 Dandan Li1†
Dandan Li1† Wei Liu
Wei Liu- 1Department of Ophthalmology, Tianjin TEDA Hospital, Tianjin, China
- 2Department of Ophthalmology, Lanzhou Huaxia Eye Hospital, Lanzhou, Gansu, China
- 3Department of Ophthalmology, Taihe Hospital, Hubei University of Medicine, Shiyan, Hubei, China
- 4Tianjin Key Laboratory of Retinal Functions and Diseases, Tianjin Branch of National Clinical Research Center for Ocular Disease, Eye Institute and School of Optometry, Tianjin Medical University Eye Hospital, Tianjin, China
Background: To explore the genetic defects of two families with autosomal dominant Marfan syndrome (MFS).
Methods: Two families with MFS were enrolled in this study. The detailed ocular presentations of the patients were recorded. Whole exome sequencing was performed to explore the pathogenic variants and Sanger sequencing was performed to confirm the gene mutations. Segregation analysis among the family members was made and bioinformatics analysis was performed to predict the functional impact of the mutations.
Results: The main ocular presentations of the probands were increased axial length and ectopia lentis. Using whole exome sequencing and Sanger sequencing, a novel heterozygous missense mutation (c.5060G > C, p.Cys1687Ser) and a recurrent missense mutation (c.2168A > T, p.Asp723Val) were identified within FBN1, which were co-segregated with the MFS phenotype in the families. Evolutionary conservation analysis showed that codons 723 and 1,687 were highly conserved among several species. Functional impact predictions made using several online programs suggested that the mutations were pathogenic.
Conclusion: We identified a novel and a recurrent missense mutation in FBN1 in two Chinese families with MFS using whole exome sequencing, and our bioinformatics analysis indicated that the mutations were disease-causing. Our results expand the mutation spectrum of FBN1 and could help us better understand the genetic defects of the patients with MFS.
1 Introduction
Marfan syndrome (MFS) is a rare disorder that affects the connective tissues of the body. It mainly involves the ocular, cardiovascular, and musculoskeletal systems, and has diverse clinical presentations. The incidence of MFS is about 2–3 per 10,000 individuals (1). MFS is autosomal dominant inherited, and FBN1 is implicated in most MFS cases. FBN1, located on 15q15-q21.1, comprises 65 exons and encodes the 2,871-amino acid fibrillin-1 protein (2), which is widely expressed in the aorta, tendons, periosteum of the bones, and ciliary zonules of the eye (3). Although the risk of developing MFS is higher in patients with a family history, the condition does not discriminate between different genders and ethnicities (1, 2). In a cohort of 131 Chinese patients with MFS, mutations in FBN1 were detected in 82 patients (4), suggesting that FNB1 mutations are the predominant cause of MFS. Although several mutations in FBN1 have been reported to be responsible for MFS, the exact phenotype-genotype correlation remains unclear (3, 4).
In this study, we investigated two Chinese families with MFS. We identified a novel missense mutation (c.5060G > C, p.Cys1687Ser) and a recurrent missense mutation (c.2168A > T, p.Asp723Val) in FBN1 using whole exome sequencing and Sanger sequencing.
2 Materials and methods
2.1 Patients
This study was approved by the ethics committee of Tianjin Medical University Eye Hospital (2021-KY03) and followed the tenets of the Declaration of Helsinki. Two Chinese families were enrolled, and written informed consent was obtained from the participants. A peripheral venous blood sample was collected from each enrolled family member for further analysis.
2.2 Whole exome sequencing and bioinformatics analysis
The whole exome sequencing and data analysis procedures were previously reported (5–7). Briefly, we first extracted genomic DNA from the blood samples according to the standard procedure of the manufacturer (MagPure Buffy Coat DNA Midi KF Kit, Magen, China), and the qualified genomic DNA was then sequenced with PE100 + 100 on MGISEQ-2000. We applied the BGI MGIEasy V4 chip, which contains exons of all human genes and their adjacent ± 20 bp introns, to capture the targeted sequences. Bioinformatics processing and data analysis were then performed to explore the potential variants after we received the primary sequencing data. Several databases, including the 1,000 Genomes Project, HapMap, NCBI dbSNP, and a database of 200 normal Chinese adults, were used to filter and estimate all the SNVs and indels. Finally, Sanger sequencing was used to validate all mutations and potential pathogenic variants. The Human Gene Mutation Database (HGMD)1 was introduced to screen previously reported mutations. To rule out the possibility of a polymorphism, the mutations were also blasted in the 1,000 Genomes Project,2 ExAC,3 HapMap,4 ESP6500,5 NCBI dbSNP,6 GnomAD,7 and a database of 200 normal Chinese adults.
2.3 Functional impact prediction
Calibrated predictions of the possible functional impacts of the FBN1 mutations were made using several online programs, including VARSOME,8 PolyPhen2,9 PROVEAN,10 and MutationTaster,11 etc. Based on the standards proposed by the American College of Medical Genetics and Genomics (ACMG) (8), the variants were also defined as “likely benign or benign,” “of uncertain significance,” or “likely pathogenic or pathogenic.”
2.4 Evolutionary conservation analysis
Evolutionary conservation analyses among several species were performed using Clustal Omega.12
2.5 Protein secondary structure analysis
Secondary structure analyses of the wild-type and the mutant proteins were performed using Network Protein Sequence Analysis.13
2.6 Protein structural effect evaluation
Protein structure homology modeling and protein structural effect evaluation were performed using HOPE14 (9). Firstly, UniProt ID of P35555 was input to obtain the amino acid sequence of FBN1. The residues and mutations were then selected to finalize the analysis.
3 Results
3.1 Clinical evaluation
For Family 1, two patients (II:3, III:2) and one normal individual (II:4) were enrolled in our study (Figure 1A). The proband (III:2) was 30 years old and had been diagnosed with MFS in his childhood. On presentation, the proband was tall and had long fingers (Figure 1B). The vision was 20/200 in both eyes. In the right eye and left eye, the axial length was 30.61 and 34.01 mm, the steep K was 40.26 D and 40.74 D, the flat K was 39.06 D and 38.75 D, and the intraocular pressure was 17.8 and 15.7 mmHg, respectively. The medical records of the proband were reviewed. The patient accepted bilateral clear lens extraction because of bilateral nasal superiorly dislocated lens when he was 14 years old. Three months after the surgery, retinal detachment occurred in the right eye, and he underwent vitrectomy and silicone oil tamponade. The oil in the right eye was removed 6 months later. His mother and grandfather were diagnosed with MFS at early ages. His mother (II:3) was 53 years old and underwent clear lens extraction because of ectopia lentis when she was young. His mother’s vision was 20/200 and 20/300, her intraocular pressure was 20.7 and 19.9 mmHg, and her axial length was 28.45 and 27.87 mm in the right eye and left eye, respectively. There was no consanguineous marriage in this family.
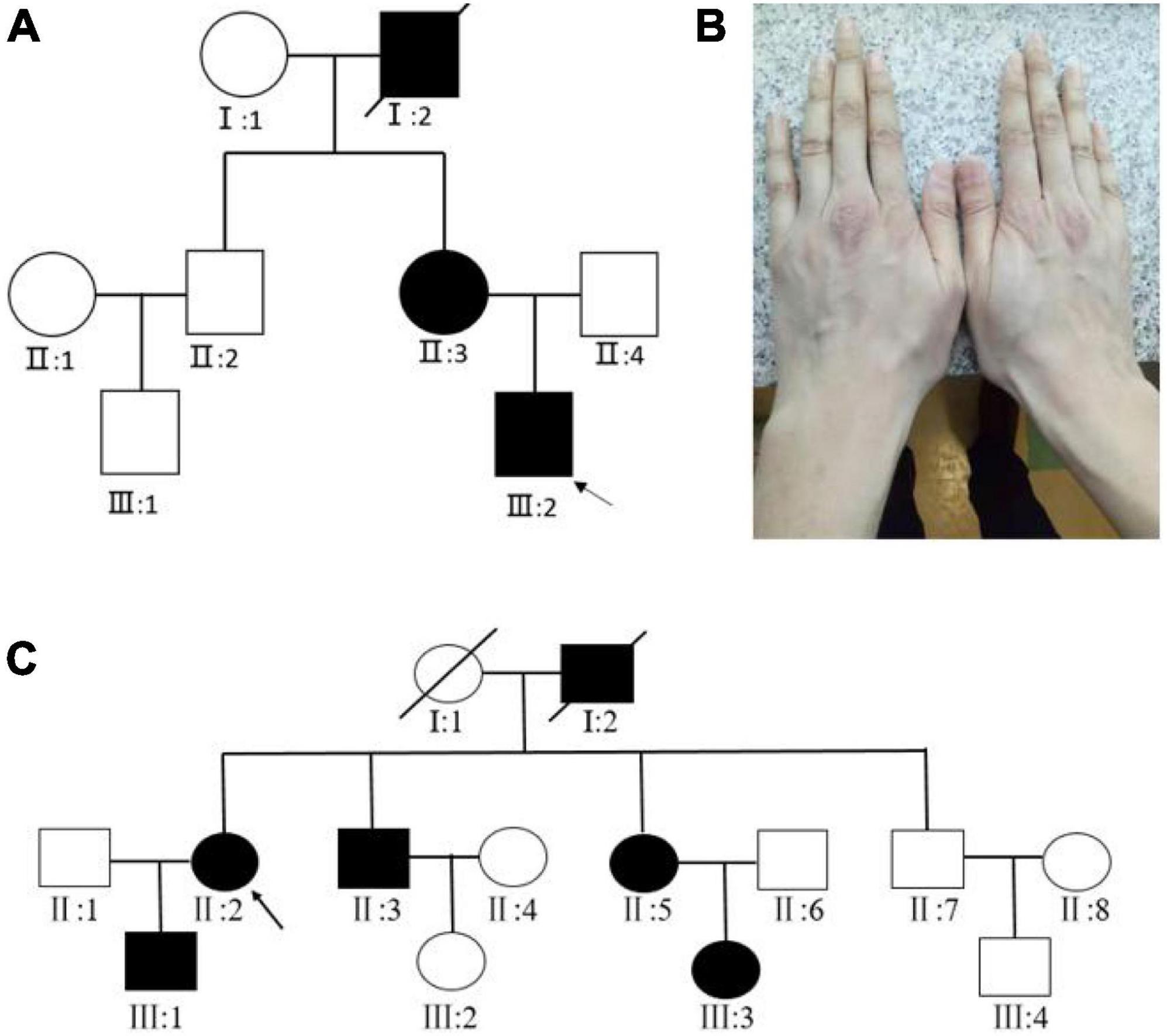
Figure 1. (A) Pedigree map of Family 1. The arrow indicates the proband. Squares and circles symbolize males and females, respectively. Black and white denote affected and unaffected individuals, respectively. (B) The long fingers of the proband of Family 1. (C) Pedigree map of Family 2.
For Family 2, four affected (III:1, II:2, II:3, III:5) and three unaffected individuals (III:4, II:1, II:7) were enrolled in our study (Figure 1C). The proband (II:2) was 50 years old, and she was diagnosed with MFS when she was six. The patient had accepted bilateral clear lens extraction for bilateral temporally dislocated lens 10 years ago. On presentation, her visual acuity was finger-counting and 20/200, her best-corrected visual acuity was 20/200 and 20/50, her axial length was 27.15 and 28.10 mm, and her intraocular pressure was 18.9 and 19.1 mmHg for the right eye and left eye, respectively. Her son (III:1) was 25 years old and was diagnosed with MFS when he was five. He also underwent bilateral clear lens extraction for ectopia lentis several years ago. His visual acuity was 20/100 and 20/80, his axial length was 25.23 and 26.37 mm, and his intraocular pressure was 15.1 and 13.5 mmHg for the right eye and left eye, respectively. The other two enrolled patients in this family (II:3, II:5) reported that they both had high myopia and had accepted surgery for ectopia lentis, but unfortunately, the detailed clinical data were not available.
3.2 Mutation identification in FBN1
Whole exome sequencing of the proband of family 1 (III:2) revealed a heterozygous transversion in exon 19 (c.2168A > T) of FBN1, which has been reported previously (10). The mutation changes wild-type aspartic acid to valine at codon 723 (p.Asp723Val; Figure 2A). The mutation was found in all affected individuals (II:3, II:2) but was not detected in the unaffected individual (II:4) in this family using further Sanger sequencing.
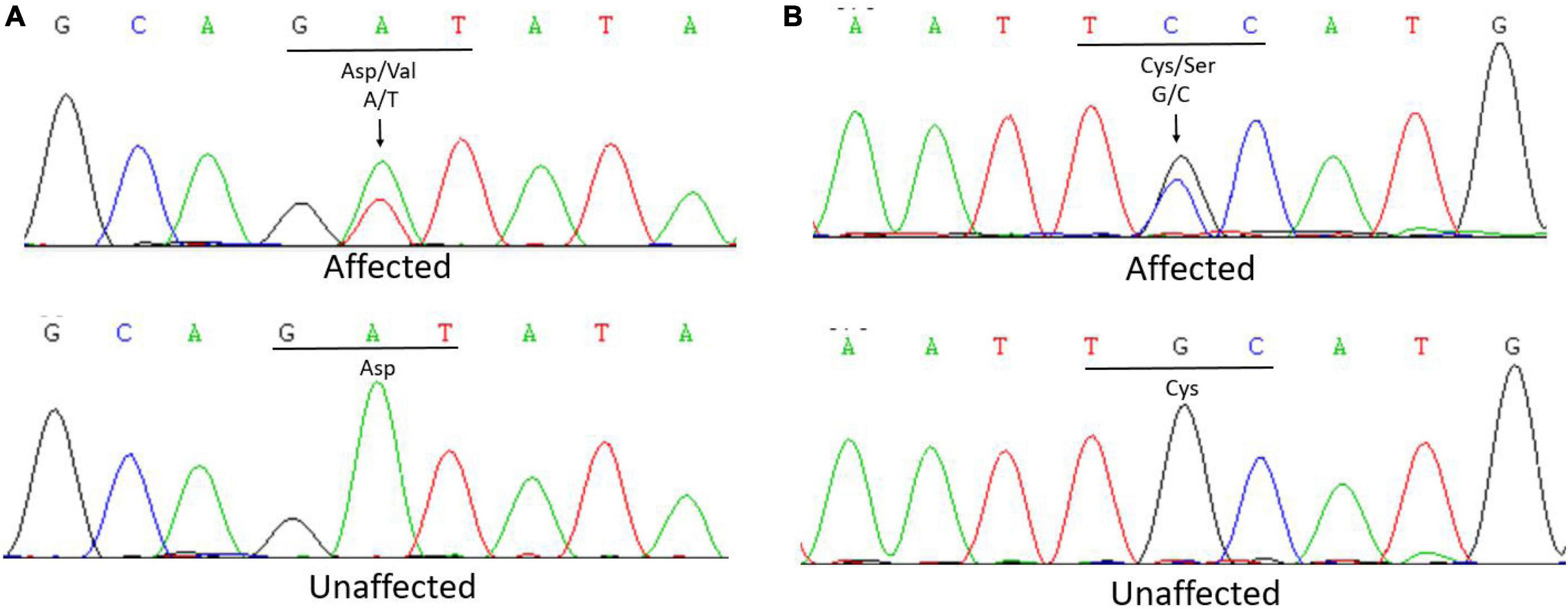
Figure 2. (A) Sanger sequencing of FBN1 detected a c.2168A > T transversion in affected patients that caused the replacement of a wild-type aspartic acid with valine at codon 723 in Family 1. (B) Sanger sequencing of FBN1 detected a c.5060G > C transversion in affected patients that caused the replacement of a wild-type cysteine with serine at codon 1,687 in Family 2.
In Family 2, the whole exome sequencing of the proband revealed a novel heterozygous transversion in exon 41 (c.5060G > C) of FBN1. This mutation changes a wild-type cysteine to serine at codon 1,687 (p.Cys1687Ser; Figure 2B). Using Sanger sequencing, the mutation was found in all affected individuals (II:3, II:5, III:1) and was not detected in the unaffected individuals (II:1, II:7, III:4) in this family, indicating that the mutation was co-segregated with the phenotype. The mutation was not found in the 1,000 Genomes Project, ExAC, GnomAD-EAS_exome_ALL, HapMap, ESP6500, NCBI dbSNP, GnomAD_exome_ALL, or the database of 200 normal Chinese adults.
No other rare variants classified as pathogenic or likely pathogenic were identified in the other genes (FBN2, TGFBR1, TGFBR2, LTBP-1, LTBP-2, LTBP-3, SKI, etc.) related to MFS in either family.
3.3 Functional impact prediction
Most of the online bioinformatics programs produced a result of “Pathogenic” (Table 1). According to ACMG guidelines and standards, the c.2168A > T, p.Asp723Val mutation was defined as “Pathogenic” (PM5 + PP3 + PM1 + PM2 + PP5) and the c.5060G > C, p.Cys1687Ser mutation as “Likely pathogenic” (PP3 + PM5 + PM1 + PM2).

Table 1. Calibrated prediction of the possible functional impact of the FBN1 mutations by online programs.
3.4 Evolutionary conservation analysis
Evolutionary conservation analysis revealed that codons 723 and 1,687 were located within a highly conserved region among several species (Figure 3).

Figure 3. Multiple-sequence alignments of FBN1 showed that codons 723 and 1,687 were highly conserved among several species.
3.5 Protein secondary structure analysis
The predicted secondary structure indicated that an original, flexible, random coil in the wild-type FBN1 was replaced by a more stable extended strand or alpha helix in the p.Asp723Val mutant FBN1 (Figure 4A). However, the mutant p.Cys1687Ser did not cause much change to the predicted secondary structure (Figure 4B).
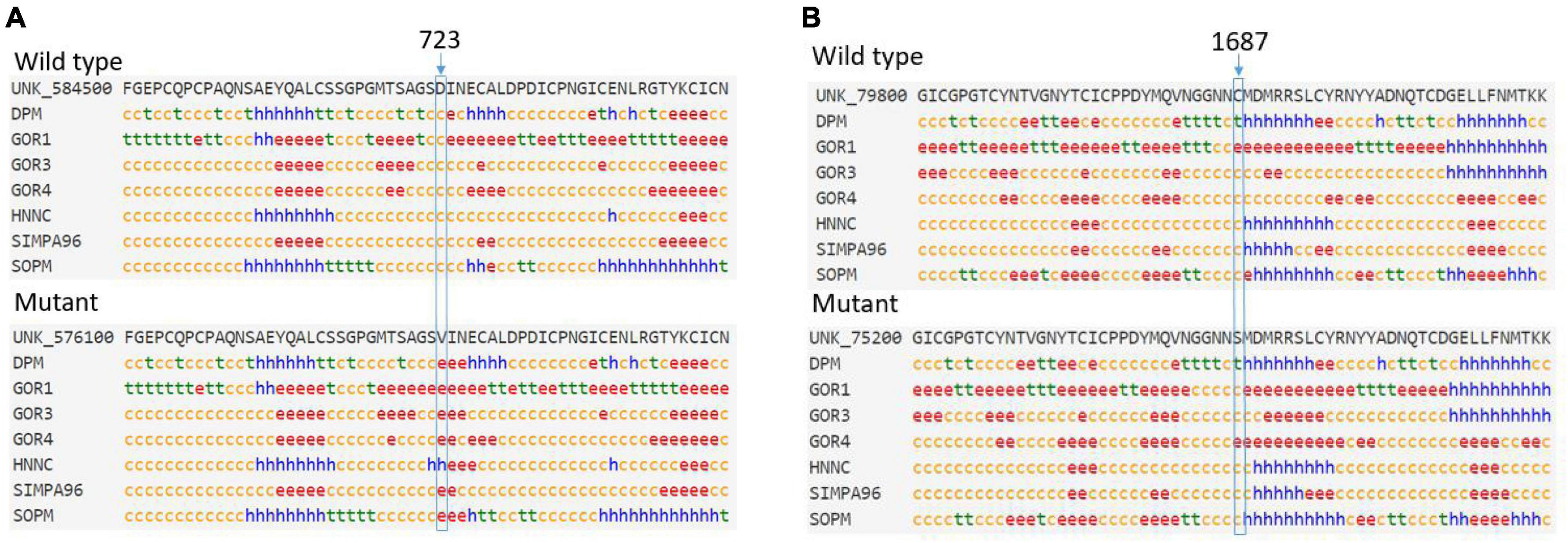
Figure 4. (A) The predicted secondary structure indicated that an original random coil in the wild-type FBN1 was replaced by an extended strand or alpha helix in the p.Asp723Val mutant FBN1. (B) The p.Cys1687Ser mutant did not cause much change in the predicted secondary structure. c, random coil; e, extended strand; h, alpha helix; t, beta turn.
3.6 Protein structural effect evaluation
In structure homology modeling, the p.Asp723Val mutation changed the side chain of the protein (Figure 5A). The Asp723 residue is involved in a metal-ion (Sm) contact (Figure 5A), and the differences in both size and charge caused by mutation can make the metal-ion interactions of Asp723 less stable. The hydrophobic difference can further interfere with the formation of hydrogen bonds, and the mutation might also disturb the function of the protein by interfering with the normal interaction of Asp723 with other molecules.
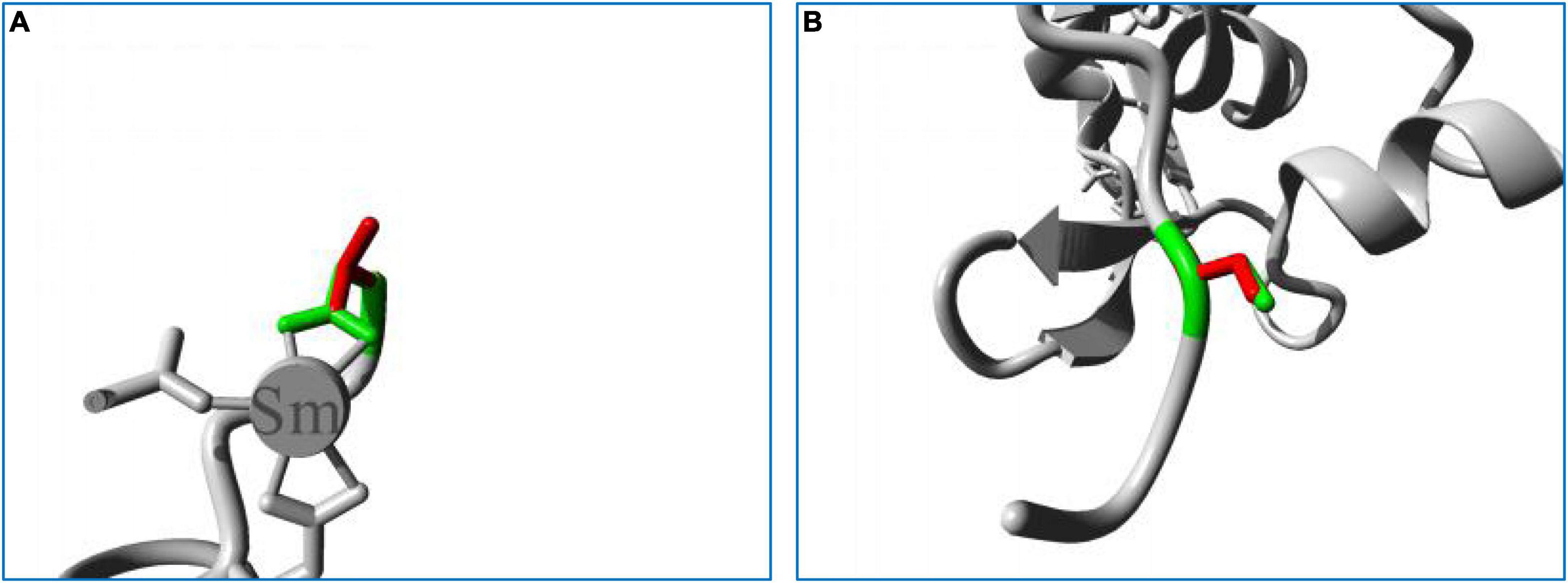
Figure 5. Close-up image of the superimposed structures of wild-type and mutant residues. (A) Asp723Val. (B) Cys1687Ser. The protein core is shown in gray, while the amino acid side chains of the wild-type (green) and the mutant (red) residues are represented as sticks.
The p.Cys1687Ser mutation also changed the side chain of the protein (Figure 5B). The replacement of Cys with Ser can sever the bonds formed by cysteines, impairing the stability of the protein, and impede the normal hydrophobic interactions of Cys with other molecules on the surface of the protein. The Cys1687 residue is located within an Egf-like calcium-binding domain, and the p.Cys1687Ser mutation can disturb this domain and affect its function by introducing an amino acid with different properties.
Together, all these observations indicate that the mutations found in this study were pathogenic and disease-causing.
4 Discussion
In this study, we evaluated two Chinese families with MFS, and the main ocular presentations were increased axial length and ectopia lentis. We also identified a novel missense mutation (c.5060G > C, p.Cys1687Ser) and a recurrent missense mutation (c.2168A > T, p.Asp723Val) in FBN1 using whole exome sequencing and Sanger sequencing, and our bioinformatics analysis indicated that both the mutations were disease-causing.
Marfan syndrome is a rare disease with manifestations mainly involving the ocular, musculoskeletal, and cardiovascular systems. Cardiovascular manifestations represent the major morbidity and mortality factors, with aortic root dissection being the main cause of death in MFS patients. Other cardiovascular manifestations include left ventricular dilation, mitral valve prolapse, and pulmonary artery enlargement (11–14). The main musculoskeletal presentation of MFS is overgrowth of the long bones (15), which leads to altered ratios among the body’s segments (overgrowth of arms and legs), anterior chest deformity (overgrowth of the ribs), and arachnodactyly (overgrowth of the fingers) (1). The typical ocular features of MFS are ectopia lentis and myopia, consistent with the results of our study. Ectopia lentis has been shown to occur in 45–87% of patients with MFS (16–18) and is a major criterion for diagnosis (19). The lens dislocation is resulted from the insufficiency of the ciliary zonules. Lens dislocation in MFS can occur in any direction, but typically superiorly (3), as demonstrated in our study. Myopia is another common ocular presentation in patients with MFS, which often begins at an early age and has a rapid progression. Other manifestations of the ocular system include an elongated globe, an abnormally flat cornea, hypoplasia of the ciliary muscle and iris, amaurosis, an increased risk of early cataract and glaucoma, and a predisposition for retinal detachment (19–21). In patients who have not developed typical cardiovascular symptoms, ocular symptoms may comprise the initial presentations of MFS, necessitating a more comprehensive diagnostic workup.
Up to now, about 3,000 different FBN1 mutations for MFS have been included in the UMD-FBN1 database.15 Although so many FBN1 mutations have been reported to be responsible for MFS, the exact phenotype-genotype correlation in MFS is still unclear because of the inter- and intra-family clinical variability. However, a few phenotype-genotype relationships have been firmly established so far, such as mutations in exons 24–32 of FBN1 being associated with neonatal MFS and a severer phenotype (22–24), while mutations in exons 43–65 have been associated with a substantial increase in cardiovascular manifestations (25–27). Meanwhile, patients with haploinsufficiency mutations tended to have more severe cardiovascular involvements than patients with dominant negative mutations (28). For ocular manifestations, in a large cohort including 1,013 probands with pathogenic FBN1 mutations, missense mutations producing or substituting cysteines were found to be related with more frequent ectopia lentis when compared with other missense mutations (22). Mutations introducing premature termination codons were connected to severer skin and skeletal presentations but less common ectopia lentis and retinal detachment (3, 22, 23, 29). This is in accordance with the results of our study; lens dislocation was the major ocular manifestation of the proband with the p.Cys1687Ser mutation in Family 2. Two other missense mutations in codon 1,687 have been reported previously: p.Cys1687Arg (30) and p.Cys1687Phe (31). The patient who carried the p.Cys1687Arg mutation was diagnosed with incomplete MFS, but the detailed clinical data were not available (30). The patient with the p.Cys1687Phe mutation was a 3-year-old Caucasian boy whose ocular presentation was mainly ectopia lentis (31). However, although the mutation identified in Family 1 (p.Asp723Val) was located in exon 19 and was not a cysteine mutation, the patient presented with bilateral ectopia lentis, myopia, and retinal detachment, indicating the highly heterogeneous nature of MFS. The p.Asp723Val mutation was first reported in a German 8-year-old girl whose ocular involvement also included ectopia lentis and myopia (10). All of these suggest that the phenotype-genotype correlations of MFS are not yet clear and need further study to confirm.
Although our bioinformatics analysis indicated that the mutations detected in our study were disease-causing, the exact molecular pathogenesis of MFS remains unknown (1, 2). A traditional dominant-negative mechanism has been proposed to be implicated in the pathogenesis of MFS. In a dominant-negative model, Eldadah et al. illustrated that an experimentally introduced mutant FBN1 allele in the presence of two endogenous wild-type alleles is sufficient to reproduce the MFS cellular phenotype by reducing fibrillin-1 accumulation in the extracellular matrix and disrupting normal microfibrillar assembly (32). On the other hand, in a transgenic mice model, Judge et al. demonstrated that selected mutations (e.g., C1039G) caused a disorganized microfibril architecture and that the introduction of exogenous wild-type fibrillin-1 can rescue phenotypes associated with the C1039G mutation (33), indicating that wild-type fibrillin-1 haploinsufficiency, rather than mutant proteins production, might be the predominant determinant of failed microfibrillar assembly.
5 Conclusion
In this study, we evaluated two Chinese families with MFS whose main ocular presentations were increased axial length and ectopia lentis. Using whole exome sequencing and Sanger sequencing, we identified a novel missense mutation (c.5060G > C, p.Cys1687Ser) and a recurrent missense mutation (c.2168A > T, p.Asp723Val) in FBN1, and our bioinformatics analysis indicated that both the mutations were disease-causing. Our results expanded the mutation spectrum of FBN1 and could help broaden the phenotype-genotype relationships of MFS. However, the exact disease-causing mechanism of the mutations needs further functional experiments to confirm.
Data availability statement
The genetic sequencing data in this manuscript were not deposited to a public repository to protect patient privacy/confidentiality. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Tianjin Medical University Eye Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
WL and JJ designed and supervised the study. JQ collected the pedigree. DL and RG drafted the manuscript. WL, RG, DH, and JJ analyzed the data. All authors read and approved the final manuscript.
Funding
This research was funded by the Open Project of Tianjin Key Laboratory of Retinal Functions and Diseases (2020tjswmq003), the Youth Special Fund of Clinical Research of Tianjin Medical University Eye Hospital (2020QN02), and the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-037A).
Acknowledgments
We thank the subjects for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
MFS, marfan syndrome.
Footnotes
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
- ^ https://www.internationalgenome.org/data
- ^ https://gnomad.broadinstitute.org/
- ^ http://hapmap.ncbi.nlm.nih.gov
- ^ http://evs.gs.washington.edu/EVS/
- ^ https://www.ncbi.nlm.nih.gov/snp/
- ^ http://www.gnomad-sg.org/
- ^ http://varsome.com
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://provean.jcvi.org/index.php
- ^ http://mutationtaster.org/
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo/
- ^ https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html
- ^ https://www3.cmbi.umcn.nl/hope/
- ^ http://www.umd.be/
References
1. Judge D, Dietz H. Marfan’s syndrome. Lancet. (2005) 366:1965–76. doi: 10.1016/S0140-6736(05)67789-6
2. Ramachandra C, Mehta A, Guo K, Wong P, Tan J, Shim W. Molecular pathogenesis of Marfan syndrome. Int J Cardiol. (2015) 187:585–91. doi: 10.1016/j.ijcard.2015.03.423
3. Latasiewicz M, Fontecilla C, Millá E, Sánchez A. Marfan syndrome: ocular findings and novel mutations-in pursuit of genotype-phenotype associations. Can J Ophthalmol. (2016) 51:113–8. doi: 10.1016/j.jcjo.2015.12.019
4. Wu Y, Sun H, Wang J, Wang X, Gong M, Han L, et al. Marfan syndrome: whole-exome sequencing reveals de novo mutations, second gene and genotype-phenotype correlations in the Chinese population. Biosci Rep. (2020) 40:BSR20203356. doi: 10.1042/BSR20203356
5. Li D, Xu C, Huang D, Guo R, Ji J, Liu W. Identification and functional analysis of a novel missense mutation in GJA8, p.Ala69Thr. BMC Ophthalmol. (2020) 20:461. doi: 10.1186/s12886-020-01725-1
6. Liu W, Guo R, Hao H, Ji J. Identification of a novel RHO heterozygous nonsense mutation in a Chinese family with autosomal dominant retinitis pigmentosa. BMC Ophthalmol. (2021) 21:360. doi: 10.1186/s12886-021-02125-9
7. Guo R, Huang D, Ji J, Liu W. A novel mutation GJA8 NM_005267.5: c.124G>A, p.(E42K) causing congenital nuclear cataract. BMC Ophthalmol. (2022) 22:172. doi: 10.1186/s12886-022-02386-y
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
9. Venselaar H, Te Beek T, Kuipers R, Hekkelman M, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. (2010) 11:548. doi: 10.1186/1471-2105-11-548
10. Katzke S, Booms P, Tiecke F, Palz M, Pletschacher A, Türkmen S, et al. TGGE screening of the entire FBN1 coding sequence in 126 individuals with marfan syndrome and related fibrillinopathies. Hum Mutat. (2002) 20:197–208. doi: 10.1002/humu.10112
11. Pyeritz R. The Marfan syndrome. Annu Rev Med. (2000) 51:481–510. doi: 10.1146/annurev.med.51.1.481
12. Pyeritz R. Marfan syndrome and related disorders. Ann Thorac Surg. (2008) 86:335–6. doi: 10.1016/j.athoracsur.2006.10.023
13. Pepe G, Giusti B, Sticchi E, Abbate R, Gensini G, Nistri S. Marfan syndrome: current perspectives. Appl Clin Genet. (2016) 9:55–65. doi: 10.2147/TACG.S96233
14. Bitterman A, Sponseller P. Marfan syndrome: a clinical update. J Am Acad Orthop Surg. (2017) 25:603–9. doi: 10.5435/JAAOS-D-16-00143
15. Shirley ED, Sponseller P. Marfan syndrome. J Am Acad Orthop Surg. (2009) 17:572–81. doi: 10.5435/00124635-200909000-00004
16. Drolsum L, Rand-Hendriksen S, Paus B, Geiran O, Semb S. Ocular findings in 87 adults with Ghent-1 verified Marfan syndrome. Acta Ophthalmol. (2015) 93:46–53. doi: 10.1111/aos.12448
18. Rand-Hendriksen S, Lundby R, Tjeldhorn L, Andersen K, Offstad J, Semb S, et al. Prevalence data on all Ghent features in a cross-sectional study of 87 adults with proven Marfan syndrome. Eur J Hum Genet. (2009) 17:1222–30. doi: 10.1038/ejhg.2009.30
19. Loeys B, Dietz H, Braverman A, Callewaert B, De Backer J, Devereux R, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. (2010) 47:476–85. doi: 10.1136/jmg.2009.072785
20. von Kodolitsch Y, De Backer J, Schüler H, Bannas P, Behzadi C, Bernhardt A, et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl Clin Genet. (2015) 8:137–55. doi: 10.2147/TACG.S60472
21. Chandra A, Ekwalla V, Child A, Charteris D. Prevalence of ectopia lentis and retinal detachment in Marfan syndrome. Acta Ophthalmol. (2014) 92:e82–3. doi: 10.1111/aos.12175
22. Faivre L, Collod-Beroud G, Loeys B, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. (2007) 81:454–66. doi: 10.1086/520125
23. Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet. (2002) 71:223–37. doi: 10.1086/341581
24. Peng Q, Deng Y, Yang Y, Liu H. A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum. BMC Pediatr. (2016) 16:60. doi: 10.1186/s12887-016-0598-6
25. Gao L, Tian T, Zhou X, Fan L, Wang R, Wu H. Detection of ten novel FBN1 mutations in Chinese patients with typical or incomplete Marfan syndrome and an overview of the genotype-phenotype correlations. Int J Cardiol. (2019) 293:186–91. doi: 10.1016/j.ijcard.2019.06.066
26. Arbustini E, Grasso M, Ansaldi S, Malattia C, Pilotto A, Porcu E, et al. Identification of sixty-two novel and twelve known FBN1 mutations in eighty-one unrelated probands with Marfan syndrome and other fibrillinopathies. Hum Mutat. (2005) 26:494. doi: 10.1002/humu.9377
27. Chung B, Lam S, Tong T, Li S, Lun K, Chan D, et al. Identification of novel FBN1 and TGFBR2 mutations in 65 probands with Marfan syndrome or Marfan-like phenotypes. Am J Med Genet A. (2009) 149A:1452–9. doi: 10.1002/ajmg.a.32918
28. Franken R, Teixido-Tura G, Brion M, Forteza A, Rodriguez-Palomares J, Gutierrez L, et al. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart. (2017) 103:1795–9. doi: 10.1136/heartjnl-2016-310631
29. Dietz H, McIntosh I, Sakai L, Corson G, Chalberg S, Pyeritz R, et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics. (1993) 17:468–75. doi: 10.1006/geno.1993.1349
30. Groth K, Von Kodolitsch Y, Kutsche K, Gaustadnes M, Thorsen K, Andersen N, et al. Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet Med. (2017) 19:772–7. doi: 10.1038/gim.2016.181
31. Baudhuin L, Kotzer K, Lagerstedt S. Decreased frequency of FBN1 missense variants in Ghent criteria-positive Marfan syndrome and characterization of novel FBN1 variants. J Hum Genet. (2015) 60:241–52. doi: 10.1038/jhg.2015.10
32. Eldadah Z, Brenn T, Furthmayr H, Dietz H. Expression of a mutant human fibrillin allele upon a normal human or murine genetic background recapitulates a Marfan cellular phenotype. J Clin Invest. (1995) 95:874–80. doi: 10.1172/JCI117737
Keywords: Marfan syndrome, FBN1, bioinformatics analysis, mutation, phenotype
Citation: Li D, Qiao J, Huang D, Guo R, Ji J and Liu W (2022) Novel and recurrent FBN1 mutations causing Marfan syndrome in two Chinese families. Front. Med. 9:1086844. doi: 10.3389/fmed.2022.1086844
Received: 01 November 2022; Accepted: 28 November 2022;
Published: 13 December 2022.
Edited by:
Dario Rusciano, Consultant, Catania, ItalyReviewed by:
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, IranOscar Campuzano, University of Girona, Spain
Copyright © 2022 Li, Qiao, Huang, Guo, Ji and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, d2VpbGl1MDVAdG11LmVkdS5jbg==
†These authors have contributed equally to this work