAF mutation occurs in exon 15 is a thymidine to adenosine transversion at the level of T1799A, leading to the valine to glutamate substitution at codon 600 (V600E), which contribute to approximately half of the BRAF-mutant NSCLC (65, 66). This alteration could result in the activation of B-RAF kinase and constitutive MAPK/ERK cascade signal transduction, which leads to 500 folds BRAF activity compared with WT (67).
Mutant BRAF plays a positive key player in the tumorigenesis (68). Li et al. reported that ARHGEF19, one of Rho guanine nucleotide exchange factors (RhoGEFs), could interact with BRAF and promote MEK1/2 phosphorylation during the NSCLC formation (69). Tumor necrosis factor receptor (TNFR)–associated factors (TRAF) is a kinases modulator of TNFR family. Wang et al. firstly proposed the relationship between BRAF and TRAF1. The study explained that overexpressed TRAF1 could regulate BRAF/MAPK/ERK axis to promote NSCLC cells’ viability (70). C-RAF, which could promote adenoma initiation and growth, belongs to RAF family as well. Zanucco et al. reported that elimination of BRAF in oncogenic C-RAF expressed alveolar epithelial type II cells inactivates MAPK signal and lung Tumor growth (71). Upregulated terminal differentiation-induced non-coding RNA (TINCR) is associated with poor survival in NSCLC patients, Zhu et al. pointed out that TINCR could target BRAF and mediate downstream MAPK pathway to promote NSCLC tumorigenesis (72).
Abundant evidence indicates that inactivation of BRAF could cause cancer cell apoptosis, thus demonstrating the necessity of mutant BRAF in tumor cells (73). Lin et al. comprehensively evaluated the molecular determinants of BRAF mutant in lung cancer. Their study summarized that inhibition of MEK/ERK signaling targeting p61VE could suppress the cell escape from BRAFV600E oncogenic inhibitions in NSCLC. Besides, they found the MAPK pathway could mediate EGFR signaling and alleviate the dependence on BRAFV600E (74). Kotani et al. analyzed the role of MAPK signaling in BRAFV600E mutant lung cancer. The results showed that EGFR signaling could govern MEK/ERK pathway more strongly, however, the situation is not the same in BRAFV600E mutant lung cancer, which induce the receptor tyrosine kinases (RTKs) resistance problem (75). However, a previous study claimed that although BRAFV600E could initiate some benign tumors, lung cancer is seldomly induced. Trejo et al. suggested that co-mutation of PIK3CAH1047R and BRAFV600E could promote lung cancer progression, including transformation of tumor phenotype into malignant and accelerate tumor growth rate (76). The team of Mcmahon also demonstrated similar conclusion. Their research confirmed that the co-mutant BRAFV600E and PIK3CAH1047R in alveolar type 2 pneumocytes accelerate cell dedifferentiation (77). Tumor suppressor STK11 (LKB1) gene is frequently deficient in lung cancer. A study revealed that Lkb1 loss could promote tumorigenesis in BRAFV600E induced LUAD (78).
2.4 ALK rearrangements
ALK gene locates on the short arm of chromosome 2 (2p23), and belongs to the insulin receptor superfamily (79). As a tyrosine kinase receptor, ALK generally expresses in the brain and spinal cord during embryo genesis and dominantly decreased following maturation (79). The high correlation between ALK and tumorigenesis was firstly identified in 1994 as a fusion partner of nucleophosmin (NPM) in anaplastic large-cell lymphoma (80). Further research revealed underlying mechanism of NSCLC genesis with ALK translocation, as well (81, 82). ALK rearrangement presented potent oncogenic drivers in approximately 5–6% of NSCLC patients population characterized with young age, barely smoking and adenocarcinoma histology (83, 84).
The most common fusion partner of ALK is EML4 (echinoderm microtubule associated protein like 4) (85). Heat shock protein 90 (Hsp90) is a novel cancer therapy target owning to its positive role in controlling oncogenic signaling proteins. Normant et al. proposed that inhibition of Hsp90 could decrease EML4-ALK thereby inducing tumor regression in ALK-driven NSCLC (86). The crosstalk between Programmed cell death-ligand 1 (PD-L1) and its receptor programmed cell death-1 (PD-1) conferred profound strength in immunotherapy. A study examined the role of ALK rearrangement in affecting PD-L1 expression, their result showed that EML4-ALK fusion could decrease the PD-L1 expression through suppression of PI3K–AKT or MEK–ERK signaling pathway, which contributes to the immune escape in NSCLC (87). Shen et al. reported that EML4-ALK G1202R mutation could increase the invasion and migration ability of A549 cells. Besides, their research proved the EML4-ALK G1202R mutation could lead to EMT phenotype transformation in NSCLC cells by activating STAT3/Slug pathway (88).
2.5 ROS1 rearrangements
The ROS1 gene, which is located on chromosome 6 (6q22.1), is a widely known proto-oncogenic gene that belongs to the sevenless subfamily of tyrosine kinase insulin receptor. ROS1 rearrangements were primarily described in glioblastoma, the close correlation with NSCLC was discovered in 2012 (89). The ROS1 tyrosine kinase has been discovered to play a vital role in plenty of intracellular signaling pathways (90).
The (c-ros oncogene1) ROS1 rearrangements, which drive malignant transformation of NSCLC, affect approximately 0.7–1.7% NSCLC patients (91). The fusion partners include CD74, SLC34A2, FIG, TPM3, SLC12A2, CCDC6, and SDC4, while CD74-ROS1 fusion is the most prevalent phenotype in NSCLC (92, 93). Gou et al. demonstrated that CD74-ROS1 mutation could lead to EMT and enhance the NSCLC invasion and migration ability by upregulating Twist1 (94). Chromosomal rearrangement of the Solute Carrier Family 34 Member 2 (SLC34A2)-ROS1 fusion accounts for more than 14% of all ROS1 fusion in NSCLCs. Cai et al.’s study revealed that BA/F3 fusion NSCLC cells (harboring SLC34A2-ROS1) could activate ROS1-SHP2 signaling to elevate PD-L1 expression and mediate immunogenicity (95). Interestingly, another study supplemented that ROS1-fusion positive NSCLC cells could target MEK/ERK signaling pathway to upregulate PD-L1 expression significantly (96).
2.6 MET mutations
The proto-oncogene MET, located on chromosome 7q31, is one of the tyrosine kinase receptors. HGF is a common ligand of MET, upon their binding, MET will be dimerized and auto-phosphorylate, thus leading to the activation of activity of intracellular tyrosine kinase. Activation of MET could lead to the modulation of multiple downstream signaling pathways including RAS/RAF/ERK/MAPKA, PI3K/AKT/mTOR, Wnt/β-catenin, STAT and so on, which play vital roles in regulating tumor growth, progression and migration (97). Aberrant activation of MET signaling pathway may contribute to the tumorigenesis process of lung cancer.
About 3–5% of NSCLC patients have MET mutations, most of which are adenocarcinoma. Up to date, various mutations of MET have been identified, including MET amplification, MET point mutations, exon 14 skipping mutation, fusions and overexpression, all of which are oncogenic in lung cancer (98). The skipping of MET exon 14 mutation occurs in 3% LUAD and 13–22% sarcomatoid lung cancer (99). Studies revealed that the exon 14 skipping could lead to the lack of Y1003-Cbl, a ligand mediating c-MET degradation and ubiquitination, which subsequently prolong the activation of c-MET, downstream proliferation, and tumorigenesis (100). MET amplification is another major subtype of MET mutations which occur mainly in TKI-resistance lung cancer. Because single amplification of MET rarely contributes significantly to cancer development, co-mutant of MET and other cancer drivers, such as EGFR, appear more commonly (101, 102). A study indicated that only high amplification level of MET could display an oncogenic effect (103). However, there’s still a lack of research to fully elucidate the mechanism of MET-driven tumor development.
Recently, more and more target therapies are focusing on mutant genes to improve the prognosis for lung cancer. However, disappointedly, clinical outcomes are not positive as expected. Firstly, the development of drug resistance is another unavoidable theme in lung cancer. Jackman et al. primarily defined acquired resistance when patients receive target therapy over 6 months but the disease progression keeps evolving (104). Accumulating evidence has elucidated that, although the acquired resistance mechanism varies, the main reasons are from three aspects: mutant gene modification, phenotypic transformation and alternative signal pathway activation (105). EGFR TKIs are the most commonly used target drugs in EGFR mutant lung cancer patients. Previous studies have revealed that the EGFR T790M mutation, MET amplification, epithelial-to-mesenchymal transition (EMT), activation of the NF-κB pathway, and so on are common foundations of later developed EGFR TKIs resistance (105–108). These various factors could induce signaling pathway cross talk in lung cancer. Hepatocyte growth factor (HGF) could induce acquired resistance to TKIs by restoring the PI3K/Akt signaling pathway via phosphorylation of MET in EGFR mutant NSCLC (109). Activation of Hedgehog (Hh) pathway is a common feature of TKIs resistance. A study fully illustrated the role of Hh signaling in EGFR mutant lung cancer, their result showed that, through the induction of mesenchymal properties, Hh could mediate the resistance of EGFR inhibitors (110). Several clinical studies reported acquired resistance to targeted drugs, such as Osimertinib, a MET tyrosine kinase inhibitor (111). The advance in K-RAS targeted medicine faced huge challenge (112). On one hand, clinical trial of K-RAS targeting drugs revealed disappointing results. It was found that K-ras could activate an alternative pathway via geranylation with resistant farnesyl transferase inhibitors (113). Blumenschein et al. reported that the downstream MEK signaling pathway inhibitor failed to achieve significant positive effect on improving the survival of lung cancer patients (114). ALK- independent resistance occurs as well. Activating bypass signaling pathways compromising KIT amplification, MAPK, MET amplification, EGFR and BRAF V600E leads to the ALK TKIs resistance (115–117). Additionally, more drugs targeting other mutant genes are under research, which is not warranted to have a certain therapeutic effect. What’s more, the narrow therapeutic window is worrying. For example, the second-generation EGFR TKI afatinib displayed an apparent adverse effect in clinical trials (118). Besides, recurrence or metastasis of lung cancer occurs frequently. Previous research has elucidated that brain relapse, bone relapse, and relapse of other organs can seriously affect the prognosis process of lung cancer (119, 120).
Taken together, gene alteration is a common and significant phenomenon in tumorigenesis progression. Slight changes in these central cancer-driven genes can lead to dysregulate in downstream signal pathways, which dramatically affect lung cancer from multiple perspectives (Figure 2). However, the therapeutic effect is not always as positive as considered, clinical feedback suggested that the developed drug resistance is uncontrollable (5). Appropriate drug concentration is still in preclinical trials and prognostic recurrence/metastasis is beyond expectation. Therefore, a novel way of overcoming existing dilemma is urgently needed.
3 ICD in lung cancer cells
Immunogenic cell death is a novel type of cell death which primes an adaptive immune response (Figure 3). Its main feature is that dying cells can secrete DAMPs, including HMGB1, CRT, ATP, and Type I IFN, which can enhance the antigen presentation capability of APCs, activate T cells, and enhance the immunogenicity of tumor cells, ultimately trigger the arise of ICD (9). In addition, induction of ER stress and ROS accumulation are indispensable components for ICD which increase the DAMPs (13, 14).
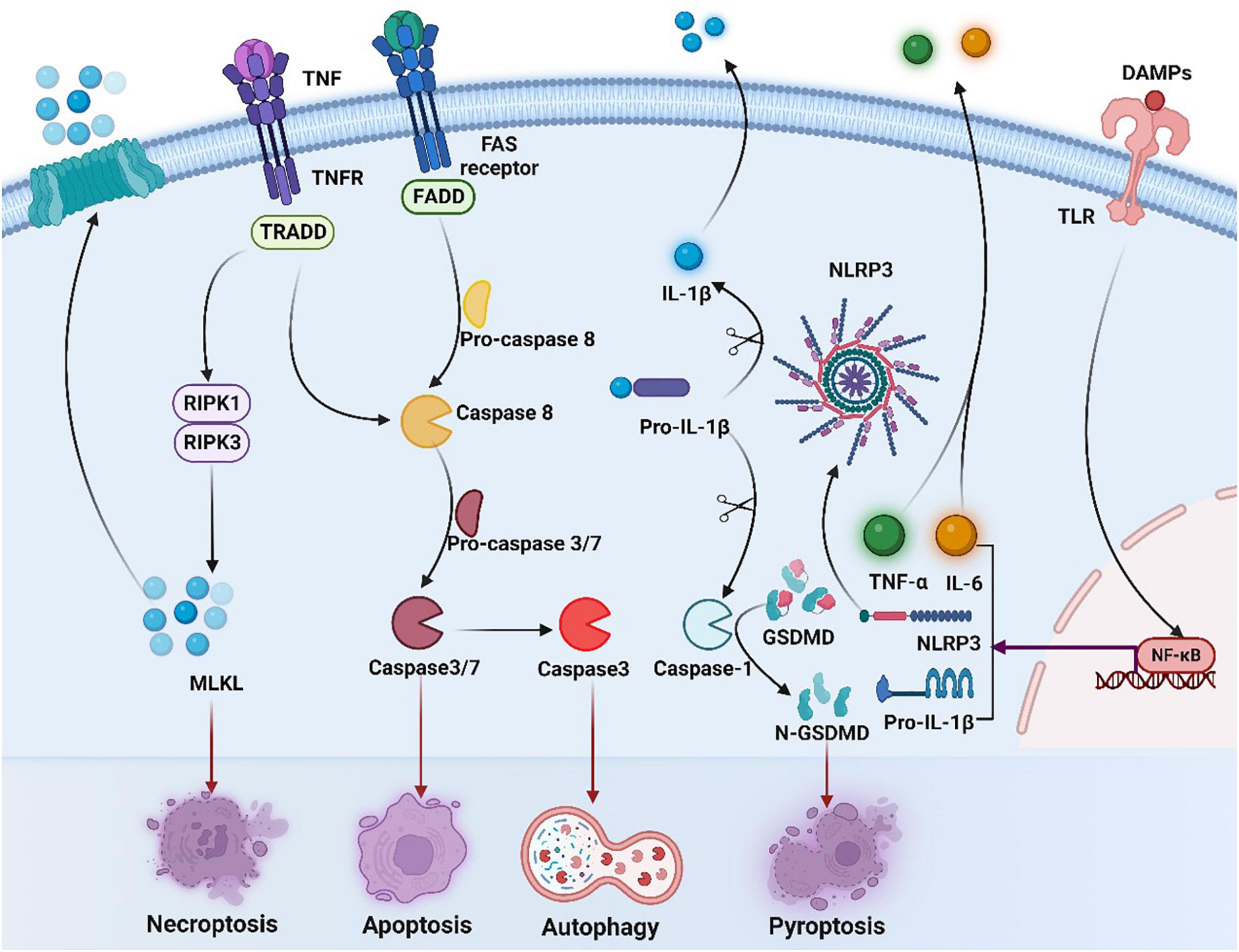
Figure 3. Overview of necroptosis, apoptosis, and pyroptosis signaling pathways. Binding of tumor necrosis factor (TNF) to its receptor (TNFR) activates RIPK3 via RIPK1, leading to the formation of necrosome, which activates mixed-lineage kinase-like (MLKL), contributing to necrosis and membrane permeation. Activation of caspase-8 induces exogenous apoptosis by triggering caspase-3 and caspase-7 activation. Meanwhile, caspase-3 induces autophagy. The occurrence of ICD in tumor cells is accompanied by the production of a series of signaling molecules in which released DAMPs can bind to pattern recognition receptors such as toll-like receptors (TLRs) on the surface of DC cells, activating NF-κB and inducing expression of NLRP3 and IL-1β/IL-18 precursors. NLRP3 recognizes various DAMPs and becomes oligomerized, contributing to activation of caspase-1 and production of mature IL-1β/IL-18. Activated caspase-1 cleaves gasdermin D (GSDMD) and releases the n-terminal (GSDMDNT) pore-forming fragments, thereby resulting in membrane permeation and pyroptosis. DAMPs, damage-associated molecular patterns; TLR, toll-like receptors; NF-κB, nuclear factor-kappa B; IL-6, interleukin-6; TNF, tumor necrosis factor, TNFR, tumor necrosis factor receptor; TNF-α, tumor necrosis factor alpha; NLRP3, NOD-like receptor thermal protein domain associated protein 3; IL-1β, interleukin-1 beta; GSDMD, gasdermin D; FADD, Fas-associating protein with a novel death domain; TRADD, TNF receptor 1 associated via death domain; RIPK1, receptor interacting serine/threonine kinase 1; RIPK3, receptor interacting serine/threonine kinase; MLKL, mixed lineage kinase domain-like protein.
Apoptosis is one of the most researched forms of ICD. Apoptosis, mediated by the activity of caspases, is regulated by both endogenous and exogenous factors at the same time, and both of them rely on the activation of caspase-3 and caspase-7 (121). Yet, it has also emerged that non-apoptotic cell death can also be immunogenic, such as necrosis and pyroptosis. Necroptosis can be initiated firstly after activating the extended tumor necrosis factor alpha (TNF-α) receptor family on the surface of the cell, and transmitted in virtue of the serine/threonine kinases, RIP1 or RIP3 interact with receptors. Then dead cells release immunogenic DAMPs to greatly activate both innate and adaptive immune systems. Pyroptosis is a lytic pro-inflammatory modality of regulated cell death (RCD), which leads to the formation of plasma membrane pores with the help of members of the gasdermin protein family, particularly gasdermin D (GSDMD) (122). In addition, the anticancer immune response can be also triggered by autophagy, while autophagy was found to be related to the resistance of cancer cells to anti-cancer therapy (12).
3.1 Extra-cellular DAMP release
One of the main features of ICD is the release of molecular signals, which are usually called “DAMPs” (14). DAMPs can recognize specific receptors and attract adaptive immune cells like neutrophils, macrophages and dendritic cells (DCs). Then DAMPs promote activation and maturation of these immune cells, including dead cell removal, antigen uptake, processing, and presentation, and cytokine production (123). Some of the most widely researched ICD-linked DAMPs include HMGB1, CRT, ATP, and Type I IFN (124).
High mobility group box 1, which can trigger strongly inflammatory response when released from nucleus of dead cells, is an abundant nuclear non-histone chromatin-binding protein (125). HMGB1 binds to several receptors, such as Toll-like receptor 4 (TLR4), a type of receptor for advanced glycation end products (RAGE) to activate MAPKs and NF-κB in DCs, which are widely expressed in lungs (126–129). After CRT exposure, dead lung cancer cells will secrete HMGB1, which has a dual effect depending on whether it is extracellular or intracellular (14). Extracellular HMGB1 can facilitate the processing and presentation of antigens by DCs (128, 130), while intracellular HMGB1 can promote cancer cell growth and invasion, and resist therapy (129). Studies found that high level of HMGB1 is associated with poor prognosis in NSCLC (131). Moreover, Łagiedo et al. discovered that the levels of HMGB1 in NSCLC patients’ serum had a significant positive correlation with the size of the tumor (132).
Adenosine triphosphate is dependent on autophagy to be released from dying cancer cells in virtue of the active exocytosis of ATP-containing vesicles through pannexin channels (13, 133, 134). After being secreted out of cells and binding to purinergic receptor P2Y2 on the target cells, ATP will send a “find-me” signal to DCs and macrophages to promote DC maturation and macrophage expansion (135, 136). Moreover, ATP can mediate immune stimulation by activating the NLRP3 inflammasome and the subsequent secretion of interleukin 1 beta (IL-1β) (127, 137).
Calreticulin, a soluble protein in ER lumen, is exposed on the cell surface at a premortem stage and confers an “eat me” signal (122). After that, CRT interacts with the CD91 receptor in phagocytes to effectively engulf dead cells, thus providing abundant antigenic substance (13, 138). CRT also induces increased expression of endothelial cell adhesion molecules to promote infiltration of specific lymphocytes in the TME (139). Research has found that high CRT levels were shown to be in association with eIF2α phosphorylation in biopsies from NSCLC patients, which is independently relevant to better prognosis in NSCLC (140). Besides, in the treatment of lung cancer, CRT plays a similar role as HMGB1 so that it can be used to assess the extent of ICD induced by the treatment (141).
Type I IFNs, which can be driven by RNA or DNA species, are actively synthesized and activate other downstream genes including genes coding for chemokines to favor an immune response (142, 143). In RNA species, the receptor is endosomal TLR3, whereas the latter setting mainly works through cytosolic cyclic GMP-AMP synthase (CGAS) and its signal transducer stimulator of IFN response cGAMP interactor 1 (STING1) (144–146). Moreover, type I IFN can trigger macrophages to secrete pro-inflammatory mediators and inhibit the immunosuppressive functions of regulatory T cells (147, 148). Apart from these direct immunostimulatory functions, type I IFN can also elicit the synthesis of the CX-C motif chemokine ligand 10 (CXCL10) by tumor cells in ICD via an autocrine signaling loop (149).
3.2 ER stress
Immunogenic cell death can be divided into two modes according to its induction mechanism (13). Instead of inducing ROS and ER stress directly, type I ICD is stimulated by indirect signals. Quite the opposite, type II ICD targets the ER, inducing ER stress and immunogenic cell death (150). The process of ER stress activation is termed as unfolded protein response (UPR), featured with phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) by Protein Kinase RNA-activated (PKR)-like ER Kinase (PERK) (151, 152). Several studies have shown that ER stress is the core of the occurrence of ICD (153). Moderate ER stress may be conducive to creating an immunosuppressive environment, while severe ER stress can stimulate immune response, as what happens in ICD (154). The more concentrated the ER stress is, the higher the immunogenicity of cell death is (155). ER stress is the main cellular mechanism for the cell surface exposure of CRT, which is closely linked to the phosphorylation of eIF2α. In Fucikova et al.’s study, they stated a subgroup of NSCLC associated with strong ER stress, which erupts in CRT expression and exposure (156). The high CRT driven by ER stress response has a positive prognostic value for NSCLC patients, however, its specific molecular mechanism remains to be further studied. ER stress also has an influence on levels of intracellular ATP by stimulating mitochondrial respiration, and cells can fill their bioenergy reserves in this way to restore cellular homeostasis (157).
4 Induction of ICD for lung cancer therapy
Immunogenic cell death can be caused by different types of stimulation and antitumor therapy, such as chemotherapy and radiation, some targeted drugs, oxygen-boosted PDT and ICIs. A lot of evidence shows that ICD can stimulate anticancer immune responses in vivo, and provide an opportunity to improve the cancer treatment and outcomes (158, 159). However, recently, only a few ICD inducers have been successfully translated into clinical practice. Here we elaborated on the role of ICD in the therapy of lung cancer.
4.1 Targeted therapy
Most anti-cancer drugs kill cancer cells in a non-immunogenic way. However, many studies demonstrated that ICD can be induced by different targeted agents. We will review these targeted drugs in detail to illustrate the important role of targeted therapy in the treatment of lung cancer (Table 1).
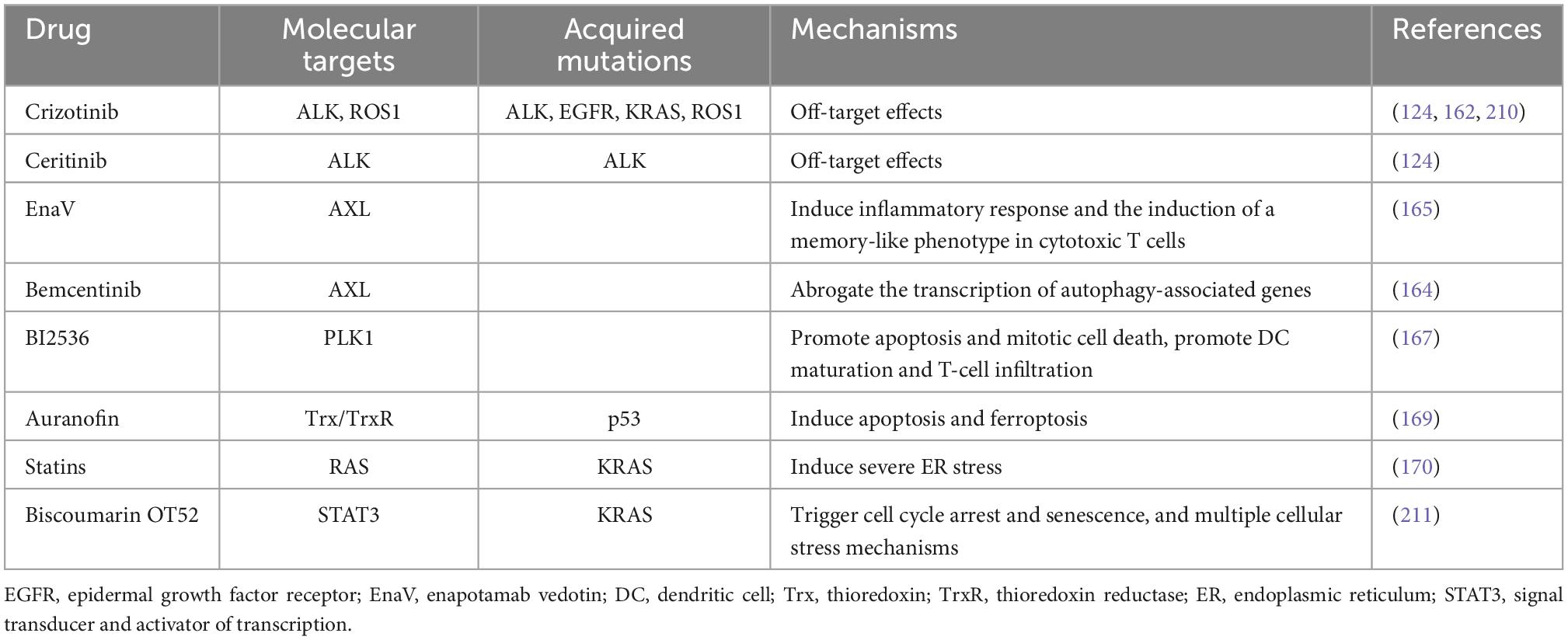
Table 1. Targeted therapies inducing immunogenic cell death (ICD) and their molecular targets in lung cancer treatment.
Crizotinib, a TKI used to treat NSCLC carrying activated ALK, ROS1, and MET, serves as an ICD stimulator via off-target effects (160). Drewry et al. have provided preclinical evidence that crizotinib can be expediently in combination with non-ICD inducing chemotherapeutics, as much with immune checkpoint blockade, to treat NSCLC in an effective way (161). One research has shown that the combination of cisplatin and high-dose crizotinib brings about an increase of PD-1 and PD-L1, and induces greatly ICD in NSCLC cells (12, 162). Hence, a sequential combination treatment including conventional chemotherapy together with crizotinib and immune checkpoint blockade may be effective for NSCLC. Besides, another ALK inhibitor ceritinib also has the function of targeting which can induce ICD in ALK-dependent NSCLC cell lines (124). For the past few years, the receptor tyrosine kinase AXL has been considered as a promising target for tumor treatment. The AXL signaling can promote a pro-survival pathway and reduce reactivation of the MAPK pathway to develop acquired resistance to EGFR TKI in NSCLC cells. And there is a positive correlation between AXL and autophagy (163, 164). Boshuizen et al. showed that an antibody–drug conjugate targeting AXL called enapotamab vedotin (EnaV) not only has direct tumor killing, but also induces inflammation and ICD of tumor cells in melanoma and lung cancer models (165). Lotsberg et al. reported that a small molecule inhibitor bemcentinib inhibits the transcription of autophagy-associated genes, releases DAMPs and then gives rise to ICD in NSCLC cells by targeted inhibition of the AXL signaling pathway (164). Erlotinib, a kind of TKI, can inhibit tumor development by inhibiting intracellular phosphorylation of EGFR-related tyrosine kinases, which commonly used in the treatment of NSCLC and pancreatic cancer. Studies have shown that targeting AXL in ER cells induces massive autophagic vacuolation before death in erlotinib-resistant cancer cells and triggers ICD (164). Hence, we suggest that ICD induction may have an unexpected effect on AXL-targeted NSCLC with drug-resistant EGFR mutations. Besides, PLK1 is a member of polo-like kinase family associated with cell division. PLK1 is overexpressed in NSCLC and it often predicts a poor prognosis (166). A selective PLK1 inhibitor BI2536 can act as an ICD inducer to cause apoptosis and alter the tumor immune microenvironment by promoting DC maturation and increasing T-cell infiltration (167).
In addition, several drugs widely used for other diseases have been found to have the ability to induce ICD in lung cancer cells. Auranofin (AF), a thioredoxin reductase 1 (TrxR) inhibitor, is known as an antirheumatic drug (168). TrxR is considered as a potential target in NSCLC on account of its high expression in NSCLC patients. A recent study indicated that AF can initiate release of DAMPs and DC maturation, then trigger apoptotic and ferroptotic cell death by targeting TrxR and launching ICD. It may provide new ideas for the treatment of NSCLC (169). Moreover, statins are one of the most frequently used drugs to treat hyperlipidemia. Statins can stimulate CD8 + T cells and provoke severe ER stress by inhibiting RAS prenylation in KRAS-mutant (KRASmut) lung tumor models, thereby leading to the ICD effects (170). Besides, coumarin is a kind of natural compounds with anti-inflammatory and anti-cancer function. Lee et al. found that biscoumarin OT52 strongly inhibited the proliferation of KRASmut NSCLC cells via ICD pathways (171). Mechanistically, biscoumarin OT52 suppresses STAT3 transactivation and expression of its target genes. Altogether, these drugs may become novel candidates in the future for more effective treatment of lung cancer. However, the specific signaling pathway in ICD inducing and other mechanisms still need to be explored in-depth.
4.2 Chemotherapy and radiation therapy
Chemotherapeutic drugs are considered to kill cancer cells selectively via direct cytotoxicity (12). A main mechanism of immunity stimulation by chemotherapy involves the induction of ICD (121). Unfortunately, only a little part of anti-cancer drugs can effectively trigger ICD (172). Some kinds of chemotherapeutic agents have been tried to modulate activity of DCs, such as cyclophosphamide, doxorubicin, oxaliplatin and anthracyclines, which can make tumor antigens be vaccines to the immune system and induce ICD, consequently provoking robust adaptive immune response (173–175). Chemotherapy with a combination regimen of oxaliplatin with cyclophosphamide is approved in clinical practice for lung cancer. Previous studies found that in a lung mouse cancer model, this regimen can foster CD8+ T cell infiltration and increase TLR4+ DCs in tumor tissues, and further increase tumor sensitivity to immune checkpoint therapy (176). But the research by Fileswasser et al. has shown that oxaliplatin does not induce ICD in NSCLC cells (4). Pemetrexed, a multi-targeting antifolate antagonist which is established as the main chemotherapy drug for the first-line treatment of advanced non-squamous NSCLC (NSq-NSCLC) and mesothelioma, has also been shown to induce ICD and to increase of immune-regulatory genes (177, 178). Liu et al. found that crizotinib, a kind of drugs used to treat NSCLC patients which carries activated ALK/ROS1, is an efficient ICD stimulator via off-target effects (162). Wang et al.’s results show that trametinib also has the ability to induce ICD by sensitizing lung cancer cells to endoplasmic reticulum stress and triggering the release of DAMPs, and can be effective in treating KRAS-mutant LUAD when used in combination with interleukin-12 (IL-12) (179). A study by Gao et al. showed that, DOC, a kind of tubulin stabilizer belonging to the taxane family, can induce DAMPs and significantly upregulated release of HMGB1 in human NSCLC cell line (180). In addition, the results of Furuwaka et al. suggest that osimertinib induced NSCLC tumor cell death may lead to exposure and release of CRT to induce ICD, and then improve the anti-tumor immunity (181).
As a topical treatment approach, radiation therapy (RT) is widely used in clinical cancer treatment. RT can induce ICD, which promotes DCs activation and the presentation of tumor antigen to prime CD8+ T cells (182). The CD8+ T cells then enter the unirradiated tumor area and attack cancer cells (14, 183). RT and many traditional chemotherapeutic agents give rise to DNA damage and multiform cell death ultimately (184). In various preclinical settings, similar to chemotherapy, induction of ICD by RT has been shown associated with increased sensitivity to immune checkpoint blockade (144, 185), and many clinical trials have proven that (186). In the same way, ablative RT can induce necroptosis in NSCLC and mediate HMGB1-driven immunological response (187).
4.3 PDT
Photodynamic therapy is able to kill cancer cells by manipulating photosensitizers and generating reactive ROS, which triggers ER stress and induces the anti-tumor immunity to eliminate residual or metastatic tumors effectively and selectively (155, 171, 188). After accumulating selectively in the tumor area, the photosensitizer (PS) is activated by illumination with visible light of appropriate wavelength, and then illuminated by red light (690 nm), which can induce local ICD at the tumor sites and strong anti-tumor immunity (10, 189). In recent years, the concept of PDT has been actively pursued. The binding of near-infrared PS to antibodies or nanocarriers improves the efficiency of PDT (190). One typical PS shown to induce ICD is hypericin, which is an anthraquinone derivative of natural origin with specific ER localization (155, 171). The other promising non-porphyrin PS is benzophenazine, OR141, which also has specific location in the ER (191). OR141 induces cell death mainly via the mammalian target of rapamycin signaling pathway and by inhibition of proteasomal deubiquitinases, leading to ER stress (192). One study reveals that in a prophylactic tumor vaccination model using PDT-treated TC1 lung cancer cells, redaporfin acts as an ICD inducer that can trigger eIF2a phosphorylation, DAMPs release and inhibit tumor growth (193). ICD can also be induced by PDT based on 8-methoxypsoralen (8- MOP) (194). But it is worth noting that it doesn’t need oxygen but intercalates into DNA and forms cross-links with one or two DNA strands under UVA irradiation (195). Furthermore, it has been shown that photofrin-based PDT of Lewis lung carcinoma cells induced release of HSPs, and surface exposure of CRT in vitro and in vivo in an hour after PDT, as well as an increase of HMGB1 (196). These data also indicate that photofrin is a potential inducer of ICD.
4.4 ICIs
The antitumor effect of ICIs works by interfering with immune tolerance (178). The most clinically common immune checkpoints include: PD-1/PD-L1, cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), indoleamine 2.3-dioxygenase (IDO), and CD47 (197, 198). ICIs have established a new model of lung cancer treatment and improved patients’ survival benefits (199, 200). It also has revolutionized the prognosis of multiple lung cancers, especially NSCLC, which have a high sensitivity to the immunotherapy against PD-1 (201). The combination therapy of platinum, PEM and ICIs has been proposed as a standard first-line treatment for advanced LUAD (202, 203). Also, numerous studies have shown platinum-based combination chemotherapy and combination ICIs, like PD-1 or for its ligand PD-L1, can markedly prolong survival in patients with stage III unresectable NSCLC (204–207). In clinical application, the combination of ICIs and chemotherapy can improve the efficacy of anti-tumor therapy, this may be because chemotherapy drugs increase tumor sensitivity to ICIs (208). Pemetrexed and ICIs targeting PD-1/PD-L1 are applied widely for the treatment of advanced NSq-NSCLC (178). Moreover, lurbinectedin is a kind of DNA-binding inhibitors of transcription, which is efficient at inducing ICD (209). The combination therapy of lurbinectedin and ICIs targeting PD-1/PD-L1 is supposed to be a salvage therapy for relapsed SCLC be over the years (172).
5 Conclusions and perspectives
Worldwide, lung cancer is one of the most common cancers and the leading cause of cancer-related deaths. For decades, researchers are exploring the pathogenesis of lung cancer and trying to find more effective treatments, such as by finding oncogenic driver gene mutations to improve targeted therapy. However, it is disappointing that clinical results have not been as positive as we expected. In recent years, ICD was noticed for evoking adaptive immune response of cancer cells. ICD can be induced by a variety of anticancer therapies, including chemotherapy, radiotherapy, targeted drugs, PDT and ICIs, etc. And it is becoming increasingly evident that ICD may offer a new idea in the anti-cancer therapeutic approaches in the future, especially for lung cancer.
In summary, it is a breakthrough to harness ICD to elevate the immunogenicity of tumor cells to maintain the efficacy of anti-tumor therapies for lung cancer. ICD induction is a promising area to explore and the mechanism of function and regulatory networks of ICD deserve further investigation. Finally, due to the limitations of current study, there are still many unanswered questions, such as whether ICD is associated with ferroptosis or cuproptosis, whether ICD is associated with anti-angiogenic drugs, and so on.
Author contributions
XZ: conceptualization and funding acquisition. JX and YX: writing – original draft preparation. ZX and HX: adapt the text and figures. XZ and LZ: writing – review and editing and project administration. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Domestic Cooperation Project of the Science and Technology Department of Jiangxi Province (No. 20212BDH81021 to XZ), Project of the Science and Technology Department of Jiangxi Province (No. 20203BBGL73148 to XZ), and Project of Health Commission of Jiangxi Province (No. 202210038 to XZ).
Acknowledgments
The graphical abstracts were created with BioRender software (www.biorender.com). We sincerely appreciated the guidance from our and every member in our team.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
TME, the tumor microenvironment; ICD, immunogenic cell death; NSCLC, non-small cell lung carcinoma; LUAD, lung adenocarcinoma; SqCC, lung squamous cell carcinoma; EGFR, epidermal growth factor receptor; TKI, tyrosine kinase inhibitors; IL-6, interleukin-6; DAMPs, damage-associated molecular patterns; HGMB1, high mobility group protein B1; CRT, calreticulin; ATP, adenosine triphosphate; Type I IFN, Type I interferon; PRRs, pattern recognition receptors; APCs, antigen-presenting cells; ER, endoplasmic reticulum; ROS, reactive oxygen species; PDT, photodynamic therapy; ICIs, immune checkpoints inhibitors; PI3K, phosphatidylinositol 3-kinase; BAD, Bcl-xL/Bcl-2 associated death promoter; RTK, receptor tyrosine kinases; ROR1, orphan receptor 1; SEMA7A, semaphorin 7A; PHPT1, phosphohistidine phosphatase 1; FBXO32, F-box protein 32; BTC, betacellulin; STAT3, signal transducer and activator of transcription 3; Grp94, glucose-regulated protein 94; YB-1, Y-box Binding protein; HGF, hepatic growth factor; TNF-α, tumor necrosis factor alpha; EMT, epithelial-to-mesenchymal transition; MVP, major vault protein; STAT3, signal transducer and activator of transcription; HGF, hepatocyte growth factor; Hh, hedgehog; Ral, Ras-like; RalGEF, Ras-like 2 guanine nucleotide exchange factor; PRC1, protein required for cytokinesis 1; BRAF, V-raf murine sarcoma viral oncogene homolog B; V600E, valine to glutamate substitution at codon 600; RhoGEFs, Rho guanine nucleotide exchange factors; TNFR, tumor necrosis factor receptor; TRAF, tumor necrosis factor receptor–associated factors; TINCR, terminal differentiation-induced non-coding RNA; RTKs, receptor tyrosine kinases; NPM, nucleophosmin; EML4, echinoderm microtubule associated protein like 4; PD-L1, programmed cell death-ligand 1; PD-1, programmed cell death-1; SLC34A2, solute carrier family 34 Member 2; NF-κ B, nuclear factor-kappa B; GSDMD, gasdermin D; DCs, dendritic cells; TLR4, toll-like receptor 4; RAGE, receptor for advanced glycation end products; RAC1, Rac family small GTPase 1; CGAS, cyclic GMP-AMP synthase; STING1, signal transducer stimulator of IFN response cGAMP interactor 1; CXCL10, CX-C motif chemokine ligand 10; IL-1β, interleukin-1 beta; UPR, unfolded protein response; eIF2α, eukaryotic translation initiation factor 2α ; PKR, protein kinase RNA; PERK, Protein Kinase RNA-activated-like ER Kinase; IRE1, inositol-requiring transmembrane kinase/endonuclease; ATE6, activating transcription factor 6; IL-12, interleukin-12; EnaV, enapotamab vedotin; AF, auranofin; TrxR, thioredoxin reductase 1; KRASmut, KRAS-mutant; NSq-NSCLC, non-squamous NSCLC; RT, radiation therapy; CTLA-4, cytotoxic T lymphocyte-associated antigen-4; IDO, indoleamine 2.3-dioxygenase.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Rudin C, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. (2021) 7:3. doi: 10.1038/s41572-020-00235-0
3. Rami-Porta R, Bolejack V, Giroux D, Chansky K, Crowley J, Asamura H, et al. The IASLC lung cancer staging project: the new database to inform the eighth edition of the TNM classification of lung cancer. J Thorac Oncol. (2014) 9:1618–24. doi: 10.1097/JTO.0000000000000334
4. Flieswasser T, Van Loenhout J, Freire Boullosa L, Van den Eynde A, De Waele J, Van Audenaerde J, et al. Clinically relevant chemotherapeutics have the ability to induce immunogenic cell death in non-small cell lung cancer. Cells. (2020) 9:1474. doi: 10.3390/cells9061474
5. Tan A, Tan D. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J Clin Oncol. (2022) 40:611–25. doi: 10.1200/JCO.21.01626
6. Duma N, Santana-Davila R, Molina J. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. (2019) 94:1623–40. doi: 10.1016/j.mayocp.2019.01.013
7. Ruiz-Cordero R, Devine W. Targeted therapy and checkpoint immunotherapy in lung cancer. Surg Pathol Clin. (2020) 13:17–33. doi: 10.1016/j.path.2019.11.002
8. Galluzzi L, Vitale I, Aaronson S, Abrams J, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
9. Garg A, Galluzzi L, Apetoh L, Baert T, Birge R, Bravo-San Pedro J, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. (2015) 6:588. doi: 10.3389/fimmu.2015.00588
10. Alzeibak R, Mishchenko T, Shilyagina N, Balalaeva I, Vedunova M, Krysko D. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer. (2021) 9:e001926. doi: 10.1136/jitc-2020-001926
11. Duan X, Chan C, Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem Int Ed Engl. (2019) 58:670–80. doi: 10.1002/anie.201804882
12. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. (2020) 11:1013. doi: 10.1038/s41419-020-03221-2
13. Krysko D, Garg A, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. (2012) 12:860–75. doi: 10.1038/nrc3380
14. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
15. Passiglia F, Commendatore O, Vitali M, Conca R. Immunotherapy in non-small-cell lung cancer: a bridge between research and clinical practice. Future Oncol. (2018) 14:41–60. doi: 10.2217/fon-2018-0098
16. To K, Fong W, Cho W. Immunotherapy in treating EGFR-mutant lung cancer: current challenges and new strategies. Front Oncol. (2021) 11:635007. doi: 10.3389/fonc.2021.635007
17. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. (2012) 12:553–63. doi: 10.1038/nrc3309
18. Cho J, Chen L, Sangji N, Okabe T, Yonesaka K, Francis J, et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. (2013) 73:6770–9. doi: 10.1158/0008-5472.CAN-13-1145
19. Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: perspectives for individualized treatment strategy. Cancer Sci. (2016) 107:1179–86. doi: 10.1111/cas.12996
20. Pacini L, Jenks A, Vyse S, Wilding C, Arthur A, Huang P. Tackling drug resistance in EGFR exon 20 insertion mutant lung cancer. Pharmacogenomics Pers Med. (2021) 14:301–17. doi: 10.2147/PGPM.S242045
21. Lynch T, Bell D, Sordella R, Gurubhagavatula S, Okimoto R, Brannigan B, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
22. Takeuchi K, Ito F. EGF receptor in relation to tumor development: molecular basis of responsiveness of cancer cells to EGFR-targeting tyrosine kinase inhibitors. FEBS J. (2010) 277:316–26. doi: 10.1111/j.1742-4658.2009.07450.x
23. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. (2014) 79:34–74. doi: 10.1016/j.phrs.2013.11.002
24. Rothenberg S, Concannon K, Cullen S, Boulay G, Turke A, Faber A, et al. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. Elife. (2015) 4:e06132. doi: 10.7554/eLife.06132
25. Bivona T, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. (2011) 471:523–6. doi: 10.1038/nature09870
26. Karachaliou N, Gimenez-Capitan A, Drozdowskyj A, Viteri S, Moran T, Carcereny E, et al. ROR1 as a novel therapeutic target for EGFR-mutant non-small-cell lung cancer patients with the EGFR T790M mutation. Transl Lung Cancer Res. (2014) 3:122–30.
27. Kinehara Y, Nagatomo I, Koyama S, Ito D, Nojima S, Kurebayashi R, et al. Semaphorin 7A promotes EGFR-TKI resistance in EGFR mutant lung adenocarcinoma cells. JCI Insight. (2018) 3:e123093. doi: 10.1172/jci.insight.123093
28. Zhang N, Liao Y, Lv W, Zhu S, Qiu Y, Chen N, et al. FBXO32 targets PHPT1 for ubiquitination to regulate the growth of EGFR mutant lung cancer. Cell Oncol. (2022) 45:293–307. doi: 10.1007/s13402-022-00669-6
29. Chava S, Bugide S, Zhang X, Gupta R, Wajapeyee N. Betacellulin promotes tumor development and EGFR mutant lung cancer growth by stimulating the EGFR pathway and suppressing apoptosis. iScience. (2022) 25:104211. doi: 10.1016/j.isci.2022.104211
30. Niu M, Xu J, Liu Y, Li Y, He T, Ding L, et al. FBXL2 counteracts Grp94 to destabilize EGFR and inhibit EGFR-driven NSCLC growth. Nat Commun. (2021) 12:5919. doi: 10.1038/s41467-021-26222-x
31. Tsai M, Chang T, Wu S, Yang H, Hsu Y, Yang P, et al. EGFR-L858R mutant enhances lung adenocarcinoma cell invasive ability and promotes malignant pleural effusion formation through activation of the CXCL12-CXCR4 pathway. Sci Rep. (2015) 5:13574. doi: 10.1038/srep13574
32. Feng J, Wei X, Li C, Guo M, Peng M, Song Q, et al. [Mechanism of EGFR over-expression and mutations leading to biological characteristics changes of human lung adenocarcinoma cells through CXCR4/CXCL12 signaling pathway]. Zhongguo Fei Ai Za Zhi. (2018) 21:503–12.
33. Li H, Tong F, Meng R, Peng L, Wang J, Zhang R, et al. E2F1-mediated repression of WNT5A expression promotes brain metastasis dependent on the ERK1/2 pathway in EGFR-mutant non-small cell lung cancer. Cell Mol Life Sci. (2021) 78:2877–91. doi: 10.1007/s00018-020-03678-6
34. Lou L, Wang J, Lv F, Wang G, Li Y, Xing L, et al. Y-box binding protein 1 (YB-1) promotes gefitinib resistance in lung adenocarcinoma cells by activating AKT signaling and epithelial-mesenchymal transition through targeting major vault protein (MVP). Cell Oncol. (2021) 44:109–33. doi: 10.1007/s13402-020-00556-y
35. Cao W, Liu Y, Zhang R, Zhang B, Wang T, Zhu X, et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal pathway in gefitinib-resistant lung cancer cells. Sci Rep. (2015) 5:8477. doi: 10.1038/srep08477
36. Han Y, Lee T, He Y, Raman R, Irizarry A, Martin M, et al. The regulation of CD73 in non-small cell lung cancer. Eur J Cancer. (2022) 170:91–102. doi: 10.1016/j.ejca.2022.04.025
37. Griesing S, Liao B, Yang J. CD73 Is regulated by the EGFR-ERK signaling pathway in non-small cell lung cancer. Anticancer Res. (2021) 41:1231–42. doi: 10.21873/anticanres.14880
38. Le X, Negrao M, Reuben A, Federico L, Diao L, McGrail D, et al. Characterization of the immune landscape of EGFR-mutant NSCLC identifies CD73/adenosine pathway as a potential therapeutic target. J Thorac Oncol. (2021) 16:583–600. doi: 10.1016/j.jtho.2020.12.010
39. Passarelli A, Aieta M, Sgambato A, Gridelli C. Targeting immunometabolism mediated by CD73 pathway in EGFR-mutated non-small cell lung cancer: a new hope for overcoming immune resistance. Front Immunol. (2020) 11:1479. doi: 10.3389/fimmu.2020.01479
40. Chen X, Gao A, Zhang F, Yang Z, Wang S, Fang Y, et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics. (2021) 11:3392–416. doi: 10.7150/thno.52435
42. Murugan A, Grieco M, Tsuchida N. RAS mutations in human cancers: roles in precision medicine. Semin Cancer Biol. (2019) 59:23–35. doi: 10.1016/j.semcancer.2019.06.007
43. Cox A, Fesik S, Kimmelman A, Luo J, Der C. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. (2014) 13:828–51. doi: 10.1038/nrd4389
44. Ricciuti B, Leonardi G, Metro G, Grignani F, Paglialunga L, Bellezza G, et al. Targeting the KRAS variant for treatment of non-small cell lung cancer: potential therapeutic applications. Expert Rev Respir Med. (2016) 10:53–68. doi: 10.1586/17476348.2016.1115349
45. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3:459–65. doi: 10.1038/nrc1097
46. Ostrem J, Peters U, Sos M, Wells J, Shokat K. MK-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. (2013) 503:548–51. doi: 10.1038/nature12796
47. Kanno E, Kawasaki O, Takahashi K, Takano K, Endo T. DA-Raf, a dominant-negative antagonist of the Ras-ERK pathway, is a putative tumor suppressor. Exp Cell Res. (2018) 362:111–20. doi: 10.1016/j.yexcr.2017.11.008
48. Costa-Machado L, Martín-Hernández R, Sanchez-Luengo M, Hess K, Vales-Villamarin C, Barradas M, et al. Sirt1 protects from K-Ras-driven lung carcinogenesis. EMBO Rep. (2018) 19:e43879. doi: 10.15252/embr.201643879
49. Schmidt M, Hobbing K, Donninger H, Clark G. RASSF1A deficiency enhances RAS-driven lung tumorigenesis. Cancer Res. (2018) 78:2614–23. doi: 10.1158/0008-5472.CAN-17-2466
50. Kharbanda A, Walter D, Gudiel A, Schek N, Feldser D, Witze E. Blocking EGFR palmitoylation suppresses PI3K signaling and mutant KRAS lung tumorigenesis. Sci Signal. (2020) 13:eaax2364. doi: 10.1126/scisignal.aax2364
51. Kadry Y, Lee J, Witze E. Regulation of EGFR signalling by palmitoylation and its role in tumorigenesis. Open Biol. (2021) 11:210033. doi: 10.1098/rsob.210033
52. Sieber B, Lu F, Stribbling S, Grieve A, Ryan A, Freeman M. iRhom2 regulates ERBB signalling to promote KRAS-driven tumour growth of lung cancer cells. J Cell Sci. (2022) 135:jcs259949. doi: 10.1242/jcs.259949
53. Lee Y, Lee J, Song S, Kim D, Lee J, Chi X, et al. K-ras-activated cells can develop into lung tumors when runx3-mediated tumor suppressor pathways are abrogated. Mol Cells. (2020) 43:889–97.
54. Hanahan D, Coussens L. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
55. Mohrherr J, Haber M, Breitenecker K, Aigner P, Moritsch S, Voronin V, et al. JAK-STAT inhibition impairs K-RAS-driven lung adenocarcinoma progression. Int J Cancer. (2019) 145:3376–88. doi: 10.1002/ijc.32624
56. Caetano M, Zhang H, Cumpian A, Gong L, Unver N, Ostrin E, et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-mutant lung cancer. Cancer Res. (2016) 76:3189–99. doi: 10.1158/0008-5472.CAN-15-2840
57. Clowers M, Moghaddam S. Cell type-specific roles of STAT3 signaling in the pathogenesis and progression of K-ras mutant lung adenocarcinoma. Cancers. (2022) 14:1785. doi: 10.3390/cancers14071785
58. Caetano M, Hassane M, Van H, Bugarin E, Cumpian A, McDowell C, et al. Sex specific function of epithelial STAT3 signaling in pathogenesis of K-ras mutant lung cancer. Nat Commun. (2018) 9:4589. doi: 10.1038/s41467-018-07042-y
59. Moghaddam S, Li H, Cho S, Dishop M, Wistuba I, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. (2009) 40:443–53. doi: 10.1165/rcmb.2008-0198OC
60. Bassères D, Ebbs A, Cogswell P, Baldwin AS. IKK is a therapeutic target in KRAS-Induced lung cancer with disrupted p53 activity. Genes Cancer. (2014) 5:41–55. doi: 10.18632/genesandcancer.5
61. Unni A, Harbourne B, Oh M, Wild S, Ferrarone J, Lockwood W, et al. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife. (2018) 7:e33718. doi: 10.7554/eLife.33718
62. Wang L, Zhao Y, Xiong Y, Wang W, Fei Y, Tan C, et al. K-ras mutation promotes ionizing radiation-induced invasion and migration of lung cancer in part via the Cathepsin L/CUX1 pathway. Exp Cell Res. (2018) 362:424–35. doi: 10.1016/j.yexcr.2017.12.006
63. Hsu P, Miao J, Huang Z, Yang Y, Xu Z, You J, et al. Inhibition of yes-associated protein suppresses brain metastasis of human lung adenocarcinoma in a murine model. J Cell Mol Med. (2018) 22:3073–85. doi: 10.1111/jcmm.13582
64. Leicht D, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, et al. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. (2007) 1773:1196–212. doi: 10.1016/j.bbamcr.2007.05.001
65. Wan P, Garnett M, Roe S, Lee S, Niculescu-Duvaz D, Good V, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. (2004) 116:855–67. doi: 10.1016/S0092-8674(04)00215-6
66. Pratilas C, Hanrahan A, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. (2008) 68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223
67. Chambard J, Lefloch R, Pouysségur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. (2007) 1773:1299–310. doi: 10.1016/j.bbamcr.2006.11.010
68. Hall R, Kudchadkar RR. BRAF mutations: signaling, epidemiology, and clinical experience in multiple malignancies. Cancer Control. (2014) 21:221–30. doi: 10.1177/107327481402100307
69. Li Y, Ye Z, Chen S, Pan Z, Zhou Q, Li Y, et al. ARHGEF19 interacts with BRAF to activate MAPK signaling during the tumorigenesis of non-small cell lung cancer. Int J Cancer. (2018) 142:1379–91. doi: 10.1002/ijc.31169
70. Wang Q, Gao G, Zhang T, Yao K, Chen H, Park M, et al. TRAF1 is critical for regulating the BRAF/MEK/ERK pathway in non-small cell lung carcinogenesis. Cancer Res. (2018) 78:3982–94. doi: 10.1158/0008-5472.CAN-18-0429
71. Zanucco E, El-Nikhely N, Götz R, Weidmann K, Pfeiffer V, Savai R, et al. Elimination of B-RAF in oncogenic C-RAF-expressing alveolar epithelial type II cells reduces MAPK signal intensity and lung tumor growth. J Biol Chem. (2014) 289:26804–16. doi: 10.1074/jbc.M114.558999
72. Zhu Z, He JK. TINCR facilitates non-small cell lung cancer progression through BRAF-activated MAPK pathway. Biochem Biophys Res Commun. (2018) 497:971–7. doi: 10.1016/j.bbrc.2018.02.059
73. Rustgi AK. BRAF: a driver of the serrated pathway in colon cancer. Cancer Cell. (2013) 24:1–2. doi: 10.1016/j.ccr.2013.06.008
74. Lin L, Asthana S, Chan E, Bandyopadhyay S, Martins M, Olivas V, et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci USA. (2014) 111:E748–57. doi: 10.1073/pnas.1320956111
75. Kotani H, Adachi Y, Kitai H, Tomida S, Bando H, Faber A, et al. Distinct dependencies on receptor tyrosine kinases in the regulation of MAPK signaling between BRAF V600E and non-V600E mutant lung cancers. Oncogene. (2018) 37:1775–87. doi: 10.1038/s41388-017-0035-9
76. Trejo C, Green S, Marsh V, Collisson E, Iezza G, Phillips W, et al. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res. (2013) 73:6448–61. doi: 10.1158/0008-5472.CAN-13-0681
77. van Veen J, Scherzer M, Boshuizen J, Chu M, Liu A, Landman A, et al. Mutationally-activated PI3’-kinase-α promotes de-differentiation of lung tumors initiated by the BRAF(V600E) oncoprotein kinase. Elife. (2019) 8:e43668. doi: 10.7554/eLife.43668
78. González-Sánchez E, Martín-Caballero J, Flores J, Hernández-Losa J, Cortés J, Mares R, et al. Lkb1 loss promotes tumor progression of BRAF(V600E)-induced lung adenomas. PLoS One. (2013) 8:e66933. doi: 10.1371/journal.pone.0066933
79. Duyster J, Bai R, Morris S. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene. (2001) 20:5623–37. doi: 10.1038/sj.onc.1204594
80. Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. (1994) 263:1281–4. doi: 10.1126/science.8122112
81. Toyokawa G, Seto T. ALK inhibitors: what is the best way to treat patients with ALK+ non-small-cell lung cancer? Clin Lung Cancer. (2014) 15:313–9. doi: 10.1016/j.cllc.2014.05.001
82. Shaw A, Kim D, Nakagawa K, Seto T, Crinó L, Ahn M, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. (2013) 368:2385–94. doi: 10.1056/NEJMoa1214886
83. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. (2015) 16:e342–51. doi: 10.1016/S1470-2045(15)00077-7
84. Shaw A, Yeap B, Mino-Kenudson M, Digumarthy S, Costa D, Heist R, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. (2009) 27:4247–53. doi: 10.1200/JCO.2009.22.6993
85. Tsao A, Scagliotti G, Bunn P Jr., Carbone D, Warren G, Bai C, et al. Scientific advances in lung cancer 2015. J Thorac Oncol. (2016) 11:613–38. doi: 10.1016/j.jtho.2016.03.012
86. Normant E, Paez G, West K, Lim A, Slocum K, Tunkey C, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. (2011) 30:2581–6. doi: 10.1038/onc.2010.625
87. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. (2015) 21:4014–21. doi: 10.1158/1078-0432.CCR-15-0016
88. Shen J, Meng Y, Wang K, Gao M, Du J, Wang J, et al. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell Signal. (2022) 92:110264. doi: 10.1016/j.cellsig.2022.110264
89. Davies K, Le A, Theodoro M, Skokan M, Aisner D, Berge E, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. (2012) 18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550
90. Alrifai D, Forster M, Janes S. Emerging resistance pathways in lung cancer: what has ROS-1 taught us? Transl Lung Cancer Res. (2018) 7(Suppl. 1):S9–12. doi: 10.21037/tlcr.2017.11.13
91. Bergethon K, Shaw A, Ou S, Katayama R, Lovly C, McDonald N, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. (2012) 30:863–70. doi: 10.1200/JCO.2011.35.6345
92. Yoshida A, Tsuta K, Wakai S, Arai Y, Asamura H, Shibata T, et al. Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol. (2014) 27:711–20. doi: 10.1038/modpathol.2013.192
93. Rodríguez-Antolín C, Rosas-Alonso R, Cruz P, Higuera O, Sánchez-Cabrero D, Esteban-Rodríguez I, et al. Novel SLC12A2-ROS1 fusion in non-small cell lung cancer with a significant response to crizotinib: the importance of choosing the appropriate next-generation sequencing assay. Oncologist. (2021) 26:e908–12. doi: 10.1002/onco.13745
94. Gou W, Zhou X, Liu Z, Wang L, Shen J, Xu X, et al. CD74-ROS1 G2032R mutation transcriptionally up-regulates Twist1 in non-small cell lung cancer cells leading to increased migration, invasion, and resistance to crizotinib. Cancer Lett. (2018) 422:19–28. doi: 10.1016/j.canlet.2018.02.032
95. Cai L, Duan J, Qian L, Wang Z, Wang S, Li S, et al. ROS1 fusion mediates immunogenicity by upregulation of PD-L1 after the activation of ROS1-SHP2 signaling pathway in non-small cell lung cancer. Front Immunol. (2020) 11:527750. doi: 10.3389/fimmu.2020.527750
96. Liu Z, Zhao K, Wei S, Liu C, Zhou J, Gou Q, et al. ROS1-fusion protein induces PD-L1 expression via MEK-ERK activation in non-small cell lung cancer. Oncoimmunology. (2020) 9:1758003. doi: 10.1080/2162402X.2020.1758003
97. Blaquier J, Recondo G. Non-small-cell lung cancer: how to manage MET exon 14 skipping mutant disease. Drugs Context. (2022) 11:2022–2–2. doi: 10.7573/dic.2022-2-2
98. Terlecka P, Krawczyk P, Grenda A, Milanowski J. MET gene dysregulation as a promising therapeutic target in lung cancer-a review. J Pers Med. (2021) 11:1370. doi: 10.3390/jpm11121370
99. Tong J, Yeung S, Chan A, Chung L, Chau S, Lung R, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. (2016) 22:3048–56. doi: 10.1158/1078-0432.CCR-15-2061
100. Frampton G, Ali S, Rosenzweig M, Chmielecki J, Lu X, Bauer T, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. (2015) 5:850–9. doi: 10.1158/1538-7445.AM2015-1118
101. Rosell R, Chaib I, Santarpia M. Targeting MET amplification in EGFR-mutant non-small-cell lung cancer. Lancet Respir Med. (2020) 8:1068–70. doi: 10.1016/S2213-2600(20)30171-5
102. Song Y, Li G, Ju K, Ran W, Zhao H, Liu X, et al. Mesenchymal-epithelial transition exon 14 skipping mutation and amplification in 5,008 patients with lung cancer. Front Oncol. (2021) 11:755031. doi: 10.3389/fonc.2021.755031
103. Noonan S, Berry L, Lu X, Gao D, Barón A, Chesnut P, et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J Thorac Oncol. (2016) 11:1293–304. doi: 10.1016/j.jtho.2016.04.033
104. Jackman D, Pao W, Riely G, Engelman J, Kris M, Jänne P, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. (2010) 28:357–60. doi: 10.1200/JCO.2009.24.7049
105. Wu S, Shih J. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. (2018) 17:38. doi: 10.1186/s12943-018-0777-1
106. Lin Y, Higashisaka K, Shintani T, Maki A, Hanamuro S, Haga Y, et al. Progesterone receptor membrane component 1 leads to erlotinib resistance, initiating crosstalk of Wnt/β-catenin and NF-κB pathways, in lung adenocarcinoma cells. Sci Rep. (2020) 10:4748. doi: 10.1038/s41598-020-61727-3
107. Frederick B, Helfrich B, Coldren C, Zheng D, Chan D, Bunn P Jr., et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. (2007) 6:1683–91. doi: 10.1158/1535-7163.MCT-07-0138
108. Zhong W, Zhou Q, Wu Y. The resistance mechanisms and treatment strategies for EGFR-mutant advanced non-small-cell lung cancer. Oncotarget. (2017) 8:71358–70. doi: 10.18632/oncotarget.20311
109. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. (2008) 68:9479–87. doi: 10.1158/0008-5472.CAN-08-1643
110. Della Corte C, Malapelle U, Vigliar E, Pepe F, Troncone G, Ciaramella V, et al. Efficacy of continuous EGFR-inhibition and role of Hedgehog in EGFR acquired resistance in human lung cancer cells with activating mutation of EGFR. Oncotarget. (2017) 8:23020–32. doi: 10.18632/oncotarget.15479
111. F Smit E, Dooms C, Raskin J, Nadal E, Tho L, Le X. INSIGHT 2: a phase II study of tepotinib plus osimertinib in MET-amplified NSCLC and first-line osimertinib resistance. Future Oncol. (2022) 18:1039–54. doi: 10.2217/fon-2021-1406
112. Adderley H, Blackhall F, Lindsay C. KRAS-mutant non-small cell lung cancer: converging small molecules and immune checkpoint inhibition. EBioMedicine. (2019) 41:711–6. doi: 10.1016/j.ebiom.2019.02.049
113. Adjei A, Mauer A, Bruzek L, Marks R, Hillman S, Geyer S, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. (2003) 21:1760–6. doi: 10.1200/JCO.2003.09.075
114. Blumenschein G Jr., Smit E, Planchard D, Kim D, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Ann Oncol. (2015) 26:894–901. doi: 10.1093/annonc/mdv072
115. Crystal A, Shaw A, Sequist L, Friboulet L, Niederst M, Lockerman E, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. (2014) 346:1480–6. doi: 10.1126/science.1254721
116. Shi R, Filho S, Li M, Fares A, Weiss J, Pham N, et al. BRAF V600E mutation and MET amplification as resistance pathways of the second-generation anaplastic lymphoma kinase (ALK) inhibitor alectinib in lung cancer. Lung Cancer. (2020) 146:78–85. doi: 10.1016/j.lungcan.2020.05.018
117. Katayama R, Shaw A, Khan T, Mino-Kenudson M, Solomon B, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. (2012) 4:120ra17. doi: 10.1126/scitranslmed.3003316
118. Park K, Tan E, O’Byrne K, Zhang L, Boyer M, Mok T, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. (2016) 17:577–89. doi: 10.1016/S1470-2045(16)30033-X
119. Biswas A, Han S, Tai Y, Ma W, Coker C, Quinn S, et al. Targeting S100A9-ALDH1A1-retinoic acid signaling to suppress brain relapse in EGFR-mutant lung cancer. Cancer Discov. (2022) 12:1002–21. doi: 10.1158/2159-8290.CD-21-0910
120. Wu Y, Herbst R, Mann H, Rukazenkov Y, Marotti M, Tsuboi M. ADAURA: phase III, double-blind, randomized study of osimertinib versus placebo in EGFR mutation-positive early-stage NSCLC after complete surgical resection. Clin Lung Cancer. (2018) 19:e533–6. doi: 10.1016/j.cllc.2018.04.004
121. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez A, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. (2020) 8:e000337. doi: 10.1136/jitc-2019-000337corr1
122. Ahmed A, Tait S. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
123. Ruan H, Leibowitz B, Zhang L, Yu J. Immunogenic cell death in colon cancer prevention and therapy. Mol Carcinog. (2020) 59:783–93. doi: 10.1002/mc.23183
124. Petrazzuolo A, Perez-Lanzon M, Liu P, Maiuri M, Kroemer G. Crizotinib and ceritinib trigger immunogenic cell death via on-target effects. Oncoimmunology. (2021) 10:1973197. doi: 10.1080/2162402X.2021.1973197
125. Scaffidi P, Misteli T, Bianchi M. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
126. Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. (2016) 128:3113–24. doi: 10.1182/blood-2016-08-731737
127. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
128. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
129. Wu X, Chen Y, Gong C, Pei D. The role of high-mobility group protein box 1 in lung cancer. J Cell Biochem. (2018) 119:6354–65. doi: 10.1002/jcb.26837
130. Wu L, Yang L. The function and mechanism of HMGB1 in lung cancer and its potential therapeutic implications. Oncol Lett. (2018) 15:6799–805.
131. Solari J, Filippi-Chiela E, Pilar E, Nunes V, Gonzalez E, Figueiro F, et al. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer. (2020) 20:474. doi: 10.1186/s12885-020-06964-5
132. Lagiedo M, Sikora J, Kaczmarek M. Damage-associated molecular patterns in the course of lung cancer–A review. Scand J Immunol. (2015) 82:95–101. doi: 10.1111/sji.12308
133. Anderson C, Macleod K. Autophagy and cancer cell metabolism. Int Rev Cell Mol Biol. (2019) 347:145–90. doi: 10.1016/bs.ircmb.2019.06.002
134. Follo C, Cheng Y, Richards W, Bueno R, Broaddus V. Autophagy facilitates the release of immunogenic signals following chemotherapy in 3D models of mesothelioma. Mol Carcinog. (2019) 58:1754–69. doi: 10.1002/mc.23050
135. Kronlage M, Song J, Sorokin L, Isfort K, Schwerdtle T, Leipziger J, et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci Signal. (2010) 3:ra55. doi: 10.1126/scisignal.2000588
136. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. (2010) 40:280–93. doi: 10.1016/j.molcel.2010.09.023
137. Swanson K, Deng M, Ting J. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
138. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
139. Wang H, Lee H, Guo J, Chen S, Liao Z, Huang K, et al. Calreticulin promotes tumor lymphocyte infiltration and enhances the antitumor effects of immunotherapy by up-regulating the endothelial expression of adhesion molecules. Int J Cancer. (2012) 130:2892–902. doi: 10.1002/ijc.26339
140. Thul P, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. (2018) 27:233–44. doi: 10.1002/pro.3307
141. Inoue H, Tsutsumi H, Tanaka K, Iwama E, Shiraishi Y, Hirayama A, et al. Increased plasma levels of damage-associated molecular patterns during systemic anticancer therapy in patients with advanced lung cancer. Transl Lung Cancer Res. (2021) 10:2475–86. doi: 10.21037/tlcr-21-92
142. Rodriguez-Ruiz M, Buque A, Hensler M, Chen J, Bloy N, Petroni G, et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. (2019) 8:e1655964. doi: 10.1080/2162402X.2019.1655964
143. Khodarev N. Intracellular RNA sensing in mammalian cells: role in stress response and cancer therapies. Int Rev Cell Mol Biol. (2019) 344:31–89. doi: 10.1016/bs.ircmb.2018.08.005
144. Yamazaki T, Kirchmair A, Sato A, Buque A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. (2020) 21:1160–71. doi: 10.1038/s41590-020-0751-0
145. Hopfner K, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
146. McLaughlin M, Patin E, Pedersen M, Wilkins A, Dillon M, Melcher A, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1
147. Muller E, Christopoulos P, Halder S, Lunde A, Beraki K, Speth M, et al. Toll-like receptor ligands and interferon-gamma synergize for induction of antitumor M1 macrophages. Front Immunol. (2017) 8:1383. doi: 10.3389/fimmu.2017.01383
148. Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale A, Glass D, et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. (2018) 14:e1006985. doi: 10.1371/journal.ppat.1006985
149. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot D, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
150. Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi M, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. (2012) 18:250–9. doi: 10.2119/molmed.2011.00389
151. Rufo N, Garg A, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer. (2017) 3:643–58. doi: 10.1016/j.trecan.2017.07.002
152. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. (2011) 13:184–90. doi: 10.1038/ncb0311-184
154. Cubillos-Ruiz J, Mohamed E, Rodriguez P. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer. (2017) 5:5. doi: 10.1186/s40425-016-0203-4
155. Garg A, Krysko D, Verfaillie T, Kaczmarek A, Ferreira G, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. (2012) 31:1062–79. doi: 10.1038/emboj.2011.497
156. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. (2016) 76:1746–56. doi: 10.1158/0008-5472.CAN-15-1142
157. Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez A, Pedrozo Z, et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell Biol. (2012) 44:16–20. doi: 10.1016/j.biocel.2011.10.012
158. Deutsch E, Chargari C, Galluzzi L, Kroemer G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. (2019) 20:e452–63. doi: 10.1016/S1470-2045(19)30171-8
159. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. (2020) 17:725–41. doi: 10.1038/s41571-020-0413-z
160. Garrido G, Rabasa A, Sanchez B, Lopez M, Blanco R, Lopez A, et al. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J Immunol. (2011) 187:4954–66. doi: 10.4049/jimmunol.1003477
161. Drewry D, Wells C, Andrews D, Angell R, Al-Ali H, Axtman A, et al. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One. (2017) 12:e0181585. doi: 10.1371/journal.pone.0181585
162. Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nature Commun. (2019) 10:1486. doi: 10.1038/s41467-019-09838-y
163. Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. (2014) 5:5712. doi: 10.1038/ncomms6712
164. Lotsberg M, Wnuk-Lipinska K, Terry S, Tan T, Lu N, Trachsel-Moncho L, et al. AXL targeting abrogates autophagic flux and induces immunogenic cell death in drug-resistant cancer cells. J Thorac Oncol. (2020) 15:973–99. doi: 10.1016/j.jtho.2020.01.015
165. Boshuizen J, Pencheva N, Krijgsman O, Altimari D, Castro P, de Bruijn B, et al. Cooperative targeting of immunotherapy-resistant melanoma and lung cancer by an AXL-targeting antibody-drug conjugate and immune checkpoint blockade. Cancer Res. (2021) 81:1775–87. doi: 10.1158/0008-5472.CAN-20-0434
166. Gheghiani L, Wang L, Zhang Y, Moore X, Zhang J, Smith S, et al. PLK1 induces chromosomal instability and overrides cell-cycle checkpoints to drive tumorigenesis. Cancer Res. (2021) 81:1293–307. doi: 10.1158/0008-5472.CAN-20-1377
167. Zhou J, Yang Q, Lu L, Tuo Z, Shou Z, Cheng J. PLK1 Inhibition induces immunogenic cell death and enhances immunity against NSCLC. Int J Med Sci. (2021) 18:3516–25. doi: 10.7150/ijms.60135
168. Saei A, Gullberg H, Sabatier P, Beusch C, Johansson K, Lundgren B, et al. Comprehensive chemical proteomics for target deconvolution of the redox active drug auranofin. Redox Biol. (2020) 32:101491. doi: 10.1016/j.redox.2020.101491
169. Freire Boullosa L, Van Loenhout J, Flieswasser T, De Waele J, Hermans C, Lambrechts H, et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. (2021) 42:101949. doi: 10.1016/j.redox.2021.101949
170. Nam G, Kwon M, Jung H, Ko E, Kim S, Choi Y, et al. Statin-mediated inhibition of RAS prenylation activates ER stress to enhance the immunogenicity of KRAS mutant cancer. J Immunother Cancer. (2021) 9:e002474. doi: 10.1136/jitc-2021-002474
171. Garg A, Krysko D, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. (2012) 61:215–21. doi: 10.1007/s00262-011-1184-2
172. Kepp O, Zitvogel L, Kroemer G. Lurbinectedin: an FDA-approved inducer of immunogenic cell death for the treatment of small-cell lung cancer. Oncoimmunology. (2020) 9:1795995. doi: 10.1080/2162402X.2020.1795995
173. Bezu L, Gomes-de-Silva L, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. (2015) 6:187. doi: 10.3389/fimmu.2015.00187
174. Rodriguez-Ruiz M, Vitale I, Harrington K, Melero I, Galluzzi L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol. (2020) 21:120–34. doi: 10.1038/s41590-019-0561-4
175. Krombach J, Hennel R, Brix N, Orth M, Schoetz U, Ernst A, et al. Priming anti-tumor immunity by radiotherapy: dying tumor cell-derived DAMPs trigger endothelial cell activation and recruitment of myeloid cells. Oncoimmunology. (2019) 8:e1523097. doi: 10.1080/2162402X.2018.1523097
176. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. (2016) 44:343–54. doi: 10.1016/j.immuni.2015.11.024
177. Mathew M, Enzler T, Shu C, Rizvi N. Combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol Ther. (2018) 186:130–7. doi: 10.1016/j.pharmthera.2018.01.003
178. Sumiyoshi I, Okabe T, Togo S, Takagi H, Motomura H, Ochi Y, et al. High lymphocyte population-related predictive factors for a long-term response in non-small cell lung cancer patients treated with pemetrexed: a retrospective observational study. J Transl Med. (2021) 19:92. doi: 10.1186/s12967-021-02761-1
179. Wang D, Cong J, Fu B, Zheng X, Sun R, Tian Z, et al. Immunogenic chemotherapy effectively inhibits KRAS-Driven lung cancer. Cancer Lett. (2020) 492:31–43. doi: 10.1016/j.canlet.2020.07.043
180. Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer. (2019) 7:42. doi: 10.1186/s40425-019-0511-6
181. Furukawa R, Inoue H, Yoneshima Y, Tsutsumi H, Iwama E, Ikematsu Y, et al. Cytotoxic chemotherapeutic agents and the EGFR-TKI osimertinib induce calreticulin exposure in non-small cell lung cancer. Lung Cancer. (2021) 155:144–50. doi: 10.1016/j.lungcan.2021.03.018
182. Wang Y, Shen N, Wang Y, Li M, Zhang W, Fan L, et al. Cisplatin nanoparticles boost abscopal effect of radiation plus anti-PD1 therapy. Biomater Sci. (2021) 9:3019–27. doi: 10.1039/D1BM00112D
183. Ma S, Song W, Xu Y, Si X, Lv S, Zhang Y, et al. Rationally designed polymer conjugate for tumor-specific amplification of oxidative stress and boosting antitumor immunity. Nano Lett. (2020) 20:2514–21. doi: 10.1021/acs.nanolett.9b05265
184. Strasser A, Cory S, Adams J. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. (2011) 30:3667–83. doi: 10.1038/emboj.2011.307
185. Akamatsu H, Teraoka S, Hayashi H, Fujimoto D, Hayata A, Haratani K, et al. Pembrolizumab plus amrubicin in patients with relapsed SCLC: multi-institutional, single-arm phase 2 study. JTO Clin Res Rep. (2021) 2:100184. doi: 10.1016/j.jtocrr.2021.100184
186. Altorki N, McGraw T, Borczuk A, Saxena A, Port J, Stiles B, et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: a single-centre, randomised phase 2 trial. Lancet Oncol. (2021) 22:824–35. doi: 10.1016/S1470-2045(21)00149-2
187. Sprooten J, De Wijngaert P, Vanmeerbeerk I, Martin S, Vangheluwe P, Schlenner S, et al. Necroptosis in immuno-oncology and cancer immunotherapy. Cells. (2020) 9:1823. doi: 10.3390/cells9081823
188. Chen C, Kono H, Golenbock D, Reed G, Akira S, Rock K. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. (2007) 13:851–6. doi: 10.1038/nm1603
189. Qi J, Jin F, Xu X, Du Y. Combination cancer immunotherapy of nanoparticle-based immunogenic cell death inducers and immune checkpoint inhibitors. Int J Nanomed. (2021) 16:1435–56. doi: 10.2147/IJN.S285999
190. Josefsen L, Boyle R. Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics. (2012) 2:916–66. doi: 10.7150/thno.4571
191. Pinto A, Mace Y, Drouet F, Bony E, Boidot R, Draoui N, et al. A new ER-specific photosensitizer unravels (1)O2-driven protein oxidation and inhibition of deubiquitinases as a generic mechanism for cancer PDT. Oncogene. (2016) 35:3976–85. doi: 10.1038/onc.2015.474
192. Doix B, Trempolec N, Riant O, Feron O. Low photosensitizer dose and early radiotherapy enhance antitumor immune response of photodynamic therapy-based dendritic cell vaccination. Front Oncol. (2019) 9:811. doi: 10.3389/fonc.2019.00811
193. Gomes-da-Silva L, Zhao L, Bezu L, Zhou H, Sauvat A, Liu P, et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. EMBO J. (2018) 37:e98354. doi: 10.15252/embj.201798354
194. Tatsuno K, Yamazaki T, Hanlon D, Han P, Robinson E, Sobolev O, et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis. (2019) 10:578. doi: 10.1038/s41419-019-1819-3
195. Ventura A, Vassall A, Robinson E, Filler R, Hanlon D, Meeth K, et al. Extracorporeal photochemotherapy drives monocyte-to-dendritic cell maturation to induce anticancer immunity. Cancer Res. (2018) 78:4045–58. doi: 10.1158/0008-5472.CAN-18-0171
196. Korbelik M, Zhang W, Merchant S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol Immunother. (2011) 60:1431–7. doi: 10.1007/s00262-011-1047-x
197. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. (2019) 50:181–94.e6. doi: 10.1016/j.immuni.2018.11.014
198. Xiao Y, Freeman G. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. (2015) 5:16–8. doi: 10.1158/2159-8290.CD-14-1397
199. Ferrara R, Mezquita L, Besse B. Progress in the management of advanced thoracic malignancies in 2017. J Thorac Oncol. (2018) 13:301–22. doi: 10.1016/j.jtho.2018.01.002
200. Doroshow D, Herbst R. Treatment of advanced non-small cell lung cancer in 2018. JAMA Oncol. (2018) 4:569–70. doi: 10.1001/jamaoncol.2017.5190
201. Chabanon R, Muirhead G, Krastev D, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. (2019) 129:1211–28. doi: 10.1172/JCI123319
202. Belani C, Wu Y, Chen Y, Kim J, Yang S, Zhang L, et al. Efficacy and safety of pemetrexed maintenance therapy versus best supportive care in patients from East Asia with advanced, nonsquamous non-small cell lung cancer: an exploratory subgroup analysis of a global, randomized, phase 3 clinical trial. J Thorac Oncol. (2012) 7:567–73. doi: 10.1097/JTO.0b013e31823d4f9d
203. Gridelli C, Casaluce F. Frontline immunotherapy for NSCLC: alone or not alone? Nat Rev Clin Oncol. (2018) 15:593–4. doi: 10.1038/s41571-018-0070-7
204. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
205. Socinski M, Jotte R, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
206. West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter H, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. (2019) 20:924–37. doi: 10.1016/S1470-2045(19)30167-6
207. Martinez P, Peters S, Stammers T, Soria J. Immunotherapy for the first-line treatment of patients with metastatic non-small cell lung cancer. Clin Cancer Res. (2019) 25:2691–8. doi: 10.1158/1078-0432.CCR-18-3904
208. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
209. Santamaria Nunez G, Robles C, Giraudon C, Martinez-Leal J, Compe E, Coin F, et al. Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol Cancer Ther. (2016) 15:2399–412. doi: 10.1158/1535-7163.MCT-16-0172
210. Nagano T, Tachihara M, Nishimura Y. Molecular mechanisms and targeted therapies including immunotherapy for non-small cell lung cancer. Curr Cancer Drug Targets. (2019) 19:595–630. doi: 10.2174/1568009619666181210114559
Keywords: immunogenic cell death, lung cancer, immunotherapy, damage-associated molecular patterns, genetic alteration
Citation: Xu J, Xiong Y, Xu Z, Xing H, Zhou L and Zhang X (2022) From targeted therapy to a novel way: Immunogenic cell death in lung cancer. Front. Med. 9:1102550. doi: 10.3389/fmed.2022.1102550
Received: 19 November 2022; Accepted: 12 December 2022;
Published: 23 December 2022.
Edited by:
Shuibang Wang, National Institutes of Health (NIH), United StatesReviewed by:
Salina Gairhe, National Institutes of Health (NIH), United StatesMariaenrica Tinè, University of Padua, Italy
Copyright © 2022 Xu, Xiong, Xu, Xing, Zhou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyun Zhou, ✉ MzUyNTA1NTA0QHFxLmNvbQ==; Xinyi Zhang, ✉ emhhbmd4aW55aTgwQDE2My5jb20=
†These authors share first authorship