
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 22 December 2022
Sec. Pathology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1070828
 Christopher Cronin1*†
Christopher Cronin1*† Ronan McLaughlin2Louise Lane3Francesca M. Brett3Michael Jansen4
Ronan McLaughlin2Louise Lane3Francesca M. Brett3Michael Jansen4 Niamh Bermingham4Gerald Wyse5Liam Grogan2Patrick G. Morris2
Niamh Bermingham4Gerald Wyse5Liam Grogan2Patrick G. Morris2 Seamus O’Reilly1†
Seamus O’Reilly1†BRAF V600E oncogene mutations have been reported in multiple central nervous system (CNS) tumor types, and emerging evidence supports the use of targeted therapy in BRAF-mutated gliomas. BRAF oncogene mutations have been recently identified in Rosai-Dorfman disease (RDD)—a rare non-Langerhans cell histiocytosis. This series describes three patients from two neurosurgical centers in Ireland with BRAF V600E-mutated CNS tumors. The study participants include a 19-year-old male patient with ganglioglioma with anaplastic features, a 21-year-old male patient with CNS involvement of RDD, and a 28-year-old female patient with ganglioglioma with anaplastic features. Two patients received radiation with concurrent temozolomide before BRAF-targeted therapy. This case series describes clinical and radiological responses to BRAF-targeted therapy in BRAF V600E-mutated gliomas across multiple tumor grades and is only the second published report of response to targeted therapy in BRAF-mutated RDD. The durability of disease control with BRAF-targeted therapy was generally superior to that achieved with chemoradiation; one patient has experienced ongoing disease control for 5 years. The reported case of treatment response in BRAF-mutated RDD supports the strategy of genotyping and utilization of targeted therapy in this rare disease. The optimal sequencing of BRAF-targeted therapy in BRAF-mutated gliomas/glioneuronal tumors remains unclear, and further prospective studies are required to guide the use of genome-matched therapy in this patient population.
Activating mutations in the BRAF oncogene are well described in many central nervous system (CNS) tumors. A large study assessing more than 1,000 CNS tumors of various types identified a 7% incidence of BRAF mutations (most commonly in pleomorphic xanthoastrocytoma (PXA) and World Health Organization (WHO) grade 1 ganglioglioma subtypes) (1).
Multiple case reports and series have described therapeutic responses to BRAF-targeted therapy in BRAF V600E-mutated gliomas across all WHO tumor grades in both pediatric and adult populations, including some cases of complete tumor regression (2–5). Phase II data from the VE-BASKET trial and the ROAR basket trial have also demonstrated the activity of BRAF-targeted therapy in this patient population (6, 7).
Rosai-Dorfman disease (RDD) is a rare non-Langerhans cell histiocytosis. CNS involvement occurs in less than 5% of cases (8). BRAF V600 mutations have recently been identified in patients with RDD (9). Response to BRAF-targeted therapy has been reported in one patient with mixed RDD and Langerhans cell histiocytosis harboring a BRAF V600E mutation (10).
This series describes the cases of three patients with BRAF V600-mutated CNS tumors in whom BRAF-targeted therapy was utilized, including one of the first reported cases of response to targeted therapy in BRAF V600E-mutated RDD. We describe the duration of treatment, the response achieved, and any relevant treatment-associated toxicity.
An 18-year-old male patient was referred to the neurology service with a history of slowly progressive left lower limb ataxia. Magnetic resonance imaging (MRI) of the brain demonstrated a 4.7-cm cystic lesion within the roof of the right lateral ventricle with a 3-cm solid component involving the corpus callosum. The patient proceeded to surgical resection, and operative histology demonstrated a mixed tumor with glial and neuronal components. Cells were largely NF52 positive on immunostaining with occasional positive staining for GFAP and CD34. The appearances were consistent with a ganglioglioma, WHO grade 1.
Approximately 8 months post-surgical resection, the patient presented with increasing headaches. An MRI of brain with contrast was performed, which demonstrated a significant increase in the size of an enhancing mass lesion on the inferomedial aspect of the right frontal lobe consistent with tumor progression. The patient underwent further tumor debulking. Histology demonstrated cells with piloid morphology with regularly interspersed dysplastic neurons (see Figure 1). Glial cells were mitotically active and remained positive for NF52 on immunostaining. CD34 labeled tumor cells only and IDH staining was negative (see Figure 2). The histopathological findings were deemed consistent with high-grade tumour transformation to ganglioglioma with anaplastic features. Immunohistochemistry (IHC) demonstrated BRAF V600E expression, and further genotyping using the Idylla™ BRAF mutation test confirmed the presence of a BRAF V600E mutation.
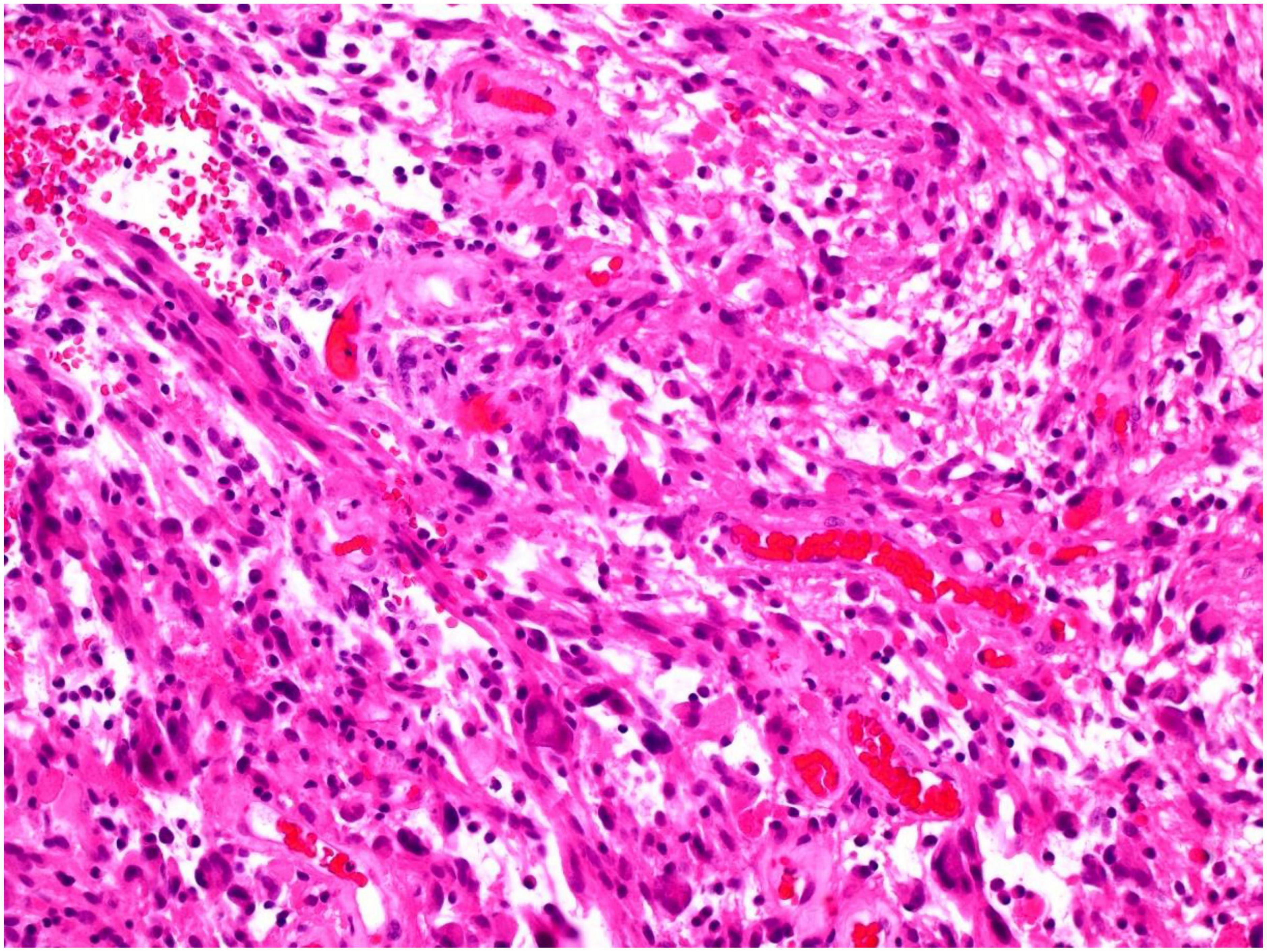
Figure 1. Tumor showing piloid morphology with regularly interspersed dysplastic neurons (H&E 100x).
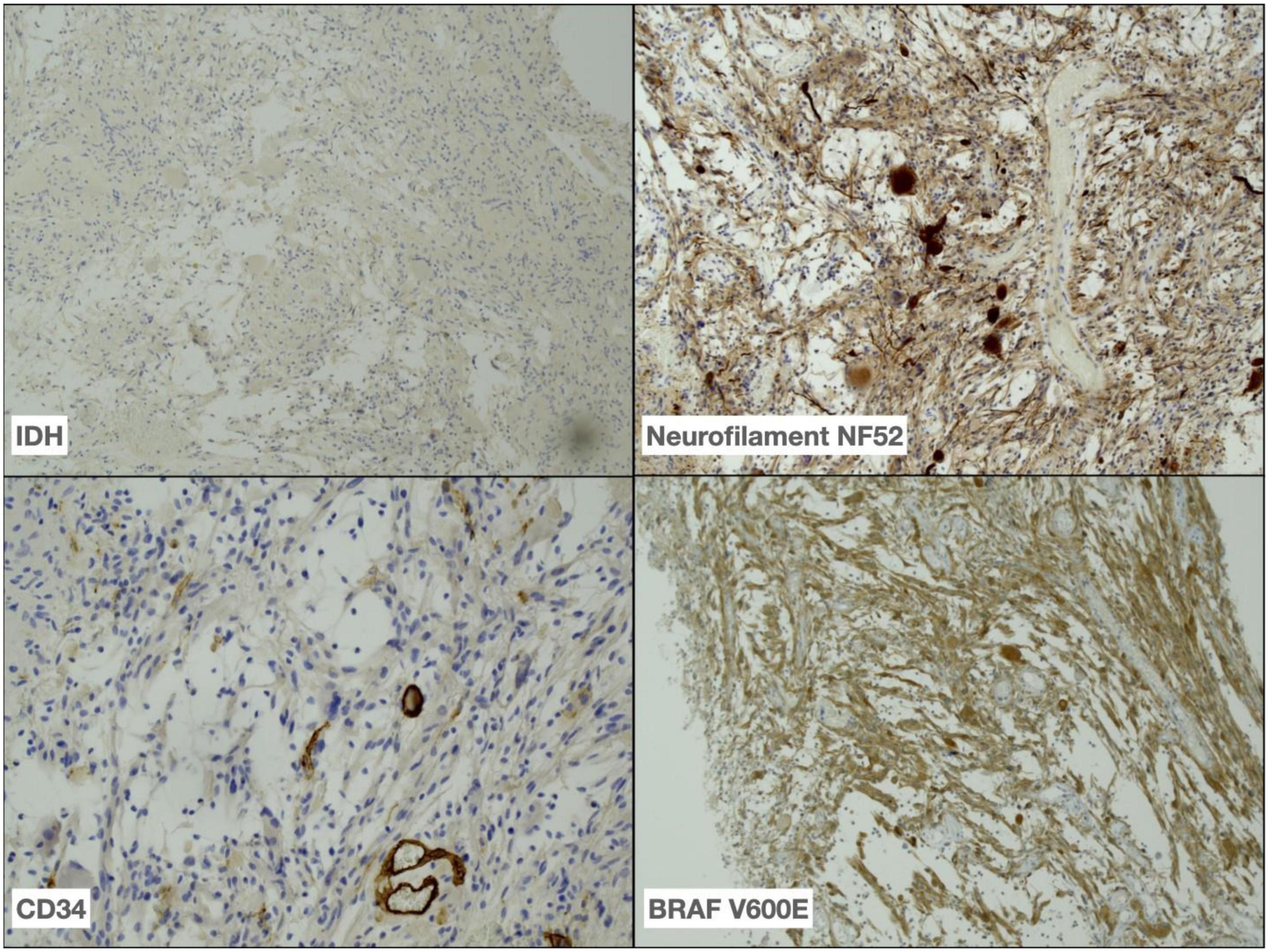
Figure 2. Immunohistochemistry performed on tumor demonstrating absence of IDH R132H expression (40x), positive neurofilament NF52 (40x) and less frequent CD34 (100x) expression in tumour as well as BRAF V600E (40x) expression in tumour cells.
The patient proceeded to concurrent chemoradiation, receiving 60 Gray in 33 fractions with concurrent oral temozolomide as part 1 of the STUPP regimen (11). He subsequently commenced oral temozolomide as part 2 of the STUPP regimen. An MRI of the brain 2 months post-completion of radiotherapy demonstrated further progression of the disease. The patient was commenced on the oral BRAF inhibitor dabrafenib. Further neuroimaging 4 months post-commencement of BRAF-targeted therapy demonstrated a significant reduction in the size of the right cerebral mass lesion consistent with partial response to BRAF-targeted therapy (see Figure 3). The patient has remained on dabrafenib therapy for more than 5 years, and interval neuroimaging studies have demonstrated ongoing disease control.

Figure 3. Axial T1 MRI images ganglioglioma pre-BRAF therapy (A) and 4 months post-BRAF therapy (B).
A 12-year-old male patient presented with failure to thrive and short stature on a background of childhood hypopituitarism resulting in diabetes insipidus, requiring desmopressin therapy. MRI of the brain revealed a large, enhancing suprasellar mass extending into the middle fossa bilaterally with encasement of both carotid arteries.
The patient underwent craniotomy and a biopsy of the suprasellar lesion. Operative histology demonstrated the nodular distribution of large histiocytes within a background of fibrous tissue. The cells within nodules were cytoplasm rich with occasional large hypochromatic nuclei and demonstrated emperipolesis (see Figure 4). By IHC, cells were positive for KP-1 and CD68 and variably positive for S100. Staining was negative for CD1a, GFAP, Desmin, and SMSA. The histology was sent for external review, and the appearances were deemed most consistent with a CNS manifestation of RDD, a non-Langerhans cell histiocytosis.
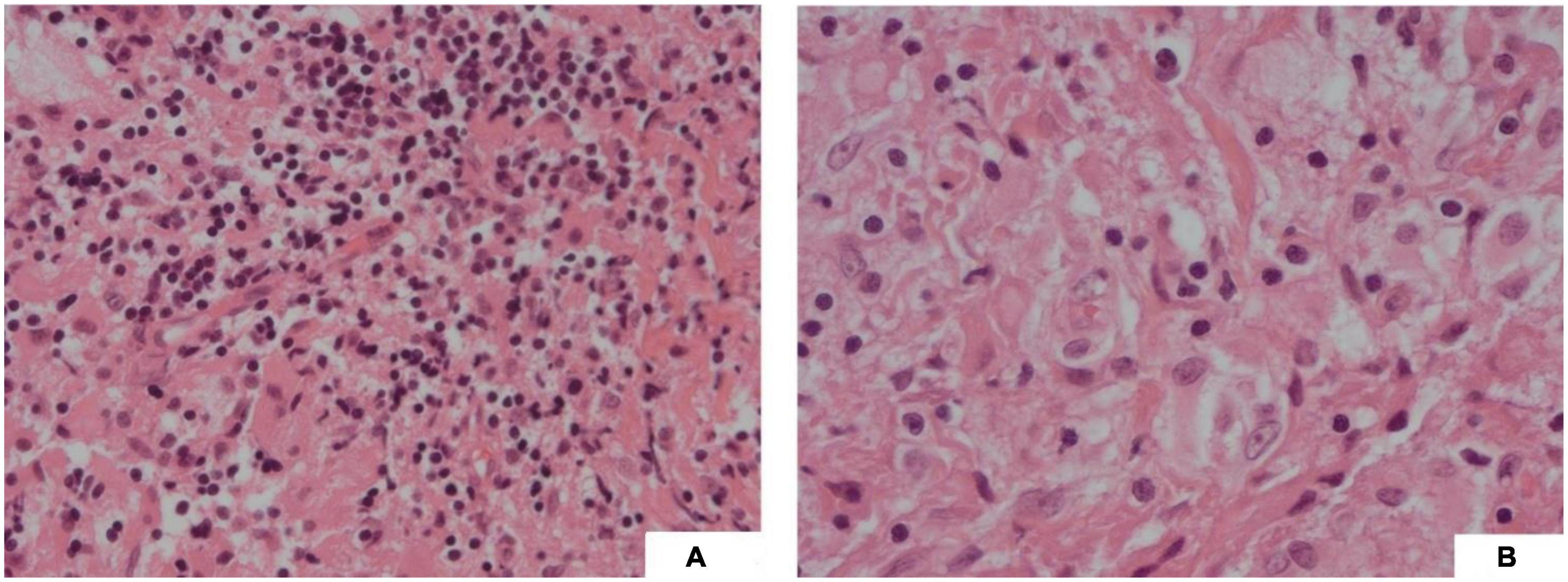
Figure 4. (A) (H&E, 20x) Fibrous type tissue with lymphocytic infiltration and vacuolated histiocytes; (B) (H&E, 40x) Vacuolated cells showing emperipolesis.
Further MRI of the brain approximately 4 years post-initial diagnosis demonstrated significant disease progression. A 6-week trial of high-dose dexamethasone did not result in any reduction in tumor burden. Further small-volume disease progression was noted over the following 12 months, and the patient subsequently underwent whole-brain radiotherapy (10 Gray in five fractions) approximately 5 years following initial diagnosis.
Approximately 3 years post-completion of whole-brain radiotherapy, the patient developed a new left-sided visual disturbance. MRI of the brain revealed the progression of the previously visualized pituitary tumor. The patient proceeded to endoscopic transsphenoidal debulking and biopsy of the suprasellar tumor. Morphological appearance remained consistent with RDD. BRAF genotyping using the Cobas® 4800 BRAF V600 mutation test assay demonstrated a BRAF V600 mutation.
The patient was commenced on BRAF/MEK inhibitor combination therapy with encorafenib and binimetinib. Interval MRI of the brain was performed 4 months post-commencement of BRAF/MEK inhibitor therapy, which demonstrated treatment response (see Figure 5). After approximately 16 months of therapy, the patient’s BRAF/MEK inhibitors were held due to treatment-associated uveitis requiring topical and systemic steroid therapy, and the patient’s therapy remains on hold. MRI of the brain approximately 20 months post-commencement of targeted therapy demonstrated ongoing disease control.
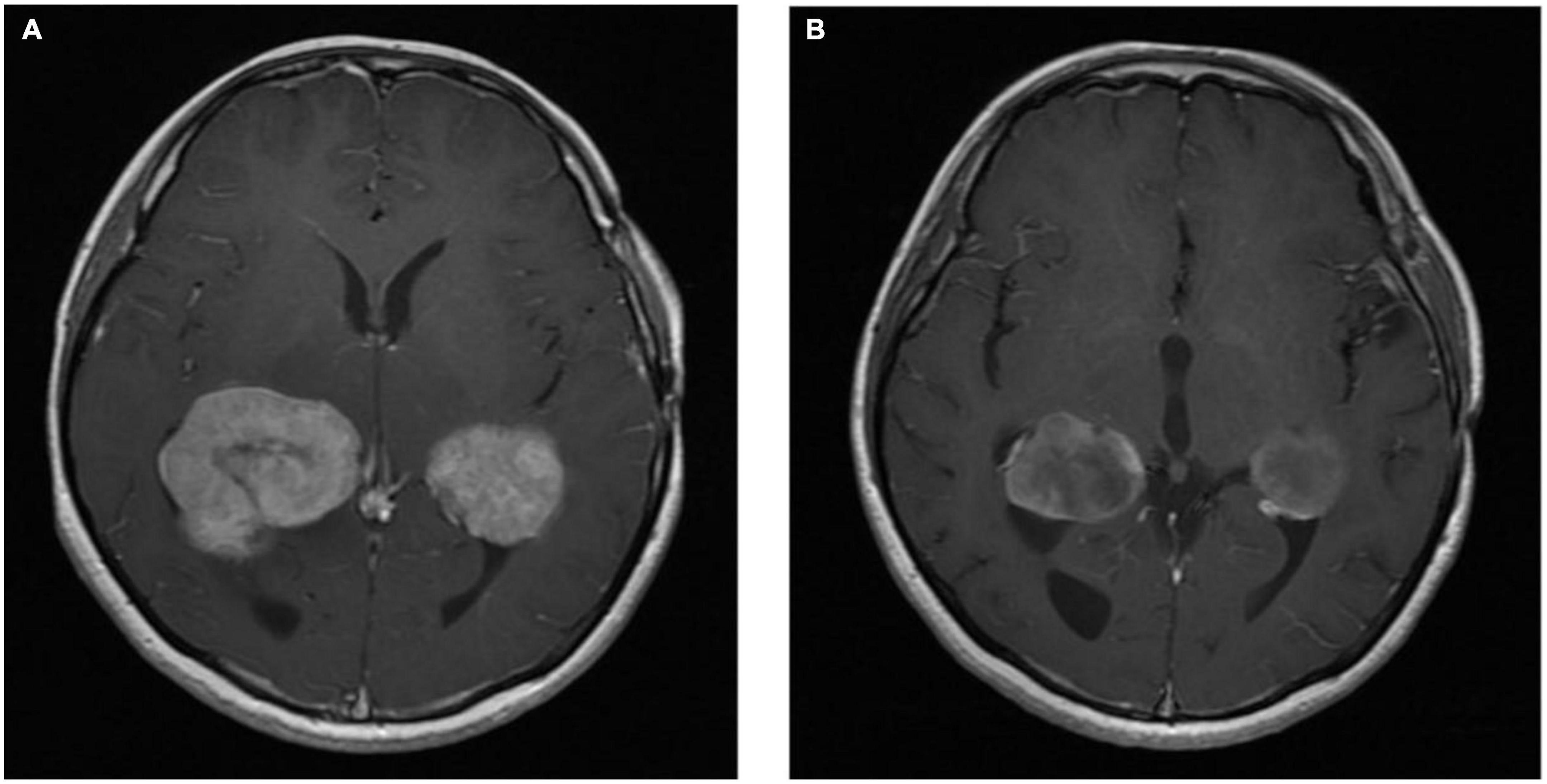
Figure 5. Axial T1 post-contrast MRI images Rosai-Dorfman disease pre-BRAF therapy (A) and 3 months post-BRAF therapy (B).
A 21-year-old female patient presented with a history of headaches, visual disturbance, and left-sided facial weakness. MRI of the brain demonstrated a large mass involving the right thalamic region measuring 5.7 cm with mixed solid and cystic components. The patient underwent right frontotemporal craniotomy and subtotal excision of the tumor. Histology demonstrated tumor cells with marked cellular and nuclear pleomorphism. Visible nuclei were mostly irregular and hyperchromatic with occasional pseudoinclusions. A small proportion of tumor cells display ganglion cell morphology. Mitotic figures were not readily identified. Immunostaining was densely positive for GFAP in pilocytic areas, with more global positive staining for MAP2, synaptophysin, and neurofilament protein in the pleomorphic cells. Histology was consistent with a ganglioglioma (WHO grade 1).
Approximately 4 years post-resection, the patient developed headaches and progressive ataxia. MRI of the brain revealed a significant increase in the size of the medial temporal lesion. The patient underwent near-complete tumour debulking. Histology demonstrated increased nuclear atypia within the pleiomorphic cell population, and mitotic figures visible within the ganglion cell-derived population consistent with tumor transformation to ganglioglioma with anaplastic features (IDH wild type; ATRX wildtype). BRAF genotyping was performed using the Cobas® 4800 BRAF V600 mutation test assay and demonstrated a BRAF V600 mutation. Sequencing did not identify a BRAF fusion. She proceeded to adjuvant radiotherapy with concurrent temozolomide as per the STUPP regimen. She completed a further 6 months of temozolomide therapy. Three months post-completion of temozolomide MRI of the brain demonstrated re-accumulation of the cystic changes within the right basal ganglia/thalamus and right brain stem consistent with disease progression.
The patient was commenced on BRAF-targeted therapy in the form of the BRAF inhibitor encorafenib in combination with the MEK inhibitor binimetinib. MRI of the brain 3 months post-initiation of BRAF-targeted therapy demonstrated interval reduction in the size of the cystic tumor lesions in the right basal ganglia and midbrain suggestive of treatment response (see Figure 6). The patient remains on therapy, and MRI of the brain 10 months post-treatment initiation demonstrates ongoing disease control.

Figure 6. Axial T1 MRI images ganglioglioma pre-BRAF therapy (A) and 3 months post-BRAF therapy (B).
Gliomas demonstrate significant heterogeneity in terms of molecular characteristics, as well as disease biology and natural history (12, 13). Systemic therapies for recurrent or unresectable gliomas have demonstrated only modest clinical benefit (14–17). BRAF mutation testing may be carried out in such cases to guide treatment. NCCN guidelines recommend sequencing methods to detect BRAF mutations even though immunohistochemical techniques are also widely utilized in this context. A study assessing IHC testing for BRAF V600E mutations in PXA demonstrated 100% concordance with sequencing-based molecular analysis (18). In other studies, small numbers of BRAF mutations were identified in primary CNS tumors using sequencing methods that were not identified on IHC. IHC methods may be limited in cases where low variant allele frequency is present, while sequencing methods may be limited by the volume of tissue available for molecular analysis (19, 20). A combined-modality testing strategy may be considered where available.
The VE-BASKET trial (an open-label non-randomized trial assessing the BRAF inhibitor vemurafenib in BRAF V600-mutated non-melanoma cancers) reported an objective response rate of 25% across all gliomas studied (6). An interim analysis from the phase II, open-label ROAR basket trial assessing the combination of BRAF/MEK inhibitor therapy with dabrafenib and trametinib in BRAF V600E-mutated gliomas reported objective response rates of 33% and 69% in the high-grade and low-grade cohorts, respectively (7). Emerging evidence highlights a degree of genetic heterogeneity in BRAF mutations across age groups. A recently published molecular analysis of more than 200 BRAF-mutant adult gliomas described distinct molecular features compared to pediatric populations, including a lower prevalence of BRAF V600E mutations in adults (21).
Gangliogliomas are rare CNS tumors, most often occurring in children and young adults, which themselves compromise a broad spectrum of disease biology and natural history. Retrospective data have demonstrated 15-year overall survival of 94% in low-grade gangliogliomas (22). Transformation, including the development of anaplastic features, was associated with a median survival of less than 1 year (23). The prevalence of BRAF V600 mutations in gangliogliomas has been reported as up to 40% in a systematic review (24). A next-generation sequencing study of a cohort of 40 gangliogliomas demonstrated mutations involved in the MAP-kinase pathway activation in 36 cases (25). BRAF V600E mutations were detected in 50% of these cases, with multiple rare variants also being reported.
Rosai-Dorfman disease is a rare non-Langerhans cell histiocytosis characterized by the accumulation of histiocytes within affected tissues. RDD demonstrates significant heterogeneity in terms of the clinical phenotype (8). Extra-nodal manifestations are observed in 43% of cases, with < 5% of cases involving the CNS. Most cases of RDD are self-limiting. Corticosteroids and immunomodulatory therapies, such as sirolimus, have shown benefits in cases of progressive disease. KRAS and MEK gene alterations are present in approximately 40% of RDD cases. A recent retrospective cohort study demonstrated clinically significant response rates using the MEK inhibitor cobimetinib in KRAS and MEK-variant RDD (26). BRAF oncogene mutations are a well-established molecular driver and treatment target in other histiocytoses such as Langerhans cell histiocytosis and Erdheim-Chester disease (27, 28). Such molecular alterations have recently been identified in RDD (10). To our knowledge, there is only one previously published case report of response to BRAF-targeted therapy in RDD, which describes the clinical and radiological response to the BRAF inhibitor dabrafenib in a pediatric patient with mixed Langerhans cell histiocytosis and RDD harboring a BRAF V600E mutation (10).
Our case series describes clinically meaningful responses to BRAF-targeted therapy in BRAF V600E-mutated CNS tumors, including the second published case of response to targeted therapy in BRAF-mutated RDD and a durable response of more than 5 years in a patient with BRAF-mutated ganglioglioma. Compared with targeted therapy, the duration of disease control was shorter for the two patients in our series who underwent concurrent chemoradiation with temozolomide. Comparison of the duration of disease control between these groups and individual responses to BRAF-targeted therapy are described in our swimmers’ plot (see Figure 7).
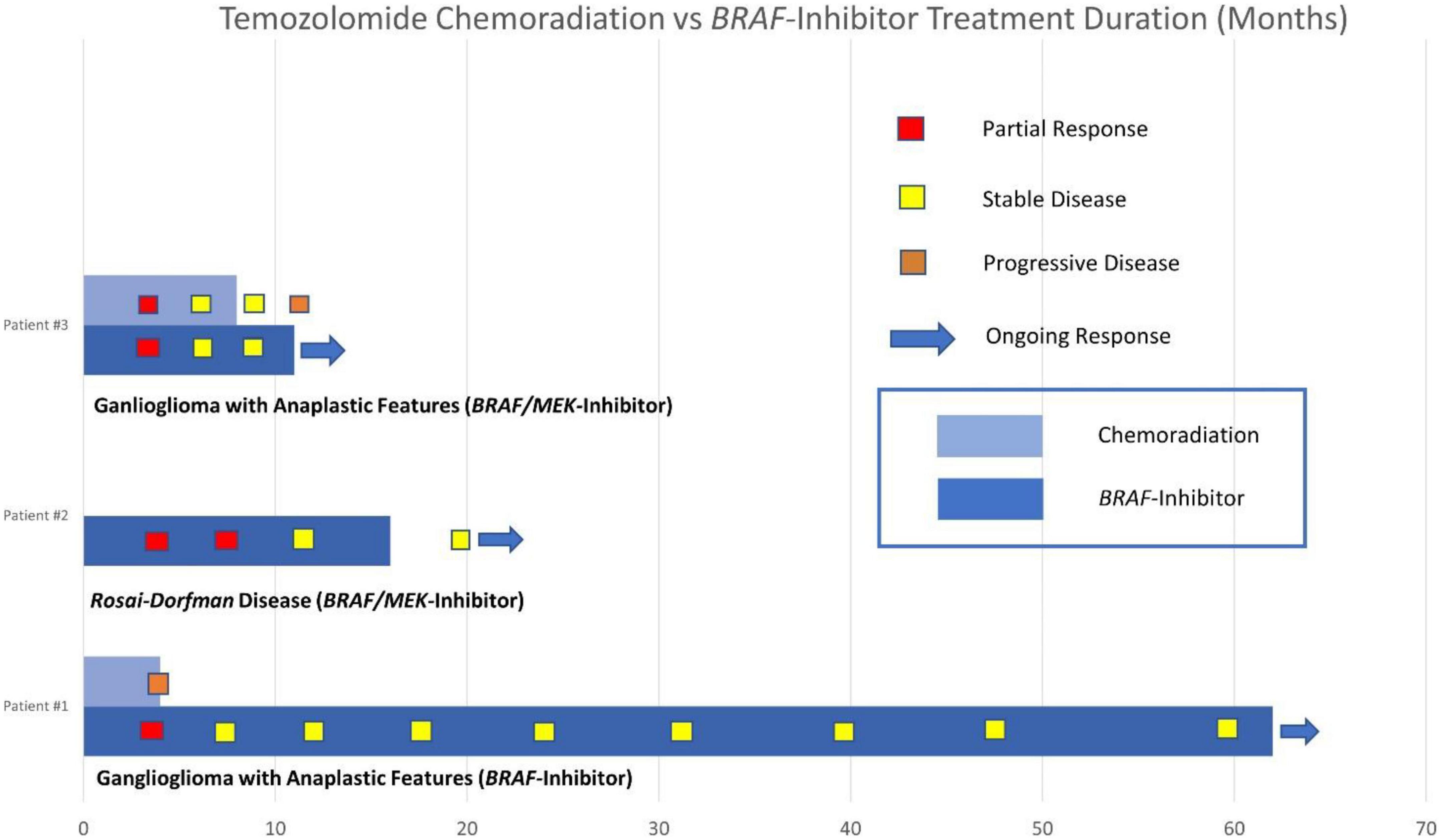
Figure 7. Swimmers plot duration of disease control with temozolomide chemoradiation vs. BRAF inhibitor therapy.
This series contributes to the limited published data on targeted therapy in BRAF-mutated CNS tumors and includes an exceptional case describing the response to BRAF inhibitor therapy in BRAF-mutated RDD. The molecular and genetic characterization of RDD and downstream implications for the use of targeted therapy require further investigation and collaboration (9). Our case of CNS RDD demonstrates the utility of BRAF-targeted therapy and supports the strategy of genotyping in this rare condition. Our series describes more durable disease control in gangliogliomas with high-grade transformation treated with BRAF-targeted therapy compared with chemoradiation with temozolomide and raises questions regarding the optimal sequencing of treatment in recurrent/unresectable disease, particularly in cases where molecular characterization is predictive of a lesser response to alkylating chemotherapy. Prospective studies are required to further establish the optimal role of genome-matched therapy in this phenotypically and genetically diverse patient population.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
CC and SO’R: conceptualization and methodology. CC: data curation and writing of the original draft. CC, RM, FB, MJ, NB, and LL: investigation. CC, FB, MJ, NB, and LL: visualization. CC, SO’R, RM, FB, MJ, NB, LL, GW, LG, and PM: writing—reviewing and editing. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. (2011) 121:397–405. doi: 10.1007/s00401-011-0802-6
2. Fusco M, Piña Y, Macaulay R, Sahebjam S, Forsyth P, Peguero E, et al. Durable progression-free survival with the use of BRAF and MEK inhibitors in four cases with BRAF V600E-mutated gliomas. Cancer Control. (2021) 28:10732748211040013. doi: 10.1177/10732748211040013
3. Toll S, Tran H, Cotter J, Judkins A, Tamrazi B, Biegel J, et al. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget. (2019) 10:551–7. doi: 10.18632/oncotarget.26560
4. Woo P, Lam T, Pu J, Li L, Leung R, Ho J, et al. Regression of BRAFV600E mutant adult glioblastoma after primary combined BRAF-MEK inhibitor targeted therapy: a report of two cases. Oncotarget. (2019) 10:3818–26.
5. Chamberlain M. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neuro Oncol. (2013) 114:237–40. doi: 10.1007/s11060-013-1176-5
6. Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart A, et al. BRAF inhibition in BRAFV600-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. (2018) 36:3477–84. doi: 10.1200/jco.2018.78.9990
7. Wen P, Stein A, van den Bent M, De Greve J, Wick A, de Vos F, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. (2021) 23:53–64. doi: 10.1016/s1470-2045(21)00578-7
8. Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. (2018) 131:2877–90. doi: 10.1182/blood-2018-03-839753
9. Fatobene G, Haroche J, Hélias-Rodzwicz Z, Charlotte F, Taly V, Ferreira A, et al. BRAF V600E mutation detected in a case of Rosai-Dorfman disease. Haematologica. (2018) 103:e377–9. doi: 10.3324/haematol.2018.190934
10. Mastropolo R, Close A, Allen S, McClain K, Maurer S, Picarsic JBRAF-. V600E–mutated Rosai-Dorfman-Destombes disease and Langerhans cell histiocytosis with response to BRAF inhibitor. Blood Adv. (2019) 3:1848–53. doi: 10.1182/bloodadvances.2019000093
11. Stupp R, Mason W, van den Bent M, Weller M, Fisher B, Taphoorn M, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/nejmoa043330
12. Nabors L, Portnow J. NCCN Clinical Practice Guidelines in Oncology: Central Nervous System Cancers. (2021). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed December 7, 2021).
13. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
14. Friedman H, Prados M, Wen P, Mikkelsen T, Schiff D, Abrey L, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. (2009) 27:4733–40. doi: 10.1200/jco.2008.19.8721
15. Kreisl T, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. (2009) 27:740–5. doi: 10.1200/jco.2008.16.3055
16. Taal W, Oosterkamp H, Walenkamp A, Dubbink H, Beerepoot L, Hanse M, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a andomized controlled phase 2 trial. Lancet Oncol. (2014) 15:943–53. doi: 10.1016/s1470-2045(14)70314-6
17. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. (2017) 377:1954–63. doi: 10.1056/nejmoa1707358
18. Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM, et al. Immunohistochemistry is highly sensitive and specific for detection of Braf V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathol Commun. (2013) 1:20. doi: 10.1186/2051-5960-1-20
19. Durślewicz J, Klimaszewska-Wiśniewska A, Antosik P, Kasperska A, Grzanka D, Szylberg T, et al. Detection of braf V600E mutation in ganglioglioma and pilocytic astrocytoma by immunohistochemistry and real-time PCR-based Idylla Test. Dis Mark. (2020) 2020:8880548. doi: 10.1155/2020/8880548
20. Tirrò E, Massimino M, Broggi G, Romano C, Minasi S, Gianno F, et al. A custom DNA-based NGS panel for the molecular characterization of patients with diffuse gliomas: diagnostic and therapeutic applications. Front Oncol. (2022) 12:861078. doi: 10.3389/fonc.2022.861078
21. Schreck K, Langat P, Li T, Bhave V, Pratilas C, Eberhart C, et al. Integrated molecular and clinical analysis of BRAF-mutant glioma in adults. Eur J Cancer. (2022) 174:S3–4. doi: 10.1016/S0959-8049(22)00816-4
22. Compton JJ, Issa Laack NN, Eckel LJ, Schomas DA, Giannini C, Meyer FB. Long-term outcomes for low-grade intracranial ganglioglioma: 30-year experience from the Mayo Clinic. J Neurosurg. (2012) 117:825–30. doi: 10.3171/2012.7.JNS111260
23. Zaky W, Patil SS, Park M, Liu D, Wang W-L, Wani KM, et al. Ganglioglioma in children and young adults: single institution experience and review of the literature. J Neuro Oncol. (2018) 139:739–47. doi: 10.1007/s11060-018-2921-6
24. Andrews LJ, Thornton ZA, Saincher SS, Yao IY, Dawson S, McGuinness LA, et al. Prevalence of brafV600 in glioma and use of braf inhibitors in patients with brafV600 mutation-positive glioma: systematic review. Neuro Oncol. (2021) 24:528–40. doi: 10.1093/neuonc/noab247
25. Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP, et al. The genetic landscape of Ganglioglioma. Acta Neuropathol Commun. (2018) 6:47. doi: 10.1186/s40478-018-0551-z
26. Abeykoon JP, Rech KL, Young JR, Ravindran A, Ruan GJ, Dasari S, et al. Outcomes after treatment with Cobimetinib in patients with Rosai-Dorfman disease based on kras and mek alteration status. JAMA Oncol. (2022) [Online ahead of print]. doi: 10.1001/jamaoncol.2022.4432.
27. Rodriguez-Galindo C, Allen C. Langerhans cell histiocytosis. Blood. (2020) 135:1319–31. doi: 10.1182/blood.2019000934
Keywords: BRAF, CNS tumors, brain tumor, glioma, Rosai-Dorfman disease, BRAF-inhibitor, ganglioglioma
Citation: Cronin C, McLaughlin R, Lane L, Brett FM, Jansen M, Bermingham N, Wyse G, Grogan L, Morris PG and O’Reilly S (2022) Case report: BRAF-inhibitor therapy in BRAF-mutated primary CNS tumours including one case of BRAF-mutated Rosai-Dorfman disease. Front. Med. 9:1070828. doi: 10.3389/fmed.2022.1070828
Received: 24 October 2022; Accepted: 29 November 2022;
Published: 22 December 2022.
Edited by:
Lorenzo Memeo, The Mediterranean Institute of Oncology (IOM), ItalyReviewed by:
Giuseppe Broggi, University of Catania, ItalyCopyright © 2022 Cronin, McLaughlin, Lane, Brett, Jansen, Bermingham, Wyse, Grogan, Morris and O’Reilly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Cronin, ✉ ZHJjaHJpc3RvcGhlcmNyb25pbkBnbWFpbC5jb20=
†ORCID: Christopher Cronin, orcid.org/0000-0002-2715-7273; Seamus O’Reilly, orcid.org/0000-0002-2887-7336
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.