 Qingqing Gu1†
Qingqing Gu1† Chithra D. Palani2,3†
Chithra D. Palani2,3† Alana Smith1Biaori Li2,3
Alana Smith1Biaori Li2,3 Ernestine Kubi Amos-Abanyie1Ugochi Ogu4
Ernestine Kubi Amos-Abanyie1Ugochi Ogu4 Lu Lu1
Lu Lu1 Betty S. Pace2,3
Betty S. Pace2,3 Athena Starlard-Davenport1,5*
Athena Starlard-Davenport1,5*- 1Department of Genetics, Genomics, and Informatics, College of Medicine, The University of Tennessee Health Science Center, Memphis, TN, United States
- 2Department of Pediatrics, Division of Hematology/Oncology, Augusta University, Augusta, GA, United States
- 3Department of Biochemistry and Molecular Biology, Augusta University, Augusta, GA, United States
- 4Center for Sickle Cell Disease, Department of Medicine-Hematology, The University of Tennessee Health Science Center, Memphis, TN, United States
- 5Center for Sickle Cell Disease, The University of Tennessee Health Science Center, Memphis, TN, United States
Introduction: Therapeutic strategies aimed at reactivating HBG gene transcription and fetal hemoglobin (HbF) synthesis remain the most effective strategy to ameliorate the clinical symptoms of sickle cell disease (SCD). We previously identified microRNA29B (MIR29B) as a novel HbF inducer via targeting enzymes involved in DNA methylation. We provided further evidence that the introduction of MIR29B into KU812 leukemia cells significantly reduced MYB protein expression. Therefore, the aim of this study was to determine the extent to which MIR29B mediates HbF induction via targeting MYB in KU812 leukemia cells and human primary erythroid progenitors and to investigate the role of MIR29B in HbF induction in vivo in the humanized Townes SCD mouse model.
Materials and methods: Human KU812 were cultured and normal CD34 cells (n = 3) were differentiated using a two-phase erythropoiesis culturing system and transfected with MIR29B (50 and 100 nM) mimic or Scrambled (Scr) control in vitro. A luciferase reporter plasmid overexpressing MYB was transfected into KU812 cells. Luciferase activity was quantified after 48 h. Gene expression was determined by quantitative real-time PCR. In vivo studies were conducted using Townes SCD mice (6 per group) treated with MIR29B (2, 3, and 4 mg/kg/day) or Scr control by 28-day continuous infusion using subcutaneous mini osmotic pumps. Blood samples were collected and processed for complete blood count (CBC) with differential and reticulocytes at weeks 0, 2, and 4. Flow cytometry was used to measure the percentage of HbF-positive cells.
Results: In silico analysis predicted complementary base-pairing between MIR29B and the 3′-untranslated region (UTR) of MYB. Overexpression of MIR29B significantly reduced MYB mRNA and protein expression in KU812 cells and erythroid progenitors. Using a luciferase reporter vector that contained the full-length MYB 3′-UTR, we observed a significant reduction in luciferase activity among KU812 cells that co-expressed MIR29B and the full-length MYB 3′-UTR as compared to cells that only expressed MYB 3′-UTR. We confirmed the inhibitory effect of a plasmid engineered to overexpress MYB on HBG activation and HbF induction in both KU812 cells and human primary erythroid progenitors. Co-expression of MIR29B and MYB in both cell types further demonstrated the inhibitory effect of MIR29B on MYB expression, resulting in HBG reactivation by real-time PCR, Western blot, and flow cytometry analysis. Finally, we confirmed the ability of MIR29B to reduce sickling and induce HbF by decreasing expression of MYB and DNMT3 gene expression in the humanized Townes sickle cell mouse model.
Discussion: Our findings support the ability of MIR29B to induce HbF in vivo in Townes sickle cell mice. This is the first study to provide evidence of the ability of MIR29B to modulate HBG transcription by MYB gene silencing in vivo. Our research highlights a novel MIR-based epigenetic approach to induce HbF supporting the discovery of new drugs to expand treatment options for SCD.
Introduction
Sickle cell disease (SCD) is a common genetic red blood cell disorder that affects over 20 million individuals worldwide (1). People with SCD have abnormal hemoglobin S (HbS) molecules, resulting from an A to T mutation in codon 6 of the HBB gene (2, 3). Under low oxygen conditions, HbS molecules polymerize and erythrocytes sickle, which can result in vaso-occlusion. Therapeutic strategies aimed at reactivating HBG gene transcription and fetal hemoglobin (HbF) synthesis remains the most effective strategy to ameliorate the clinical symptoms of SCD, including vaso-occlusive crises (4, 5).
Recently microRNAs (miRNAs) have emerged as a novel class of potential therapeutics due to their ability to restore expression of genes involved in tumor suppression (6), aging (7), and various human diseases (8–11). MiRNAs are endogenous, small (∼22 nt in size) regulatory RNA molecules that function to modulate post-transcriptional gene silencing through complimentary base-pair binding to their target mRNAs (12). MiRNAs are naturally expressed at varying levels in mammalian tissues, including blood plasma, and serum (13). Studies also provide evidence for a role of miRNAs in directly targeting HBG gene expression or transcriptional repressors of HBG gene expression during hemoglobin switching (14, 15). Thus, miRNAs that target genes involved in regulating HBG gene expression may serve as attractive therapeutic candidates for HbF induction.
Previously, we demonstrated that microRNA29B (MIR29B) reactivated HBG gene transcription and induced HbF expression in vitro by inhibiting the de novo DNA methyltransferases, DNMT3A and DNMT3B (16). Our findings further suggested that MIR29B may also target the HBG transcriptional repressor protein MYB in KU812 leukemia cells (16). Here, we have expanded our findings and show for the first time that MIR29B reactivates HBG gene transcription and induces HbF expression by silencing the HBG repressor protein MYB in vitro in KU812 leukemia cells and normal human erythroid progenitors generated from CD34+ stem cells and in the preclinical Townes SCD mouse model. Our preclinical findings highlight the therapeutic potential of MIR29B as a promising treatment for inducing HbF in SCD and other β-hemoglobinopathies.
Materials and methods
In vitro cell culture with microRNA29B and co-transfection with MYB DNA
Human KU812 leukemia cells were cultured in Iscove’s Modified Dulbecco Medium with 10% fetal bovine serum as previously published by our lab (16). Cells were harvested for cell count and viability using 0.4% Trypan blue exclusion assay. Cells were seeded at a density of 0.5 × 106 viable cells per 100 mm plate for different treatments. During log phase growth, KU812 cells were transfected with 50 and 100 nmol/l of pre-MIR29B (Applied Biosystems, Waltham, MA, USA) or Scrambled (Scr) oligonucleotide control (100 nmol/l) for 48 h in three independent replicates using Opti-MEM media (Gibco, Waltham, MA, USA) and Lipofectamine™ 2000 transfection reagent (Invitrogen Carlsbad, CA, USA) according to the manufacturer’s instructions, then harvested for subsequent analyses. KU812 cells were also co-transfected with pGL3-MYB-3′-untranslated region (UTR) (Addgene plasmid # 25,798, Watertown, MA, USA), which contains the full length MYB 3′UTR cloned into the XbaI/SalI restriction sites of the pGL3-control luciferase reporter vector (17). The human tagged ORF clone engineered to overexpress c-MYB (Origene, Rockville, MD, USA) was used alone or co-transfected with MIR29B.
Erythroid differentiation of human CD34+ stem cells and microRNA29B co-electroporation with MYB DNA
Human bone marrow CD34+ stem cells (ReachBio, Seattle, WA, USA) were cultured in a modified two-phase liquid culture system as previously published (18). During phase I, stem cells were grown in minimum essential medium-α (αMEM) containing AB serum, interleukin-3 (10 ng/ml), stem cell factor (10 ng/ml), and erythropoietin (2 IU/ml). On day 7, cells transitioned to Phase II media where they remained under erythropoietin (2 IU/ml) stimulation. Erythroid progenitors were electroporated on day 8 with human mature MIR29B or Scr mimic or with c-MYB overexpression plasmid using the Amaxa® Human CD34+ Cell Nucleofector® Kit. After 48 h, cells were harvested for reverse transcription-quantitative PCR (RT-qPCR), Western blot, and flow cytometry analysis. Giemsa staining was used to monitor cell morphology and cell counts; viability was monitored using 0.4% Trypan blue exclusion assay (Gibco, Carlsbad CA, USA).
RNA isolation and RT-qPCR analysis
Total RNA was isolated as previously published (16). To quantify mRNA levels for MYB, HBG, and HBB and the internal control β-actin, gene specific primers were used (Supplementary Table 1). All mRNA levels were normalized to β-actin before analysis. Quantification of MIR29B was performed using the TaqMan miRNA assay (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions and RNU48 was used as endogenous control. The 2–ΔΔCt method was used for calculating the relative amount of target mRNA. All RT-qPCR reactions were performed in triplicate, repeated at least three times, and always included a no-template sample as a negative control. RT-qPCR results are presented as average fold change of target gene in cells relative to Scr control, which was normalized to one.
Western blot analysis
Total protein was isolated and Western blot analysis was performed as previously published (16). Primary antibodies against MYB (59995S), HbF (39386S), and HbA (84934S) were purchased from Cell Signaling Technology (Danvers, MA, USA) and diluted in the range of 1:250 to 1:2000, incubated overnight and then followed by treatment with secondary antibody. The primary antibody against β-actin (AM4302), the internal control, was purchased from Invitrogen (Waltham, MA, USA).
Animal models and drug treatment
The humanized Townes SCD transgenic mouse (B6; 129-Hbatm1 [HBA] Tow Hbbtm2 [HBG1, HBB*] Tow/Hbbtm3 [HBG1, HBB] Tow/J), which completes hemoglobin switching from human γ-globin to βs-globin shortly after birth (19), were purchased from Jackson Laboratories. Mice were maintained and genotyping was performed with gene specific primers. All animal studies were approved by the Augusta University Institutional Animal Care and Use Committee. Townes SCD mice ages 4–6 months old, 4–10 mice per group (equal males and females), were treated by 28 days of continuous infusion using surgically implanted subcutaneous Alzet mini-osmotic pumps (DURECT corporation, Alzet Osmotic Pumps, Cupertino, CA, USA). Drug treatment groups in SCD mice included: (1) 2 mg/kg/day Scr control, (2) 2 mg/kg/day MIR29B mimic, (3) 3 mg/kg/day Scr control, (4) 3 mg/kg/day MIR29B mimic, (5) 4 mg/kg/day Scr control, and (6) 4 mg/kg/day MIR29B mimic. MIR29B mimics were purchased from Dharmacon (Lafayette, CO, USA). Mice were weighed at week 0 before and after pump placement and then again at weeks 2 and 4. Blood samples were collected in BD Microtainer Capillary blood collection K2-EDTA tubes at weeks 0, 2, and 4 and were processed for complete blood count (CBC) with differential. The percentage of reticulocytes (acridine orange) was determined by flow cytometry. To quantify MIR29B levels, total RNA was isolated using TRIzol and RT-qPCR analysis was performed.
Alzet mini-osmotic pump implantation
Before surgery, Buprenorphine SR was given for pain and then mice anesthetize with Isoflurane by inhalation. Once the animal was anesthetized, the area where pump was placed was shaved and then cleaned with a surgical scrub consisting of alternating betadine and alcohol wipes. Lidocaine was injected locally followed by a half-inch mid-scapular incision for pump placement on the back of the animal. A hemostat was used to spread the incision and subcutaneous tissue to create a pocket for the pump. Scr control or MIR29B filled Alzet mini-osmotic pump was inserted with the delivery portal first and wound closed with wound clips. Mice were monitored daily and pain was addressed with an additional dose of Buprenorphine SR. Once the mice wound was healed, clips were removed, and the blood samples were collected at weeks 0, 2, and 4 for analysis.
Complete blood count with differential
Peripheral blood from 4–6 months old mice was collected in BD Microtainer Lithium heparin tubes by tail vein bleeding. Automated CBC and differentials were completed on the Micros 60 CS/CT machine (HORIBA Medical/ABX Diagnostics, Irvine, CA, USA) according to the manufacturer’s protocol.
In vitro RBC sickling analysis
In a BD Vacutainer EDTA tube, peripheral blood was collected through tail bleeding and washed with 1X phosphate buffered saline. Cells were suspended 1:300 ratio in Iscove’s Modified Dulbecco’s Medium containing 10% heat inactivated fetal bovine serum, and incubated at 37°C in normoxic (21%) and hypoxic (1%) conditions for 12 h in a O2 hypoxic chamber (Coy Laboratory Products, Grass Lake, MI, USA). Subsequently, blood samples were treated with 4% formaldehyde for 10 min and then transferred to room temperature. Using light microscopy, sickling of red blood cells were quantified by changes in cell morphology. 20X magnification of bright field images were attained on an EVOS Cell Imaging systems (Thermo Fisher Scientifics, Waltham, MA, USA).
Flow cytometry analysis
To measure the percentage of HbF positive cells (F-cells), mouse peripheral blood samples were washed using cold PBS containing 0.5% BSA and fixed with 4% paraformaldehyde. Centrifuged to discard the supernatant, cells were washed and permeabilized with 1:1 of Acetone and Methanol and then stained with FITC conjugated sheep anti-human HbF antibody (Cat. #A80-136) purchased from Bethyl Laboratories, Inc. (Montgomery, TX, USA). Sheep IgG Isotype control was used to detect non-specific staining. The cells were washed once with phosphate buffered saline containing 0.5% bovine serum albumin buffer, and F-cells were measured and quantified by flow cytometry (20) using AttuneTM NxT Flow Cytometer and Attune and Novex software (ThermoFisher Scientific, Rockland, IL, USA).
Statistical analysis
Data from at least 3–6 replicates of independent experiments were reported as the mean ± standard error of the mean (SEM). Data was analyzed by ANOVA or by a two-tailed Student’s t-test to determine statistical significance. Statistical analysis was completed by unpaired student’s t-test and ANOVA for N = 6 mice per group for two independent experiments. P < 0.05 was considered statistically significant.
Results
We previously showed that overexpression of MIR29B resulted in a significant decrease in expression of MYB, a known HBG repressor protein, in KU812 leukemia cells (16). To determine the effect of exogenous MIR29B expression on MYB gene silencing, we initially introduced MIR29B mimic (50 and 100 nM) into KU812 cells, which expresses the HBG and HBB genes, and measured MYB mRNA expression using RT-qPCR (Figure 1). We observed that transfection of MIR29B mimic (100 nM) for 48 h significantly decreased MYB mRNA expression by 25% as compared to Scr control (100 nM) cells (p < 0.01) (Figure 1A). Therefore, we utilized KU812 cells to determine whether MIR29B alters MYB expression as a mechanism of HbF induction.
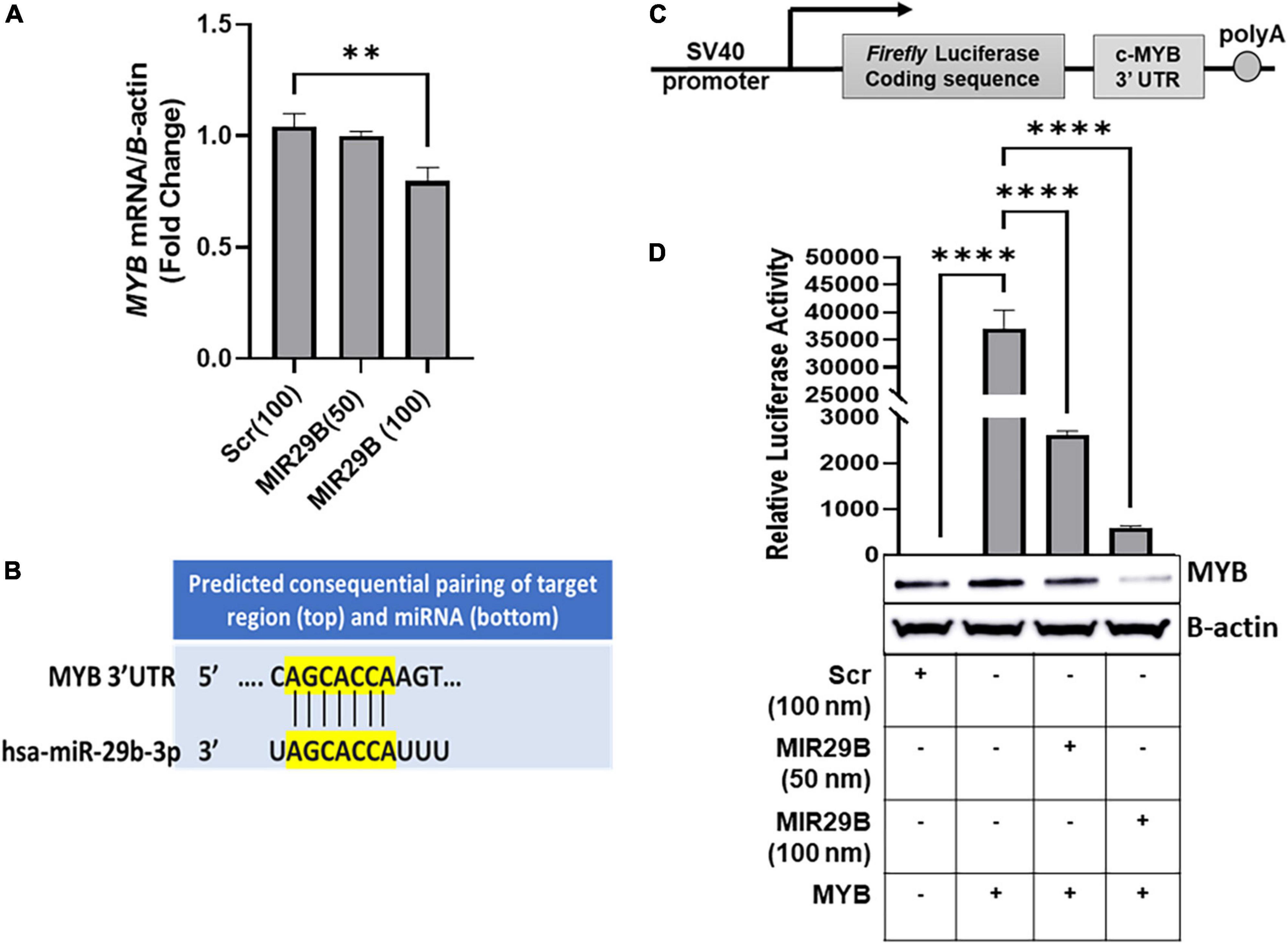
Figure 1. MicroRNA29B (MIR29B) targets 3′-untranslated region (UTR) of MYB in KU812 cells. (A) Real-time PCR analysis of MYB mRNA relative to β-actin internal control in KU812 cells transfected with MIR29B mimic (50 and 100 nM) or scrambled (Scr) control (100 nm). (B) Complementary sequence for MIR29B and the 3 – UTR of MYB. (C) Luciferase reporter construct containing the c-MYB 3′-UTR. (D) Luciferase activity in KU812 cells 48 h after transfection with MIR29B mimic (50 and 100 nM) and MYB 3′-UTR reporter plasmid. Accompanied Western blot showing expression of MYB protein in cells co-transfected with MYB overexpression plasmid and MIR29B mimic or Scr control (100 nm). The + and – signs indicate the presence or absence of MYB or MIR29B overexpression in KU812 cells. Data are shown at the mean ± standard error of the mean (SEM). **P < 0.005, ***P < 0.0005, and ****P < 0.0001 is statistically significant.
Because miRNAs are known to post-transcriptionally regulate expression of their target mRNAs via binding to the 3′-UTR to repress protein production, we hypothesized that MIR29B might target the 3′-UTR of MYB. Using in silico analysis, we discovered that MIR29B has a consensus sequence complimentary to the 3′-UTR of MYB (Figure 1B). Thus, we wanted to determine whether MYB is regulated by MIR29B in KU812 cells. We transfected a luciferase reporter plasmid containing the full-length MYB 3′-UTR sequence. The sequence replaces the SV40 enhancer and SV40 poly (A) signal of pGL3-control (17) (Figure 1C) into KU812 cells alone or in combination with MIR29B mimic (50 and 100 nM) or Scr control (100 nM) and assessed luciferase activity (Figure 1D). In cells that overexpressed the full length MYB 3′-UTR alone, luciferase activity was significantly increased (P < 0.0001) as compared to Scr control cells (Figure 1D). However, when exogenous MIR29B mimic (50 and 100 nM) was co-expressed with the full length MYB 3′-UTR, luciferase activity was significantly reduced by up to 16-fold (MIR29B 50 nM, p < 0.001) and 80-fold (MIR29B 100 nM, p < 0.001) as compared to KU812 cells transfected with the full length MYB 3′-UTR alone (Figure 1D). Western blot analysis further confirmed the inhibitory effect of introducing MIR29B (100 nM) on MYB protein expression (Figure 1D). Specifically, co-transfection of MIR29B mimic (100 nM) and a plasmid engineered to overexpress c-MYB into KU812, resulted in a significant decrease in MYB protein expression in KU812 cells (Figure 1D). Thus, based on these findings we postulated that MIR29B might activate HBG transcription, in part via binding the 3′-UTR of MYB.
To determine the extent to which MIR29B reactivates HBG gene expression via MYB gene silencing, we used a human tagged c-MYB ORF clone to overexpress MYB alone or when co-transfected with MIR29B mimic (50 or 100 nM) or Scr control (100 nM) into KU812 cells (Figure 2). Western blot analysis revealed that transfection of MYB plasmid DNA at a concentration of 2 μg resulted in the highest expression of MYB protein and lowest expression of HbF when compared to control cells (Figure 2A). Therefore, we used a concentration of 2 μg of MYB plasmid DNA to overexpress MYB in the remaining experiments. We further confirmed efficient transfection of MYB alone or when co-transfected with MIR29B (50 and 100 nM) into KU812 cells after 48 h by RT-PCR (Figure 2). Specifically, in KU812 cells co-transfected with MIR29B (50 and 100 nM) and MYB, we observed a significant increase in MYB mRNA expression as compared to Scr (100 nM) control cells and cells transfected with MIR29B alone (Figure 2B). Furthermore, we observed a drastic decrease, up to two-fold, in MYB mRNA expression in cells that were co-transfected with MIR29B (50 and 100 nM), which further suggests that MIR29B targets MYB resulting in its gene silencing.
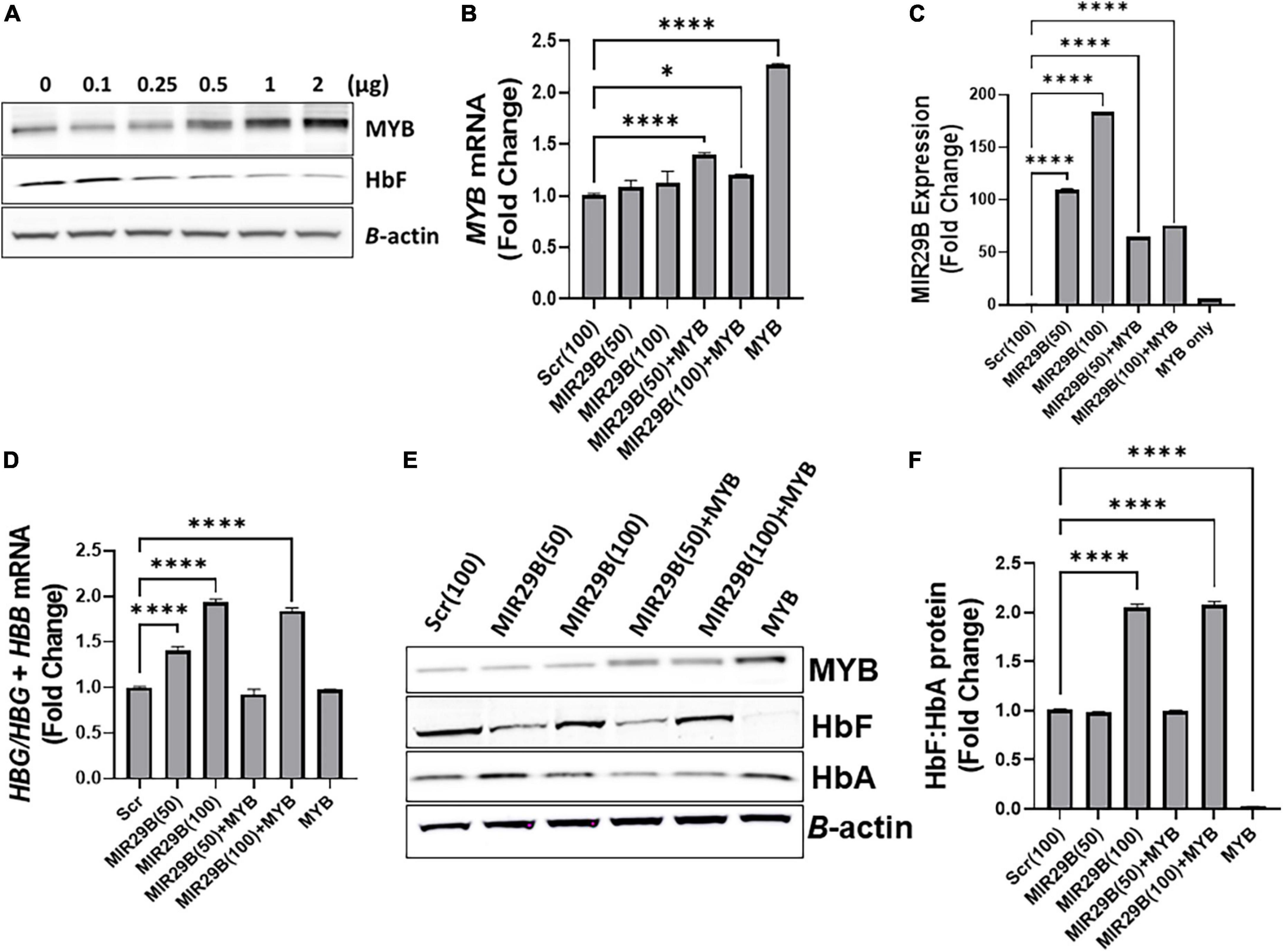
Figure 2. MicroRNA29B (MIR29B) mediates HBG reactivation via inhibiting MYB in KU812 cells. (A) Western blot analysis of MYB and HbF protein expression relative to β-actin protein in KU812 cells transfected with increasing concentrations of MYB plasmid DNA (μg). Fold change of (B) MYB mRNA relative to β-actin, (C) MIR29B expression relative to RNU48, and (D) HBG-to-HBB mRNA ratio as a function of HBG/HBG + HBB mRNA expression in KU812 cells following transfection for 48 h with MIR29B mimic (50 or 100 nm) alone or in combination with MYB overexpression plasmid vector (2 μg). (E) Western blot of MYB, HbF, and HbA protein expression with β-actin internal control. (F) Bar graphs showing quantification of the ratio of HbF-to-HbA protein in KU812 cells transfected with MIR29B alone or in combination with MYB. *P < 0.05 **P < 0.005, ***P < 0.0005, and ****P < 0.0001 is statistically significant.
Therefore, to determine whether MIR29B mediates HBG reactivation, in part via MYB gene silencing, we next quantified the ratio of HBG-to-HBB mRNA expression as a function of HBG/HBG + HBB using RT-qPCR analysis (Figure 2D). Based on our previous findings in KU812 cells, we observed a significant, up to a two-fold, increase in the ratio of HBG-to-HBB mRNA in cells transfected with MIR29B (50 and 100 nM) alone as compared to Scr control cells (p < 0.001). Furthermore, there was no significant difference in the ratio of HBG-to-HBB mRNA in KU812 cells transfected with MYB alone as compared to Scr control cells, which further confirms the role of MYB as an HBG repressor protein. Interestingly, in cells co-transfected with both MIR29B (100 nM) and MYB, the ratio of HBG-to-HBB mRNA yielded close to a two-fold increase which is similar to that of cells transfected with MIR29B alone (Figure 2D). In support of these findings, we further demonstrated a significant increase in HbF protein expression as a ratio of HbF-to-HbA in KU812 cells transfected with MIR29B (100 nM) alone or in combination with MYB as compared to Scr control cells (Figures 2E,F). We also confirmed the positive expression of MYB protein in cells transfected with MYB alone or in combination with MIR29B (50 and 100 nM). Flow cytometry analysis further confirmed the ability of MIR29B (50 and 100 nM) alone and in combination with MYB to increase the% of HbF-positive cells by up to 4.8-fold as compared to Scr control cells (Supplementary Figure 1). These findings further support our hypothesis that MIR29B mediates HBG reactivation and HbF induction in part via MYB gene silencing.
We next performed studies under physiological conditions to further determine the extent to which MIR29B mediates HBG reactivation via inhibiting MYB using normal erythroid progenitors generated from human CD34+ stem cells in a liquid culture system as previously described (16, 21). Cell morphology examination by Giemsa staining confirmed erythroid lineage commitment by day 7 followed by the appearance of mature red blood cells by day 12 (Figure 3A). To compliment cell morphology, we also measured expression of the erythroid differentiation markers CD235a and CD71 by flow cytometry (Supplementary Figures 2A,B) to gain a better understanding of the effect of ectopic expression of MIR29B and MYB on erythroid maturation. The expression of CD71 did not significantly change following introduction of MIR29B alone; however, levels of CD71 decreased in cells that expressed MYB as compared to Scr control (Supplementary Figure 2A). Conversely, the percentage of CD235a positive cells significantly increased following MIR29B alone or in combination with MYB when compared to Scr control cells (Supplementary Figure 2B). Together, these data suggest that elevated levels of MIR29B and MYB may enhance erythroid maturation.
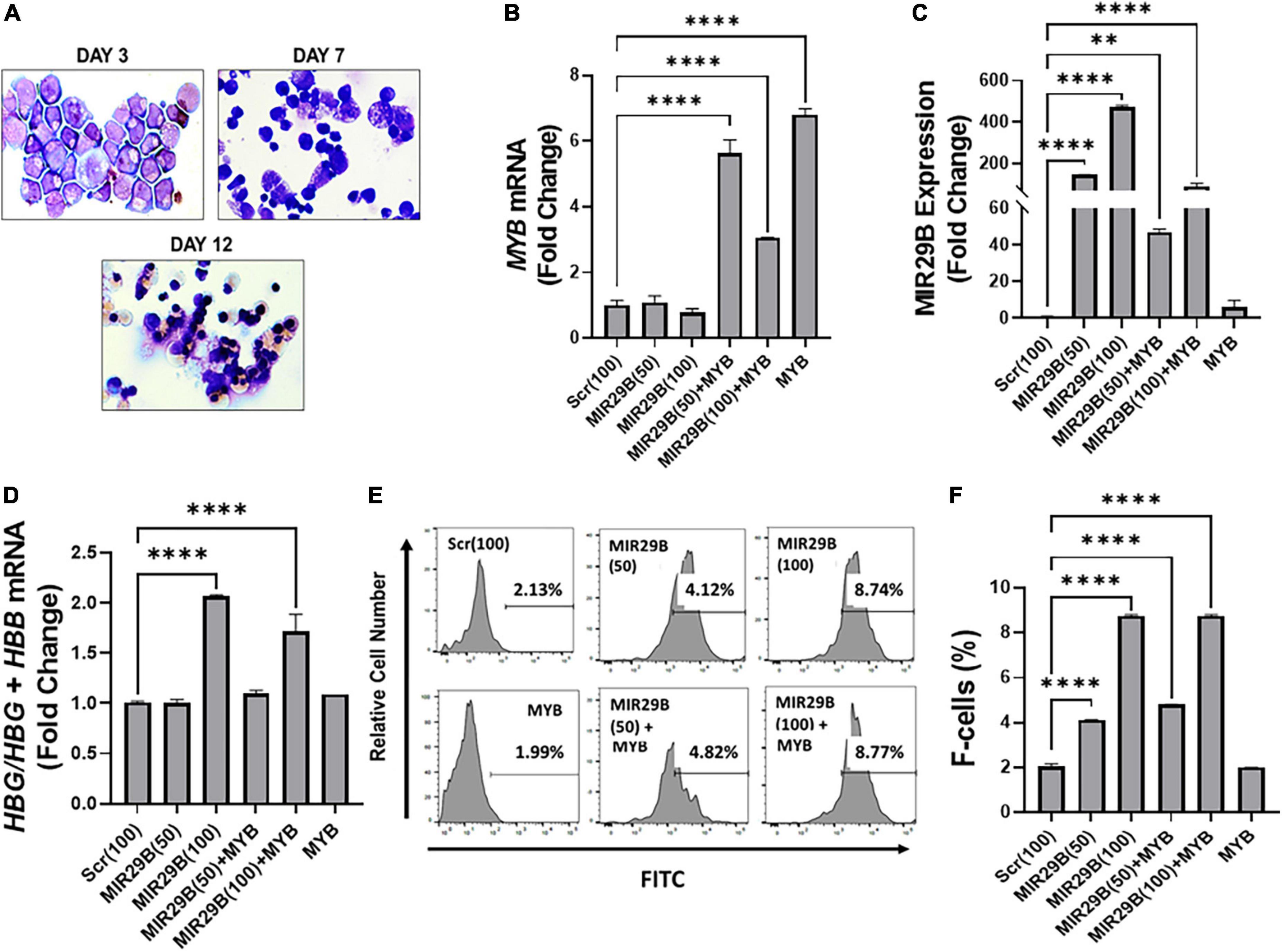
Figure 3. MicroRNA29B (MIR29B) mediates HBG reactivation via MYB gene silencing in normal human erythroid progenitors. (A) Wright-Giemsa-stained images of human erythroid progenitors (n = 3) undergoing differentiation at day 3, day 7, and day 12 in vitro. Quantitative RT-qPCR analysis of (B) MYB mRNA relative to β-actin, (C) MIR29B relative to RNU48, and (D) HBG-to-HBB mRNA ratio as a function of HBG/HBG + HBB, (E) Representative histograms from flow cytometry analysis of the percentage of HbF-positive cells following transfection of MIR29B alone or in combination with MYB into primary erythroid progenitors (n = 3) stained with FITC anti-HbF antibody. (F) Bar graphs showing quantification of the ratio of HbF-to-HbA in erythroid progenitors (n = 3) transfected with MIR29B alone or in combination with MYB. *P < 0.05, **P < 0.005, ***P < 0.0005, and ****P < 0.0001 is statistically significant.
To further determine whether MIR29B and MYB were effectively delivered into the cells by electroporation, we measured MIR29B and MYB mRNA expression by RT-qPCR (Figures 3B,C). We observed a significant seven-fold increase in erythroid progenitors electroporated with MYB alone as compared to Scr control cells (Figure 3B). MYB mRNA expression was also confirmed in erythroid progenitors following co-electroporation with MIR29B (50 and 100 nM). Co-electroporation of erythroid progenitors with MIR29B (100 nM) and MYB significantly decreased MYB mRNA expression by 2.3-fold when compared to MYB alone (Figure 3B). In addition, we observed a dose-dependent increase up to 400-fold in MIR29B expression in cells transfected with MIR29B (50 and 100 nM) alone as compared to Scr control cells (p < 0.0001)(Figure 3C). This finding further supports the role of MIR29B in inhibiting MYB gene expression. This difference in expression of MIR29B between cells electroporated with MIR29B alone or co-electroporated with MYB is likely due to overexpression of MYB.
We further determined whether MIR29B mediates HBG activation via silencing MYB under physiological conditions. We co-expressed MIR29B (50 and 100 nM) and MYB into normal erythroid progenitors and measured the expression of HBG-to-HBB mRNA by RT-qPCR (Figure 3D). Real-time PCR analysis revealed a significantly higher HBG-to-HBB mRNA ratio by up to two-fold in erythroid progenitors electroporated with MIR29B (100 nM) alone or up to 1.5-fold when co-expressed with MYB as compared to Scr control cells (Figure 3D). Furthermore, there was no change in the ratio of HBG-to-HBB mRNA in erythroid progenitors electroporated with MYB alone when compared to Scr control cells (Figure 3D). In support of these findings, we further demonstrated by flow cytometry analysis the inhibitory effect of overexpressing MIR29B on MYB function (Figure 3E). Specifically, we confirmed a significant, up to four-fold, increase (P < 0.0001) in the percentage of HbF-positive cells among erythroid progenitors whether they overexpressed MIR29B alone (50 and 100 nM) or in combination with MYB (P < 0.0001) (Figures 3E,F). Furthermore, the percentage of HbF-positive cells in erythroid progenitors electroporated with MYB alone was similar to that of Scr control cells (Figures 3E,F). These studies support an important role for MIR29B in MYB and HBG gene regulation in vitro.
To advance the field and move novel small molecules from bench to bedside requires evidence of in vivo efficacy of HbF induction. Therefore, our final preclinical studies evaluated the potential of MIR29B to induce HbF using the humanized sickle cell mouse model. Townes sickle cell mice are an excellent model since they express human α-globin, γ-globin, and βS-globin genes and exhibit phenotypes including chronic hemolysis and sickling similar to human SCD. Mice 4–6 months old were administered MIR29B (2, 3, and 4 mg/kg/day) or Scr control (same concentration) to establish optimal dosing by continuous 4 weeks infusion using subcutaneous mini-osmotic pumps with six mice per treatment group (Figure 4A). At week 0, 2, and 4, mice were weighed and blood samples collected by tail bleed for automated CBC and reticulocyte percent, percentage of F-cells by flow cytometry. Over 4 weeks of treatment, no drug toxicity occurred or death (Supplementary Figure 3). Scr control mice had no significant change in blood counts over the 4 weeks treatment period, except for platelets at 3 mg/kg after 2 weeks (Supplementary Figure 3A). Treatment of mice with MIR29B (2–4 mg/kg/day) after 2 weeks caused a significant increase in the number of platelets 1.6-fold (p < 0.0005) and reticulocytes 1.7-fold (p < 0.05), suggesting that MIR29B stimulated erythropoiesis. By contrast, levels of total hemoglobin, hematocrit, and lymphocytes did not significantly change after MIR29B treatment (Supplementary Figures 3B,C). During MIR29B treatment, all mice exhibited normal behavior and had steady weight gain (Supplementary Figure 3D). At the end of treatment, pumps were removed to confirm that all medications were delivered.
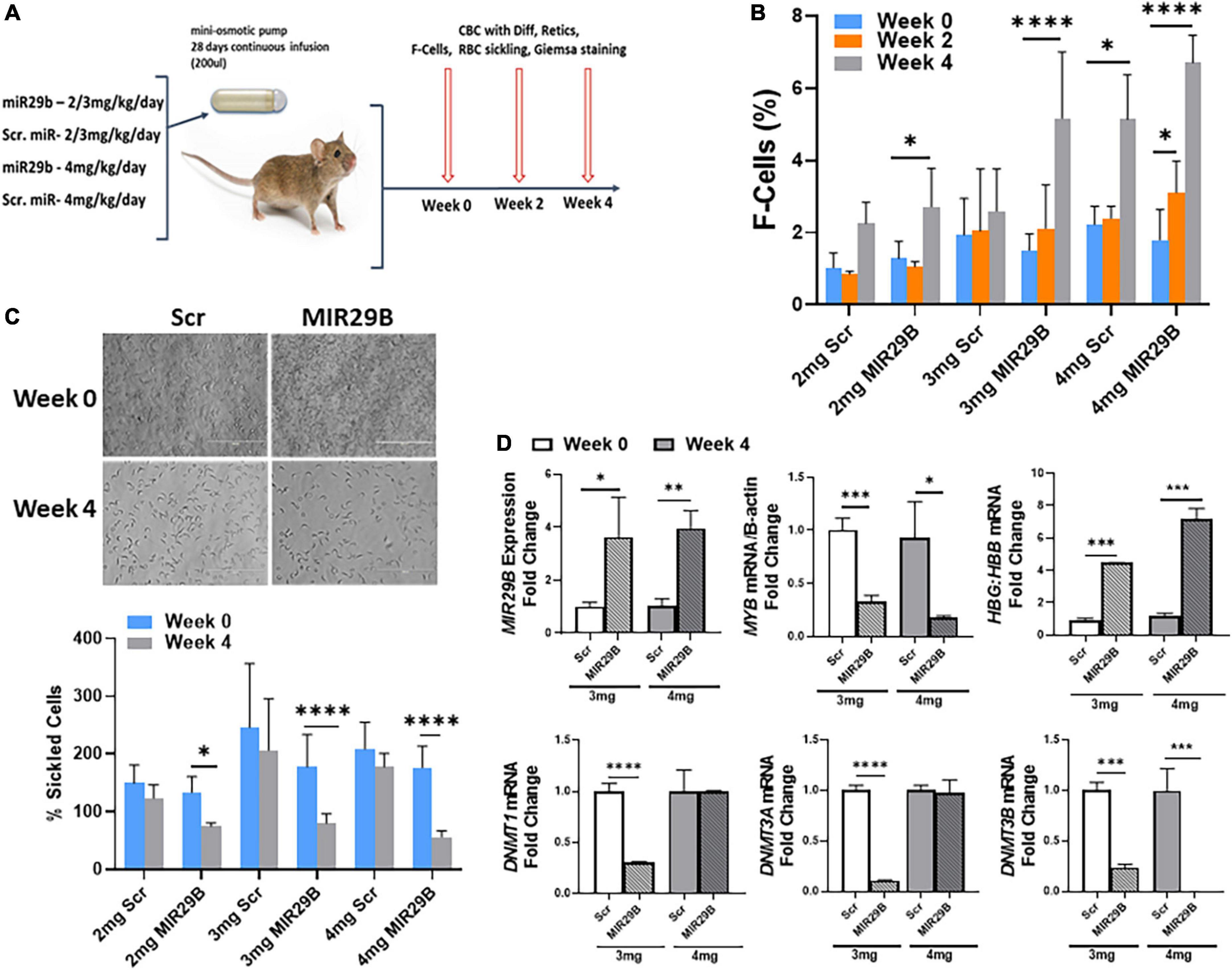
Figure 4. MicroRNA29B (MIR29B) induces HbF in humanized Townes sickle cell mice. (A) Townes SCD mice, which express human α-globin, γ-globin, and βS-globin genes and exhibit phenotypes including chronic hemolysis and sickling similar to human SCD, were surgically implanted with a mini-osmotic pump (Alzet) that continuously released MIR29B mimic at the concentration shown over 28 days. Age-and sex-matched mice (n = 4–10 per group) were treated and blood was collected at weeks 0, 2, and 4 for complete blood counts (CBC) with differential, reticulocytes, %F-cells, and red blood cell sickling under hypoxia conditions. Treatments consisted of 4 mice per group (2 males and 2 females) for MIR29B or SCR mimic at 2, 3, and 4 mg/kg/day and water vehicle and HU (100 mg/kg/day) controls. Tail–vein blood samples were collected in EDTA blood tubes at week 0, 2, and 4 along with weights. Evaluation of the effects of MIR29B on bone marrow hematopoiesis, were conducted by (B) Shown are bar graphs of the percentage of HbF-positive cells (F–cells) in erythroid progenitors isolated from Townes SCD mice and stained with fluorescein isothiocyanate-labeled anti-HbF antibody under the different treatment conditions. (C) Shown are bar graphs of the percentage of sickled cells obtained from freshly collected EDTA blood from sickle mice incubated under 1% hypoxia conditions for 12 h following the treatment conditions. (D) RT-qPCR analysis of MIR29B, MYB, DNMT gene expression and the HBG-to-HBB mRNA ratio as a function of HBG/HBG + HBB in mice treated with MIR29B or Scr control; *P < 0.05, **P < 0.005, ***P < 0.0005, and ****P < 0.0001 is statistically significant.
We next analyzed the ability of MIR29B to induce HbF expression in vivo. As shown in Figure 4B, the percentage of F-cells significantly increased 2.1 and 3.4-fold in mice treated with 2 and 3 mg/kg MIR29B, respectively. We further demonstrated the anti-sickling effect mediated by MIR29B under hypoxic conditions (Figure 4C). Sickle erythroid precursors from Townes mice were incubated in 1% hypoxia conditions overnight, fixed with formaldehyde and examined by light microscopy. As shown in Figure 4D, MIR29B reduced the percentage of sickled erythroid precursors by up to 68% (p < 0.0005) after 4 weeks treatment, supporting the anti-sickling effects mediated by MIR29B. These findings support the ability of MIR29B to induce HbF in vivo in preclinical Townes sickle cell mice.
We previously showed that MIR29B functions as a DNA methyltransferase inhibitor by targeting DNMT gene expression in vitro (16). Since treatment of mice with 3 and 4 mg/kg MIR29B at 4 weeks significantly reduced the percentage of sickle cells, we measured mRNA expression from mice spleen tissue at week 0 and week 4 (Figure 4D). We confirmed a significant increase up to 3.8-fold in MIR29B expression in mice treated with 3 and 4 mg/kg MIR29B, respectively, compared to Scr control. Treatment of mice with 3 and 4 mg/kg MIR29B resulted in up to a 68% significant decrease in MYB mRNA expression (p < 0.005) and up to 76% for DNMT3B (Figure 4D). Similarly, treatment of mice with 3 mg/kg significantly decreased DNMT1 mRNA up to 70% and DNMT3A mRNA expression by 90%, while HBG mRNA shown as a function of HBG-to-HBB mRNA ratio significantly increased (Figure 4D), suggesting that MIR29B reactivated HBG gene expression in part via inhibiting DNMT3 and MYB gene expression. Collectively, our findings support the ability of MIR29B to function as an HbF inducer for the treatment of β-hemoglobinopathies in part via targeting MYB.
Discussion
MicroRNAs represent a novel class of small molecules that have gained much attention for their diagnostic and therapeutic potential to treat a wide variety of clinical diseases via targeting oncogenes and their gene products (22–25). MiRNAs are short (∼22 nucleotides) non-coding RNA molecules that associate with the miRNA-induced silencing complex (mRISC) and guide mRISC to silence specific mRNA in the cytoplasm (26). MicroRNAs facilitate mRNA degradation or suppression of translation by base-pairing to complementary sequences in the 3′-UTR of target mRNA (12, 26). We previously showed that MIR29B functions as an HbF inducer in KU812 cells and normal human erythroid progenitors by targeting DNMT3 gene silencing (16). The discovery of novel small miRNA molecules, such as MIR29B, that target genes involved in HBG gene silencing to induce HbF levels will expand strategies to develop therapeutic options for SCD and other β-hemoglobinopathies.
Indeed, several miRNAs have been associated with HbF induction. In earlier studies by Bianchi et al. (27) they reported the upregulation of MIR210 in erythroid precursors of a thalassemia patient with high HbF levels. Additionally, they demonstrated that mithramycin targeted MIR210 as a mechanism of HBG activation in KU812 cells (27). In a subset of patients with SCD treated with hydroxyurea, expression of MIR26B, and MIR-151-3p was associated with HbF levels at maximum tolerated doses (28). Sangokoya et al. (29) were the first to report a correlation between MIR144 expression and the level of anemia in sickle cell patients and oxidative stress in sickle red blood cells due to reduced levels of the transcription factor NRF2. Following genome-wide miRNA expression profiling using reticulocytes isolated from SCD patients with extremes of HbF levels, our group confirmed increased MIR144 and reduced NRF2 levels in SCD patients with low HbF levels (21). Our findings provided additional supporting evidence for indirect mechanisms of HbF regulation by miRNAs (21). Previous studies by our group also demonstrated that MIR34A mediated HbF induction in KU812 cells by repression of STAT3 expression, another known repressor of HBG (30). Previously, Lee et al. (31) confirmed overexpression of LIN28B decreased miR-Let7 expression and increased HbF levels in primary erythroid cells. As further evidence of the therapeutic potential of miRNAs, MIR-486-3p induced HbF in adult erythroid progenitors by inhibiting BCL11A expression (32). MIR15A and MIR16-1 also induced HbF by targeting the HBG repressor MYB in infants with human trisomy 13 (33). Our recently published work demonstrated that MIR29B functions as an HbF inducer in KU812 cells and normal human erythroid progenitors by targeting DNMT3 gene silencing (16). This is the first study to provide evidence of a miRNA that targets DNA methylation machinery as a mechanism of HbF induction. In that same study, we showed that introduction of MIR29B into KU812 cells further decreased MYB protein expression (16).
MYB is a well-characterized oncogene protein that also functions as an HBG repressor protein (34). In a genome-wide association study consisting of SCD and β-Thalassemia patients, the HBS1L-MYB intergenic region was associated with ∼17% of the inherited HbF variance in those patients (35). The transcriptional activator MYB is known to be essential for definitive hematopoiesis (36) and is highly expressed in immature hematopoietic cells and downregulated during erythropoiesis (37). Overexpression of MYB has been demonstrated to reduce HbF levels in KU812 cells (34). Knockdown of MYB in primary human erythroid progenitors has been reported to also induce HbF expression (33). Moreover, MYB regulates HbF expression in quantitative trait locus studies and functional assays (20, 28, 29, 33–35). MYB also indirectly silences HBG expression by activating KLF1 (Krüppel-like factor 1), a transcription factor that directly binds the HBB gene and BCL11A promoters during adult erythropoiesis (38).
Considering the function of MYB as an HBG repressor protein and our published study suggesting that MIR29B targets MYB in KU812 cells, we hypothesized that overexpression of MIR29B to silence MYB would mediate HbF induction. Using in silico analysis, we discovered that MIR29B has a consensus sequence complimentary to the 3′-UTR of MYB. We confirmed the ability of MIR29B to interact with the 3′-UTR of MYB using a luciferase reporter construct that expresses the MYB 3′-UTR. Introduction of MIR29B into KU812 cells and erythroid progenitors resulted in decreased MYB mRNA and protein, accompanied by an increase in HbF expression. Our findings support the ability of MIR29B to silence MYB through 3′UTR interactions. Collectively, our published studies support an essential role of MIR29B in modulating HBG transcription by two mechanisms involving DNA methylation and MYB gene silencing.
An excellent candidate for clinical development is MIR29B since it is a well-characterized tumor suppressor gene that functions as a DNMT inhibitor. To our knowledge, the only miRNA mimic that has previously undergone investigation in clinical trials was for liposomal injection of MIR34A mimic MRX43 for the treatment of advanced melanoma cancer (ClinicalTrials.gov Identifier: NCT01829971). Although this phase I clinical trial has now been terminated due to five immune related serious adverse events, the authors provided proof-of-concept for a miRNA-based therapy for cancer (39). MIR34A, similarly to MIR29B, regulates a broad number of genes involved in proliferation, metastasis, and chemo-resistance (40). Moreover, small RNA inhibitor molecules and antisense oligonucleotides, such as Patisiran (41) and Nusinersen (42), have been used in clinical trials for the treatments of hereditary transthyretin amyloidosis and infantile Spinal Muscular Atrophy, respectively. In the present study, we demonstrated for the first time the ability of MIR29B to increase HbF levels via inhibiting MYB in KU812 cells and normal human erythroid progenitors. We further showed the ability of MIR29B to induce HbF and reduce red blood cell sickling without producing adverse side effects in Townes sickle mice. Our findings further support the development of MIR29B as an HbF inducer in preclinical sickle transgenic mice.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Augusta University Institutional Animal Care and Use Committee.
Author contributions
AS-D designed the research study. QG, AS, EA-A, LL, UO, and BL performed the research. AS-D and BP contributed essential reagents and tools. AS-D, QG, AS, CP, and BL analyzed the data. AS-D and BP wrote several drafts of the manuscript. All authors reviewed the final draft of the manuscript.
Funding
This work was funded through an NIH grant R01HL144641 (AS-D) and R01HL069234 (BP) and The University of Tennessee Health Science Center Office of Research CORNET award (AS-D).
Acknowledgments
We thank Yu Yao and Mitch Weiss at the St. Jude Children’s Research Hospital in Memphis, TN for providing the research support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.1043686/full#supplementary-material
References
1. Weatherall DJ. The role of the inherited disorders of hemoglobin, the first “molecular diseases,” in the future of human genetics. Annu Rev Genomics Hum Genet. (2013) 14:1–24. doi: 10.1146/annurev-genom-091212-153500
2. Rees DC, Gibson JS. Biomarkers in sickle cell disease. Br J Haematol. (2012) 156:433–45. doi: 10.1111/j.1365-2141.2011.08961.x
3. Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. (2004) 364:1343–60. doi: 10.1016/S0140-6736(04)17192-4
4. Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. (1984) 63:921–6. doi: 10.1182/blood.V63.4.921.bloodjournal634921
5. Estepp JH, Smeltzer MP, Kang G, Li C, Wang WC, Abrams C, et al. A clinically meaningful fetal hemoglobin threshold for children with sickle cell anemia during hydroxyurea therapy. Am J Hematol. (2017) 92:1333–9. doi: 10.1002/ajh.24906
6. Starlard-Davenport A, Kutanzi K, Tryndyak V, Word B, Lyn-Cook B. Restoration of the methylation status of hypermethylated gene promoters by microRNA-29b in human breast cancer: a novel epigenetic therapeutic approach. J Carcinog. (2013) 12:15. doi: 10.4103/1477-3163.115720
7. Jaber VR, Zhao Y, Sharfman NM, Li W, Lukiw WJ. Addressing Alzheimer’s Disease (AD) Neuropathology Using Anti-microRNA (AM) strategies. Mol Neurobiol. (2019) 56:8101–8. doi: 10.1007/s12035-019-1632-0
8. Wang V, Wu W. MicroRNA-based therapeutics for cancer. BioDrugs. (2009) 23:15–23. doi: 10.2165/00063030-200923010-00002
9. Shah V, Shah J. Restoring ravaged heart: molecular mechanisms and clinical application of miRNA in heart regeneration. Front Cardiovasc Med. (2022) 9:835138. doi: 10.3389/fcvm.2022.835138
10. Sobolewski C, Dubuquoy L, Legrand N. MicroRNAs, tristetraprolin family members and HuR: a complex interplay controlling cancer-related processes. Cancers. (2022) 14:3516. doi: 10.3390/cancers14143516
11. Contiliani DF, Ribeiro YA, de Moraes VN, Pereira TC. MicroRNAs in prion diseases-from molecular mechanisms to insights in translational medicine. Cells. (2021) 10:1620. doi: 10.3390/cells10071620
12. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
13. Duttagupta R, Jiang R, Gollub J, Getts RC, Jones KW. Impact of cellular miRNAs on circulating miRNA biomarker signatures. PLoS One. (2011) 6:e20769. doi: 10.1371/journal.pone.0020769
14. Starlard-Davenport A, Gu Q, Pace BS. Targeting genetic modifiers of HBG gene expression in sickle cell disease: the miRNA option. Mol Diagn Ther. (2022) 26:497–509. doi: 10.1007/s40291-022-00589-z
15. Starlard-Davenport A, Fitzgerald A, Pace BS. Exploring epigenetic and microRNA approaches for gamma-globin gene regulation. Exp Biol Med. (2021) 246:2347–57. doi: 10.1177/15353702211028195
16. Starlard-Davenport A, Smith A, Vu L, Li B, Pace BS. MIR29B mediates epigenetic mechanisms of HBG gene activation. Br J Haematol. (2019) 186:91–100. doi: 10.1111/bjh.15870
17. Navarro F, Gutman D, Meire E, Caceres M, Rigoutsos I, Bentwich Z, et al. miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood. (2009) 114:2181–92. doi: 10.1182/blood-2009-02-205062
18. Li B, Ding L, Yang C, Kang B, Liu L, Story MD, et al. Characterization of transcription factor networks involved in umbilical cord blood CD34+ stem cells-derived erythropoiesis. PLoS One. (2014) 9:e107133. doi: 10.1371/journal.pone.0107133
19. Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. (2006) 108:1183–8. doi: 10.1182/blood-2006-02-004812
20. Kodeboyina S, Balamurugan P, Liu L, Pace BS. cJun modulates Ggamma-globin gene expression via an upstream cAMP response element. Blood Cells Mol Dis. (2010) 44:7–15. doi: 10.1016/j.bcmd.2009.10.002
21. Li B, Zhu X, Ward CM, Starlard-Davenport A, Takezaki M, Berry A, et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp Hematol. (2019) 70:85–96.e5. doi: 10.1016/j.exphem.2018.11.002
22. Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics – challenges and potential solutions. Nat Rev Drug Discov. (2021) 20:629–51. doi: 10.1038/s41573-021-00219-z
23. Fyfe I. MicroRNAs – diagnostic markers in Parkinson disease? Nat Rev Neurol. (2020) 16:65. doi: 10.1038/s41582-019-0305-y
24. Bhatnagar B, Garzon R. Clinical applications of MicroRNAs in acute myeloid leukemia: a mini-review. Front Oncol. (2021) 11:679022. doi: 10.3389/fonc.2021.679022
25. Geisler L, Mohr R, Lambrecht J, Knorr J, Jann H, Loosen SH, et al. The role of miRNA in the pathophysiology of neuroendocrine tumors. Int J Mol Sci. (2021) 22:8569. doi: 10.3390/ijms22168569
27. Bianchi N, Zuccato C, Lampronti I, Borgatti M, Gambari R. Expression of miR-210 during erythroid differentiation and induction of gamma-globin gene expression. BMB Rep. (2009) 42:493–9. doi: 10.5483/BMBRep.2009.42.8.493
28. Walker AL, Steward S, Howard TA, Mortier N, Smeltzer M, Wang YD, et al. Epigenetic and molecular profiles of erythroid cells after hydroxyurea treatment in sickle cell anemia. Blood. (2011) 118:5664–70. doi: 10.1182/blood-2011-07-368746
29. Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. (2010) 116:4338–48. doi: 10.1182/blood-2009-04-214817
30. Ward CM, Li B, Pace BS. Original research: stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp Biol Med. (2016) 241:719–29. doi: 10.1177/1535370216636725
31. Lee YT, de Vasconcellos JF, Yuan J, Byrnes C, Noh SJ, Meier ER, et al. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. (2013) 122:1034–41. doi: 10.1182/blood-2012-12-472308
32. Lulli V, Romania P, Morsilli O, Cianciulli P, Gabbianelli M, Testa U, et al. MicroRNA-486-3p regulates gamma-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One. (2013) 8:e60436. doi: 10.1371/journal.pone.0060436
33. Sankaran VG, Menne TF, Scepanovic D, Vergilio JA, Ji P, Kim J, et al. MicroRNA-15a and –16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U.S.A. (2011) 108:1519–24. doi: 10.1073/pnas.1018384108
34. Jiang J, Best S, Menzel S, Silver N, Lai MI, Surdulescu GL, et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood. (2006) 108:1077–83. doi: 10.1182/blood-2006-01-008912
35. Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U.S.A. (2007) 104:11346–51. doi: 10.1073/pnas.0611393104
36. Sheiness D, Gardinier M. Expression of a proto-oncogene (proto-myb) in hemopoietic tissues of mice. Mol Cell Biol. (1984) 4:1206–12. doi: 10.1128/mcb.4.7.1206-1212.1984
37. Vegiopoulos A, Garcia P, Emambokus N, Frampton J. Coordination of erythropoiesis by the transcription factor c-Myb. Blood. (2006) 107:4703–10. doi: 10.1182/blood-2005-07-2968
38. Tallack MR, Perkins AC. Three fingers on the switch: kruppel-like factor 1 regulation of gamma-globin to beta-globin gene switching. Curr Opin Hematol. (2013) 20:193–200. doi: 10.1097/MOH.0b013e32835f59ba
39. Hong DS, Kang YK, Borad M, Sachdev J, Ejadi S, Lim HY, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. (2020) 122:1630–7. doi: 10.1038/s41416-020-0802-1
40. Saki N, Abroun S, Hajizamani S, Rahim F, Shahjahani M. Association of chromosomal translocation and MiRNA expression with the pathogenesis of multiple myeloma. Cell J. (2014) 16:99–110.
41. Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. (2018) 379:11–21.
Keywords: MIR29B, fetal hemoglobin (HbF), MYB, HBG, sickle cell
Citation: Gu Q, Palani CD, Smith A, Li B, Amos-Abanyie EK, Ogu U, Lu L, Pace BS and Starlard-Davenport A (2022) MicroRNA29B induces fetal hemoglobin via inhibition of the HBG repressor protein MYB in vitro and in humanized sickle cell mice. Front. Med. 9:1043686. doi: 10.3389/fmed.2022.1043686
Received: 13 September 2022; Accepted: 10 November 2022;
Published: 25 November 2022.
Edited by:
Robert W. Maitta, Case Western Reserve University, United StatesReviewed by:
J. Peter Ray Pelletier, UF Health Shands Hospital, United StatesLauire Steiner, University of Rochester Medical Center, United States
Copyright © 2022 Gu, Palani, Smith, Li, Amos-Abanyie, Ogu, Lu, Pace and Starlard-Davenport. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Athena Starlard-Davenport, YXN0YXJsYXJAdXRoc2MuZWR1
†These authors share first authorship