 Yvette C. Tanhehco
Yvette C. Tanhehco Ghazala Nathu2†
Ghazala Nathu2† Ljiljana V. Vasovic
Ljiljana V. Vasovic- 1Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, NY, United States
- 2Department of Clinical Pathology, Bassett Healthcare Network—Cobleskill Regional Hospital, Cobleskill, NY, United States
- 3Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY, United States
Recent advances in managing Sickle Cell Disease (SCD) significantly improved patient survival and quality of life. Disease-modifying drug therapies such as hydroxyurea, L-glutamine, voxelotor, and crizanlizumab reduce pain crises and severe complications. Allogeneic hematopoietic stem cell transplantation using matched-sibling donors is currently the only standard curative option; however, only a small proportion of patients have such donors. Cord blood and haploidentical transplantation with a modified conditioning regimen have expanded the allogeneic donor pool, making the therapy available to more patients. Gene therapy is a promising cure that is currently undergoing clinical trials and different approaches have demonstrated efficacy. Multidisciplinary expertise is needed in developing the best treatment strategy for patients with SCD.
Introduction
Sickle Cell Disease (SCD) is caused by a single point mutation in the hemoglobin beta gene that codes for a valine in the sixth amino acid position instead of the wild type glutamic acid creating hemoglobin S (HbS) (1). Under deoxygenated conditions, HbS polymerizes causing erythrocytes to assume a sickle shape (2). These HbS-containing erythrocytes are insufficient for transporting oxygen. The conformational change of erythrocytes and increased surface expression of adhesion molecules can lead to blood vessel occlusion, painful vaso-occlusive crises (VOC), and long-term damage to tissues and organs (3).
SCD is among the most prevalent inherited monogenic disorders worldwide. It affects Black Americans more than any other racial group. It is believed that more than 20 million people worldwide and 80,000–100,000 Americans battling SCD, with an average of 3,000 children being born with it annually in the United States (US) (4). Newborn screening programs, early interventions, and preventive care improved pediatric patients' survival. Nevertheless, the life span of patients with SCD is about 20 years shorter than the general population. The National Institutes of Health Cure Sickle Cell Initiative established a collaborative, patient-centric research program aimed at discovering effective genetic treatments for SCD. This is a systematic effort to support the development of curative therapies and empower understanding of basic research, clinical trials, and patient support which have wide implications for educational and community engagement activities (5).
Traditional therapies for SCD include pain and symptom management. In certain cases, physicians might recommend a blood transfusion in order to instill an appropriate amount of hemoglobin A containing erythrocytes, prevent strokes, and treat anemia (6, 7). Simple transfusion, chronic transfusion or red blood cell exchange protocols have been utilized. As with any transfusion, there is a risk for alloimmunization due to incomplete phenotypic matches, iron accumulation, and other transfusion-related adverse events.
The first disease modifying drug therapy that was used to reduce complications is hydroxyurea (Table 1). Subsequently, the US Food and Drug administration (FDA) approved L-glutamine, voxelotor, and crizanlizumab-tmca to reduce pain crises, sickling, and VOC which have all significantly improved the quality of life of patients with SCD (7).
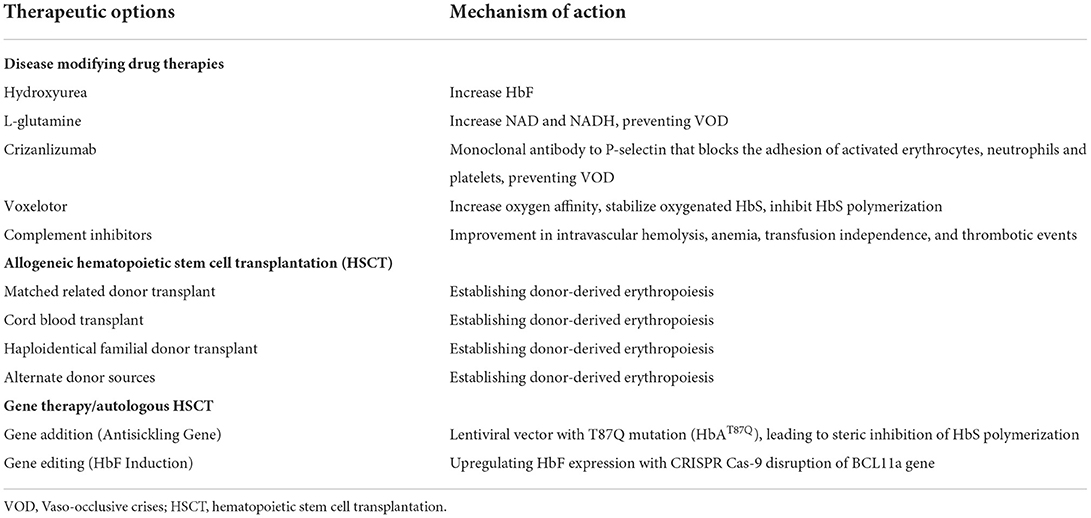
Table 1. Sickle cell disease therapeutic options.
Thus, far, the only curative approach to SCD has been a hematopoietic stem cell (HSC) transplantation. Allogeneic stem cells lacking the genetic mutation will yield healthy erythrocytes in the recipient (8). However, the potential complications of an allogenic transplant, such as rejection or graft-vs. host disease (GVHD), are considerable potentially causing morbidity and death. The major hurdle of procuring a fully matched HLA donor can be circumvented by using cord blood and haploidentical donors.
There is a new treatment paradigm on the horizon that aims to cure SCD by correcting the core issue (i.e., the genetic mutation) through gene therapy. Gene therapy approaches are variable and may involve introducing a new gene into the patient to be integrated and expressed or to modify the native gene itself through gene editing. All of these methodologies require that the HSCs, the targets of these therapies, be collected and harvested from the patient. Genetic modification then ensues in-vitro followed by transplantation back to the patient after the residual marrow population is ablated using chemotherapy (9).
This minireview discusses the available therapeutic modalities for SCD including gene therapy.
Disease modifying drug therapies
Hydroxyurea
Hydroxyurea (HU) is a disease modifying drug (DMD) therapy that was approved by the US FDA in 1998 for the treatment of adults (>18 years) with SCD who have had at least 3 painful crises in the past year (10). HU increases the level of fetal hemoglobin (HbF) and reduces the tendency for HbS to polymerize, preventing RBCs from sickling and causing VOC (11). HU also reduces circulating leukocytes and inflammatory mediators. A double-blind, randomized clinical trial enrolled 148 men and 151 women at least 18 years of age with sickle cell anemia at 21 clinics. Study subjects must have had at least 3 crises in the year before entry into the study. Study results showed that the 152 patients who received HU treatment had lower annual rates of crises than the 147 patients who received placebo (median, 2.5 vs. 4.5 crises per year, P < 0.001). The median times to the first crisis (3.0 vs. 1.5 months, P = 0.01) and the second crisis (8.8 vs. 4.6 months, P < 0.001) were longer with HU treatment. Fewer patients who were treated with HU had chest syndrome (25 vs. 51, P < 0.001), and fewer received transfusions (48 vs. 73, P = 0.001). No significant adverse effects were observed but its use must be carefully monitored. The beneficial effects of HU did not manifest for several months and the maximally tolerated doses of HU may not be necessary to achieve a therapeutic effect (12). HU was subsequently approved in 2017 for the reduction in the frequency of painful crises and the need for blood transfusions in pediatric patients (>2 years) with SCD with recurrent moderate to severe painful crises.
L-glutamine
1In 2017, L-glutamine received approval from the FDA for the treatment of SCD. L-glutamine is an amino acid required to synthesize nicotinamide adenine dinucleotide (NAD+) and its reduced form NADH. NAD+ is an important antioxidant in RBCs, so the rationale for using L-glutamine in patients with SCD is to improve the redox ratio to reduce oxidative stress, RBC sickling and hemolysis, and decrease the frequency of VOC. A multicenter, randomized, placebo-controlled, double-blind, phase 3 trial enrolled patients with sickle cell anemia (HbSS or HbSβ0-thalassemia) at least 5 years old at 31 sites across the United States who had a history of at least 2 pain crises during the previous year (13). This clinical trial studied the efficacy of pharmaceutical-grade L-glutamine (0.3 per kilogram of body weight per dose) administered twice daily compared to placebo in reducing the incidence of pain crises. Patients who were receiving HU at least 3 months prior to screening continued that therapy throughout the 48-week treatment period. Two hundred and thirty patients (age range, 5–58 years; 53.9% female) were randomly assigned in a 2:1 ratio to receive L-glutamine (n = 152) or placebo (n = 78). Patients receiving L-glutamine had significantly fewer pain crises than those receiving placebo (median, 3 vs. 4, P = 0.005) and fewer hospitalizations (2 vs. 3, P = 0.005). The adverse effects of low-grade nausea, non-cardiac chest pain, fatigue, and musculoskeletal pain occurred more frequently in the L-glutamine group compared to the placebo group (13).
Crizanlizumab
Crizanlizumab is an anti-P selectin inhibitor that decreases the adhesion of white blood cells and RBCs to the endothelium that was approved by the FDA in 2019 based on a phase 2 randomized, double-blind, placebo-controlled trial. Patients with sickle cell disease (HbSS, HbSC, HbSβ0-thalassemia, HbSβ+-thalassemia, and other genotypes) who were between 16 and 65 years of age and had 2–10 sickle cell-related pain crises in the 12 months prior to enrollment received low-dose crizanlizumab (2.5 mg per kg body weight), high-dose crizanlizumab (5 mg/kg) or placebo, administered intravenously 14 times over 52 weeks. One hundred and ninety-eight patients were enrolled across 60 sites. The median rate of crises per year was significantly lower with high-dose crizanlizumab compared to placebo (median, 1.63 vs. 2.98, P = 0.01). The median time to first and the second crisis was significantly longer with high-dose crizanlizumab compared to placebo (first crisis, median, 4.07 vs. 1.38 months, P = 0.001; second crisis, median, 10.32 vs. 5.09 months, P = 0.02). The median rate of uncomplicated crises per year was lower with high-dose crizanlizumab compared with placebo (median, 1.08 vs. 2.91, P = 0.02). Adverse events such as arthralgia, diarrhea, pruritus, vomiting, and chest pain were observed (14).
Voxelotor
Voxelotor is another DMD that was approved by the FDA in 2019. It inhibits HbS polymerization by binding covalently to the N-terminal valine of the β-globin chain in Hb to stabilize the oxygenated HbS (11). A phase 3, multicenter, double-blind, randomized, placebo-controlled trial (HOPE trial), compared the efficacy and safety of 2 dose levels of voxelotor (1500 mg and 900 mg) with placebo in patients aged 12–65 years old with SCD (HbSS, HbSC, HbSβ-thalassemia, and other variants) of different genotypes (15). Participants (n = 274) were randomly assigned in a 1:1:1 ratio to receive a once-daily oral dose of 1500 mg of voxelotor, 900 mg of voxelotor, or placebo. Study results showed a significantly higher percentage of study subjects with a Hb response in the 1500 mg voxelotor group (51%; 95% confidence interval [CI], 41–61) compared to the 900 mg voxelotor group (33%; 95% CI, 23–42) and placebo group (7%; 95% CI, 1–12) at 24 weeks. The annualized adjusted incidence rate of VOC was 2.77, 2.76, and 3.19 in the 1500 mg, 900 mg and placebo groups, respectively. Furthermore, there was a trend toward reduced incidence of crises over time with voxelotor compared to placebo. Most adverse events were concluded to be unrelated to voxelotor or placebo. There was no significant difference of SCD-related adverse events among the 3 groups (76% 1500 mg voxelotor, 73% 900 mg voxelotor, and 73% placebo group) (15).
Complement inhibitors
One area of growing interest in SCD pathogenesis is complement activation, the protein-based arm of the innate immune system. The first report on complement activation in SCD was published in 1967 by Francis and Womack in which they discovered high levels of serum complement markers in patients with SCD (16). Alternative pathway (AP) of complement activation may be implicated in SCD; anaphylatoxins C3a and C5a were determined to be increased during a VOC (17, 18). In adults with SCD levels of sC5b-9, a marker of complete complement activation, was significantly elevated compared to those without SCD (19, 20). Of note is that hydroxyurea reduces the degree of complement activation: 61% of patients with SCD who did not take hydroxyurea had elevated levels of sC5b-9 compared to those on the treatment (19). To date, no complement inhibitors have been specifically FDA approved within the context of SCD nor have any large-scale clinical trials been undertaken. Nonetheless, case reports of small-scale administration of certain complement inhibitors (e.g., eculizumab and ravulizumab) within certain contexts of SCD (e.g., pregnancy, delayed hemolytic transfusion reaction, VOC) have been published (21). Further research is needed to elucidate the thorough relationship between complement and SCD.
Allogeneic hematopoietic stem cell transplantation
A significant expertise is needed to render peri-transplant care for SCD patients. In general, pediatric patients have better outcomes. Adult patients with SCD who have acquired significant organ damage due to chronic inflammation and transplant related toxicity potentially prolonging recovery. Delayed immune reconstitution with infectious complications remain significant problems. In addition, SCD patients are at risk of rejection due to immunological responses. Blood transfusion support during the recovery period might require extensive RBC antigen matching. Targeting the bone marrow microenvironment pharmacologically or by modifying the effect of mesenchymal stromal cells might improve durable engraftment with a high degree of chimerism (22).
Allogeneic hematopoietic stem cell transplantation from related donors
Allogeneic hematopoietic stem cell transplantation (HSCT) using matched sibling donors is the only standard curative option for SCD and offers a >90% cure rate (23). Unfortunately, <20% of patients have HLA-matched donors. In the absence of a matched sibling donor, HLA-matched unrelated donors and HLA-identical sibling cord blood (CB) donors are alternatives (24).
Unrelated cord blood transplantation
Unrelated CB transplantation (CBT) significantly expanded the donor pool for patients with severe SCD. These transplants are associated with better outcomes and low incidences and severity of GVHD. In a study of pediatric patients, a reduced intensity regimen supported donor engraftment in the majority of patients with 100% 1 year overall survival and 78% disease-free survival (25). Historically, a major limitation associated with CBT in adults was a high rate of graft failure due to inadequate cell dose. Therefore, transplantation with double cord blood units is increasingly used in adults who lack a matched related or unrelated donor.
An ex vivo expanded HSC from a single umbilical cord blood unit, Omidubicel, demonstrated faster hematopoietic recovery with no differences in GVHD incidences compared to the standard myeloablative CBT (p < 0.001) in a phase 3 randomized clinical study (26).
Haploidentical donor transplant
Recent data of haploidentical familial donor transplants are encouraging with high overall survival, limited toxicities, and reduced rates of acute and chronic GVHD with up to 60 months of follow-up. Expanding the donor pool to haploidentical donors such as parents, children, siblings, half-siblings, and extended family donors is a promising therapeutic option for patients with SCD. To minimize rejection and GVHD, T-cell depletion of donor grafts is performed either ex vivo using CD34+ positive cell selection methods or in vivo with high-dose post-transplant cyclophosphamide (PTCy) (27). PTCy targeted depletion of alloreactive T cells is superior to anti-thymocyte globulin or alemtuzumab rendered non-selective T-cell depletion. Increasing total body irradiation in the preparative regimen decreased the rejection rate from 43 to 6%, with most of patients achieving full donor chimerism, and 100% survival (median follow-up 705 days) (28). Addition of thiotepa in haploidentical HSCT showed an improvement in engraftment, 20% acute GVHD rates, and 100% survival after a median follow-up of 13 months (29).
Gene therapy
Autologous HSCT offers several advantages over allogeneic HSCT: lowering risk of serious complications, increasing donor availability, absence of GVHD and graft rejection, and strengthened rapid immune reconstitution. Granulocyte colony-stimulating factor is an effective mobilization agent but is contraindicated in SCD, because it triggers severe adverse events. The CXCR4 antagonist, plerixafor, has been successful in gene therapy trials for SCD (30, 31). However, the degree of mobilization in patients with SCD is often suboptimal, so the patient may require multiple cycles of mobilization and apheresis collection. As a pretransplant preparative regimen, RBC exchange transfusions is performed to prevent the occurrence of SCD-related morbidities associated with the HSC mobilization and procurement. Gene modification of the autologous stem cells is required to reverse the effects of HbS and sustain hematopoietic engraftment. To that end, the following include potential gene therapy avenues: HbS gene correction, hemoglobin F induction, and modified HbA gene addition. Toxicities may be associated with the conditioning chemotherapy and lentiviral vector; namely, insertional mutagenesis leading to secondary malignancies.
Antisickling gene therapy
The first gene therapy treatment of SCD was reported in 2017 (32). LentiGlobin BB305 is a lentiviral vector that encodes a modified β-globin gene, HbAT87Q. HbAT87Q is a modified adult hemoglobin with an amino acid substitution at position 87 from threonine to glutamine, which results in anti-sickling properties due to steric inhibition of polymerization of HbS. As of February 2021, in Group C of phase 1–2 studies, all patients (n = 35) who received LentiGlobin infusion engrafted at a median follow-up of 17.3 months (range 3.7 to 37.6 months) (33). The median total hemoglobin level increased from 8.5 g/dL at baseline to ≥11 g/dL from 6–36 months after infusion. HbAT87Q accounted for at least 40% of total Hb and lower levels of hemolysis were observed. Severe vaso-occlusive events resolved in the 25 patients who could be evaluated who experienced a median of 3.5 events per year (range, 2–13.5) in the 24 months before enrollment. Three patients experienced vaso-occlusive events after infusion. The median time to neutrophil engraftment (absolute neutrophil count, ≥500 per microliter for 3 days) was 20 days (range, 12–35) while the median time to platelet engraftment (platelet count, ≥50 x 103 per microliter for 3 days without platelet transfusion) was 36 days (range, 18–136). Adverse events such as abdominal pain, opiate withdrawal syndrome, nausea, and vomiting were reported. One case of leukopenia, one of decreased diastolic blood pressure and one of febrile neutropenia were attributed possibly or definitely to LentiGlobin infusion. No cases of hematologic malignancies were reported at 37.6 months of follow-up (33).
Gene editing to induce fetal hemoglobin
Other gene therapy approaches being studied include upregulating the expression of HbF with CRISPR Cas-9 disruption of the BCL11a gene (34), CRISPR-Cas-12 mutation of the HGB 1 and 2 enhancer sites (35, 36), and RNAi-induced suppression of BCL11a mRNA transcription through a short hairpin RNA expressed through a lentiviral-based vector (37). Very promising results of a phase I study involving six patients suggest this is a feasible approach for HbF induction in patients with SCD. All the patients evaluated achieved robust and stable HbF induction (percentage HbF/(F+S) ranged from 20.4 to 41.3%). Clinical manifestations of SCD were reduced or absent during the 6–29 month follow-up period (37).
Discussion
Number of potentially curative therapies recently become available, see Figure 1 for a suggested algorithm for evaluation of patients with SCD for novel treatments. While gene therapies are promising and may perhaps cure SCD, further research is still needed before they can be utilized on all patients. Lentiviral vectors are currently preferred over gamma retroviral vectors because of their ability to transduce non-dividing primitive cells such as HPCs. A shorter in-vitro transduction period with lentiviral vectors is preferred in order to minimize the loss of HSC “stemness.” The long-term engraftment durability needs to be evaluated and the long-term risks of conditioning regimens such as infertility and risk of secondary malignancy must be addressed.
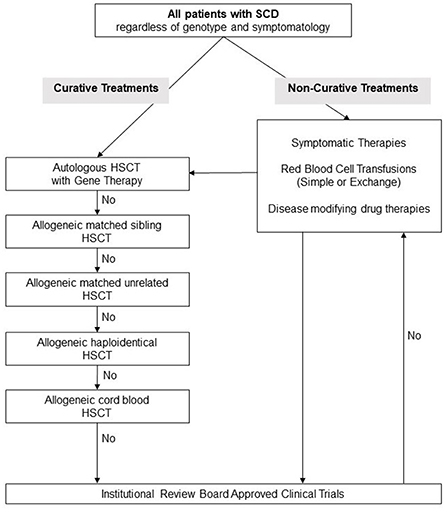
Figure 1. Suggested management algorithm for patients with sickle cell disease. SCD, Sickle Cell Disease; HSCT, hematopoietic stem cell transplantation.
The high cost of transplant and gene therapy currently limits the availability of curative treatment to primarily developed countries. The estimated cost of HSCT per patient ranges from $350,000 to $800,000, while costs for gene therapy is estimated to be as high as $1- $2 million, which severely constricting access. However, this upfront cost may be acceptable when compared to the total lifetime cost of managing a patient by age 50 with a chronic disease such as SCD that exceeds $8 million (38).
Future developments of alternative therapies include pharmacologic, biologic and genetic approaches. There has been an expansion of pharmacologic agents that target mediators of inflammation in SCD, such as cellular adhesion molecules, cytokines, complement, leukotrienes, and nuclear signaling factors (39). Development of an in vivo gene therapy delivery system that contains nucleases packaged in viral or non-viral vehicles would bypass the need for an autologous transplant and make worldwide application logistics easier. However, reactivation of HbF using pharmacologic approaches such as small molecule regulators targeting BCL11A or other genes are more likely to be broadly implemented (40).
Author contributions
YT, GN, and LV co-wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ingram VM. Abnormal human haemoglobins. I. The comparison of normal human and sickle-cell haemoglobins by “fingerprinting”. 1958. Biochim Biophys Acta. (1989) 1000:151–7.
2. Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. (1910) 6:517–21. doi: 10.1001/archinte.1910.00050330050003
3. Gardner RV. Sickle cell disease: advances in treatment. Ochsner J. (2018) 18:377–89. doi: 10.31486/toj.18.0076
4. Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Arch Dis Child. (2015) 100:48–53. doi: 10.1136/archdischild-2013-303773
5. Silberstein LE, Telen MJ. Update on the cure sickle cell initiative. Hematologist. The Hematologist Ash Publication. (2021) 18. Available online at: https://ashpublications.org/thehematologist/article/475190/Update-on-the-Cure-Sickle-Cell-Initiative
6. Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. (2014) 312:1033–48. doi: 10.1001/jama.2014.10517
7. NHLBI NIH,. Sickle Cell Disease Treatment [Online Multimedia]. (2022). Available online at: https://www.nhlbi.nih.gov/health/sickle-cell-disease/treatment (accessed July 15, 2022).
8. Bhatia M, Sheth S. Hematopoietic stem cell transplantation in sickle cell disease: patient selection and special considerations. J Blood Med. (2015) 6:229–38. doi: 10.2147/JBM.S60515
9. Tanhehco YC, Bhatia M. Hematopoietic stem cell transplantation and cellular therapy in sickle cell disease: where are we now? Curr Opin Hematol. (2019) 26:448–52. doi: 10.1097/MOH.0000000000000541
10. Ault A, US FDA. approves first drug for sickle-cell anaemia. Lancet. (1998) 351:809. doi: 10.1016/S0140-6736(05)78941-8
11. Leibovitch JN, Tambe AV, Cimpeanu E, Poplawska M, Jafri F, Dutta D, et al. l-glutamine, crizanlizumab, voxelotor, and cell-based therapy for adult sickle cell disease: Hype or hope? Blood Rev. (2022) 53:100925. doi: 10.1016/j.blre.2021.100925
12. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. (1995) 332:1317–22. doi: 10.1056/NEJM199505183322001
13. Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. (2018) 379:226–35. doi: 10.1056/NEJMoa1715971
14. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2017) 376:429–39. doi: 10.1056/NEJMoa1611770
15. Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. a phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. (2019) 381:509–19. doi: 10.1056/NEJMoa1903212
16. Francis WG, Womack CR. Serum complement activity in normal individuals and patients with sickle cell hemoglobin abnormalities. Am J Med Technol. (1967) 33:77–86.
17. Yoo JJ, Graciaa SH, Jones JA, Zuo Z, Arthur CM, Leong T, et al. Complement activation during vaso-occlusive pain crisis in pediatric sickle cell disease. Blood. (2021) 138:858. doi: 10.1182/blood-2021-154132
18. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. (2018) 68:263–99. doi: 10.3233/CH-189012
19. Roumenina LT, Chadebech P, Bodivit G, Vieira-Martins P, Grunenwald A, Boudhabhay I, et al. Complement activation in sickle cell disease: dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am J Hematol. (2020) 95:456–64. doi: 10.1002/ajh.25742
20. Vercellotti GM, Dalmasso AP, Schaid TR Jr, Nguyen J, Chen C, Ericson ME, et al. Critical role of C5a in sickle cell disease. Am J Hematol. (2019) 94:327–37. doi: 10.1002/ajh.25384
21. Varelas C, Tampaki A, Sakellari I. Anagnostopoulos, Gavriilaki E, Vlachaki E. Complement in sickle cell disease: are we ready for prime time? J Blood Med. (2021) 12:177–87. doi: 10.2147/JBM.S287301
22. Kharbanda S, Smith AR, Hutchinson SK, McKenna DH, Ball JB, Lamb LS Jr, et al. Unrelated donor allogeneic hematopoietic stem cell transplantation for patients with hemoglobinopathies using a reduced-intensity conditioning regimen and third-party mesenchymal stromal cells. Biol Blood Marrow Transplant. (2014) 20:581–6. doi: 10.1016/j.bbmt.2013.12.564
23. Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. (2017) 129:1548–56. doi: 10.1182/blood-2016-10-745711
24. Kanter J, Liem RI, Bernaudin F, Bolaños-Meade J, Fitzhugh CD, Hankins JS, et al. American society of hematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Advances. (2021) 5:3668–89. doi: 10.1182/bloodadvances.2021004394C
25. Abraham A, Cluster A, Jacobsohn D, Delgado D, Hulbert ML, Kukadiya D, et al. Unrelated umbilical cord blood transplantation for sickle cell disease following reduced-intensity conditioning: results of a phase I trial. Biol Blood Marrow Transplant. (2017) 23:1587–92. doi: 10.1016/j.bbmt.2017.05.027
26. Horwitz ME, Stiff PJ, Cutler C, Brunstein C, Hanna R, Maziarz RT, et al. Omidubicel vs standard myeloablative umbilical cord blood transplantation: results of a phase 3 randomized study. Blood. (2021) 138:1429–40. doi: 10.1182/blood.2021011719
27. Bolaños-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. (2012) 120:4285–91. doi: 10.1182/blood-2012-07-438408
28. Bolaños-Meade J, Cooke KR, Gamper CJ, Ali SA, Ambinder RF, Borrello IM, et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: a prospective clinical trial. Lancet Haematol. (2019) 6:e183–e93. doi: 10.1016/S2352-3026(19)30031-6
29. de la Fuente J, Dhedin N, Koyama T, Bernaudin F, Kuentz M, Karnik L, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. (2019) 25:1197–209. doi: 10.1016/j.bbmt.2018.11.027
30. Leonard A, Bonifacino A, Dominical VM, Luo M, Haro-Mora JJ, Demirci S, et al. Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br J Haematol. (2019) 186:286–99. doi: 10.1111/bjh.15902
31. Naoya U, Alexis L, David S, Sandhya RP, Kamille W, Eoghan M, et al. Safe and efficient peripheral blood stem cell collection in patients with sickle cell disease using plerixafor. Haematologica. (2020) 105:e497. doi: 10.3324/haematol.2019.236182
32. Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. (2017) 376:848–55. doi: 10.1056/NEJMoa1609677
33. Kanter J, Walters MC, Krishnamurti L, Mapara MY, Kwiatkowski JL, Rifkin-Zenenberg S, et al. Biologic and clinical efficacy of lentiglobin for sickle cell disease. N Engl J Med. (2022) 386:617–28. doi: 10.1056/NEJMoa2117175
34. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. (2021) 384:252–60. doi: 10.1056/NEJMoa2031054
35. Métais JY, Doerfler PA, Mayuranathan T, Bauer DE, Fowler SC, Hsieh MM, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. (2019) 3:3379–92. doi: 10.1182/bloodadvances.2019000820
36. Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. (2016) 22:987–90. doi: 10.1038/nm.4170
37. Esrick EB, Lehmann LE, Biffi A, Achebe M, Brendel C, Ciuculescu MF, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. (2021) 384:205–15. doi: 10.1056/NEJMoa2029392
38. Leonard A, Tisdale J, Abraham A. Curative options for sickle cell disease: haploidentical stem cell transplantation or gene therapy? Br J Haematol. (2020) 189:408–23. doi: 10.1111/bjh.16437
39. Hoppe CC. Inflammatory mediators of endothelial injury in sickle cell disease. Hematol Oncol Clin North Am. (2014) 28:265–86. doi: 10.1016/j.hoc.2013.11.006
Keywords: Sickle Cell Disease (SCD), gene therapy, hematopoietic stem cell transplantation (HSCT), disease modifying drugs, lentiviral vectors (LVS)
Citation: Tanhehco YC, Nathu G and Vasovic LV (2022) Development of curative therapies for sickle cell disease. Front. Med. 9:1055540. doi: 10.3389/fmed.2022.1055540
Received: 27 September 2022; Accepted: 04 November 2022;
Published: 24 November 2022.
Edited by:
Hollie Marie Reeves, University Hospitals Cleveland Medical Center, United StatesReviewed by:
Ioanna Sakellari, G. Papanikolaou General Hospital, GreeceCopyright © 2022 Tanhehco, Nathu and Vasovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ljiljana V. Vasovic, bGp2OTAwNEBtZWQuY29ybmVsbC5lZHU=
†These authors have contributed equally to this work