 Austin N. Smith
Austin N. Smith Michael Shaughness
Michael Shaughness Sean Collier2,3
Sean Collier2,3 Kimberly R. Byrnes
Kimberly R. Byrnes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 03 November 2022
Sec. Translational Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1034692
This article is part of the Research TopicImpacts of Drug-Induced Oxidative StressView all 5 articles
Inflammation is a primary component of the central nervous system injury response. Traumatic brain and spinal cord injury are characterized by a pronounced microglial response to damage, including alterations in microglial morphology and increased production of reactive oxygen species (ROS). The acute activity of microglia may be beneficial to recovery, but continued inflammation and ROS production is deleterious to the health and function of other cells. Microglial nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), mitochondria, and changes in iron levels are three of the most common sources of ROS. All three play a significant role in post-traumatic brain and spinal cord injury ROS production and the resultant oxidative stress. This review will evaluate the current state of therapeutics used to target these avenues of microglia-mediated oxidative stress after injury and suggest avenues for future research.
Traumatic injury to the central nervous system (CNS), including the brain (traumatic brain injury, TBI) and spinal cord (SCI), affects millions of people every year, adding to those already living with these injuries (1–3). Microglia are the brain’s primary immunocompetent cells; these cells’ response to CNS trauma ranges from restorative to detrimental and studies have shown microglial activity after a traumatic event shapes recovery and impacts functional and behavioral outcomes (4, 5). Post-injury phagocytosis, neurotrophic support, and release of mediators of cytotoxicity, including reactive oxygen species (ROS), fall into the category of microglial responses (6). In this review, we collect recent efforts to use pharmacological therapies to mediate microglial production of ROS in the CNS after trauma to improve functional and behavioral outcomes.
The vast functions of neurons and glia require high energy expenditure that is dependent upon oxygen for mitochondrial ATP production. However, as an electron acceptor, oxygen can form ROS. ROS include but are not limited to superoxide (O2–), peroxide (O22–), and hydroxyl radical (•OH). While low levels of ROS act as signaling molecules, high levels of ROS can cause cellular damage. To maintain homeostasis, cells increase antioxidants to offset accumulated ROS.
Within microglia, ROS are associated with proliferation (7, 8), immune defense (9–11), and redox signaling (12). ROS-sensitive pathways mediate the activation of nuclear factor-kappa B (NFκB) and other transcription factors critical to the inflammatory response of microglia and other phagocytes (13). Not only do microglia use ROS as a defense against pathogens, but they activate pathways that allow microglia to respond to CNS injury. This rapid response of microglia, including migration to a site of injury, aids the clearance of debris and dead cells, preventing secondary cell death (14, 15). However, chronic activation of microglia and the overproduction of ROS is neurotoxic, precipitating oxidative stress which contributes to distinct secondary injury cascades, such as lipid peroxidation and oxidative histone phosphorylation (16) (Figure 1). Microglia are also a source of the highly toxic peroxynitrite (17). Activated microglia produce nitric oxide (NO), which rapidly reacts with superoxide due to its unpaired electron to form peroxynitrite (ONO2–), a reactive nitrogen species (RNS) toxic to both neurons and oligodendrocytes (17, 18).
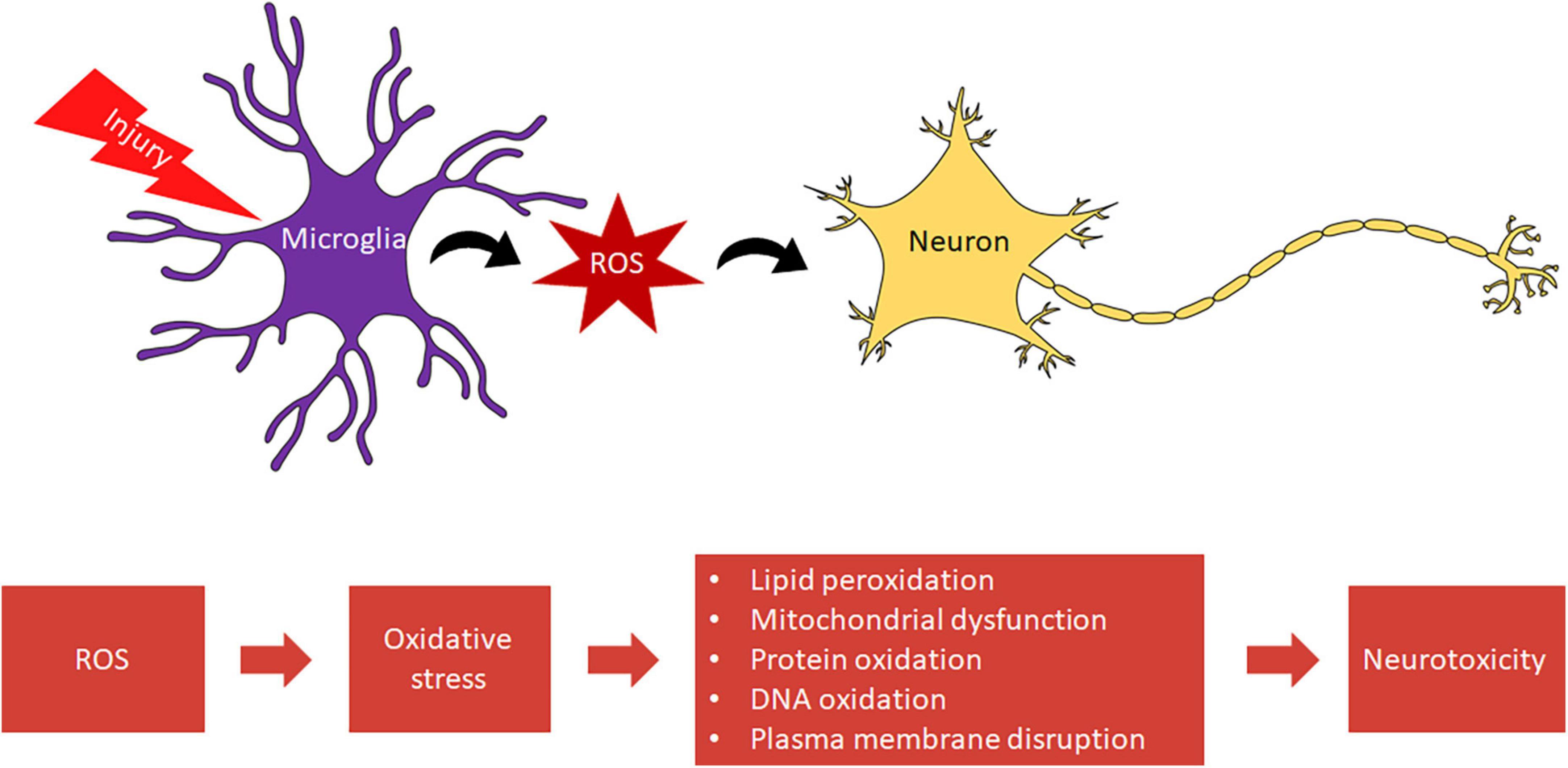
Figure 1. Microglia mediated oxidative stress leads to neurotoxicity.
Oxidative stress is an imbalance between generated ROS and innate antioxidants. Under oxidative stress, there is a greater probability that excessive ROS will react with lipids, nucleic acids, and proteins. These reactions induce lipid peroxidation and oxidation of proteins and DNA. Lipid peroxidation and DNA oxidation in particular can disrupt the plasma membrane and bring about DNA damage capable of inducing apoptosis after neurotrauma (19, 20). Thus, oxidative stress intensifies conditions of neurodegenerative disease and injury.
The production of ROS originating from multiple intracellular mechanisms must be strictly regulated to minimize excessive damage to the CNS. A number of these mechanisms are active in microglia. This review will focus on three of the most common inducers of ROS identified from microglia, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, iron, and mitochondria (Figure 2).
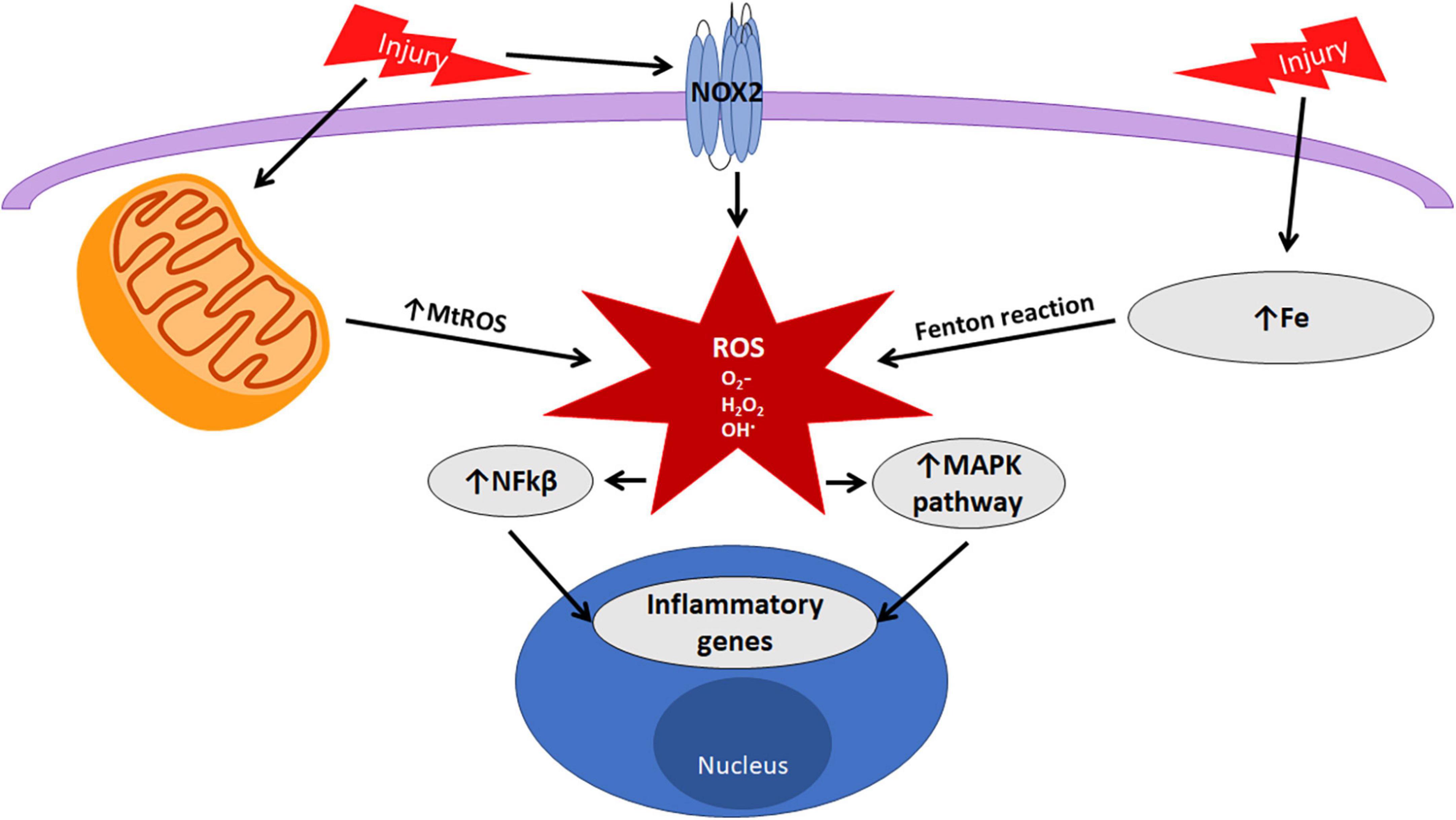
Figure 2. Oxidative stress in microglia. Microglia can produce reactive oxygen species (ROS) through NOX2, mitochondrial oxidative phosphorylation, and the Fenton reaction with iron. ROS act as signaling molecules to mediate pathways pivotal in microglial functions.
NADPH oxidase (NOX) is a primary ROS-generating enzyme that microglia express (21, 22). The NOX family consists of seven isoforms, most of which catalytically transfer one electron from NADPH to oxygen, producing superoxide. The antioxidant superoxide dismutase (SOD) can then dismutate superoxide to hydrogen peroxide (Figure 3), but imbalance in anti- and pro-oxidant components due to injury or disease can shift hydrogen peroxide concentrations and lead to oxidative stress.
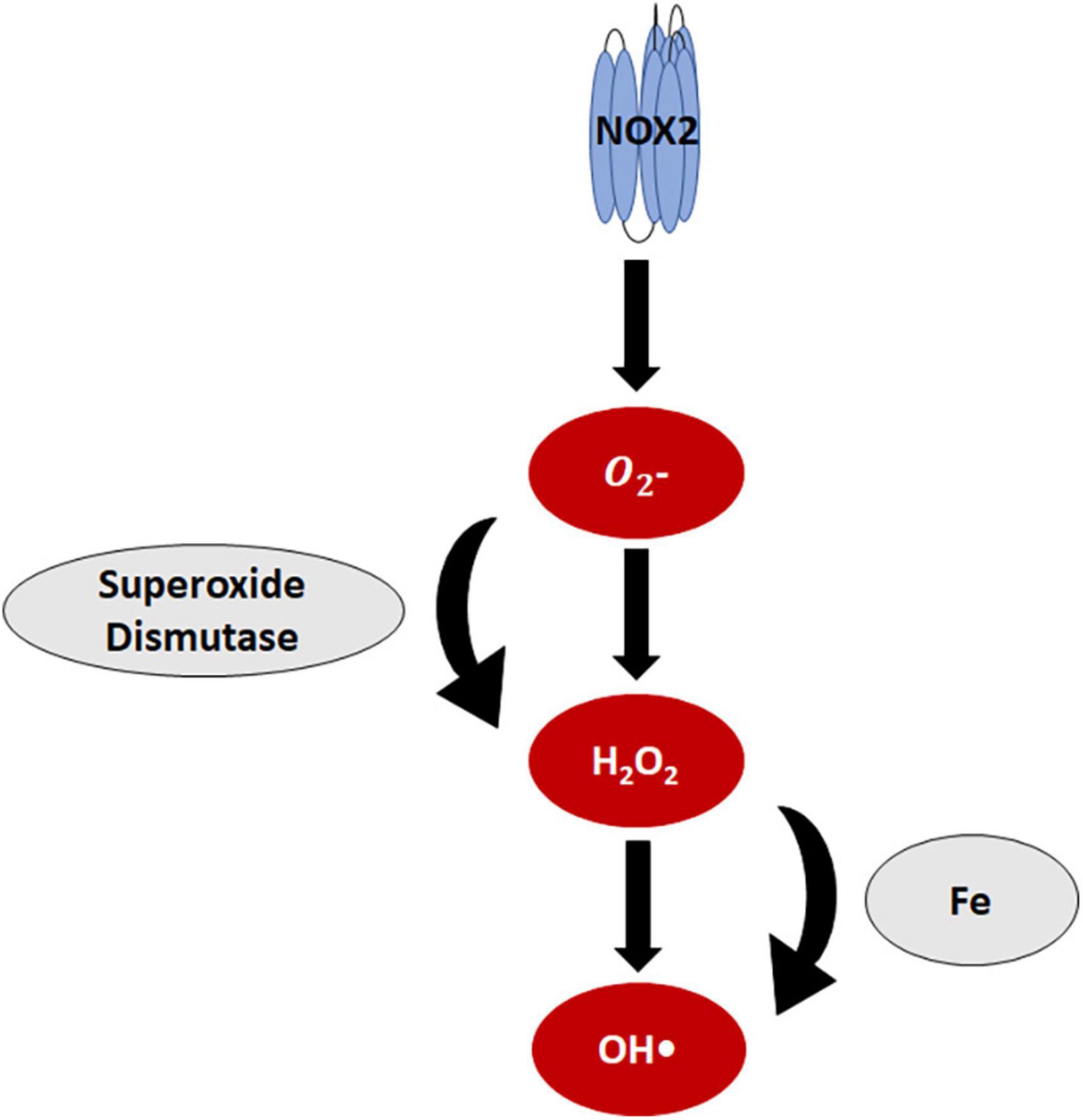
Figure 3. Reactive oxygen species in microglia. NOX2, the primary reactive oxygen species (ROS)-generating enzyme in microglia, produces superoxide. Superoxide dismutase then catalyzes the dismutation of superoxide to produce hydrogen peroxide. Iron reacts with hydrogen peroxide, generating hydroxyl radicals.
Microglia express the isoforms NOX1, NOX2, and NOX4 (23, 24). Membrane bound NOX2 (gp91phox) is requisite for microglia-mediated neuroinflammation and is considerably increased after TBI and SCI (23, 25–27). NOX2 is activated in association with membrane bound p22phox and the translocation of cytosolic Rac2, p40phox, p47phox, and p67phox.
Plasma membrane bound NOX2 is the principal generator of extracellular superoxide, as evidenced by the finding that lipopolysaccharide (LPS)-stimulated microglial cultures derived from gp91phox–/– rodents fail to increase extracellular superoxide (28). Activation of NOX2 on the phagosome membrane releases superoxide into the lumen to be converted to other reactive species, facilitating anti-microbial function (29, 30). Studies have reported that microglial phagocytosis induces NOX-dependent superoxide production in response to cellular debris and IgG antibodies (31, 32).
NOX function is also necessary to activate phosphoinositide 3-kinase, MAP kinases, and NFκB, which upregulate microglial proinflammatory gene expression (33) and control LPS-induced phagocytosis (34). NOX-dependent NLRP3 inflammasome activity regulates the secretion of pro-inflammatory cytokines, which may mediate the neurodegenerative effects of microglial oxidative stress (35). Based upon these findings, NOXs present a significant source of ROS production while NOX pathways govern microglial functions, both of which influence neuroinflammation.
Microglial activation and subsequent neurotoxicity is in many cases NOX-dependent (28, 36). Other research suggests that NOX activity alone may not cause significant neuron death, but rather requires additional cellular mechanisms such as iNOS (37). Microglial NOX activity also contributes to the breakdown of the blood brain barrier (BBB). Astrocyte and endothelial cell death increases after oxygen-glucose deprivation/reperfusion when co-culturing with microglia, an effect prevented with NOX inhibition (38).
NOX2 has been shown to be particularly responsive to injury in the CNS, including TBI and SCI, with a rapid upregulation and maximal expression within the first 7 days after injury, and chronic expression through at least 1 month (23, 24). NOX2 activity is induced by stimulation of the toll-like receptors and requires induction of the NFκB and p38 MAPK signal transduction pathways (39). NOX2 and 4 have both also been implicated in post-SCI neuropathic pain development (40, 41).
A variety of therapeutics targeting ROS and the primary inducers of ROS in microglia have been developed, and a summary of some of these therapeutic approaches is presented in Table 1. Inhibition of microglial NOX attenuates pro-inflammatory gene expression, ROS, and oxidative stress (42–44). Reduced or indirectly inhibited function of NOX explains the anti-inflammatory effects of other neuroprotective treatments, including antioxidants and non-steroidal anti-inflammatory drugs (45–47). Although NOX2 is not the sole generator of ROS within activated microglia, it presents a possible therapeutic target to reduce oxidative stress and neuroinflammation after neurotrauma. In further sections, details of NOX therapy will be discussed.
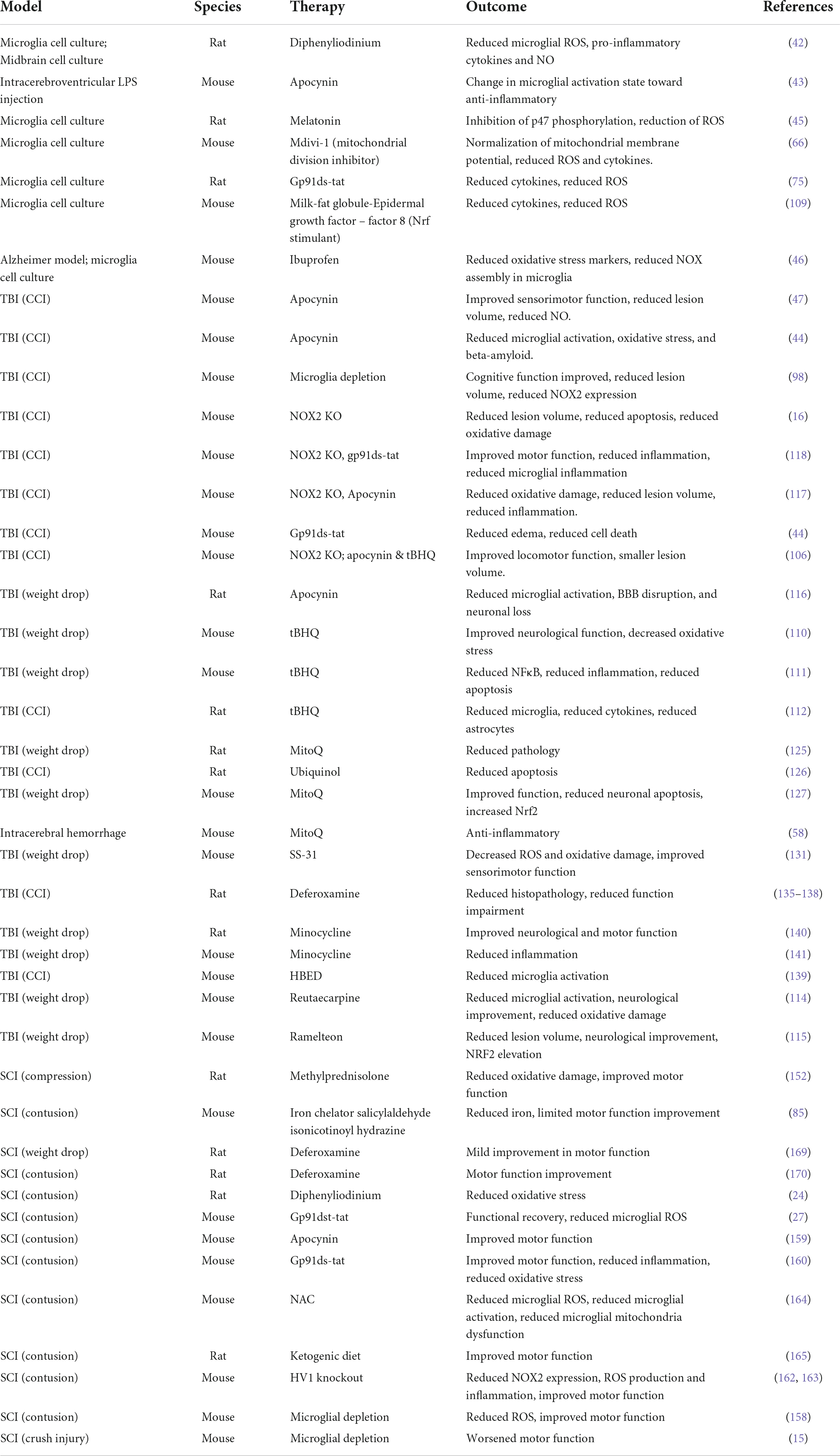
Table 1. Therapeutics targeting microglial reactive oxygen species (ROS).
Oxidative phosphorylation can generate mitochondrial ROS (mtROS) within the intermembrane space and matrix. As electrons are passed through the electron transport chain to form ATP, both complex I and complex III can release electrons to form superoxide. The rate of this ROS production normally remains low, and mitochondrial SOD reduces mtROS to form hydrogen peroxide, which glutathione peroxidase then converts to water (48). The inner mitochondrial membrane transporter, uncoupling protein 2 (UCP2), downregulates mtROS. Decreased expression of UCP2 increases ROS in other phagocytes to provide infection resistance (49). Activated microglia increase mtROS via downregulation of UCP2. In fact, the phasic down- or upregulation of UCP-2 modulates microglial phenotype, such that UCP2-silenced microglia exhibit a continuous pro-inflammatory phenotype (50).
In addition, the co-occurring increase in superoxide and nitric oxide in activated microglia heightens the probability of producing peroxynitrite, which inhibits the electron transport chain (51, 52). Experimental administration of electron transport chain inhibitors enhances mtROS production and pro-inflammatory microglial activation (53, 54).
Like NOX2-induced ROS, increased mtROS can be transported to the cytoplasm and regulate microglial pro-inflammatory gene expression via MAP kinases and NFκB (53, 55). MtROS also activate the NLRP3 inflammasome (56). These results indicate not only that mtROS govern microglial activation, but that mtROS present a positive feedback loop resulting in mitochondrial dysfunction and elevated microglia-mediated oxidative stress (Figure 2).
Mitochondrial functioning impacts the outcomes of neurotrauma. For example, chronic inflammation due to neurotrauma may cause cells to consume the antioxidant, glutathione, faster than it can be replenished. Reduced glutathione levels are associated with increased electron transport chain complex activity without increasing ATP production as demonstrated with in vivo injections of LPS (57). Conversely, increased mitochondrial antioxidants can effectively reduce ROS and attenuate microglia-induced inflammation (58).
Mitochondrial metabolism is reduced at 1 h after SCI (59), and mitochondrial dysfunction is noted at 12 h post-injury in animal models, accompanied by increases in protein nitrosylation, lipid peroxidation and protein oxidation (60). However, it is important to note that increases in 3-nitrotyrosine (3NT), 4-hydroxynonenal (4HNE) and other oxidative stress markers have been noted prior to this 12 h time point, suggesting that while mitochondrial ROS certainly plays a role, additional sources of ROS are also at work. Similar alterations are observed in TBI, with acute disruptions in mitochondrial function that are associated with elevated ROS and oxidative stress (61, 62).
Intracellular mtROS production is also associated with mitochondrial fission in activated microglia (63, 64). LPS-stimulated microglia release mitochondria fragments that lead to an increase in astrocyte ROS and mitochondrial fragmentation (65), propagating oxidative stress.
To counter unfavorable mitochondrial functioning, microglia treated with mitochondrial division inhibitor show normalized glycolysis and oxidative phosphorylation and reduced ROS (66). In addition, UCP2 overexpression is neuroprotective in brain injured mice and in vitro cortical neurons subjected to oxygen glucose deprivation (67). From these data, it is apparent that the level of mtROS determines mitochondrial activity and dysfunction in altering microglial function and oxidative stress.
Iron is an essential cofactor and molecular component for many intracellular mechanisms of neurons and glia. Iron is necessary for the function of NOX, oxidative phosphorylation, and the citric acid cycle (68, 69). The iron-binding protein, ferritin, sequesters intracellular iron, preventing iron toxicity. Free iron that is unbound to ferritin impairs phagocytosis (70) and contributes to oxidative stress. The Fe2+ iron ion reacts with the hydrogen peroxide generated from oxidative respiration or NOX to produce a hydroxyl radical in a catalytic conversion known as the Fenton reaction (Figures 2, 3).
Iron combined with mitochondrial oxidative stress can cause microglia to undergo ferroptosis, an iron-dependent programmed cell death (71, 72), or induce ferroptosis in other cells via iron dysregulation or NO release (73, 74). Recent research from our laboratory demonstrated that iron induces ROS production in microglia and amplifies ROS of stimulated microglia (75). Both NOX2 and NOX4 inhibitors prevent the effects of iron treatment on microglia.
Not only does iron exacerbate oxidative stress, but inflammatory states and ROS alter iron levels. Under physiological conditions, microglial ferritin levels are associated with levels of free iron (76). Prior studies have found mixed results determining whether microglial ferritin expression is decreased (77) or increased (78) with oxidative stress and LPS activation. Divalent metal transporter-1 (DMT1) and transferrin receptor-mediated endocytosis transport Fe2+ and Fe3+, respectively, to regulate intracellular iron levels (79). Meanwhile, ferroportin is the primary iron exporter. Pro-inflammatory stimuli cause an increase in intracellular iron accumulation and induce an upregulation of DMT1 in neurons, astrocytes, and microglia (80). Additional investigations revealed that microglia increase iron influx in response to both pro- and anti-inflammatory cytokines (81). Microglia may alter substrate preference for transferrin-bound iron under pro-inflammatory states or non-transferrin-bound iron under anti-inflammatory states (79). While Urrutia et al. (80) found neither TNF-α, IL-6, nor LPS significantly altered ferroportin expression among microglia, Holland et al. (78) found that IFNγ reduced microglial ferroportin. Microglial iron uptake is initially neuroprotective by reducing free extracellular iron and ROS (82, 83); however, the resulting interactions between iron and oxidative stress mediators within microglia contribute to microglia-mediated neurotoxicity. Future studies are essential to evaluate whether microglial uptake of iron impacts ferroptosis among other CNS cell types.
Excessive iron accumulation may also contribute to post-injury oxidative damage. Liu et al. demonstrated that iron was a primary contributor to hydroxyl radical formation within 5 h after spinal cord contusion injury (84). Iron staining has shown that there is an increase in iron phagocytosis by microglia/macrophages in the lesion site through 14 days post-injury (85). Magnetic resonance imaging showed that iron deposits are present through 35 days post-injury, both in the lesion site and distant to it, in macrophages/microglia, astrocytes and oligodendrocytes (86). In addition to uptake within cells, diffuse staining with Prussian blue suggests deposition of iron in the extracellular matrix (86). Iron homeostatic proteins are upregulated after injury, including ceruloplasmin (CP), which is increased at 1 day and remains elevated through 21 days post-SCI in astrocytes and macrophages near the lesion site and ferritin protein, which is increased from 3 days post-injury (85).
Traumatic brain injury is associated with microglial activation and oxidative stress. Oxidative damage post-TBI can impair neuronal functioning, disrupt the BBB, and potentiate cell death (87). Postmortem analysis on human TBI brains found elevated cortical expression of microglial NOX2 and increased 8-hydroxydeoxyguanosine (8-OHdG), a predominant form of ROS-induced oxidative damage to DNA (88). A rodent TBI model, using the controlled cortical impact (CCI) showed increased cortical 8-OHdG in rats within 15 min of the injury (89). Other markers of protein and lipid oxidation, 4HNE and 3NT, vary with injury models, with studies showing an increase by 3 h and resolving by 24 h after blast injury (90) and others peaking between 24-48 h after a CCI injury (91). 4HNE was observed in the brain within 6 h after blast exposure (92, 93). Human brain tissue collected within 91 h of a TBI also displayed prominent evidence of oxidative stress (94). These elevations in markers of oxidative stress were often associated with areas of microglial activation (90). Oxidative damage is not just an acute response, and has been noted through 42 days after blast exposure (95). After a CCI, mice show evidence of oxidative stress in neurons and microglia through 8 months after injury (96). However, despite temporal variations between injury models, oxidative stress is an established pathophysiological process in human and animal models of TBI.
This section will highlight recent research that examined therapies and pharmacological agents targeting microglia-mediated oxidative stress in experimental models of TBI.
Chronically activated microglia are observed in patients with a TBI up to 17 years after a single injury (97). Microglial depletion may be a mechanism to reduce chronic microglia mediated oxidative stress. Using microglial depletion, Henry et al. (98) showed, in a mouse CCI injury model, that depleting microglia during the chronic phase of TBI followed by repopulation resulted in marked improvements in downstream neurological dysfunction and reduced posttraumatic neurodegeneration. Coinciding with the behavioral and cognitive improvements, histological analysis revealed a significant reduction in the lesion volume and an increased density of neurons in the cortex and dentate gyrus. Transcriptional analysis documented various oxidative stress-related genes that were differentially expressed in the cortex 2 months post-injury, including a reduction of oxidative stress-inducing genes and an increase in antioxidant-related genes. Furthermore, repopulated microglia presented as less reactive with significantly more ramifications, less hypertrophy, and a reduction in the expression of NOX2 characteristic of a less inflammatory response. The results from this study implicate reactive microglia in oxidative stress, cognitive/behavioral dysfunction and neuropathology associated with TBI. However, microglial reduction as a therapeutic approach is limited, considering the necessity of microglia in normal physiology of the brain. Further work in this field is necessary to truly understand the overall contribution of microglia to TBI.
In addition to non-specific microglial depletion, non-microglial specific oxidant reduction has been tested as a therapeutic target. Nuclear erythroid 2-related factor 2 (Nrf2) is known to bind to the antioxidant response element to upregulate antioxidant gene expression and counter oxidative stress. Increasing evidence suggests Nrf2 also modulates the functions of both mitochondria and NOX2 (99, 100). Protein levels of Nrf2 are increased after weight drop and blast models of TBI in rats and mice (101, 102), peaking at 1 day post-injury in neurons and 7 days post-injury in microglia near the nucleus, which suggests active transcriptional activity (103). However, Nrf2 may be downregulated during chronic stages of TBI (104). Nrf2 KO mice had greater neurological impairment, increased lesion volume, and higher levels of oxidative stress markers (4HNE, 8-OHdG, and protein carbonyls) in response to a CCI injury (105, 106). Nrf2 KO mice also showed exacerbated neuroinflammation, 4HNE, and 3NT 1 day following a fluid percussion injury (107). In vitro studies have confirmed that Nrf2 suppresses the microglia pro-inflammatory phenotype (108, 109).
Oxidative stress and microglial activation post-TBI can be altered with Nrf2 activators, of which tertiary butylhydroquinone (tBHQ) is the most well studied. Three doses of tBHQ (50 mg/kg) prior to mouse weight drop TBI improved neurological function, decreased the oxidative stress marker malondialdehyde (MDA), and increased SOD activity (110). One week of a tBHQ-supplemented diet before a weight drop injury reduced NFκB, proinflammatory cytokines, and apoptotic cell death (111). TBHQ (25 mg/kg) given at 5 min or 2 h post-CCI followed by a second dose at 24 h improved motor functioning but had no effect on lesion volume. However, lesion volume was reduced by combining the treatment with apocynin (106). Daily administration of tBHQ (25 mg/kg) beginning 24 h post-CCI significantly reduced CD68 + /Iba1 + microglia immunostaining concurrent with attenuated proinflammatory cytokine levels in rats 3 days post-injury. Treatment also reduced astrocyte activation at 7 days post-injury and lesion volume at 28 days post-injury (112). The results of these studies suggest tBHQ activates Nrf2 to offset oxidative stress and provide neuroprotective effects after TBI. Unfortunately, Nrf2 affects a multitude of intracellular targets, making it difficult to pare down Nrf2-targeted treatments to a particular effector such as the mitochondria or NOX2. A limitation of tBHQ is its debated genotoxicity, but it is approved for human consumption in small concentrations (113).
More recently, additional therapeutics targeting Nrf2 have been identified that may also hold promise after TBI, although the specific cellular target remains unclear. In these studies, approaches including rutaecarpine and ramelteon, have been shown to reduce neuronal oxidative damage and microglial related inflammation via Nrf2 pathway activation after TBI in mice models (114, 115). Additional study to identify mechanism of action and cellular source is necessary to continue to refine therapies and translate to clinical trials.
NOX2 activity is involved in post-TBI neuropathology and oxidative stress damage (16, 44, 116). Rodent studies of TBI show elevated microglial NOX2 expression within days (16, 117) and sustained at 28 days (118) and 12 months post-injury (119). Activated microglia expressing NOX2 are present in the periphery of a lesion up to one year following an injury (119). To study the pathophysiological contributions of NOX2 in a TBI, multiple studies have used NOX2 knockout (KO) mice in a CCI model. Dohi et al. (16) found that 2 days post-injury, NOX2 KO mice had reduced lesion volume and peri-lesional apoptotic cells. This was associated with decreased oxidation products ethidine and 3NT in CD11b + microglia and infiltrating macrophages. Kumar et al. (118) extended these findings, showing a reduced lesion volume and neuronal cell death at 21 days post-injury. Injured NOX2 KO mice also demonstrated improved motor function at 14 and 21 days post-injury. Genomic analysis of inflammatory markers in the cortex 1-day post-injury revealed a marked reduction in proinflammatory (NOS2, TNFα, IL-6, IL-12b, IL-1β) and increased anti-inflammatory genes (IL-4Rα, SOCS3, and Ym1). Moreover, the number of Iba1 + microglia/macrophages expressing the pro-inflammatory-associated CD16/32 cell surface marker was diminished at 7 days post-injury. In a separate study, Wang et al. (117) corroborated the neuroprotective effects of knocking out the NOX2 enzyme, by showing that NOX2 KO TBI mice displayed less oxidative damage (shown by reduced 4HNE) as well as increased neuronal survival and reduced lesion volume at 4 and 7 days after injury. Furthermore, this was associated with significantly downregulated pro-inflammatory-associated genes (CD16, CD32, CD86, iNOS) and significantly upregulated anti-inflammatory-associated genes (CD206, Ym1). Interestingly, when microglia were isolated from KO and wild-type TBI mice, the microglia from the former were less pro-inflammatory and more anti-inflammatory compared to the latter, with cells expressing less pro-inflammatory associated CD86 and more anti-inflammatory associated CD206. Lastly, in vitro studies showed that healthy neurons co-cultured with microglia from injured NOX2 KO mice were healthier, displayed less neuronal apoptosis, and cytotoxicity compared to neurons co-cultures with microglia from injured wild-type mice. This suggests that the activation of the NOX2 receptor on microglia in part, mediates the neurotoxicity observed in the in vivo injury model.
A limitation of these studies is the NOX2 KO mouse is a complete knockout of the NOX2 gene and not microglia specific. Therefore, effects may be partially mediated by additional cell types. Given that neurons also express NOX2, some neuroprotective effects of NOX2 KO could be related to direct intracellular effects on neurons. Additionally, studies used markers that cannot distinguish microglia from macrophages, which could be contributing to results. In fact, Kumar et al. (118) determined that macrophages accounted for 25% of NOX2 expression post-CCI while microglia accounted for 8%. Further research is needed to investigate the contributions of microglial versus macrophage NOX2 following TBI.
Researchers have also tested the therapeutic potential of targeting the NOX2 enzyme with various pharmacological modulators. The specific peptide inhibitor, gp91ds-tat, has been utilized to target NOX2 in CCI injury models. Administration of gp91ds-tat (250 μg/mouse) 20 min prior to CCI reduced edema 1-day post-injury and cell death 4-days post-injury (44). Consistent with the aforementioned KO studies, administration of gp91ds-tat (5 mg/kg) at 24, 48, and 72 h post-CCI reduced CD16/32 expression in the cortex (120) and promoted an anti-inflammatory microglia phenotype, indicated by increased anti-inflammatory Arg1 and Ym1 expression in P2Y12 + /CD11b + microglia (118). Treatment was associated with improved performance in spatial working memory tests but had no improvement on fine motor function within the first week post-injury (118).
Apocynin is a medicinal compound isolated from the plant Picrorhiza kurroa and inhibits NOX activity in neutrophils and macrophages by preventing p47phox and p67phox subunit translocation (121). Choi et al. (116) showed that rats pre-treated with apocynin (100 mg/kg) fifteen min before a weight drop model of TBI had reduced microglial/macrophage activation in the CA1 region of the hippocampus 7 days after injury. Additionally, this was associated with reduced BBB dysfunction and fewer degenerating neurons in the hippocampus. Using a CCI mouse model of TBI, researchers found that administration of apocynin (4 mg/kg) 20 min before injury or 2 h post-injury significantly reduced immunostaining for oxidative stress markers 4HNE, 8-OHdG, and p-H2AX in the cortex and CA1 regions of hippocampus 2 days post-injury (44). The decrease in oxidative stress was followed by reduced microglia/macrophage activation in the cortex and CA1 region of the hippocampus 4 days post-injury. Another CCI mouse study found that apocynin (5 mg/kg) administered for 4 consecutive days prior and 1 day post-injury significantly decreased ROS (measured with hydroethidine) within CD11b + microglia/macrophages (117).
Although these results suggest apocynin has NOX-mediated anti-inflammatory effects on microglia, the effects of apocynin are not specific to a single NOX isoform and have off-target effects (122). For this reason, the favorable outcomes of apocynin treatment may indicate a benefit of general antioxidant treatments for TBI. In addition, global NOX2 or NOX inhibition may have negative immune suppressive effects. Reports of gastrointestinal disorders or dysfunction of neutrophils have been reported with NOX2 inhibition (123), which suggest that additional research is needed to better target NOX2 inhibitors to microglia or to the brain after injury.
Treatments aimed to alter the function of mitochondria or its electron transport chain may limit ROS production and suppress oxidative stress. Cortical mitochondria isolated post-CCI show phasic dysfunction, with significant oxidative damage beginning at 24 h, allowing for a larger therapeutic window for treatments (124). Antioxidant compounds present one treatment strategy to target mitochondrial dysfunction post-TBI. Here we will discuss just a few mitochondria-targeted antioxidants.
Coenzyme Q10 (CoQ10), or ubiquinone, is an essential part of the electron transport chain, but it also acts as an antioxidant. TBI outcomes have been assessed following treatments with CoQ10, its reduced form ubiquinol, and mitoquinone (MitoQ) which is composed of covalently bound CoQ10 and triphenylphosphonium ions. CoQ10 (10 mg/kg) administered immediately after a weight drop injury reduced MDA, generated a non-statistically significant increase in SOD, and attenuated TBI-induced histopathology (125). Ubiquinol (100 mg/kg), administered to rats 30 min before a CCI mitigated mitochondrial damage and reduced apoptosis (126). Administration of MitoQ (4 mg/kg & 8 mg/kg) 30 min after injury reduced neuronal apoptosis and improved neurobehavioral function at 1 and 3 days post-injury. MitoQ also reduced post-TBI oxidative stress, increasing the antioxidant activity of glutathione peroxidase and SOD, while decreasing MDA. MitoQ also increased Nrf2 activity and upregulated downstream proteins of Nrf2 signaling (127). Although the above studies did not confirm the association between treatments and microglial activation, MitoQ did have neuroprotective effects and anti-inflammatory effects on microglia in vivo after intracerebral hemorrhage (58). Despite these results, separate studies have found that MitoQ can cause mitochondrial damage due to increased inner membrane permeability (128) and increase mitochondrial ROS driven by complex I activity (129). Further research is necessary to corroborate the effects of CoQ10-based treatments on microglial functions and microglia-mediated oxidative stress.
SS-31, also known as elamipretide or bendavia, is a synthetic antioxidant peptide that binds to cardiolipin of the inner mitochondrial membrane where it inhibits cytochrome c peroxidase activity, inhibits mitochondrial permeability, and reduces mitochondrial ROS (130). SS-31 (5 mg/kg & 10 mg/kg) administered to mice 30 min after a weight drop injury decreased ROS, 8-OHdG, MDA, and increased SOD (131). Treatment also improved sensorimotor functioning and reduced apoptosis as well as intracellular iron loads. SS-31 (5 mg/kg) given to mice 30 min before an intracerebral LPS injection followed by three daily doses attenuated ROS, MDA, and pro-inflammatory cytokine levels while also increasing SOD in the hippocampus (132). LPS-stimulated microglia pre-treated with SS-31 showed attenuated mitochondrial fragmentation, iNOS and COX2 expression, and ROS production (133). Mice subject to brain ischemia/reperfusion immediately followed by SS-31 treatment (5 mg/kg) had reduced microglia/macrophage activation in the injured hemisphere (134). Safety or efficacy of SS-31 for the treatment of neurotrauma has not been confirmed in humans, but clinical trials may be forthcoming.
Although iron is essential to the healthy brain, iron overload can exacerbate oxidative stress. Accordingly, iron chelation therapy has been tested as a treatment of TBI. Administration of the iron chelator, deferoxamine has demonstrated some efficacy in improving histological and functional outcomes of CCI, lateral fluid percussion, and weight drop models of TBI on mice and rats, but investigations of deferoxamine’s effects on oxidative stress and microglial activation remains incomplete (135–138). Mixed findings are likely a result of deferoxamine being limited to access the CNS in areas of BBB damage.
Hydroxybenzyl ethylenediamine (HBED) is an iron chelator which can cross the BBB. HBED (100 mg/kg) followed with bidaily doses (50 mg/kg) for 3 days post-CCI had a neuroprotective effect with reduced microglial activation but had varying results on functional outcomes (139).
Minocycline, an antibiotic that has iron chelation properties and can also stabilize mitochondrial function, improved neurological and functional outcomes of a weight drop model of TBI on rats using multiple dose concentrations (140). A separate weight drop study using mice found minocycline (45 & 90 mg/kg) reduced inflammation but had no effect on the glutathione reduction ratio as a marker of oxidative stress (141). Minocycline reduced microglial activation in human TBI patients but also increased neurofilament light, a marker of neurodegeneration in humans (142).
In humans, iron chelators used for the treatment of iron overload syndromes have painful and potentially neurotoxic side effects (143). After lateral fluid percussion in rats, long-term term iron deposits and chronically activated microglia are associated with BBB leakage, which suggests treatments aimed at treating BBB dysfunction would be a more efficient target for both iron and overall neuroinflammation (144). In conclusion, although iron chelation may attenuate microglia-mediated oxidative stress, iron-targeted treatments may not be as effective or protective as other oxidative stress targets involved with TBI.
Injury to the spinal cord results in acute and long-lasting oxidative stress at both the lesion site and surrounding tissue. Interestingly, measurements of local oxidative stress have shown higher levels of ROS, lipid peroxidation and mitochondrial DNA oxidation in spinal cord after SCI than in cortical tissue after brain injury (145), suggesting that spinal cord tissue may be particularly sensitive to oxidative damage or contain cells that are primed toward a shift to oxidative stress. In blood samples from patients with cervical SCI, markers of oxidative stress were elevated acutely and up to 7 days after injury, while antioxidant levels were reduced (146). In rats with contusion SCI, markers of oxidative stress, including 3NT and 4HNE, were elevated by 3 h post-injury (147). The expression of these markers expanded throughout the tissue, into both gray and white matter, through 72 h post-injury. This expansion of expression began to decline by 2 weeks post-injury. In rabbits with spinal cord ischemia, markers of DNA oxidation (8-OHdG) were found to be elevated by 8 h after reperfusion, and peaked at 24 h (148). In a study of multiple models of SCI, contusion, dislocation and distraction injuries in rats were all found to increase oxidative stress markers within 3 h after injury, particularly in neurons, with slight differences in foci (149). Peroxynitrite after SCI shows an increase by 1 h that was sustained through 1 week, with a peak at 24 h (150).
Oxidative stress is not just an acute response to CNS injury, however. Analysis of SCI tissue at 1 month after SCI showed reductions in antioxidants (glutathione, alpha tocopherol) and increases in oxidative stress markers (epi-prostaglandin F2 alpha); these alterations remained present through 12 months after injury (151).
Treatment to reduce oxidative stress after SCI has been varied. One of the first treatments approved for SCI treatment, methylprednisolone, was found to significantly reduce oxidative damage in the injured spinal cord (152). However, methylprednisolone was found to impair systemic inflammatory responses and have other negative systemic effects. Therapies that have focused on directly scavenging oxidants after SCI have also proven somewhat successful in preclinical trials (153), but therapeutic windows for these approaches seem to be short and focused on the acute burst of ROS production (154). Therefore, treatments focusing on spinal cord specific oxidative stress may be more beneficial.
Directly reducing microglia in the injured spinal cord by microglial depletion has been met with conflicting results and appears to be heavily dependent on timing of the depletion initiation. Acute depletion or depletion prior to injury using PLX3397 or genetic manipulation was found to reduce functional recovery and impair neuronal survival after SCI (15, 155, 156). A recent study demonstrated that deletion of microglia prior to SCI led to a disruption in the glial scar (157). These data suggest that acute activity of microglia is essential to injury recovery.
However, delayed depletion using PLX5622 reduced microglial ROS and improved outcomes (158). This study demonstrated that PLX5622 administration starting at 1 day post-injury, in which microglia were depleted by 7 days post-injury, significantly improved motor function of mice after SCI and reduced long-term expression of inflammatory and apoptosis markers. Therefore, one can conclude that microglia within the injured spinal cord play a complicated and nuanced role that can not be overlooked or eliminated for therapeutic efficacy. Targeting specific microglial functions, such as ROS production, may then optimally improve outcomes.
NOX2 inhibition has been shown to reduce oxidative stress and improve functional recovery after SCI (24, 27). Using the non-specific NOX inhibitor apocynin, Zhang et al. (159) showed that acute systemic NOX inhibition improved functional recovery in aged, but not young, female mice, suggesting an age-related impact of treatment. These functional improvements were associated with significant reductions in macrophage invasion and macrophage-related ROS production. Interestingly, we have shown that in young male mice, NOX inhibition with gp91ds-tat acute intrathecal infusion did significantly improve functional outcomes in young mice (160), accompanied by significant reductions in local ROS markers and microglial numbers. This difference suggests that location (systemic vs. intrathecal) of treatment may also have a marked impact on treatment efficacy. Alternatively, sex may play a role, as systemic administration of gp91ds-tat significantly improved motor function in young adult male mice after a T10 contusion injury (27); this beneficial effect was associated with a significant reduction in ROS production in Iba1 + cells, although whether these cells were microglia or macrophages was not determined. The therapeutic approach to targeting NOX may also be the reason – apocynin is a non-specific approach; gp91ds-tat, which directly blocks NOX2 activity by binding the p47phox cytosolic component instead of allowing for p47phox to bind to the enzymatic core gp91phox, may be a more efficient method for inhibition (161).
In addition, targeting of the microglial proton channel HV1, which has been linked to NOX2 generated ROS, for genetic depletion has been shown to reduce NOX2 expression, ROS production and neuroinflammatory gene expression and improve motor function after SCI (162, 163).
Therapies targeting mitochondrial ROS have also been shown to reduce SCI oxidative stress and improve functional outcomes. Administration of n-acetylcysteine (NAC) has been shown to reduce ROS production by microglia in vitro and in SCI, accompanied by reduced microglial activation and reduced microglial mitochondrial dysfunction (164). Ketogenic diets, in which β-hydroxybutyrate metabolites are presumed to be able to bypass damaged metabolic pathways and enter the TCA cycle in mitochondria to assist in production of ATP, have been shown to improve functional outcomes after SCI (165). The ketogenic diet was also found to increase antioxidant availability and reduce cytokine production. Administration of minocycline, which is reported to stabilize mitochondria and inhibit the release of cytochrome c, has also been reported to reduce oxidative stress and improve outcomes after SCI (166–168).
Iron chelation therapy has proven effective in reducing oxidative stress in the injured spinal cord (85). Systemic administration of the lipophilic iron chelator salicylaldehyde isonicotinoyl hydrazine twice a week starting 1 h after SCI significantly reduced iron presence in the injured cord (85). However, despite this reduction, improvement in motor function was limited, with a significant but only 1 point improvement in the Basso Mouse Scale by 42 days post-injury. Deferoxamine, another iron chelator, administration after SCI was found to have similar results – depressing iron accumulation and inducing mild improvements in motor function (169). Increasing the frequency of treatment with deferoxamine may improve outcome – daily administration of deferoxamine starting 30 min prior to SCI was shown to significantly reduce iron load in the injured cord and significantly improve motor scores by at least 2 points (170). Daily administration after spinal cord compression was also found to improve recovery and induce vascularization (171). A recent study showed that deferoxamine administration after SCI improved Basso, Beattie and Bresnahan scale locomotor scores and reduced iron accumulation in the brain as well, reducing oxidative stress in the motor cortex (73). More importantly, deferoxamine administration has been shown to specifically act on microglia to reduce oxidative stress in an SCI-pain model (172). However, other studies have shown that oral administration of iron chelators, such as deferasirox, have moderate to no effect on motor function after SCI, and that high doses can be toxic (173). Therefore, additional research, particularly focused on non-systemic administration routes, is needed.
It is important to note that much of the early work in characterizing oxidative stress temporo-spatial dynamics was performed in female animals; the contribution of sex-differences in oxidative stress responses is not well understood. Sex has been shown to play a significant role in several post-injury inflammatory responses, including cytokine release (174), inflammasome formation (175), and T and B cell responses (176). In SCI, recent work by Stewart et al. demonstrated that while male mice demonstrate an elevated number of microglia in the injured spinal cord vs. females, female mice showed more NOX2 gene expression than males (177). These data suggest that oxidative stress may differ substantially between the sexes. Further work is warranted to identify how these variables affect ROS production.
Targeting of microglia mediated oxidative stress enhances behavioral and functional outcomes after both SCI and TBI and shows promise as a possible clinical therapy. While many of the studies detailed above do not specifically target just microglia, the complementary microglial depletion work demonstrates several of the same effects, suggesting that microglia may be a key target of global antioxidant therapies. The current research identifies Iba1 + cells as major ROS producers, but more studies are needed to identify the differential effects of macrophages compared with microglia. It is clear that additional research is needed to understand the best circumstances for beneficial outcomes when inhibiting microglial ROS production, including NOX, iron or mitochondrial dysfunction. Longitudinal studies are needed to identify the impact of therapies at longer time points to ensure short term improvements are not being traded for negative long-term consequences. This work would be a necessary foundation to support the expansion into clinical trials. Resolving or understanding the mechanisms that drive mixed results is integral to moving therapies into human use.
To date, several therapeutics have been met with mixed results. More research is needed to identify how to appropriately target ROS sources to reduce post-injury oxidative stress, and this remains a rich area for study and therapeutic targeting.
KB designed the overview of the manuscript and wrote the spinal cord injury section. AS researched and wrote the introduction and TBI section. MS researched and wrote the TBI section. DH designed the figures. SC wrote the abstract and conclusions. All authors contributed to the article and approved the submitted version.
This work was supported by the Defense Health Program, Congressionally Directed Medical Research Programs through the Spinal Cord Injury Research Program under Award No. SC170244.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Defense Health Program, Congressionally Directed Medical Research Programs, the Department of Defense, Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., or the Uniformed Services University.
1. Nscisc. National spinal cord injury statistical center, facts and figures at a glance. Birmingham, Al: University of Alabama at Birmingham (2016).
2. Chaychi S, Valera E, Tartaglia MC. Sex and gender differences in mild traumatic brain injury/concussion. Int Rev Neurobiol. (2022) 164:349–75. doi: 10.1016/bs.irn.2022.07.004
3. Lindberg MA, Moy Martin EM, Marion DW. Military traumatic brain injury: The history, impact, and future. J Neurotrauma. (2022) 39:1133–45. doi: 10.1089/neu.2022.0103
4. Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. (2015) 33:1732–50.
5. Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. (2010) 7:366–77. doi: 10.1016/j.nurt.2010.07.002
6. Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic Glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. (2009) 57:550–60. doi: 10.1002/glia.20783
7. Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating Tnf-alpha release and H2o2 derived from nadph oxidase: A cell culture study. J Neuroinflammation. (2006) 3:24. doi: 10.1186/1742-2094-3-24
8. Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from nadph oxidase. J Immunol. (2006) 176:1046–52.
10. Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, et al. Interactive role of the toll-like receptor 4 and reactive oxygen species in Lps-induced microglia activation. Glia. (2005) 52:78–84. doi: 10.1002/glia.20225
11. Rojo AI, McBean G, Cindric M, Egea J, Lopez MG, Rada P, et al. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid Redox Signal. (2014) 21:1766–801. doi: 10.1089/ars.2013.5745
12. Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. (2010) 7:354–65. doi: 10.1016/j.nurt.2010.05.014
13. Forman HJ, Torres M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am J Respir Crit Care Med. (2002) 166:S4–8. doi: 10.1164/rccm.2206007
14. Herzog C, Pons Garcia L, Keatinge M, Greenald D, Moritz C, Peri F, et al. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Development. (2019) 146:dev174698. doi: 10.1242/dev.174698
15. Fu H, Zhao Y, Hu D, Wang S, Yu T, Zhang L. Depletion of microglia exacerbates injury and impairs function recovery after spinal cord injury in mice. Cell Death Dis. (2020) 11:528. doi: 10.1038/s41419-020-2733-4
16. Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, et al. Gp91phox (Nox2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation. (2010) 7:41. doi: 10.1186/1742-2094-7-41
17. Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite Generated by inducible nitric oxide synthase and nadph oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U.S.A. (2005) 102:9936–41. doi: 10.1073/pnas.0502552102
18. Brown GC, Vilalta A. How microglia kill neurons. Brain Res. (2015) 1628:288–97. doi: 10.1016/j.brainres.2015.08.031
19. Davis CK, Vemuganti RDNA. Damage and repair following traumatic brain injury. Neurobiol Dis. (2021) 147:105143. doi: 10.1016/j.nbd.2020.105143
20. Hall ED. The contributing role of lipid peroxidation and protein oxidation in the course of cns injury neurodegeneration and neuroprotection: An overview. In: Kobeissy FH editor. Brain neurotrauma: Molecular, neuropsychological, and rehabilitation aspects. Frontiers in neuroengineering. Boca Raton FL: CRC Press/Taylor & Francis (2015).
21. Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: Evidence for an nadph oxidase-dependent pathway. Arch Biochem Biophys. (1998) 353:312–21. doi: 10.1006/abbi.1998.0658
22. Lavigne MC, Malech HL, Holland SM, Leto TL. Genetic requirement of P47phox for superoxide production by murine microglia. FASEB J. (2001) 15:285–7. doi: 10.1096/fj.00-0608fje
23. Cooney SJ, Bermudez-Sabogal SL, Byrnes KR. Cellular and temporal expression of nadph oxidase (Nox) Isotypes after brain injury. J Neuroinflammation. (2013) 10:155. doi: 10.1186/1742-2094-10-155
24. Cooney SJ, Zhao Y, Byrnes KR. Characterization of the expression and inflammatory activity of nadph oxidase after spinal cord injury. Free Radic Res. (2014) 48:929–39. doi: 10.3109/10715762.2014.927578
25. Cheret C, Gervais A, Lelli A, Colin C, Amar L, Ravassard P, et al. neurotoxic activation of microglia is promoted by a nox1-dependent nadph oxidase. J Neurosci. (2008) 28:12039–51. doi: 10.1523/JNEUROSCI.3568-08.2008
26. Bermudez S, Khayrullina G, Zhao Y, Byrnes KR. Nadph oxidase isoform expression is temporally regulated and may contribute to microglial/macrophage polarization after spinal cord injury. Mol Cell Neurosci. (2016) 77:53–64. doi: 10.1016/j.mcn.2016.10.001
27. Sabirzhanov B, Li Y, Coll-Miro M, Matyas JJ, He J, Kumar A, et al. Inhibition of Nox2 signaling limits pain-related behavior and improves motor function in male mice after spinal cord injury: Participation of Il-10/Mir-155 pathways. Brain Behav Immun. (2019) 80:73–87. doi: 10.1016/j.bbi.2019.02.024
28. Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, et al. Nadph oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. (2004) 279:1415–21. doi: 10.1074/jbc.M307657200
29. Minakami R, Sumimotoa H. Phagocytosis-Coupled activation of the superoxide-producing phagocyte oxidase, a member of the nadph oxidase (Nox) family. Int J Hematol. (2006) 84:193–8. doi: 10.1532/IJH97.06133
30. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. (2009) 7:355–66. doi: 10.1038/nrmicro2128
31. Ueyama T, Lennartz MR, Noda Y, Kobayashi T, Shirai Y, Rikitake K, et al. Superoxide production at phagosomal cup/phagosome through beta I protein kinase C during Fc Gamma R-mediated phagocytosis in microglia. J Immunol. (2004) 173:4582–9. doi: 10.4049/jimmunol.173.7.4582
32. Liu Y, Hao W, Letiembre M, Walter S, Kulanga M, Neumann H, et al. Suppression of microglial inflammatory activity by myelin phagocytosis: Role of P47-phox-mediated generation of reactive oxygen species. J Neurosci. (2006) 26:12904–13. doi: 10.1523/JNEUROSCI.2531-06.2006
33. Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. (2004) 77:540–51. doi: 10.1002/jnr.20180
34. Sun HN, Kim SU, Lee MS, Kim SK, Kim JM, Yim M, et al. Nicotinamide adenine dinucleotide phosphate (Nadph) oxidase-dependent activation of phosphoinositide 3-Kinase and P38 mitogen-activated protein kinase signal pathways is required for lipopolysaccharide-induced microglial phagocytosis. Biol Pharm Bull. (2008) 31:1711–5. doi: 10.1248/bpb.31.1711
35. Huang R, Hou L, Zhai X, Ruan Z, Sun W, Zhang D, et al. 2,5-Hexanedione induces Nlrp3 inflammasome activation and neurotoxicity through nadph oxidase-dependent pathway. Free Radic Biol Med. (2021) 162:561–70. doi: 10.1016/j.freeradbiomed.2020.11.013
36. Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, et al. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of nadph oxidase. J Pharmacol Exp Ther. (2006) 319:44–52.
37. Mander P, Brown GC. Activation of microglial nadph oxidase is synergistic with glial inos expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation. (2005) 2:20. doi: 10.1186/1742-2094-2-20
38. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke. (2006) 37:1087–93. doi: 10.1161/01.STR.0000206281.77178.ac
39. Lim H, Kim D, Lee SJ. Toll-like receptor 2 mediates peripheral nerve injury-induced nadph oxidase 2 expression in spinal cord microglia. J Biol Chem. (2013) 288:7572–9. doi: 10.1074/jbc.M112.414904
40. Kallenborn-Gerhardt W, Schroder K, Del Turco D, Lu R, Kynast K, Kosowski J, et al. Nadph oxidase-4 maintains neuropathic pain after peripheral nerve injury. J Neurosci. (2012) 32:10136–45. doi: 10.1523/JNEUROSCI.6227-11.2012
41. Kim J, Whyte J, Patel S, Avants B, Europa E, Wang J, et al. Resting cerebral blood flow alterations in chronic traumatic brain injury: An arterial spin labeling perfusion fmri study. J Neurotrauma. (2010) 27:1399–411. doi: 10.1089/neu.2009.1215
42. Qian L, Gao X, Pei Z, Wu X, Block M, Wilson B, et al. Nadph oxidase inhibitor Dpi is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord. (2007) 13(Suppl. 3):S316–20. doi: 10.1016/S1353-8020(08)70023-3
43. Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of nadph oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. (2012) 120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x
44. Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, et al. Critical role of nadph oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. (2012) 7:e34504. doi: 10.1371/journal.pone.0034504
45. Zhou J, Zhang S, Zhao X, Wei T. Melatonin impairs nadph oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J Pineal Res. (2008) 45:157–65. doi: 10.1111/j.1600-079X.2008.00570.x
46. Wilkinson BL, Cramer PE, Varvel NH, Reed-Geaghan E, Jiang Q, Szabo A, et al. Ibuprofen attenuates oxidative damage through Nox2 inhibition in Alzheimer’s disease. Neurobiol Aging. (2012) 33:e21–32. doi: 10.1016/j.neurobiolaging.2010.06.014
47. Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI. Activation of Mglur5 and inhibition of nadph oxidase improves functional recovery after traumatic brain injury. J Neurotrauma. (2013) 30:403–12. doi: 10.1089/neu.2012.2589
48. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
49. Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. (2000) 26:435–9. doi: 10.1038/82565
50. De Simone R, Ajmone-Cat MA, Pandolfi M, Bernardo A, De Nuccio C, Minghetti L, et al. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J Neurochem. (2015) 135:147–56. doi: 10.1111/jnc.13244
51. Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. (1994) 308:89–95. doi: 10.1006/abbi.1994.1013
52. Boczkowski J, Lisdero CL, Lanone S, Carreras MC, Aubier M, Poderoso JJ. Peroxynitrite-mediated mitochondrial dysfunction. Biol Signals Recept. (2001) 10:66–80. doi: 10.1159/000046876
53. Ye J, Jiang Z, Chen X, Liu M, Li J, Liu N. Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp Cell Res. (2016) 340:315–26. doi: 10.1016/j.yexcr.2015.10.026
54. Chausse B, Lewen A, Poschet G, Kann O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain Behav Immun. (2020) 88:802–14. doi: 10.1016/j.bbi.2020.05.052
55. Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, et al. Mitochondrial ros govern the Lps-induced pro-inflammatory response in microglia cells by regulating Mapk and Nf-Kappab pathways. Neurosci Lett. (2015) 584:191–6. doi: 10.1016/j.neulet.2014.10.016
56. Alfonso-Loeches S, Urena-Peralta JR, Morillo-Bargues MJ, Oliver-De La Cruz J, Guerri C. Role of mitochondria ros generation in ethanol-induced Nlrp3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci. (2014) 8:216. doi: 10.3389/fncel.2014.00216
57. Noh H, Jeon J, Seo H. Systemic injection of Lps induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int. (2014) 69:35–40. doi: 10.1016/j.neuint.2014.02.008
58. Chen W, Guo C, Huang S, Jia Z, Wang J, Zhong J, et al. Mitoq attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the Nlrp3 inflammasome after Ich. Pharmacol Res. (2020) 161:105122. doi: 10.1016/j.phrs.2020.105122
59. Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. (1997) 765:283–90. doi: 10.1016/s0006-8993(97)00573-8
60. Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J Neurotrauma. (2007) 24:991–9. doi: 10.1089/neu.2006.0242
61. Ahluwalia M, Kumar M, Ahluwalia P, Rahimi S, Vender JR, Raju RP, et al. Rescuing mitochondria in traumatic brain injury and intracerebral hemorrhages – a potential therapeutic approach. Neurochem Int. (2021) 150:105192. doi: 10.1016/j.neuint.2021.105192
62. Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. (2015) 350:132–8. doi: 10.1097/MAJ.0000000000000506
63. Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ, et al. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem. (2013) 127:221–32. doi: 10.1111/jnc.12361
64. Katoh M, Wu B, Nguyen HB, Thai TQ, Yamasaki R, Lu H, et al. Polymorphic regulation of mitochondrial fission and fusion modifies phenotypes of microglia in neuroinflammation. Sci Rep. (2017) 7:4942. doi: 10.1038/s41598-017-05232-0
65. Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW II, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. (2019) 22:1635–48. doi: 10.1038/s41593-019-0486-0
66. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia. (2019) 67:1047–61. doi: 10.1002/glia.23587
67. Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. (2003) 9:1062–8. doi: 10.1038/nm903
68. Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte nadph oxidase cytochrome B558. Role of heme incorporation and heterodimer formation in maturation and stability of Gp91phox and P22phox subunits. J Biol Chem. (1997) 272:27288–94. doi: 10.1074/jbc.272.43.27288
69. Gille G, Reichmann H. Iron-dependent functions of mitochondria–relation to neurodegeneration. J Neural Transm. (2011) 118:349–59. doi: 10.1007/s00702-010-0503-7
70. Mairuae N, Connor JR, Cheepsunthorn P. Increased cellular iron levels affect matrix metalloproteinase expression and phagocytosis in activated microglia. Neurosci Lett. (2011) 500:36–40. doi: 10.1016/j.neulet.2011.06.001
71. Wu T, Liang X, Liu X, Li Y, Wang Y, Kong L, et al. Induction of Ferroptosis in response to graphene quantum dots through mitochondrial oxidative stress in microglia. Part Fibre Toxicol. (2020) 17:30. doi: 10.1186/s12989-020-00363-1
72. Jiao L, Li X, Luo Y, Wei J, Ding X, Xiong H, et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front Cell Neurosci. (2022) 16:995084. doi: 10.3389/fncel.2022.995084
73. Feng Z, Min L, Chen H, Deng W, Tan M, Liu H, et al. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. (2021) 43:101984. doi: 10.1016/j.redox.2021.101984
74. Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral iron deposition in neurodegeneration. Biomolecules. (2022) 12:714. doi: 10.3390/biom12050714
75. Yauger YJ, Bermudez S, Moritz KE, Glaser E, Stoica B, Byrnes KR. Iron accentuated reactive oxygen species release by nadph oxidase in activated microglia contributes to oxidative stress in vitro. J Neuroinflammation. (2019) 16:41. doi: 10.1186/s12974-019-1430-7
76. Mehlhase J, Sandig G, Pantopoulos K, Grune T. Oxidation-induced ferritin turnover in microglial cells: Role of proteasome. Free Radic Biol Med. (2005) 38:276–85. doi: 10.1016/j.freeradbiomed.2004.10.025
77. Mehlhase J, Gieche J, Widmer R, Grune T. Ferritin levels in microglia depend upon activation: Modulation by reactive oxygen species. Biochim Biophys Acta. (2006) 1763:854–9. doi: 10.1016/j.bbamcr.2006.04.012
78. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in App/Ps1 mice. Brain Behav Immun. (2018) 68:183–96. doi: 10.1016/j.bbi.2017.10.017
79. McCarthy RC, Sosa JC, Gardeck AM, Baez AS, Lee CH, Wessling-Resnick M. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. (2018) 293:7853–63. doi: 10.1074/jbc.RA118.001949
80. Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, et al. Inflammation alters the expression of Dmt1, Fpn1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. (2013) 126:541–9. doi: 10.1111/jnc.12244
81. Rathore KI, Redensek A, David S. Iron homeostasis in astrocytes and microglia is differentially regulated by Tnf-Alpha and Tgf-Beta1. Glia. (2012) 60:738–50. doi: 10.1002/glia.22303
82. Thomsen MS, Andersen MV, Christoffersen PR, Jensen MD, Lichota J, Moos T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol Dis. (2015) 81:108–18. doi: 10.1016/j.nbd.2015.03.013
83. Nnah IC, Wessling-Resnick M. Brain iron homeostasis: A focus on microglial iron. Pharmaceuticals. (2018) 11:129. doi: 10.3390/ph11040129
84. Liu D, Liu J, Sun D, Alcock NW, Wen J. Spinal cord injury increases iron levels: Catalytic production of hydroxyl radicals. Free Radic Biol Med. (2003) 34:64–71. doi: 10.1016/s0891-5849(02)01184-x
85. Rathore KI, Kerr BJ, Redensek A, Lopez-Vales R, Jeong SY, Ponka P, et al. Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J Neurosci. (2008) 28:12736–47. doi: 10.1523/JNEUROSCI.3649-08.2008
86. Blomster LV, Cowin GJ, Kurniawan ND, Ruitenberg MJ. Detection of endogenous iron deposits in the injured mouse spinal cord through high-resolution ex vivo and in vivo mri. NMR Biomed. (2013) 26:141–50. doi: 10.1002/nbm.2829
87. O’Connell KM, Littleton-Kearney MT. The role of free radicals in traumatic brain injury. Biol Res Nurs. (2013) 15:253–63. doi: 10.1177/1099800411431823
88. Schiavone S, Neri M, Trabace L, Turillazzi E. The nadph oxidase Nox2 mediates loss of parvalbumin interneurons in traumatic brain injury: Human autoptic immunohistochemical evidence. Sci Rep. (2017) 7:8752. doi: 10.1038/s41598-017-09202-4
89. Mendez DR, Cherian L, Moore N, Arora T, Liu PK, Robertson CS. Oxidative DNA lesions in a rodent model of traumatic brain injury. J Trauma. (2004) 56:1235–40. doi: 10.1097/01.ta.0000130759.62286.0e
90. Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM, et al. Increase in Blood-Brain Barrier Permeability, Oxidative Stress, and Activated Microglia in a Rat Model of Blast-Induced Traumatic Brain Injury. J Neurosci Res (2010) 88:3530–9. doi: 10.1002/jnr.22510
91. Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. (2008) 45:443–52. doi: 10.1016/j.freeradbiomed.2008.04.038
92. Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. (2013) 60:282–91. doi: 10.1016/j.freeradbiomed.2013.02.029
93. Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. BioMed Res Int. (2014) 2014:723060. doi: 10.1155/2014/723060
94. Harish G, Mahadevan A, Pruthi N, Sreenivasamurthy SK, Puttamallesh VN, Keshava Prasad TS, et al. Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J Neurochem. (2015) 134:156–72. doi: 10.1111/jnc.13082
95. Ahmed FA, Kamnaksh A, Kovesdi E, Long JB, Agoston DV. Long-term consequences of single and multiple mild blast exposure on select physiological parameters and blood-based biomarkers. Electrophoresis. (2013) 34:2229–33. doi: 10.1002/elps.201300077
96. Ritzel RM, Li Y, He J, Khan N, Doran SJ, Faden AI, et al. Sustained neuronal and microglial alterations are associated with diverse neurobehavioral dysfunction long after experimental brain injury. Neurobiol Dis. (2020) 136:104713. doi: 10.1016/j.nbd.2019.104713
97. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
98. Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, et al. Microglial depletion with Csf1r inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci. (2020) 40:2960–74. doi: 10.1523/JNEUROSCI.2402-19.2020
99. Holmstrom KM, Kostov RV, Dinkova-Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. (2016) 1:80–91. doi: 10.1016/j.cotox.2016.10.002
100. Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ros production by mitochondria and nadph oxidase. Biochim Biophys Acta. (2015) 1850:794–801. doi: 10.1016/j.bbagen.2014.11.021
101. Yan W, Wang HD, Hu ZG, Wang QF, Yin HX. Activation of Nrf2-Are pathway in brain after traumatic brain injury. Neurosci Lett. (2008) 431:150–4. doi: 10.1016/j.neulet.2007.11.060
102. Zhou Y, Tian M, Wang HD, Gao CC, Zhu L, Lin YX, et al. Activation of the Nrf2-are signal pathway after blast induced traumatic brain injury in mice. Int J Neurosci. (2019) 129:801–7. doi: 10.1080/00207454.2019.1569652
103. Dong W, Sun Y, Cheng H, Yang B, Wang L, Jiang Z, et al. Dynamic cell type-specific expression of Nrf2 after traumatic brain injury in mice. Eur J Neurosci. (2019) 50:1981–93. doi: 10.1111/ejn.14399
104. Brooks DM, Patel SA, Wohlgehagen ED, Semmens EO, Pearce A, Sorich EA, et al. Multiple mild traumatic brain injury in the rat produces persistent pathological alterations in the brain. Exp Neurol. (2017) 297:62–72. doi: 10.1016/j.expneurol.2017.07.015
105. Hong Y, Yan W, Chen S, Sun CR, Zhang JM. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin. (2010) 31:1421–30. doi: 10.1038/aps.2010.101
106. Chandran R, Kim T, Mehta SL, Udho E, Chanana V, Cengiz P, et al. A combination antioxidant therapy to inhibit Nox2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J Cereb Blood Flow Metab. (2018) 38:1818–27. doi: 10.1177/0271678X17738701
107. Bhowmick S, D’Mello V, Caruso D, Abdul-Muneer PM. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J Mol Med. (2019) 97:1627–41. doi: 10.1007/s00109-019-01851-4
108. Wu G, Liu Z. Nuclear factor erythroid 2-related factor 2 (Nrf2) mediates neuroprotection in traumatic brain injury at least in part by inactivating microglia. Med Sci Monit. (2016) 22:2161–6. doi: 10.12659/msm.896568
109. Li J, Xu X, Cai X, Weng Y, Wang Y, Shen Q, et al. Milk fat globule-epidermal growth factor-factor 8 reverses lipopolysaccharide-induced microglial oxidative stress. Oxid Med Cell Longev. (2019) 2019:2601394. doi: 10.1155/2019/2601394
110. Lu XY, Wang HD, Xu JG, Ding K, Li T. Pretreatment with tert-butylhydroquinone attenuates cerebral oxidative stress in mice after traumatic brain injury. J Surg Res. (2014) 188:206–12. doi: 10.1016/j.jss.2013.11.1106
111. Jin W, Kong J, Wang H, Wu J, Lu T, Jiang J, et al. Protective effect of tert-butylhydroquinone on cerebral inflammatory response following traumatic brain injury in mice. Injury. (2011) 42:714–8. doi: 10.1016/j.injury.2011.03.009
112. Zhang ZW, Liang J, Yan JX, Ye YC, Wang JJ, Chen C, et al. Tbhq Improved Neurological Recovery after Traumatic Brain Injury by Inhibiting the Overactivation of Astrocytes. Brain Res (2020) 1739:146818. doi: 10.1016/j.brainres.2020.146818
113. Gharavi N, Haggarty S, El-Kadi AO. Chemoprotective and carcinogenic effects of tert-butylhydroquinone and its metabolites. Curr Drug Metab. (2007) 8:1–7. doi: 10.2174/138920007779315035
114. Xu M, Li L, Liu H, Lu W, Ling X, Gong M. Rutaecarpine attenuates oxidative stress-induced traumatic brain injury and reduces secondary injury Via the Pgk1/Keap1/Nrf2 signaling pathway. Front Pharmacol. (2022) 13:807125. doi: 10.3389/fphar.2022.807125
115. Wang J, Jiang C, Zhang K, Lan X, Chen X, Zang W, et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic Biol Med. (2019) 131:345–55. doi: 10.1016/j.freeradbiomed.2018.12.014
116. Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK, et al. Prevention of traumatic brain injury-induced neuronal death by inhibition of nadph oxidase activation. Brain Res. (2012) 1481:49–58. doi: 10.1016/j.brainres.2012.08.032
117. Wang J, Ma MW, Dhandapani KM, Brann DW. Regulatory role of nadph oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol Med. (2017) 113:119–31. doi: 10.1016/j.freeradbiomed.2017.09.017
118. Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Nox2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun. (2016) 58:291–309. doi: 10.1016/j.bbi.2016.07.158
119. Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J Neuropathol Exp Neurol. (2014) 73:14–29. doi: 10.1097/NEN.0000000000000021
120. Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. (2016) 33:1732–50.
121. Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of nadph oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. (1994) 11:95–102. doi: 10.1165/ajrcmb.11.1.8018341
122. Maraldi T. Natural compounds as modulators of nadph oxidases. Oxid Med Cell Longev. (2013) 2013:271602. doi: 10.1155/2013/271602
123. Dang PM, Rolas L, El-Benna J. The dual role of reactive oxygen species-generating nicotinamide adenine dinucleotide phosphate oxidases in gastrointestinal inflammation and therapeutic perspectives. Antioxid Redox Signal. (2020) 33:354–73. doi: 10.1089/ars.2020.8018
124. Hill RL, Singh IN, Wang JA, Hall ED. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int. (2017) 111:45–56. doi: 10.1016/j.neuint.2017.03.015
125. Kalayci M, Unal MM, Gul S, Acikgoz S, Kandemir N, Hanci V, et al. Effect of coenzyme Q10 on Ischemia and neuronal damage in an experimental traumatic brain-injury model in rats. BMC Neurosci. (2011) 12:75. doi: 10.1186/1471-2202-12-75
126. Pierce JD, Gupte R, Thimmesch A, Shen Q, Hiebert JB, Brooks WM, et al. Ubiquinol treatment for Tbi in male rats: Effects on mitochondrial integrity, injury severity, and neurometabolism. J Neurosci Res. (2018) 96:1080–92. doi: 10.1002/jnr.24210
127. Zhou J, Wang H, Shen R, Fang J, Yang Y, Dai W, et al. Mitochondrial-targeted antioxidant mitoq provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-are pathway. Am J Transl Res. (2018) 10:1887–99.
128. Gottwald EM, Duss M, Bugarski M, Haenni D, Schuh CD, Landau EM, et al. The targeted anti-oxidant mitoq causes mitochondrial swelling and depolarization in kidney tissue. Physiol Rep. (2018) 6:e13667. doi: 10.14814/phy2.13667
129. Doughan AK, Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal. (2007) 9:1825–36. doi: 10.1089/ars.2007.1693
130. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound Ss-31 Re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. (2013) 24:1250–61. doi: 10.1681/ASN.2012121216
131. Zhu Y, Wang H, Fang J, Dai W, Zhou J, Wang X, et al. Ss-31 provides neuroprotection by reversing mitochondrial dysfunction after traumatic brain injury. Oxid Med Cell Longev. (2018) 2018:4783602. doi: 10.1155/2018/4783602
132. Zhao W, Xu Z, Cao J, Fu Q, Wu Y, Zhang X, et al. Elamipretide (Ss-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice. J Neuroinflammation. (2019) 16:230. doi: 10.1186/s12974-019-1627-9
133. Mo Y, Deng S, Zhang L, Huang Y, Li W, Peng Q, et al. Ss-31 reduces inflammation and oxidative stress through the inhibition of Fis1 expression in lipopolysaccharide-stimulated microglia. Biochem Biophys Res Commun. (2019) 520:171–8. doi: 10.1016/j.bbrc.2019.09.077
134. Imai T, Matsubara H, Nakamura S, Hara H, Shimazawa M. The mitochondria-targeted peptide, bendavia, attenuated ischemia/reperfusion-induced stroke damage. Neuroscience. (2020) 443:110–9. doi: 10.1016/j.neuroscience.2020.07.044
135. Long DA, Ghosh K, Moore AN, Dixon CE, Dash PK. Deferoxamine improves spatial memory performance following experimental brain injury in rats. Brain Res. (1996) 717:109–17. doi: 10.1016/0006-8993(95)01500-0
136. Panter SS, Braughler JM, Hall ED. Dextran-coupled deferoxamine improves outcome in a murine model of head injury. J Neurotrauma. (1992) 9:47–53. doi: 10.1089/neu.1992.9.47
137. Zhang L, Hu R, Li M, Li F, Meng H, Zhu G, et al. Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol Sci. (2013) 34:639–45. doi: 10.1007/s10072-012-1090-1
138. Zhao J, Chen Z, Xi G, Keep RF, Hua Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl Stroke Res. (2014) 5:586–94. doi: 10.1007/s12975-014-0353-y
139. Khalaf S, Ahmad AS, Chamara K, Dore S. Unique properties associated with the brain penetrant iron chelator hbed reveal remarkable beneficial effects after brain trauma. J Neurotrauma. (2018) 36:43–53. doi: 10.1089/neu.2017.5617
140. Zhang L, Xiao H, Yu X, Deng Y. Minocycline attenuates neurological impairment and regulates iron metabolism in a rat model of traumatic brain injury. Arch Biochem Biophys. (2020) 682:108302. doi: 10.1016/j.abb.2020.108302
141. Homsi S, Federico F, Croci N, Palmier B, Plotkine M, Marchand-Leroux C, et al. Minocycline effects on cerebral edema: Relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. (2009) 1291:122–32. doi: 10.1016/j.brainres.2009.07.031
142. Scott G, Zetterberg H, Jolly A, Cole JH, De Simoni S, Jenkins PO, et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain. (2018) 141:459–71. doi: 10.1093/brain/awx339
143. Mobarra N, Shanaki M, Ehteram H, Nasiri H, Sahmani M, Saeidi M, et al. A review on iron chelators in treatment of iron overload syndromes. Int J Hematol Oncol Stem Cell Res. (2016) 10:239–47.
144. van Vliet EA, Ndode-Ekane XE, Lehto LJ, Gorter JA, Andrade P, Aronica E, et al. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol Dis. (2020) 145:105080. doi: 10.1016/j.nbd.2020.105080
145. Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, et al. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J Comp Neurol. (2004) 474:524–34. doi: 10.1002/cne.20130
146. Wozniak B, Wozniak A, Mila-Kierzenkowska C, Kasprzak HA. Correlation of oxidative and antioxidative processes in the blood of patients with cervical spinal cord injury. Oxid Med Cell Longev. (2016) 2016:6094631. doi: 10.1155/2016/6094631
147. Carrico KM, Vaishnav R, Hall ED. Temporal and spatial dynamics of peroxynitrite-induced oxidative damage after spinal cord contusion injury. J Neurotrauma. (2009) 26:1369–78. doi: 10.1089/neu.2008-0870
148. Sakurai M, Nagata T, Abe K, Horinouchi T, Itoyama Y, Tabayashi K. Oxidative damage and reduction of redox factor-1 expression after transient spinal cord ischemia in rabbits. J Vasc Surg. (2003) 37:446–52. doi: 10.1067/mva.2003.100
149. Choo AM, Liu J, Dvorak M, Tetzlaff W, Oxland TR. Secondary pathology following contusion, dislocation, and distraction spinal cord injuries. Exp Neurol. (2008) 212:490–506. doi: 10.1016/j.expneurol.2008.04.038
150. Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem. (2007) 100:639–49. doi: 10.1111/j.1471-4159.2006.04312.x
151. Bastani NE, Kostovski E, Sakhi AK, Karlsen A, Carlsen MH, Hjeltnes N, et al. Reduced Antioxidant Defense and Increased Oxidative Stress in Spinal Cord Injured Patients. Arch Phys Med Rehabil (2012) 93:2223–8e2. doi: 10.1016/j.apmr.2012.06.021
152. Christie SD, Comeau B, Myers T, Sadi D, Purdy M, Mendez I. Duration of lipid peroxidation after acute spinal cord injury in rats and the effect of methylprednisolone. Neurosurg Focus. (2008) 25:E5. doi: 10.3171/FOC.2008.25.11.E5
153. Bao F, Liu D. Hydroxyl radicals generated in the rat spinal cord at the level produced by impact injury induce cell death by necrosis and apoptosis: Protection by a metalloporphyrin. Neuroscience. (2004) 126:285–95. doi: 10.1016/j.neuroscience.2004.03.054
154. Scholpa NE, Schnellmann RG. Mitochondrial-based therapeutics for the treatment of spinal cord injury: Mitochondrial biogenesis as a potential pharmacological target. J Pharmacol Exp Ther. (2017) 363:303–13. doi: 10.1124/jpet.117.244806
155. Bellver-Landete V, Bretheau F, Mailhot B, Vallieres N, Lessard M, Janelle ME, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. (2019) 10:518. doi: 10.1038/s41467-019-08446-0
156. Brennan FH, Li Y, Wang C, Ma A, Guo Q, Li Y, et al. Microglia coordinate cellular interactions during spinal cord repair in mice. Nat Commun. (2022) 13:4096. doi: 10.1038/s41467-022-31797-0
157. Xia L, Qi J, Tang M, Liu J, Zhang D, Zhu Y, et al. Continual deletion of spinal microglia reforms astrocyte scar favoring axonal regeneration. Front Pharmacol. (2022) 13:881195. doi: 10.3389/fphar.2022.881195
158. Li Y, Ritzel RM, Khan N, Cao T, He J, Lei Z, et al. Delayed microglial depletion after spinal cord injury reduces chronic inflammation and neurodegeneration in the brain and improves neurological recovery in male mice. Theranostics. (2020) 10:11376–403. doi: 10.7150/thno.49199
159. Zhang B, Bailey WM, McVicar AL, Stewart AN, Veldhorst AK, Gensel JC. Reducing age-dependent monocyte-derived macrophage activation contributes to the therapeutic efficacy of Nadph oxidase inhibition in spinal cord injury. Brain Behav Immun. (2019) 76:139–50. doi: 10.1016/j.bbi.2018.11.013
160. Khayrullina G, Bermudez S, Byrnes KR. Inhibition of Nox2 reduces locomotor impairment, inflammation, and oxidative stress after spinal cord injury. J Neuroinflammation. (2015) 12:172. doi: 10.1186/s12974-015-0391-8
161. Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule nox inhibitors: Ros-generating nadph oxidases as therapeutic targets. Antioxid Redox Signal. (2009) 11:2535–52. doi: 10.1089/ARS.2009.2585
162. Li Y, Ritzel RM, He J, Cao T, Sabirzhanov B, Li H, et al. The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav Immun. (2021) 91:267–83. doi: 10.1016/j.bbi.2020.10.005
163. Murugan M, Zheng J, Wu G, Mogilevsky R, Zheng X, Hu P, et al. The voltage-gated proton channel Hv1 contributes to neuronal injury and motor deficits in a mouse model of spinal cord injury. Mol Brain. (2020) 13:143. doi: 10.1186/s13041-020-00682-6
164. Lin S, Zhou Z, Zhao H, Xu C, Guo Y, Gao S, et al. Tnf Promotes M1 polarization through mitochondrial metabolism in injured spinal cord. Free Radic Biol Med. (2021) 172:622–32. doi: 10.1016/j.freeradbiomed.2021.07.014
165. Lu Y, Yang YY, Zhou MW, Liu N, Xing HY, Liu XX, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the Nf-Kappab signaling pathways. Neurosci Lett. (2018) 683:13–8. doi: 10.1016/j.neulet.2018.06.016
166. Wells JE, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. (2003) 126(Pt 7):1628–37. doi: 10.1093/brain/awg178
167. Casha S, Zygun D, McGowan MD, Bains I, Yong VW, Hurlbert RJ. Results of a phase Ii placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain. (2012) 135(Pt 4):1224–36. doi: 10.1093/brain/aws072
168. Aras M, Altas M, Motor S, Dokuyucu R, Yilmaz A, Ozgiray E, et al. Protective effects of minocycline on experimental spinal cord injury in rats. Injury. (2015) 46:1471–4. doi: 10.1016/j.injury.2015.05.018
169. Liu J, Tang T, Yang H. Protective effect of deferoxamine on experimental spinal cord injury in rat. Injury. (2011) 42:742–5. doi: 10.1016/j.injury.2010.08.028
170. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res. (2019) 14:532–41. doi: 10.4103/1673-5374.245480
171. Tang G, Chen Y, Chen J, Chen Z, Jiang W. Deferoxamine ameliorates compressed spinal cord injury by promoting neovascularization in rats. J Mol Neurosci. (2020) 70:1437–44. doi: 10.1007/s12031-020-01564-1
172. Meng FX, Hou JM, Sun TS. In vivo evaluation of microglia activation by intracranial iron overload in central pain after spinal cord injury. J Orthop Surg Res. (2017) 12:75. doi: 10.1186/s13018-017-0578-z
173. Sauerbeck A, Schonberg DL, Laws JL, McTigue DM. Systemic iron chelation results in limited functional and histological recovery after traumatic spinal cord injury in rats. Exp Neurol. (2013) 248:53–61. doi: 10.1016/j.expneurol.2013.05.011
174. Pan MX, Li J, Ma C, Fu K, Li ZQ, Wang ZF. Sex-Dependent effects of gper activation on neuroinflammation in a rat model of traumatic brain injury. Brain Behav Immun. (2020) 88:421–31. doi: 10.1016/j.bbi.2020.04.005
175. Zendedel A, Monnink F, Hassanzadeh G, Zaminy A, Ansar MM, Habib P, et al. Estrogen attenuates local inflammasome expression and activation after spinal cord injury. Mol Neurobiol. (2018) 55:1364–75. doi: 10.1007/s12035-017-0400-2
176. Hauben E, Mizrahi T, Agranov E, Schwartz M. Sexual dimorphism in the spontaneous recovery from spinal cord injury: A gender gap in beneficial autoimmunity? Eur J Neurosci. (2002) 16:1731–40. doi: 10.1046/j.1460-9568.2002.02241.x
Keywords: iron, microglia, mitochondria, NADPH oxidase, oxidative stress, spinal cord injury, traumatic brain injury
Citation: Smith AN, Shaughness M, Collier S, Hopkins D and Byrnes KR (2022) Therapeutic targeting of microglia mediated oxidative stress after neurotrauma. Front. Med. 9:1034692. doi: 10.3389/fmed.2022.1034692
Received: 01 September 2022; Accepted: 12 October 2022;
Published: 03 November 2022.
Edited by:
Atsushi Umemura, Kyoto Prefectural University of Medicine, JapanCopyright © 2022 Smith, Shaughness, Collier, Hopkins and Byrnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly R. Byrnes, a2ltYmVybHkuYnlybmVzQHVzdWhzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.