 Wu Dawei
Wu Dawei Miao Shuangman
Miao Shuangman Huang Huiyao
Huang Huiyao Cui Dandan
Cui Dandan Tang Yu
Tang Yu Li Ning
Li Ning- Department of Clinical Trials Center, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Unblinding at disease progression in double-blinded randomized controlled cancer drug clinical trials is ethical to the patient by ensuring optimal subsequent treatment, but the effect of study treatment on overall survival may be confounded. The views of science and ethics in this issue are controversial and the unblinding procedures should be well-designed. In real world settings, a lack of use of this unblinding process in protocol was observed in the analysis of 134 double-blind randomized controlled anticancer drug clinical trials conducted in China from 2018 to 2021. Unblinding at disease progression was allowed in only 26 (18.2%) trials. Among them, Only 9 (34.6%) trials involved patient-level unblinding. None of the 134 included trials accounted for the risk of blind-maintenance after disease progression. Based on the analysis and case studies, we believe that unblinding at disease progression should be stated in the protocol when the treatment assignment directly affected the choice of subsequent regimen, in which the drug category, control group design, standard of care of further-line therapy and primary endpoint together play a role. When unblinding at disease progression is adopted, the sensitivity analytics are recommended to understand the true effect of study drug on overall survival. The notification of treatment allocation after unblinding and the informed consent also require attention. A decision-making framework is established to help understand this controversy, which should be carefully discussed by the investigator and the sponsor.
Introduction
The double-blinded randomized controlled trial (RCT) is a golden standard in the clinical assessment of novel drugs. Blinding investigators and patients decreases the likelihood of biased observations of the effectiveness outcomes and the patient drop out (1). Withholding the participants' treatment assignment may adversely affect their medical care in case of emergency (2) or after the completion of study (3, 4), thus raises ethical concerns. The unblinding procedure is well-established in trial protocol in response to these circumstances. However, in double-blinded randomized controlled cancer drug clinical trials, maintaining the blind at disease progression may also have a negative impact on the subjects' subsequent treatment, namely, preventing or delaying them from receiving approved therapy or entering into other clinical trials (1). Whether and how to set the unblinding procedure in this situation require further discussion.
From a policy perspective, this issue seems ignored by current regulations and guidelines, which are mainly aimed at unblinding under emergency or at the end of study (2–4). Only Food and Drug Administration (FDA) of the United States has issued relative guidance, which from the ethical view, recommended unblinding the patient and the investigator at disease progression or recurrence to ensure optimal subsequent management. The guidance also emphasized the informed consent of relevant risk if the sponsor intended to maintain patient-level blinding at disease progression (1). As only placebo-controlled studies are within the scope of guidance, it is unclear whether this recommendation should be adopted in other double-blinded cancer drug RCTs.
To make matters worse, it is reported that pharmaceutical industry trials most often maintain blinding until the completion of the entire study (5). For cancer clinical trials, unblinding process at disease progression or crossover design is avoided when OS is part of the primary endpoint. The sponsors, from the scientific view, claim that the reliability of OS endpoint is negatively impacted by the open-label stage beyond disease progression, and the effect of study treatment is potentially confounded by switch-over from control to study treatment or subsequent therapies (6, 7). Therefore, the rules or procedures should be established to balance the paradoxical views of science and ethics at this question.
Cross-sectional study
In order to clarify whether the blinding is maintained within disease progression at trial level, and how the procedures are designed in real world settings, a total of 134 double-blinded randomized controlled cancer drug clinical trials launched in China from 2018 to 2021 were analyzed. Based on the protocols, except for 2 (1.5%) trials without any unblinding process, unblinding at disease progression was allowed and stated in 26 (18.2%) trials. Most trials (106, 74.1%) only had an unblinding process under emergency or after the final analysis of study.
The 26 trials with unblinding process at disease progression were all testing novel drugs, which accounted for 20.1% of all the trials for this drug category. For sponsor type, a higher proportion of trials sponsored by international cooperations (13/48, 27.1%) included such procedures, compared with that of domestic cooperations (13/86, 15.1%). Placebo-controlled trials (24/112, 21.4%), without OS as the primary endpoint (20/99, 20.2%) were more likely to adopt the procedures than trials within active comparators (2/21, 9.5%), and OS as the primary endpoint (6/35, 17.1%). The χ2 test was used for subgroup comparisons, but no statistically significance was observed (Table 1).
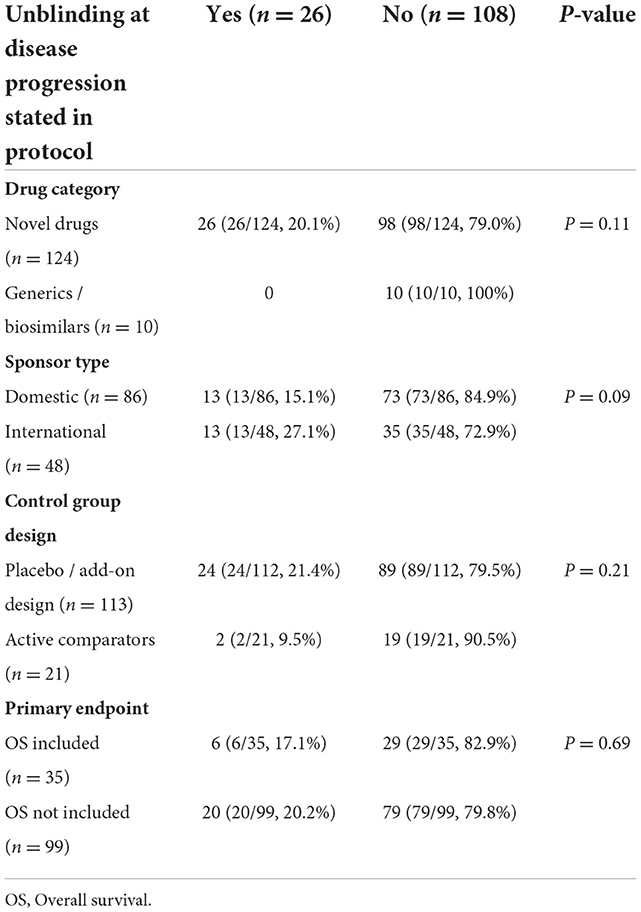
Table 1. The distribution of unblinding procedures by drug category, sponsor type, control group design and primary endpoint of the 134 trials.
Among the 26 protocols with unblinding procedures at disease progression, the patients would receive standard of care (SOC) after unblinding in 16 (61.6%) trials. The patients in control group would crossover to the study treatment after unblinding in the other 10 (38.5%) trials. As for the disclosure of treatment assignment after unblinding, there were 13 (50.0%) trials at the investigator level, 7 (26.9%) at both the investigator and the patient level, 4 (15.4%) at both the investigator and the sponsor level, 2 (7.7%) unblinded all the three parties. Only 9 (34.6%) trials in accumulation involved patient-level unblinding. Looking at the informed consent documents, unfortunately, none of the included clinical trials accounted for the risk of blind-maintenance after disease progression.
Discussion
The data demonstrated that unblinding at disease progression was not received enough attention in cancer clinical trials, though it is beneficial to patients and supported by the FDA guidance. In order to better reshape the unblinding rules, the influence of blinding on subsequent treatment is the most important factor. The analysis has given us typical cases for how blind-maintenance at disease progression affect the medical care, in which the drug category, control group design and SOC of further-line therapy together play a role (Supplementary Figure 1). In Case A: the first-line study treatment of non-small-cell lung cancer (NSCLC), when the study drug is a biosimilar, patients receive “similar” study treatment that doesn't affect the choice of the second-line therapy. Maintaining blinding at disease progression is acceptable. In Case B: the first-line study treatment of esophageal squamous cell carcinoma (ESCC), when the trial is placebo-controlled, patients receive totally “different” study treatment. Also, the SOC of the second-line shares the same target of the study drug. The treatment assignment directly affected the choice of subsequent regimen, thus unblinding should be considered.
The OS will obviously be impacted by the unblinding progress in trials like Case B. Due to patient welfare, early unblinding and crossover treatment should still be considered (8). To reduce the effect on the trial data interpretation, sometimes the primary endpoint may be carefully re-designed and a discussion with the regulatory authority is necessary. When the primary endpoint involves OS and the study treatment does not directly affect the subsequent care, unblinding at disease progression at trial level is not mandatory, but unblinding individual patient may still be considered. The sensitivity analytics with modeling of crossover impact are recommended in the above cases whenever unblinding occurs in a group of patients or an individual, to understand the true benefit of study drug and meet the requirements of regulatory authorities (9).
Simply replacing the rules of unblinding at disease progression by emergency unblinding or unblinding upon the completion of study is not recommended. Firstly, the “disease progression” is not necessarily an “emergency.” Secondly, “completion of study treatment” is far from “completion of study” for cancer patients (5), especially when overall survival (OS) is collected as a key endpoint. Finally, without a detailed standard procedure, the degree of operating freedom is too large to fully protect the welfare of patients.
The notification of treatment allocation after unblinding and the informed consent cannot be ignored. The treatment assignment must be disclosed at the patient level, especially when the subsequent care is provided by other physicians rather than the investigators (10). The risk of blinding (e.g., delaying the subsequent medical care) and the conditions of unblinding should be addressed in the written informed consent and reviewed by the ethics committee. If the patient-level blindness is maintained at disease progression, the additional risk should also be reflected in the informed consent (1).
Based on the above considerations, a framework for protocol design is established to help sponsors, investigators and ethics committees to understand the controversy (Figure 1). When the study design is a head-to-head comparison of a generic or biosimilar and its reference drug, maintaining blinding at disease progression is acceptable. For novel cancer drug clinical trials, when the study drug is part of, or share the same target of subsequent SOC, unblinding at disease progression is recommended. When the study treatment does not directly affect the subsequent therapy, and the primary endpoint includes OS, maintaining blinding at disease progression is acceptable, but the risk should be addressed in the written informed consent.
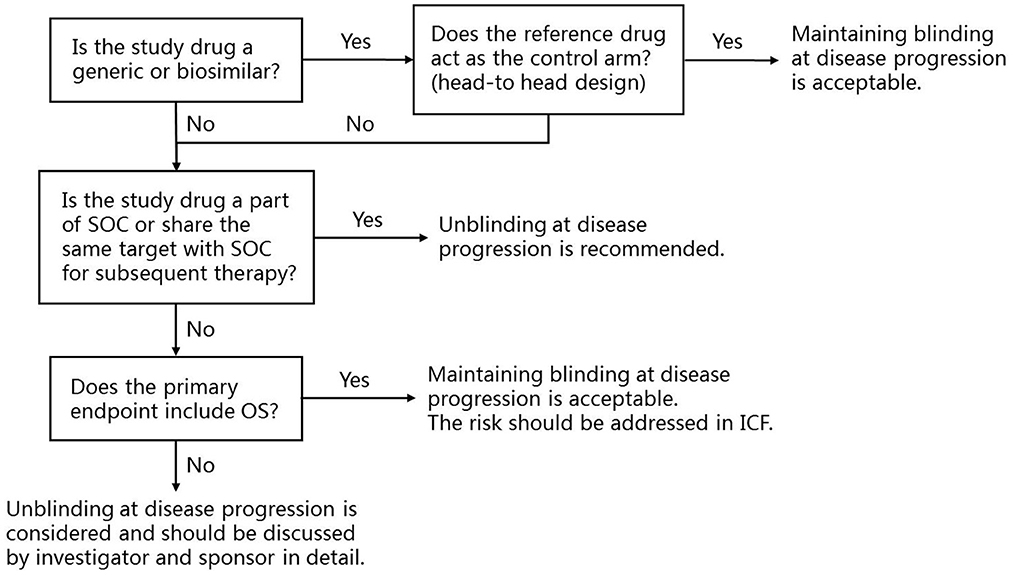
Figure 1. The framework for the application of unblinding at disease progression at trial level and recommendations. SOC, Standard of care; OS, Overall survival; ICF, Informed consent form.
In summary, the application of unblinding procedures at disease progression in double-blinded cancer drug RCTs has not been paid enough attention and should be carefully reshaped by the investigator and the sponsor under patient-centric considerations. To fully establish the decision-making mechanism in the future, additional studies are needed, such as surveys on the awareness of different stakeholders to this issue, implementation of the unblinding procedures at patient level in real world, and cohort studies exploring the effect of blinding to the prognosis of trial participants.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
The initial conceptual framework for the viewpoint was developed by WD together with TY and LN, and WD, MS, HH, CD, TY, and LN collaborated in refining it. The first draft of the article was written by WD and MS. Literature search and data collection were done by MS. The figures in this article were drawn by WD. Data analysis and data interpretation were accessed by CD and HH separately. All authors contributed to the article and approved the submitted version.
Funding
The analysis and interpretation of the manuscript were supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (Construction and Application of Clinical Trial and Institution Evaluation System) (2021-I2 M-1-045).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.1082445/full#supplementary-material
References
1. U.S Food and Drug Administration. Guidance for Industry: Placebos and Blinding in Randomized Controlled Cancer Clinical Trials for Drug and Biological Products. (2019). Available online at: https://www.fda.gov/media/130326/download (accessed May 16, 2022).
2. International Conference on Harmonization. ICH Efficacy Guidelines E6 R2. (2016). Available online at: https://database.ich.org/sites/default/files/E6_R2_Addendum.pdf (accessed May 16, 2022).
3. Van Delden JJ, van der Graaf R. Revised CIOMS international ethical guidelines for health-related research involving humans. JAMA. (2017) 317:135–6. doi: 10.1001/jama.2016.18977
4. National Medical Products Administration, and National Health Commission of China. The Announcement by National Medical Products Administration and National Health Commission on Issuing the Good Clinical Practice. (2020). Available online at: https://www.nmpa.gov.cn/zhuanti/ypzhcglbf/ypzhcglbfzhcwj/20200426162401243.html (accessed May 16, 2022).
5. Blader JC. Can keeping clinical trial participants blind to their study treatment adversely affect subsequent care? Contemp Clin Trials. (2005) 26:290–9. doi: 10.1016/j.cct.2005.01.003
6. U.S Food and Drug Administration. Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. (2018). Available online at: https://www.fda.gov/media/71195/download (accessed May 16, 2022).
7. Center of Drug Evaluation of China. Guidance for Endpoint Technology of Clinical Trials of Anticancer Drugs. (2012). Available online at: https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=24d174aa6995cf17e7aa12d6aa0317aa
8. Gyawali B, Booth CM. Cancer treatments should benefit patients: a common-sense revolution in oncology. Nat Med. (2022) 28:617–20. doi: 10.1038/s41591-021-01662-6
9. Demetri GD, Reichardt P, Kang YK, Blay JY, Joensuu H, Schaefer K, et al. Final overall survival (OS) analysis with modeling of crossover impact in the phase III GRID trial of regorafenib vs placebo in advanced gastrointestinal stromal tumors (GIST). J Clin Oncol. (2016) 34:156. doi: 10.1200/jco.2016.34.4_suppl.156
Keywords: unblinding, disease progression, ethics, double-blinded randomized controlled trials, clinical trials design
Citation: Dawei W, Shuangman M, Huiyao H, Dandan C, Yu T and Ning L (2022) Unblinding at disease progression in double-blinded randomized controlled cancer drug clinical trials: A controversy requires more attention. Front. Med. 9:1082445. doi: 10.3389/fmed.2022.1082445
Received: 07 November 2022; Accepted: 28 November 2022;
Published: 13 December 2022.
Edited by:
Beatriz S. Lima, Research Institute for Medicines (iMed.ULisboa), PortugalReviewed by:
Domenico Criscuolo, Italian Society of Pharmaceutical Medicine, ItalyCopyright © 2022 Dawei, Shuangman, Huiyao, Dandan, Yu and Ning. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tang Yu, dGFuZ3l1X2NhbXNAMTYzLmNvbQ==; Li Ning, bGluaW5nQGNpY2Ftcy5hYy5jbg==
†These authors have contributed equally to this work