
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 07 December 2022
Sec. Nuclear Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1034315
This article is part of the Research TopicTargeted Alpha Particle Therapy in OncologyView all 17 articles
 Mengqi Shi1,2
Mengqi Shi1,2 Vivianne Jakobsson1,3
Vivianne Jakobsson1,3 Lukas Greifenstein4
Lukas Greifenstein4 Pek-Lan Khong1,5Xiaoyuan Chen1,2,5,6,7
Pek-Lan Khong1,5Xiaoyuan Chen1,2,5,6,7 Richard P. Baum4
Richard P. Baum4 Jingjing Zhang1,2,5*
Jingjing Zhang1,2,5*Peptide receptor radionuclide therapy (PRRT) has over the last two decades emerged as a very promising approach to treat neuroendocrine tumors (NETs) with rapidly expanding clinical applications. By chelating a radiometal to a somatostatin receptor (SSTR) ligand, radiation can be delivered to cancer cells with high precision. Unlike conventional external beam radiotherapy, PRRT utilizes primarily β or α radiation derived from nuclear decay, which causes damage to cancer cells in the immediate proximity by irreversible direct or indirect ionization of the cells’ DNA, which induces apoptosis. In addition, to avoid damage to surrounding normal cells, PRRT privileges the use of radionuclides that have little penetrating and more energetic (and thus more ionizing) radiations. To date, the most frequently radioisotopes are β– emitters, particularly Yttrium-90 (90Y) and Lutetium-177 (177Lu), labeled SSTR agonists. Current development of SSTR-targeting is triggering the shift from using SSTR agonists to antagonists for PRRT. Furthermore, targeted α-particle therapy (TAT), has attracted special attention for the treatment of tumors and offers an improved therapeutic option for patients resistant to conventional treatments or even beta-irradiation treatment. Due to its short range and high linear energy transfer (LET), α-particles significantly damage the targeted cancer cells while causing minimal cytotoxicity toward surrounding normal tissue. Actinium-225 (225Ac) has been developed into potent targeting drug constructs including somatostatin-receptor-based radiopharmaceuticals and is in early clinical use against multiple neuroendocrine tumor types. In this article, we give a review of preclinical and clinical applications of 225Ac-PRRT in NETs, discuss the strengths and challenges of 225Ac complexes being used in PRRT; and envision the prospect of 225Ac-PRRT as a future alternative in the treatment of NETs.
Neuroendocrine tumors (NETs) are well-differentiated, low proliferating neuroendocrine neoplasms (NENs) (1), most commonly arising from gastroenteropancreatic structures and the lung, although NEN have been described in almost every tissue. Accounting for only 0.5% of all malignancies, NETs are considered rare (2), however, the incidence/prevalence has been increasing in many epidemiological studies over the last decades (with GEP NETs demonstrating the highest incidence rate with 3.56 cases per 100,000) (2–7). The WHO grading system relies extensively on the proliferation rate to classify low proliferative NETs (NET-G1) with good prognosis, intermediate grade (NET-G2), and high grade (NET-G3) that show poor prognosis (8).
As a heterogeneous disease with very diverse symptomatology, NETs require multidisciplinary treatment and care, including medical control, surgery, chemotherapy, and internal or external radiation therapy (9). The cornerstone of therapy is still surgery with curative intent, whenever possible. However, in the case of metastatic disease, total excision is generally not possible due to the infiltration of other tissues and/or blood vessels or the number of metastatic sites (10, 11).
Systemic chemotherapy provides only modest benefit in rapidly proliferating tumors (grade 3) (12, 13). Therapeutic options such as somatostatin analogs (SSAs) or interferon-α may improve symptoms caused by hormonal excess or even lengthen the time to disease progression by offering hormonal and antiproliferative control over NETs, but rarely lead to partial or complete tumor response (14, 15). External beam radiotherapy (EBRT) unfortunately is not effective for the treatment of metastasized and secondary cancer sites beyond the treatment area (16, 17).
Theranostics, the concept of combining the inevitably intertwined arts of diagnostics and therapy, is a treatment option that has gained momentum over the last two and a half decades. Peptide receptor imaging and peptide receptor radionuclide therapy (PRRT) were the first successful examples of the theranostic concept, for imaging and treating cancer. PRRT has long been considered as a palliative treatment for NETs, but is now attracting more and more attention as a very effective symptomatic and well-tolerated treatment prolonging progression-free (and possibly overall) survival. As a complement to surgery, neoadjuvant therapy can make previously difficult-to-operate tumors operable by shrinking them, and as an adjuvant therapy, it may prevent tumor re-growth after surgical manipulation and growth of pre-existing micrometastases (18, 19).
Unlike chemotherapy and EBRT, PRRT targets disease at the cellular level in the systemic treatment of non-resectable and metastasized NETs (16). The overexpression of somatostatin receptors (SSTRs) of various sub-types in about 80% of NETs provides a continuously evolving way to diagnose and treat NETs (18, 20). The working principle of PRRT is using a therapeutic radionuclide chelated to a SSTR binding peptide and as the compound binds to SSTR expressing tissue, DNA-damaging radiation is delivered nearly exclusively to tumor cells and its microenvironment while sparing the surrounding healthy tissue. Somatostatin, the native peptide, is an obvious example of SSTR-binding peptide (21). However, it is susceptible to fast enzymatic degradation and is thus not suitable for in vivo applications (22). Instead, synthetic peptides, including those based on SSAs, have been developed with the intent to optimize metabolic stability, tumor retention time, and affinity.
The first PRRT was performed in the early 1990s (Figure 1). The Rotterdam group successfully developed [111In-DTPA-D-Phe1]-octreotide (111In-pentetreotide) somatostatin scintigraphy (Octreoscan), and subsequently examined its imaging potential in more than 1,000 patients (23–25). Based on high uptake of 111In-pentetreotide by tumors as demonstrated by imaging, Krenning’s team successfully treated a patient with metastatic glucagonoma using a high dose of 111In-pentetreotide, which resulted in a decreased level of circulating glucagon as well as decreased tumor size (26).
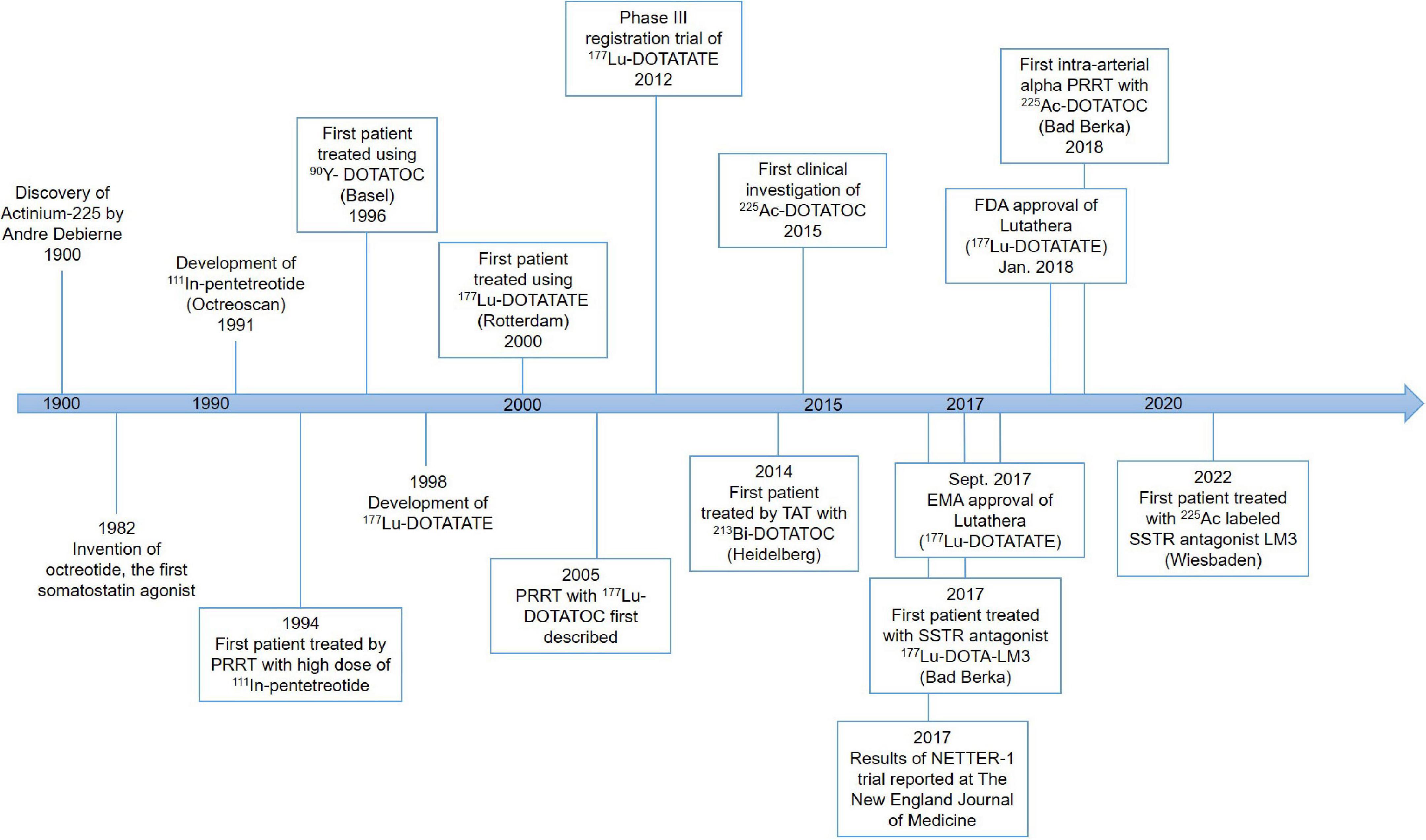
Figure 1. History of somatostatin-based peptide receptor radionuclide therapy (PRRT).
This early work set the stage for further development of this exciting new field of radiomolecular precision medicine. For example, guided by the experience with PRRT using 111In-pentetreotide, the need for more suitable radionuclides was identified because the properties of 111In (decay by electron capture with a half-life of 2.8 days) do not provide good tissue penetration which corresponds with a modest or no tumor shrinkage. Improvement of PRRT has been made tremendously since then due to the development and availability of novel peptides, chelators, and radionuclides in various combinations (27).
Derivatizing [Tyr3]-octreotide (Figure 2), which has a higher binding affinity for SSTR2 than the natural somatostatin analog (SSA) octreotide, and combining it with the chelator 1,4,7,10-tetra-azacyclododecane-tetra-acetic acid (DOTA) enabled stable radiolabeling with the high-energy beta particle-emitter Yttrium-90 (90Y-DOTATOC).
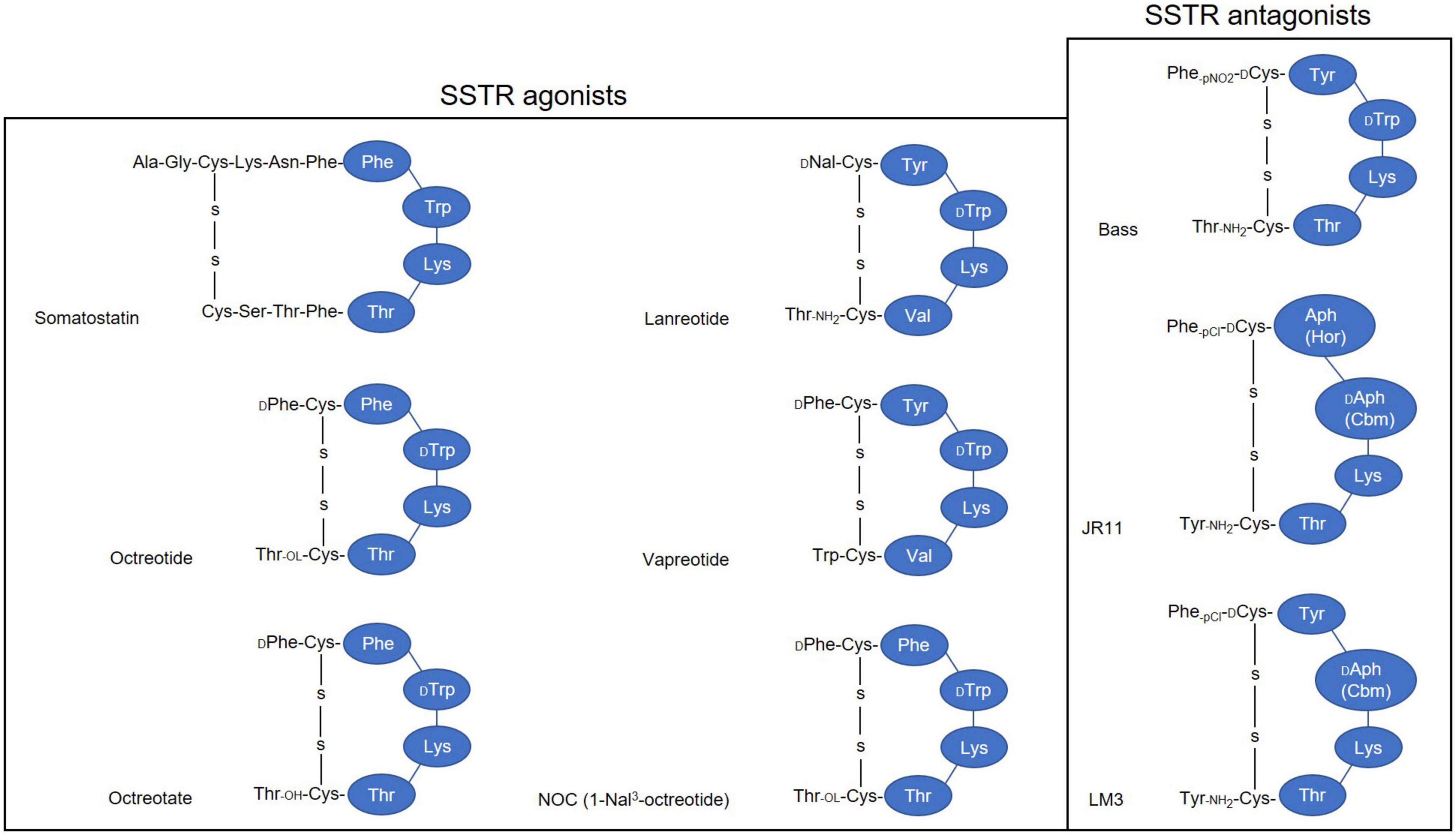
Figure 2. Simplified illustration of somatostatin receptor agonists and antagonists. 1-Nal, naphthyl-alanine; Aph(Hor), 4-amino-L-hydroorotyl -phenylalanine; D-Aph(Cbm), D-4-amino-carbamoyl-phenylalanine.
This therapeutic moiety was first applied in a pilot study for the treatment of three patients with abdominal metastases of neuroendocrine carcinoma of unknown localization (28, 29); and its therapeutic potential was evaluated subsequently with larger SSTR-positive patient numbers (30, 31). Treatment with 90Y-DOTATOC stopped rapid tumor progression, decreased the tumor marker neuron-specific enolase (NSE), and allowed disease stabilization (28, 30, 31). DOTATOC has since become a popular theranostics agent, demonstrating superior diagnostic sensitivity compared to Octreoscan, and demonstrating promising therapeutic value for treating SSTR-positive NETs when labeled with β- emitters, particularly Yttrium-90 (90Y) and Lutetium-177 (177Lu).
Another extensively studied SSA is DOTA-[Tyr3]-octreotate (DOTATATE), where the alcohol Thr(ol) of the C-terminus of DOTATOC is replaced by the natural amino acid Thr. This somatostatin analog was developed in 1998 and was found to show an even higher affinity to SSTR2 and a higher uptake in pancreatic tumor cells compared to the previously described SSAs (32).
The first clinical trial with 177Lu-DOTATATE started in 2000 in Rotterdam, The Netherlands, and led to the multinational phase three trial named NETTER-1 (33, 34). In this randomized controlled trial, a significantly higher response rate and extended progression-free survival were demonstrated in patients with advanced progressive SSTR-positive midgut NETs, compared to the double dose of long-acting repeatable (LAR 60 mg) octreotide administrations (18, 27, 34, 35). In January 2018, 177Lu-DOTATATE under the name of Lutathera was approved by the Food and Drug Administration (FDA) for the treatment of SSTR2-positive gastroenteropancreatic NETs (GEP-NETs) in adults (36). The approval of Lutathera in Europe was granted by the European Medicines Agency (EMA) already in September 2017 (37).
Other well-known somatostatin SSAs include lanreotide and vapreotide, but as these are not approved for PRRT of NETs, possibly due to their affinity pattern to SSTR subtypes and mode of action (38–40), they will not be discussed in this review.
Despite the success of [90Y]Y-DOTATOC and [177Lu]Lu-DOTATATE in the treatment of NETs in terms of progression-free survival, there were problems with 90Y concerning renal toxicity and a low rate of complete remissions, suggesting an improvement in PRRT efficacy is required (27, 41) with the main aspects of (1) identification of prognostic and predictive factors; (2) α-PRRT using 225Ac labeled ligands; and (3) shift from using SSAs to somatostatin antagonists.
Even with the success of DOTATOC and DOTATATE, clinicians need to take the individual variability in the therapy response of patients with seemingly similar profiles into account. 68Ga is a positron emitter that can be chelated to DOTATOC or DOTATATE for PET/CT imaging before therapy. SSTR-PET/CT with 68Ga-labeled DOTATATE and/or DOTATOC showed very promising results (42). All studies agreed on the important role of SSTR-PET using 68Ga for NET imaging and therapy planning, supporting the potent theranostic role of a radiolabeled DOTA-peptide (43–48). As a result, 68Ga-DOTATOC was approved by the FDA in 2019 as the first 68Ga-labeled radiopharmaceutical for imaging of SSTR positive GEP-NET using PET (49).
Table 1 lists the physical properties regarding the clinically most frequently used radioisotopes in PRRT of NETs.
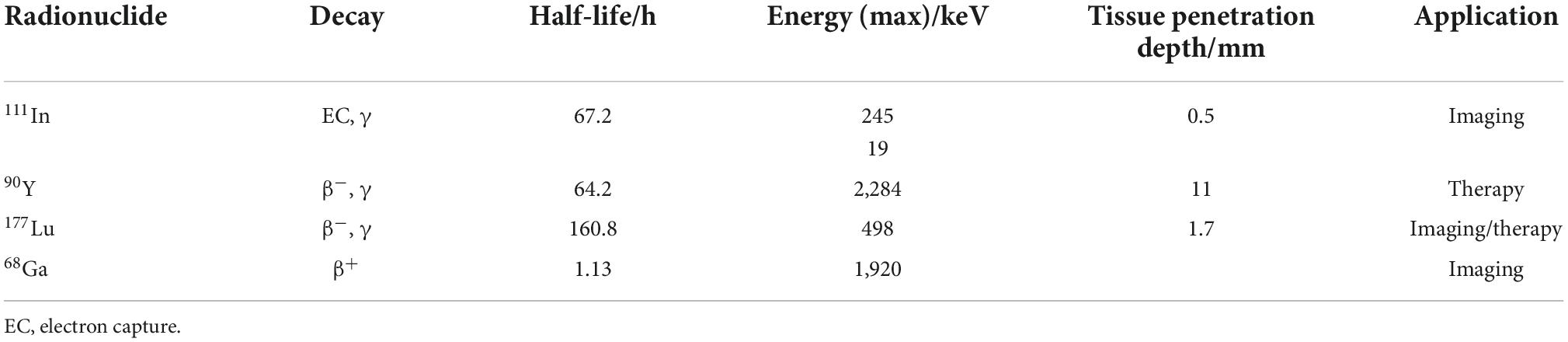
Table 1. Physical properties of selected radioisotopes used in PRRT to treat NETs.
While 68Ga is useful for imaging purposes, all other radionuclides in Table 1 are used both for therapy and for single-photon emission computed tomography (SPECT) imaging. Out of these, 90Y and 177Lu are the two favorable isotopes due to their higher particle energies compared to 111In. 90Y (t1/2 = 64.2 h) emits β–-particles with a maximum energy of 2.284 MeV, allowing penetration of soft tissue to a depth of around 11 mm (50, 51). Because of its longer range compared to 177Lu, 90Y-DOTATE/TOC is suggested to be more suitable for bigger lesions, while 177Lu-DOTATATE might be preferred for smaller lesions (52). It has been demonstrated that 90Y-PRRT has a small potential of causing renal toxicity when used without nephroprotection, especially in patients with compromised renal function (53, 54). For 177Lu-DOTATATE or 177Lu-DOTATOC no significant renal damage occurred even in long-term follow up studies. An increasing number of clinical studies suggest that the combination of 90Y and 177Lu could be better than either radionuclide alone for PRRT in NETs, with an improved overall survival (55, 56). In parallel to β–-emitters, radionuclides emitting α-particles recently are gaining special interest for the treatment of NETs.
To date, most PRRTs rely on β-emitters, especially 90Y and 177Lu, because of the availability of these radioisotopes and the proven clinical effect. However, due to the relatively large range of these radionuclides surrounding normal tissues are also exposed to radioactivity. Furthermore, hypoxic cancer tissue could be resistant to β-emitter treatment, causing radiotherapeutic failure of β-PRRT (57, 58).
Targeted α-particle therapy (TAT) offers a therapeutic option for patients resistant to β-irradiation treatments. An α-particle is a helium-4 (4He) nucleus consisting of two protons and two neutrons with an overall charge of +2 (59). Because of the double-positive charge, α-particles deliver dense ionization along a linear track, often described as high linear transfer (LET), ranging from 50 to 230 keV/μm (Figure 3) (60). This high LET renders higher target cell toxicity originating from higher probability of DNA double strand breaks (DSB) compared to β-particles with low LET (0.1–1.0 keV/μm) (61). Moreover, the primary target of high-LET α-particle is DNA, and a low number of particles can result in irreparable DSBs and lack of oxygen effects on cytotoxicity (62–64). Thus, the cytotoxicity of α-particle may be extremely effective and may also be more dose independent than β-emissions with cell death occurring from a single or a few α-particle traversals of the cell nucleus (65, 66). On the other hand, the typical tissue range of α-particles does not exceed 100 μm, which is significantly shorter than that of β-particles (0.05–12 mm) (Figure 3) (16, 58). This allows for selective ablation of the targeted tumor cells whilst minimizing the damage to surrounding healthy tissues (67).
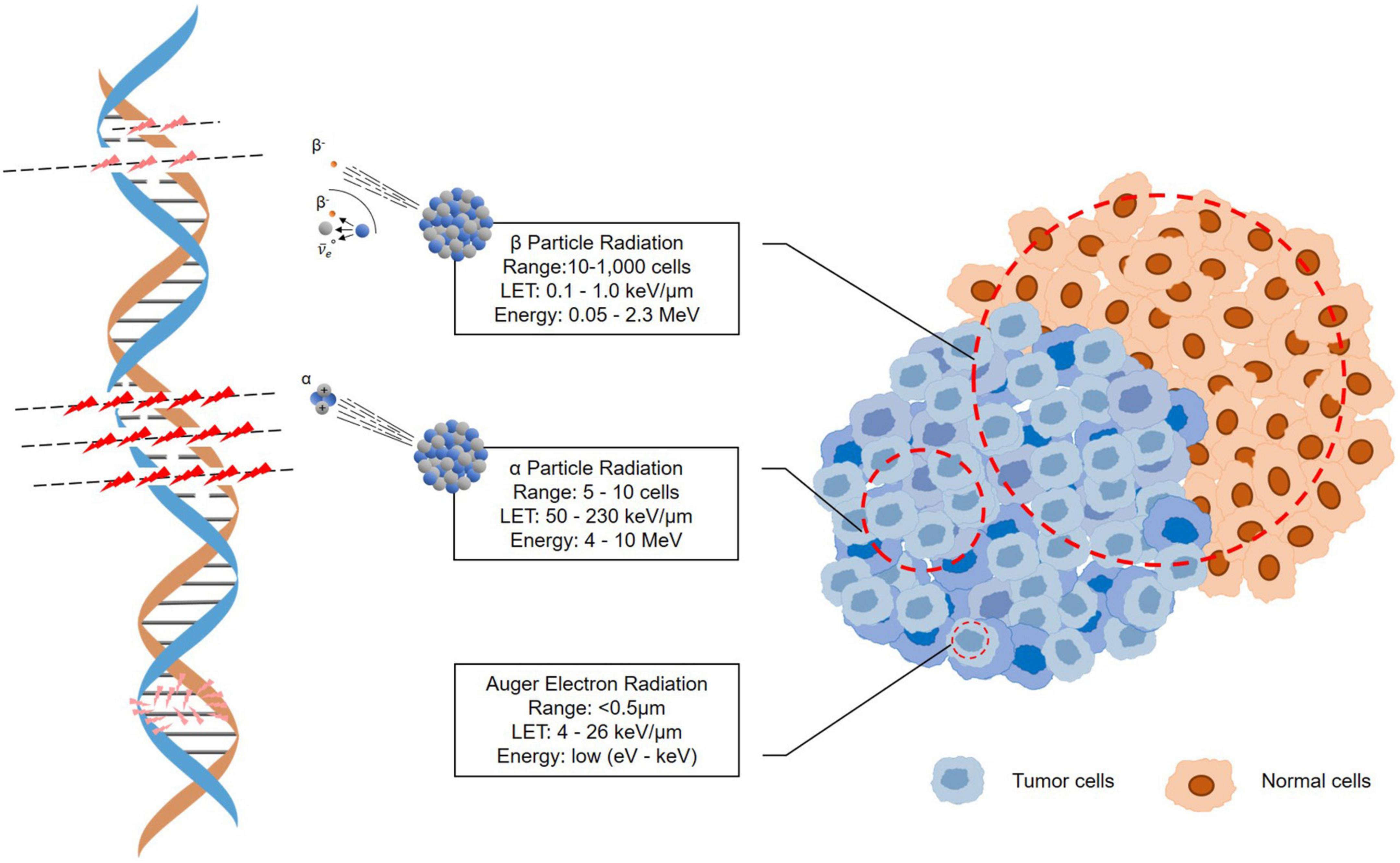
Figure 3. Illustration of the tracks of α-particle, β-particle, and auger election radiation.
Considering the half-life, production, availability, and ability to be stably incorporated into a suitable vector, only a handful α-radionuclides have potential for clinical use, including actinium-225, bismuth-213, astatin-211, thorium-227, radium-223, radium-224, lead-212, bismuth-212, and terbium-149 (Table 2) (57, 64, 68–75).
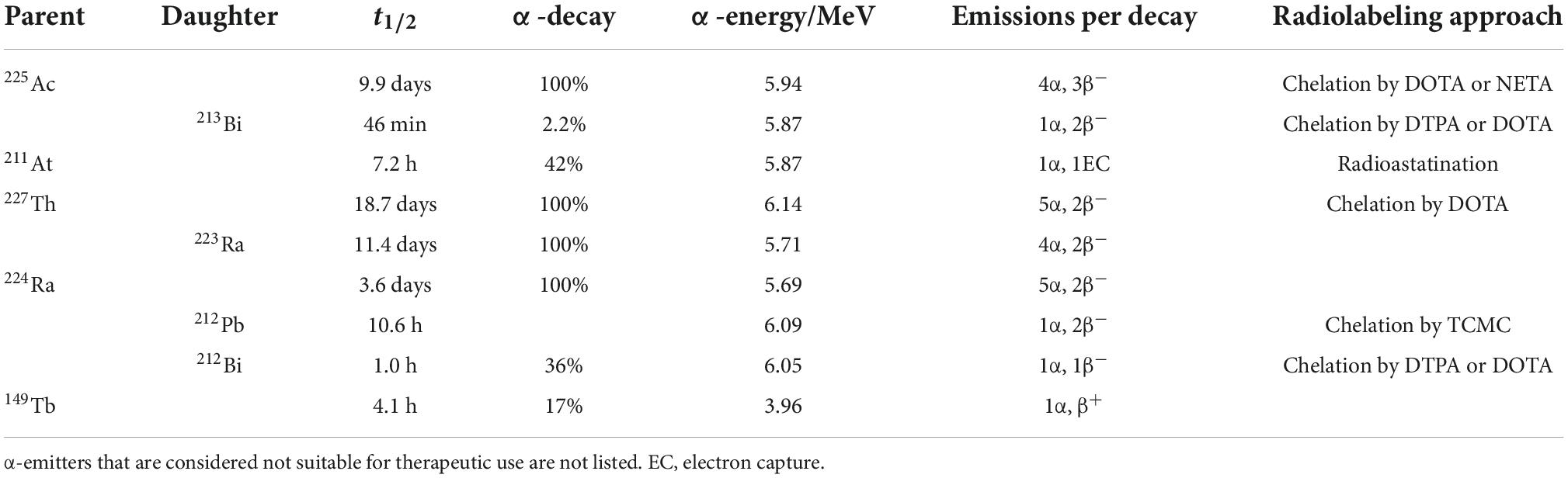
Table 2. Medically relevant α-emitters with their decay properties.
Among all medically relevant α-particles, the generator derived radionuclide 225Ac (and its daughter radionuclide 213Bi) are considered particularly promising. 225Ac was discovered by Andre Debierne in 1899 and Friedrich Giesel in 1902 (76). As a pure α-emitter with a half-life of 9.9 days, the decay of 225Ac produces seven radionuclide daughters in the decay chain to stable 209Bi (Figure 4). From this decay path, a single 225Ac decay yields a total of four α, three β– disintegrations, and two γ emissions. As such, 225Ac is classified as nanogenerator or in vivo generator (77). The relatively long half-life, the multiple α-particle emissions in the decay chain, and the rapid decay to stable 209Bi makes 225Ac a candidate of great potential for application in TAT (78). Moreover, the isomeric γ emissions with energy suitable for SPECT imaging grants 225Ac the theranostic possibility (68, 69). Although the feasibility of using 225Ac for imaging is debatable because the amount administered for therapy may not produce enough gamma emission to be effectively detected by gamma camera.
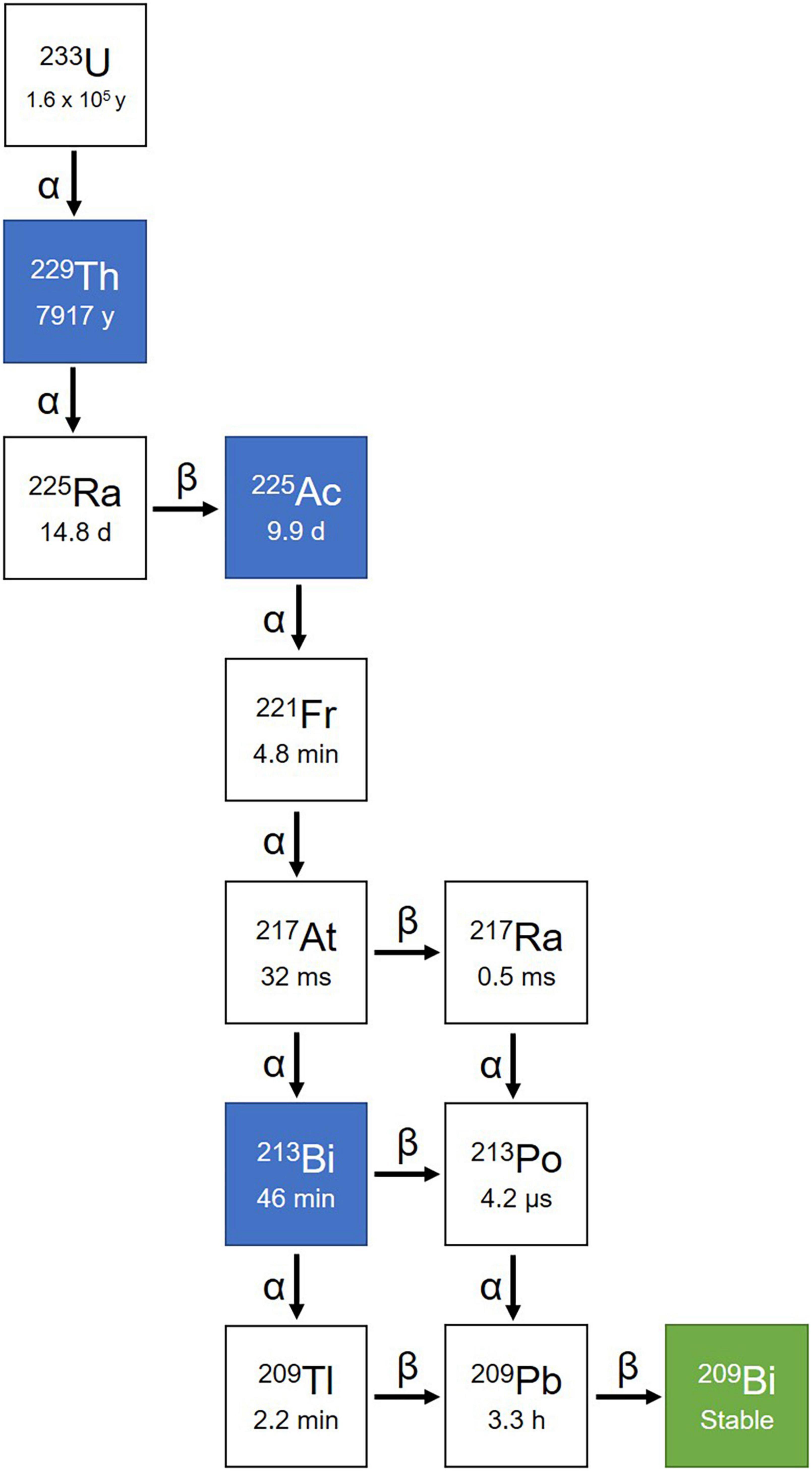
Figure 4. Decay chain of 225Ac. 225Ac decays to 209Bi with seven intermediate radionuclide progenies, including 221Fr, 217At, 213Bi, 209Tl, 217Ra, 213Po, and 209Pb. Among the decay chain 225Ac and 213Bi are medically relevant and intensively investigated.
The initial focus for harnessing the therapeutic potential of 225Ac was to identify a suitable chelating agent for in vivo delivery of 225Ac to target cells (79, 80). DOTA remains the gold standard for 225Ac labeling for all clinical work. Examples include 225Ac-PSMA-617, 225Ac-DOTATOC, 225Ac-DOTATATE, and 225Ac-DOTA-HuM195 (81). The first-in-human phase I dose escalation trial used 225Ac-DOTA-HuM195, which not only demonstrated the safety of 225Ac and its antileukemic activity, but also suggested that targeted therapy with an in vivo α-particle nanogenerator is a feasible approach in humans (82). With this proof of concept, 225Ac was then attached to PSMA-617 for prostate cancer therapy (83–86) and also bound to SSA-based pharmaceuticals for treating NETs.
Several preclinical studies tested the efficacy of α-particle emitting conjugates 225Ac-DOTATATE and 225Ac-DOTATOC in xenografted NET models and tested their toxicity as well as the biological effects of 225Ac. γH2AX was suggested as early key parameter in predicting tumor response to α-PRRT. The great reduction of growth and improved efficacy compared with 177Lu-labeled SSAs suggested that 225Ac-DOTATATE/DOTATOC has significant potential for improving PRRT in NETs and for the clinical translation in NET.
The first clinical study of 225Ac-PRRT in NET treatment was started in 2011 as a collaboration between the Joint Research Centre in Karlsruhe (Germany) and the University Hospital Heidelberg to treat patients with progressive NETs using 225Ac-DOTATOC. Based on 46 treatment cycles in 34 patients, the maximum tolerable dose was determined to be 40 MBq. The treatment was found to be safe with doses of 18.5 MBq every 2 months or 25 MBq every 4 months, and a cumulative activity of 75 MBq in regard to delayed toxicity. Despite the treatment response observed in several patients, further investigations were found to be necessary to improve patient selection and dosage regimens (87). Since then, clinical studies of 225Ac-PRRT in different NETs focused on whole-body SPECT/CT imaging possibility, efficacy and safety, therapeutic effect, as well as comparison to β-PRRT, has burgeoned (Table 3).
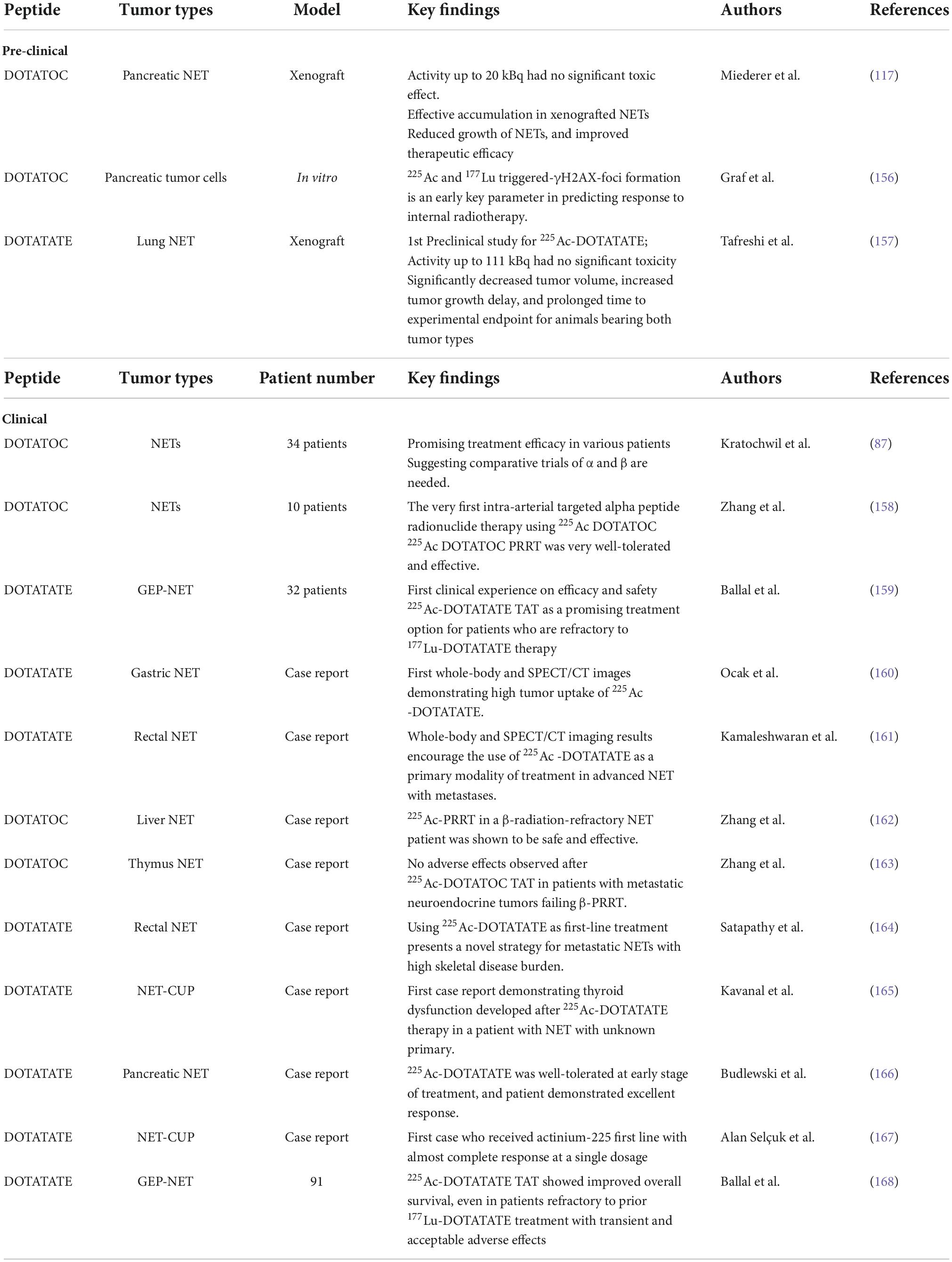
Table 3. Actinium-225-peptide receptor radionuclide therapy in NET treatment.
Considering the minimal/acceptable side effects, and the improved therapeutic efficacy and survival, one can conclude that 225Ac-PRRT not only provides an alternative in the treatment of β-radiation-refractory NETs, but also presents as possible frontline in treatment of NETs and can potentially usher a new era in radiopharmaceuticals even in tumors beyond NET.
Limited 225Ac supply poses the most important challenge for widely implementing 225Ac-based PRRT. For more than two decades, the radiochemical extraction from 229Th, which is originated from the decay of the fissile isotope 233U (Figure 4), has been the most utilized strategy for the production of 225Ac and its daughter 213Bi (88, 89). Even today, 225Ac used in all clinical and virtually all preclinical studies is still obtained from the decay of 229Th. Worldwide, only three sources of 229Th are available (81). The Directorate for Nuclear Safety and Security of the Joint Research Centre (JRC) of the European Commission in Karlsruhe, Germany, the first laboratory to prepare 225Ac/213Bi, has produced approximately 13 GBq 225Ac annually since the 1990s for their center and a wide network of clinical collaborators (89, 90). The Oak Ridge National Laboratory (ORNL), USA produces up to 33 GBq per year for extensive application of treatment (91), while the Institute of Physics and Power Engineering (IPPE) in Russia reported an estimated production of 22 GBq annually (92) with no direct clinical application reported yet to our knowledge. This amounts to a current global production of approximately 68 GBq per year. At this level of supply, 225Ac-based treatments will continue to only be available for some few 100 patients per year, which obviously is insufficient to meet the growing demand of 225Ac labeled compounds in hospitals worldwide (81, 93).
Consequently, multiple accelerator-based routes to scale up 225Ac production have been investigated, including the irradiation of 226Ra target using protons, deuterons or gamma-rays and the spallation of natTh or natU targets with highly energetic protons (Figure 5) (94). Among the routes described so far, the spallation of natTh is currently the most frequently used accelerator-based route. Even though the feasibility of the process has been demonstrated in the USA (95, 96) and Russia (97, 98), the implication of coproducing the long-lived 227Ac (t1/2 = 21.8 years) as impurity is a serious limitation in terms of clinical translation and waste management (99). In addition, issues with licensing and clinical safe handling need to be resolved (81).
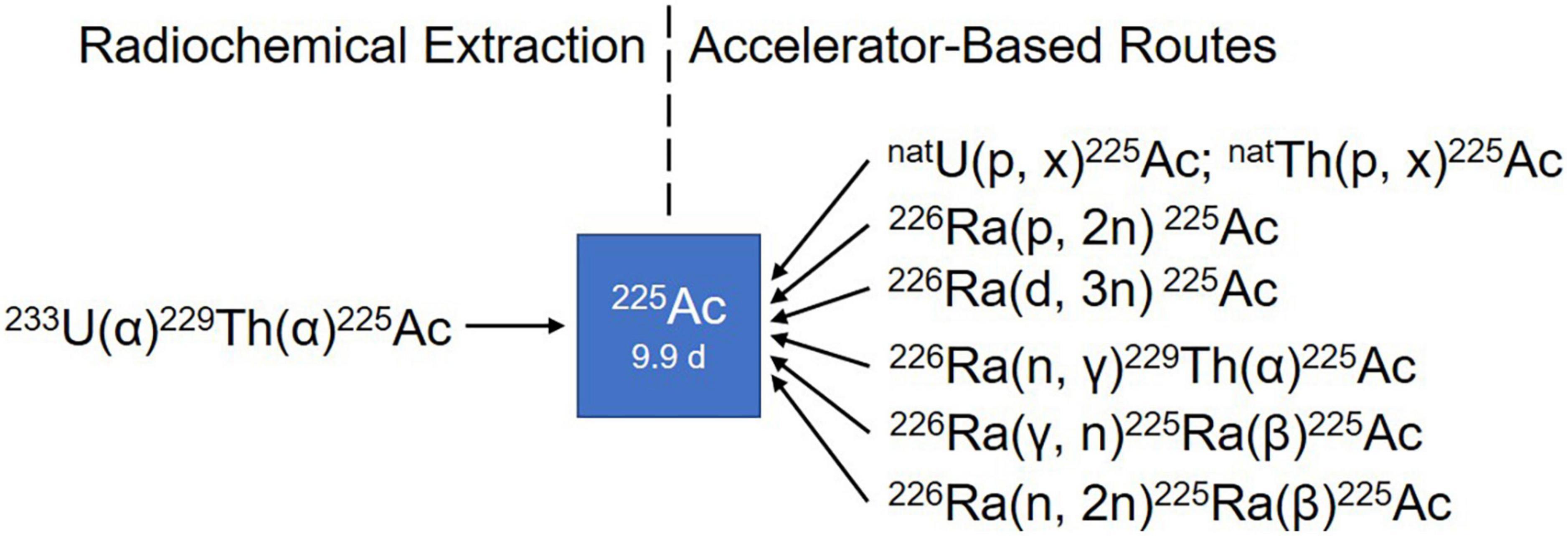
Figure 5. Production routes of 225Ac.
Medium-energy proton irradiation of 226Ra in a cyclotron using the reaction 226Ra(p,2n)225Ac offers a number of advantages over the natTh spallation, and is currently the most promising method of large-scale and cost-effective production of 225Ac. Chemical purification of the irradiated targets generates 225Ac with high isotopic purity because there are no other long-lived actinium isotopes, such as 227Ac, co-produced (100). It is important to mention that the availability of appropriate cyclotrons worldwide [energy range 15–25 MeV (101)] makes the production of 225Ac feasible for basic and applied research (94). The downsides of this approach are related to the preparation and safe handling of targets containing milligram of radioactive 226Ra and managing its highly radiotoxic gaseous decay product 222Rn. Further research on the practical implementation of this production route is required to meet the high demand in the mid-term future (81).
The reaction 226Ra(d,3n)225Ac has been suggested as an improved approach for producing 225Ac (102). Model calculations are predicting an increased production yield compared to the 226Ra(p,2n)225Ac reaction. However, deuteron irradiation leads to an enhanced co-production of 226Ac via the 226Ra(d,2n)226Ac reaction, resulting in an extended cooling time necessary to allow for 226Ac decay. Moreover, limited accelerators are available worldwide that can provide deuteron beams of sufficient energy and the complicated handling of 226Ra and its decay product remains an issue (81).
Other production routes being studied involve 226Ra(n, γ)229Th(α)225Ac, 226Ra(γ,n)225Ra(β)225Ac, and 226Ra(n,2n)225Ra(β)225Ac. It is important to point out that the main limitation of all these strategies is the handling of radium target and the generation of long-lived co-products, such as 227Ac (t1/2 = 21.77 years) and 228Ac (t1/2 = 1.9 years). However, preliminary results demonstrated by these above-mentioned methods are promising (94, 100, 103–105). Unfortunately, but understandably, these supply limitations bring about a high cost that is considered unaffordable by many researchers.
Clinical imaging using 225Ac also presents challenges. Therefore, post-therapy imaging is usually not done after 225Ac administration for tracer localization. The use of two photopeaks at 218 keV and 440 keV has long been suggested for clinical imaging of α-particles (106), until recently, Rasheed et al. focusing on gamma-ray spectrum for 225Ac showed an additional third photopeak at 78 keV, with higher counting density (107). Although imaging using the additional 78 keV photopeak was suggested to yield higher counts, better images, and more lesion delineations, the literature showing the feasibility of using the three photopeaks is limited to only few clinical case reports (108, 109).
The short mean free path of 225Ac (110), as well as the complexity and timing of 225Ac decay in relation to its radiopharmaceutical stability, uptake and clearance makes measurement of 225Ac activity very difficult. A number of preclinical studies have estimated 225Ac activity using measurements related to γ-emissions (111). However, a low probability of γ-emission and overlapping Bremsstrahlung due to β-emitters in the 225Ac decay chain preclude simultaneous treatment and dosimetry measurement in a clinical setting (84). Thus, current clinical TAT research relies greatly on indirect approximation by extrapolating pre-existing 177Lu-labeled pharmaceuticals (84, 112, 113). Preclinical studies focusing on 225Ac have used the standard approach described by the Medical Internal Radiation Dose (MIRD) committee. Total dosimetry was calculated from the summation of doses of 225Ac, 221Fr, 217At, 213Bi, and 213Po recorded in a biodistribution study (111). Unfortunately, collection of biodistribution data for dosimetry estimation is not possible for most α-emitters with therapeutic potential. This makes a direct and accurate preclinical dosimetry measurement for 225Ac to be extrapolated into and to guide clinical trials, as well as standardizing α-dosimetry measurement highly demanded in clinical application.
Finding a chelator to accommodate 225Ac and its progenies with sufficient stability was proven a great challenge given the range of different periodic properties of the daughters of 225Ac. The recoil effect associated with α-decay of 225Ac imparts an energy that is thousands of times greater than the binding energy of any chemical bond (75), resulting in an inevitable release of the daughter nuclide from the chelate moiety. Subsequently, the unbound α-emitting daughter nuclides are redistributed in vivo causing substantial harm to normal tissue and reducing the therapeutic effect. For example, the renal toxicity induced by 213Bi (in small animals) is considered a critical constraint to clinical use of 225Ac (114). However, recent results indicated that the release of free metals from DOTA may be not as evident as expected shown by fractioned radio-HPLC (115).
Finding a new chelator is one of the strategies to improve 225Ac-TAT. McDevitt et al. presented a comparison of 225Ac radiolabeling efficiency and in vitro stability of multiple chelators including DTPA, DOTA, TETA, DOTPA, TETPA, and DOTMP (116), and concluded that only DOTA and DOTMP showed chelation of 225Ac after 2 h at 37°C with radiochemical yields of >99 and 78%, respectively. Further in vitro serum stability testing showed that the 225Ac-DOTA complex outperformed the rest with >90% of complexes still intact after 10 days (104). Due to its outstanding stability, DOTA remains the gold standard for 225Ac-radiolabeling for all clinical research. However, DOTA has a decreased thermodynamic stability when used with larger metal ions. Moreover, chelation of 225Ac is a slow reaction demanding extensive heating and high amount of ligand for an adequate yield (77, 116, 117). Macropa, crown, and py4pa are new chelators with improved radiochemical yield and specific activity, as well as achievable labeling conditions. However, none of the new chelators was investigated enough so far to confirm in vivo stability, and their potential to translate into clinical application is yet to be assessed (118–122).
In addition to complicated handling, standardized quality control of radiopharmaceuticals remains a problem in routine clinical production. Because of the complicated decay chain of Ac-225 (Figure 4), special methods must be used for both measurements of the activities and for quality control by radio-thin-layer chromatography (radio-TLC) and radio-high-performance-liquid chromatography (radio-HPLC). Since alpha emitters are difficult to detect directly, indirect measurements are usually used for all measurements by detecting the daughters Fr-221 or Bi-213 (gamma emitters). Fr-221, with its short half-life of 4.9 min, is nearly in radioactive equilibrium after 60–120 min (123). In contrast, Bi-213, with a half-life of 45.6 min, takes hours to reach equilibrium. Thus, measurements of Fr-221 are often used for a faster quality control and release within a clinical environment. When separating free metals and labeled compounds on a TLC plate, the plate is equilibrated for an hour after development before being measured on a TLC scanner. During this period, a constant amount of Fr-221 has formed for both possible species, so comparison is possible, and a labeling yield can be determined. Afterward, an additional distinction must be made between the activity caused by the decay of Fr-221 and the activity of Bi-213. This can typically be differentiated in a gamma spectrometer by splitting the plate. Fr-221 has a line at 218 keV and Bi-213 at 440 keV (122, 124). However, the measurement with radio-TLC is not a valid method to determine the yield and purity of a radiopharmaceutical and additional radio-HPLC methods are required. Two possibilities are conceivable here. Direct detection of Ac-225 by liquid scintillation detection is relatively easy to implement, but may require large activities for injection, which is not always practical and can be very expensive. Therefore, indirect detection of the daughters can be used again by the fractionated collection of the measured sample and subsequent analysis of their components in a gamma spectrometer. In this way, the retention behavior of the compounds on the column can be investigated even when injecting small quantities, and possibly more precise statements can be made about side- or decomposition-products (115).
Today, the development of novel SSTR antagonists holds promise for enhanced diagnostic accuracy and efficacy of SSTR-mediated imaging and therapy. SSTR2-selective 111In-DOTA-BASS (125) and SSTR3-selective ODN-8 (126, 127), the first generation of radiolabeled SSTR antagonists all recognized a larger number of binding sites and revealed an in general twofold higher uptake level in vitro than their agonist counterparts (128). Despite this, BASS labeled with 64Cu via the chelator 4, 11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo [6.6.2]hexadecane (CB-TE2A) (64Cu-CB-TE2A-BASS) showed a compromised tumor uptake compared to the agonist 64Cu-CB-TE2A-octreotate in SSTR2-positive AR42J xenografts (129).
This suboptimal tumor uptake triggered the second generation of radiolabeled SSTR antagonists. In 2006, Ginj et al. presented the idea that radiolabeled SSTR antagonists may perform better than agonists despite the lack of receptor-mediated internalization (127). Despite its lack of internalization, SSTR antagonist demonstrated higher tumor uptake with a higher tumor-to-normal ratio and longer tumor retention time than that of agonist, possibly due to its capability to bind larger variety of receptor conformations, than that of SSTR agonists (130). The initial evidence that antagonists are superior to agonists guided the design and development of more potent SSTR2 antagonists with improved affinity (127, 131), with some of the potential antagonists studied as radiotracers. Among all the potential SSTR2 antagonists, LM3 and JR11 were the most interesting, having the highest hydrophilicity and best affinity. These two were evaluated in a comprehensive study, in combination with two chelators DOTA and NODAGA, and multiple radiometals (132, 133). Interestingly, 68Ga-DOTA-JR11 and -LM3, which have drastically lower affinities for SSTR2 (approximately 150-fold and 60-fold, respectively) than 68Ga-DOTATATE, showed higher tumor uptake (132). Likewise, the therapeutic counterpart 177Lu-DOTA-JR11 exhibited a higher tumor uptake, a longer tumor retention time, and an improved tumor-to-kidney ratio compared to 177Lu-DOTATATE (134), and hence led to a delayed tumor growth and an extended median survival period (135).
Lutetium-177-DOTA-JR11 was first compared in a pilot study with 177Lu-DOTATATE. In the same four patients with grade 1–3 metastatic NET, it showed a 1.7–10.6 times higher tumor dose, and at the same time a 1.1–7.2 times higher tumor-to-kidney and tumor-to-bone marrow ratio, compared with 177Lu-DOTATATE (136, 137). A phase I study with 20 grade 1–3 patients reported a best overall response of 45% (RECIST 1.1 criteria) and a median progression-free survival of 21 months (95% CI: 13.6-not reported). Unfortunately, grade 4 hematotoxicity was reported in 4 out of 7 patients after two cycles of 177Lu-DOTA-JR11, resulting in the suspension of this therapy protocol (138). The protocol was under subsequent modification to limit cumulative absorbed bone marrow dose. A phase I/II open-label study (Clinical trial identification: EudraCT: 2015-002867-41; NCT02592707) is currently ongoing to evaluate the safety and efficacy with the adjusted treatment regimen. Hitherto, only one abstract summarizing the promising efficacy and low toxicity is available. In 20 patients with adequate follow-up, no grade 3/4 renal toxicities and a 90% (95% CI: 68.3–98.8%) disease control rate was reported (139).
In parallel to 177Lu-DOTA-JR11, 177Lu-DOTA-LM3 was evaluated in 51 patients with metastatic neuroendocrine neoplasm at the Theranostics Center for Molecular Radiotherapy and Precision Oncology in Wiesbaden, Germany. 177Lu-DOTA-LM3 was reported to have a 3–5.1 times higher absorbed doses and a 22 h longer whole-body effective half-life than the agonist 177Lu-DOTATOC. All patients tolerated therapy well without any serious acute adverse effects, in particular, there was no nephrotoxicity observed (130).
Here we would like to showcase our recent therapeutic result of PRRT using 225Ac-DOTA LM3 to demonstrate the exciting potential of 225Ac labeled SSTR antagonist in NET treatment (Figure 6). A 40-year-old pancreatic-NET patient suffered from bilobar liver metastases and extensive bone metastasis even after multiple cycles of PRRT with 177Lu-DOTATATE, and compromised therapeutic response after one cycle of 177Lu-DOTA-LM3 treatment, was suggested for PRRT with 225Ac-DOTA-LM3. This case with β-radiation refractory metastatic NET demonstrated incredibly auspicious therapeutic response after two cycles of 225Ac-DOTA-LM3, especially concerning the bone metastases. With this successful case reported for the first time, we would like to suggest that PRRT with 225Ac-labeled SSRT antagonist can potentially be a game changer in therapeutic nuclear medicine, and a promising cure for metastasized tumor.

Figure 6. A 40-year-old patient was diagnosed with poorly differentiated non-functioning pancreatic-NET with bilobar liver and extensive bone metastases, Ki-67 index of 25% NEN-G3 without known mutations. The patients had previously undergone laparoscopic subtotal pancreatic resection and splenectomy, CAPTEM chemotherapy, transarterial chemoembolization (TACE) of right liver lobe and resection of abdominal lesion. In addition, the patient had previous ineffective treatment with Lanreotide, Everolimus, and Sunitinib. In total, the patient had received eight cycles of PRRT, and had a very poor prognosis, extensive bone metastases, even after multiple cycles treatment with 177Lu-DOTATATE (A,B, 68Ga-DOTATOC PET/CT before and after 177Lu-DOTATATE treatment). The 9th PRRT cycle was performed with 177Lu-DOTA-LM3 (B,C, PET/CT imaging before and after 177Lu-DOTA-LM3 treatment), and the last two cycles with 225Ac DOTA-LM3 (C,D, PET/CT imaging before and after 225Ac-DOTA-LM3 treatment), 10 MBq in March and 15 MBq in May 2022, respectively. The cumulative administered radio activities are 66.7 GBq of 177Lu and 25 MBq 225Ac. Restaging result in July 2022 showed excellent response to 225Ac-DOTA-LM3 treatment with partial remission according to the THERCIST. As shown in the most recent PET/CT, there is dramatic improvement, especially concerning the previous innumerable bone metastases in the spine, ribs, and pelvis. The primary tumor in the pancreas, and the liver and bone metastases further decreased in size and number, and no new metastatic lesions were noted. The patient felt dramatically better in comparison to the previous treatments. After the last PRRT, he only experienced mild alopecia and mild pain in the upper right abdomen over 1 week, and has been physically active and gained 3 kg body weight over the past 2 months.
Moreover, the pharmacodynamics of SSTR antagonists have been studied as the clinical interest for them is rapidly growing. The antagonist showed faster association, but slower dissociation, as well as longer cellular retention time compared to the agonist. Moreover, antagonists recognize more binding sites than agonists, providing more targeting opportunities (140). Taking the proposed mechanism, preclinical and clinical studies into consideration, it is obvious that using radiolabeled SSTR antagonists, especially LM3 and JR11, may provide more successful imaging and PRRT strategies for neuroendocrine tumors, even those with relatively low SSTR expression (130, 137, 141) compared to agonists.
Most PRRTs using radiolabeled SSAs and antagonists has until now depended substantially on the most prominently expressed SSTR2 (142). However, NET expression of SSTR subtypes is heterogeneous, and things are made even more difficult by contradictory expression profiles due to different detection methods (20, 143–153). Also, the downregulation or loss of SSTR2 in advanced stages is inherently associated with worse disease prognosis, compromised image sensitivity, and suboptimal therapy with SSTR2-specific radiolabeled SSAs. Using SSTR agonists and antagonists with affinity to more SSTR subtypes is therefore a proposed method—a cocktail approach—of great clinical interest (154). An impressive clinical case showing the application of this cocktail approach, where six tracers were given to a single patient with prostate cancer, was reported at the 2016 SNMMI Highlights Lecture (155). Inspired by the cocktail approach in treating prostate cancer, using SSTR analogs and antagonists targeting various SSTR subtypes in combination with 225Ac-PRRT is worth exploring.
Peptide receptor radionuclide therapy in NET patients has come a long way since Krenning’s first treatments in the 1990s. Novel radionuclides are constantly being developed and tested, in a race to find the perfect theranostic pair. Modified chelators and new ligands including SSTR antagonists are gaining more and more attention, the latter in particular as they have been revealed to give great tumor uptake, retention time and tumor-to-background ratio. 225Ac in particular is very worth investigating.
The rise of the α-emitters as a compliment or replacement to β-emitters is one of the most exciting recent developments. The shorter range gives “precision” in precision medicine a new meaning: Even less damage to surrounding healthy tissue and even more powerful damage to tumor cells.
To this date, confirmed literature on PRRT using an α-emitter with a SSTR antagonist has yet to be published, and obviously, there are still many hurdles to overcome. Technical ones, such as how to best combine chemical moieties into a stable, pharmacokinetically feasible drug; economical ones, such as how to best implement a global mass production of radionuclides for research and clinical use; clinical ones, such as how to set a dosage of the existing theranostic pairs that minimizes toxicity whilst maximizing tumor uptake. Yet, to have the recent promising clinical studies on 177Lu-DOTA-LM3 and -JR11 in mind at the same time as the potentials of TAT is intriguing; hopefully a new and promising era for NET therapy will see daylight in the foreseeable future.
JZ, XC, and RB: study conception and design. MS, VJ, and RB: data collection. MS and JZ: analysis and interpretation of results. MS and VJ: writing—original draft preparation. JZ, LG, P-LK, RB, and XC: writing—review and editing. All authors reviewed the results and approved the final version of the manuscript.
This study was supported by the National University of Singapore Start-up grant (NUHSRO/2021/097/Startup/13 and NUHSRO/2020/133/Startup/08), NUS School of Medicine, Nanomedicine Translational Research Program (NUHSRO/202 1/034/TRP/09/Nanomedicine), and the International Centers for Precision Oncology (ICPO) Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rindi G, Inzani F. Neuroendocrine neoplasm update: toward universal nomenclature. Endocr Relat Cancer. (2020) 27:R211–8. doi: 10.1530/ERC-20-0036
2. Taal BG, Visser O. Epidemiology of neuroendocrine tumours. Neuroendocrinology. (2004) 80(Suppl. 1):3–7. doi: 10.1159/000080731
3. Kulke MH, Siu LL, Tepper JE, Fisher G, Jaffe D, Haller DG, et al. Future directions in the treatment of neuroendocrine tumors: consensus report of the National Cancer Institute Neuroendocrine Tumor clinical trials planning meeting. J Clin Oncol. (2011) 29:934–43. doi: 10.1200/JCO.2010.33.2056
4. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. (2017) 3:1335–42. doi: 10.1001/jamaoncol.2017.0589
5. Oberg K, Akerström G, Rindi G, Jelic S. Neuroendocrine gastroenteropancreatic tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2010) 21(Suppl. 5):V223–7. doi: 10.1093/annonc/mdq192
6. Kos-Kudła B, Blicharz-Dorniak J, Handkiewicz-Junak D, Jarząb B, Jarząb M, Kunikowska J, et al. Diagnostic and therapeutic guidelines for gastro-entero-pancreatic neuroendocrine neoplasms (recommended by the Polish Network of Neuroendocrine Tumours). Endokrynol Pol. (2013) 64:418–43.
7. Trofimiuk-Müldner M, Lewkowicz E, Wysocka K, Pach D, Kiełtyka A, Stefańska A, et al. Epidemiology of gastroenteropancreatic neuroendocrine neoplasms in Krakow and Krakow district in 2007-2011. Endokrynol Pol. (2017) 68:42–6.
8. Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. (2010) 39:707–12. doi: 10.1097/MPA.0b013e3181ec124e
9. Metz DC, Choi J, Strosberg J, Heaney AP, Howden CW, Klimstra D, et al. A rationale for multidisciplinary care in treating neuroendocrine tumours. Curr Opin Endocrinol Diabetes Obes. (2012) 19:306–13. doi: 10.1097/MED.0b013e32835570f1
10. Oberg K, Kvols L, Caplin M, Delle Fave G, de Herder W, Rindi G, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. (2004) 15:966–73. doi: 10.1093/annonc/mdh216
11. Sowa-Staszczak A, Hubalewska-Dydejczyk A, Tomaszuk M. PRRT as neoadjuvant treatment in NET. Recent Results Cancer Res. (2013) 194:479–85. doi: 10.1007/978-3-642-27994-2_27
12. Das S, Al-Toubah T, Strosberg J. Chemotherapy in neuroendocrine tumors. Cancers. (2021) 13:4872. doi: 10.3390/cancers13194872
13. Papamichail DG, Exadaktylou PE, Chatzipavlidou VD. [Neuroendocrine tumors: peptide receptors radionuclide therapy (PRRT)]. Hell J Nucl Med. (2016) 19:75–82.
14. Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. (2009) 27:4656–63. doi: 10.1200/JCO.2009.22.8510
15. Faiss S, Pape U-F, Böhmig M, Dörffel Y, Mansmann U, Golder WA. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors–the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. (2003) 14:2689–96. doi: 10.1200/JCO.2003.12.142
16. Dash A, Knapp FF, Pillai MR. Targeted radionuclide therapy–an overview. Curr Radiopharm. (2013) 6:152–80. doi: 10.2174/18744710113066660023
17. Volkert WA, Hoffman TJ. Therapeutic radiopharmaceuticals. Chem Rev. (1999) 99:2269–92. doi: 10.1021/cr9804386
18. Hirmas N, Jadaan R, Al-Ibraheem A. Peptide receptor radionuclide therapy and the treatment of gastroentero-pancreatic neuroendocrine tumors: current findings and future perspectives. Nucl Med Mol Imaging. (2018) 52:190–9. doi: 10.1007/s13139-018-0517-x
19. Partelli S, Bertani E, Bartolomei M, Perali C, Muffatti F, Grana CM, et al. Peptide receptor radionuclide therapy as neoadjuvant therapy for resectable or potentially resectable pancreatic neuroendocrine neoplasms. Surgery. (2018) 163:761–67. doi: 10.1016/j.surg.2017.11.007
20. Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. (2003) 30:781–93. doi: 10.1007/s00259-003-1184-3
21. Krenning EP, Bakker WH, Breeman WA, Koper JW, Kooij PP, Ausema L, et al. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet. (1989) 1:242–4. doi: 10.1016/S0140-6736(89)91258-0
22. Patel YC, Wheatley T. In vivo and in vitro plasma disappearance and metabolism of somatostatin-28 and somatostatin-14 in the rat. Endocrinology. (1983) 112:220–5. doi: 10.1210/endo-112-1-220
23. Bakker WH, Albert R, Bruns C, Breeman WAP, Hofland LJ, Marbach P, et al. [111In-DTPA-D-Phe1]-octreotide, a potential radiopharmaceutical for imaging of somatostatin receptor-positive tumors: synthesis, radiolabeling and in vitro validation. Life Sci. (1991) 49:1583–91. doi: 10.1016/0024-3205(91)90052-D
24. Krenning EP, Bakker WH, Kooij PP, Breeman WA, Oei HY, de Jong M, et al. Somatostatin receptor scintigraphy with indium-111-DTPA-D-Phe-1-octreotide in man: metabolism, dosimetry and comparison with iodine-123-Tyr-3-octreotide. J Nucl Med. (1992) 33:652–8.
25. Krenning EP, Kwekkeboom DJ, Bakker WH, Breeman WA, Kooij PP, Oei HY, et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. (1993) 20:716–31. doi: 10.1007/BF00181765
26. Krenning EP, Kooij PP, Bakker WH, Breeman WA, Postema PT, Kwekkeboom DJ, et al. Radiotherapy with a radiolabeled somatostatin analogue, [111In-DTPA-D-Phe1]-octreotide. A case history. Ann N Y Acad Sci. (1994) 733:496–506. doi: 10.1111/j.1749-6632.1994.tb17300.x
27. Feijtel D, de Jong M, Nonnekens J. Peptide receptor radionuclide therapy: looking back, looking forward. Curr Top Med Chem. (2020) 20:2959–69. doi: 10.2174/1568026620666200226104652
28. Otte A, Jermann E, Behe M, Goetze M, Bucher HC, Roser HW, et al. DOTATOC: a powerful new tool for receptor-mediated radionuclide therapy. Eur J Nucl Med. (1997) 24:792–5. doi: 10.1007/BF00879669
29. de Jong M, Bakker WH, Krenning EP, Breeman WA, van der Pluijm ME, Bernard BF, et al. Yttrium-90 and indium-111 labelling, receptor binding and biodistribution of [DOTA0, d-Phe1,Tyr3]octreotide, a promising somatostatin analogue for radionuclide therapy. Eur J Nucl Med. (1997) 24:368–71. doi: 10.1007/BF00881807
30. Otte A, Herrmann R, Heppeler A, Behe M, Jermann E, Powell P, et al. Yttrium-90 DOTATOC: first clinical results. Eur J Nucl Med. (1999) 26:1439–47. doi: 10.1007/s002590050476
31. Otte A, Mueller-Brand J, Dellas S, Nitzsche EU, Herrmann R, Maecke HR. Yttrium-90-labelled somatostatin-analogue for cancer treatment. Lancet. (1998) 351:417–8. doi: 10.1016/S0140-6736(05)78355-0
32. de Jong M, Breeman WA, Bakker WH, Kooij PP, Bernard BF, Hofland LJ, et al. Comparison of 111In-labeled somatostatin analogues for tumor scintigraphy and radionuclide therapy. Cancer Res. (1998) 58:437–41.
33. de Jong M, Breeman WA, Bernard BF, Bakker WH, Schaar M, van Gameren A, et al. [177Lu-DOTA(0),Tyr3] octreotate for somatostatin receptor-targeted radionuclide therapy. Int J Cancer. (2001) 92:628–33. doi: 10.1002/1097-0215(20010601)92:5<628::AID-IJC1244>3.0.CO;2-L
34. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. (2017) 376:125–35. doi: 10.1056/NEJMoa1607427
35. Levine R, Krenning EP. Clinical history of the theranostic radionuclide approach to neuroendocrine tumors and other types of cancer: historical review based on an interview of eric P. krenning by rachel levine. J Nucl Med. (2017) 58(Suppl. 2):3S–9S. doi: 10.2967/jnumed.116.186502
36. Nelson KL, Sheetz MA. Radiation safety observations associated with 177lu dotatate patients. Health Phys. (2019) 117:680–7. doi: 10.1097/HP.0000000000001122
37. Hennrich U, Kopka K. Lutathera§: The First FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals. (2019) 12:114. doi: 10.3390/ph12030114
38. Fortune BE, Jackson J, Leonard J, Trotter JF. Vapreotide: a somatostatin analog for the treatment of acute variceal bleeding. Expert Opin Pharmacother. (2009) 10:2337–42. doi: 10.1517/14656560903207019
39. Kvols LK, Woltering EA. Role of somatostatin analogs in the clinical management of non-neuroendocrine solid tumors. Anticancer Drugs. (2006) 17:601–8. doi: 10.1097/01.cad.0000210335.95828.ed
40. Prasad V, Srirajaskanthan R, Toumpanakis C, Grana CM, Baldari S, Shah T, et al. Lessons from a multicentre retrospective study of peptide receptor radionuclide therapy combined with lanreotide for neuroendocrine tumours: a need for standardised practice. Eur J Nucl Med Mol Imaging. (2020) 47:2358–71. doi: 10.1007/s00259-020-04712-2
41. Lewington VJ. Targeted radionuclide therapy for neuroendocrine tumours. Endocr Relat Cancer. (2003) 10:497–501. doi: 10.1677/erc.0.0100497
42. Laudicella R, Albano D, Annunziata S, Calabrò D, Argiroffi G, Abenavoli E, et al. Theragnostic use of radiolabelled dota-peptides in meningioma: from clinical demand to future applications. Cancers. (2019) 11:1412. doi: 10.3390/cancers11101412
43. Sommerauer M, Burkhardt JK, Frontzek K, Rushing E, Buck A, Krayenbuehl N, et al. 68Gallium-DOTATATE PET in meningioma: a reliable predictor of tumor growth rate? Neuro Oncol. (2016) 18:1021–7. doi: 10.1093/neuonc/now001
44. Acker G, Kluge A, Lukas M, Conti A, Pasemann D, Meinert F, et al. Impact of 68Ga-DOTATOC PET/MRI on robotic radiosurgery treatment planning in meningioma patients: first experiences in a single institution. Neurosurg Focus FOC. (2019) 46:E9. doi: 10.3171/2019.3.FOCUS1925
45. Gehler B, Paulsen F, Öksüz MÖ, Hauser T-K, Eschmann SM, Bares R, et al. [68Ga]-DOTATOC-PET/CT for meningioma IMRT treatment planning. Radiat Oncol. (2009) 4:56. doi: 10.1186/1748-717X-4-56
46. Henze M, Schuhmacher J, Hipp P, Kowalski J, Becker DW, Doll J, et al. PET imaging of somatostatin receptors using [68GA]DOTA-D-Phe1-Tyr3-octreotide: first results in patients with meningiomas. J Nucl Med. (2001) 42:1053–6.
47. Ivanidze J, Roytman M, Lin E, Magge RS, Pisapia DJ, Liechty B, et al. Gallium-68 DOTATATE PET in the evaluation of intracranial meningiomas. J Neuroimaging. (2019) 29:650–6. doi: 10.1111/jon.12632
48. Rachinger W, Stoecklein VM, Terpolilli NA, Haug AR, Ertl L, Pöschl J, et al. Increased 68Ga-DOTATATE uptake in PET imaging discriminates meningioma and tumor-free tissue. J Nucl Med. (2015) 56:347–53. doi: 10.2967/jnumed.114.149120
49. Hennrich U, Benešová M. [68Ga]Ga-DOTA-TOC: The First FDA-Approved 68Ga-Radiopharmaceutical for PET Imaging. Pharmaceuticals. (2020) 13:38. doi: 10.3390/ph13030038
50. Wu T-J, Chiu H-Y, Yu J, Cautela MP, Sarmento B, das Neves J, et al. Nanotechnologies for early diagnosis, in situ disease monitoring, and prevention. In: Uskoković V, Uskoković DP editors. Nanotechnologies in Preventive and Regenerative Medicine. Amsterdam: Elsevier (2018). 1–92. doi: 10.1016/B978-0-323-48063-5.00001-0
51. Chinol M, Franceschini R, Paganelli G, Pecorale A, Paiano A. Simple Production of Yttrium-90 in a Chemical form Suitable to Clinical Grade Radioconjugates. In: Bergmann H, Kroiss A, Sinzinger H editors. Radioactive Isotopes in Clinical Medicine and Research. Basel: Birkhäuser Basel (1997). 327–32. doi: 10.1007/978-3-0348-7772-5_49
52. Kunikowska J, Królicki L, Hubalewska-Dydejczyk A, Mikołajczak R, Sowa-Staszczak A, Pawlak D. Clinical results of radionuclide therapy of neuroendocrine tumours with 90Y-DOTATATE and tandem 90Y/177Lu-DOTATATE: which is a better therapy option? Eur J Nucl Med Mol Imaging. (2011) 38:1788–97. doi: 10.1007/s00259-011-1833-x
53. Bodei L, Cremonesi M, Ferrari M, Pacifici M, Grana CM, Bartolomei M, et al. Long-term evaluation of renal toxicity after peptide receptor radionuclide therapy with 90Y-DOTATOC and 177Lu-DOTATATE: the role of associated risk factors. Eur J Nucl Med Mol Imaging. (2008) 35:1847–56. doi: 10.1007/s00259-008-0778-1
54. Cremonesi M, Ferrari ME, Bodei L, Chiesa C, Sarnelli A, Garibaldi C, et al. Correlation of dose with toxicity and tumour response to 90Y- and 177Lu-PRRT provides the basis for optimization through individualized treatment planning. Eur J Nucl Med Mol Imaging. (2018) 45:2426–41. doi: 10.1007/s00259-018-4044-x
55. Todorović-Tirnanić M, Kaemmerer D, Prasad V, Hommann M, Baum RP. Intraoperative Somatostatin Receptor Detection After Peptide Receptor Radionuclide Therapy with 177Lu- and 90Y-DOTATOC (Tandem PRRNT) in a Patient with a Metastatic Neuroendocrine Tumor. Theranostics, Gallium-68, and Other Radionuclides. Berlin: Springer (2013). doi: 10.1007/978-3-642-27994-2_28
56. Baum RP, Kulkarni HR, Singh A, Kaemmerer D, Mueller D, Prasad V, et al. Results and adverse events of personalized peptide receptor radionuclide therapy with (90)Yttrium and (177)Lutetium in 1048 patients with neuroendocrine neoplasms. Oncotarget. (2018) 9:16932–50. doi: 10.18632/oncotarget.24524
57. Kunikowska J, Królicki L. Targeted α-emitter therapy of neuroendocrine tumors. Semin Nucl Med. (2020) 50:171–6. doi: 10.1053/j.semnuclmed.2019.11.003
58. Kassis AI, Adelstein SJ. Radiobiologic principles in radionuclide therapy. J Nucl Med. (2005) 46(Suppl. 1):4S–12S.
59. Sgouros G. Alpha-particles for targeted therapy. Adv Drug Deliv Rev. (2008) 60:1402–6. doi: 10.1016/j.addr.2008.04.007
60. Pouget JP, Constanzo J. Revisiting the radiobiology of targeted alpha therapy. Front Med. (2021) 8:692436. doi: 10.3389/fmed.2021.692436
61. Brechbiel MW. Targeted alpha-therapy: past, present, future? Dalton Trans. (2007) 43:4918–28. doi: 10.1039/b704726f
62. Zalutsky MR, Bigner DD. Radioimmunotherapy with alpha-particle emitting radioimmunoconjugates. Acta Oncol. (1996) 35:373–9. doi: 10.3109/02841869609101654
63. Zalutsky MR, Schuster JM, Garg PK, Archer GE Jr, Dewhirst MW, Bigner DD. Two approaches for enhancing radioimmunotherapy: alpha emitters and hyperthermia. Recent Results Cancer Res. (1996) 141:101–22. doi: 10.1007/978-3-642-79952-5_7
64. Poty S, Francesconi LC, McDevitt MR, Morris MJ, Lewis JS. α-Emitters for Radiotherapy: From Basic Radiochemistry to Clinical Studies-Part 1. J Nucl Med. (2018) 59:878–84. doi: 10.2967/jnumed.116.186338
65. Nikula TK, McDevitt MR, Finn RD, Wu C, Kozak RW, Garmestani K, et al. Alpha-emitting bismuth cyclohexylbenzyl DTPA constructs of recombinant humanized anti-CD33 antibodies: pharmacokinetics, bioactivity, toxicity and chemistry. J Nucl Med. (1999) 40:166–76.
66. Raju MR, Eisen Y, Carpenter S, Inkret WC. Radiobiology of alpha particles. III. Cell inactivation by alpha-particle traversals of the cell nucleus. Radiat Res. (1991) 128:204–9. doi: 10.2307/3578139
67. Koh TT, Bezak E, Chan D, Cehic G. Targeted alpha-particle therapy in neuroendocrine neoplasms: a systematic review. World J Nucl Med. (2021) 20:329–35. doi: 10.4103/wjnm.wjnm_160_20
68. Nelson BJB, Andersson JD, Wuest F. Targeted alpha therapy: progress in radionuclide production, radiochemistry, and applications. Pharmaceutics. (2021) 13:49. doi: 10.3390/pharmaceutics13010049
69. Tafreshi NK, Doligalski ML, Tichacek CJ, Pandya DN, Budzevich MM, El-Haddad G, et al. Development of targeted alpha particle therapy for solid tumors. Molecules. (2019) 24:4314. doi: 10.3390/molecules24234314
70. Wadas TJ, Pandya DN, Solingapuram Sai KK, Mintz A. Molecular targeted α-particle therapy for oncologic applications. AJR Am J Roentgenol. (2014) 203:253–60. doi: 10.2214/AJR.14.12554
71. Elgqvist J, Frost S, Pouget JP, Albertsson P. The potential and hurdles of targeted alpha therapy - clinical trials and beyond. Front Oncol. (2014) 3:324. doi: 10.3389/fonc.2013.00324
72. Kim YS, Brechbiel MW. An overview of targeted alpha therapy. Tumour Biol. (2012) 33:573–90. doi: 10.1007/s13277-011-0286-y
73. Jadvar H, Colletti PM. Targeted α-therapy in non-prostate malignancies. Eur J Nucl Med Mol Imaging. (2021) 49:47–53. doi: 10.1007/s00259-021-05405-0
74. Imam SK. Advancements in cancer therapy with alpha-emitters: a review. Int J Radiat Oncol Biol Phys. (2001) 51:271–8. doi: 10.1016/S0360-3016(01)01585-1
75. de Kruijff RM, Wolterbeek HT, Denkova AGA. Critical review of alpha radionuclide therapy-how to deal with recoiling daughters? Pharmaceuticals. (2015) 8:321–36. doi: 10.3390/ph8020321
76. Adloff JP. The centenary of a controversial discovery: actinium. Radiochim Acta. (2000) 88:123–8. doi: 10.1524/ract.2000.88.3-4.123
77. McDevitt MR, Ma D, Lai LT, Simon J, Borchardt P, Frank RK, et al. Tumor therapy with targeted atomic nanogenerators. Science. (2001) 294:1537–40. doi: 10.1126/science.1064126
78. Scheinberg DA, McDevitt MR. Actinium-225 in targeted alpha-particle therapeutic applications. Curr Radiopharm. (2011) 4:306–20. doi: 10.2174/1874471011104040306
79. Chen X, Ji M, Fisher DR, Wai CM. Monofunctionalization of Calix[4]arene Tetracarboxylic Acid at the Upper Rim with Isothiocyanate Group: first bifunctional chelating agent for Alpha-Emitter Ac-225. Synlett. (1999) 11:1784–6. doi: 10.1055/s-1999-2944
80. Deal KA, Davis IA, Mirzadeh S, Kennel SJ, Brechbiel MW. Improved in vivo stability of actinium-225 macrocyclic complexes. J Med Chem. (1999) 42:2988–92. doi: 10.1021/jm990141f
81. Morgenstern A, Apostolidis C, Bruchertseifer F. Supply and clinical application of Actinium-225 and Bismuth-213. Semin Nucl Med. (2020) 50:119–23. doi: 10.1053/j.semnuclmed.2020.02.003
82. Rosenblat TL, McDevitt MR, Pandit-Taskar N, Carrasquillo JA, Chanel S, Frattini MG, et al. Phase I trial of the targeted alpha-particle nano-generator Actinium-225 (225Ac)-HuM195 (Anti-CD33) in Acute Myeloid Leukemia (AML). Blood. (2007) 110:910. doi: 10.1182/blood.V110.11.910.910
83. Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F, et al. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation therapy of metastatic castration-resistant prostate cancer. J Nucl Med. (2016) 57:1941–4. doi: 10.2967/jnumed.116.178673
84. Kratochwil C, Bruchertseifer F, Rathke H, Bronzel M, Apostolidis C, Weichert W, et al. Targeted α-Therapy of metastatic castration-resistant prostate cancer with (225)Ac-PSMA-617: dosimetry estimate and empiric dose finding. J Nucl Med. (2017) 58:1624–31. doi: 10.2967/jnumed.117.191395
85. Sathekge M, Bruchertseifer F, Knoesen O, Reyneke F, Lawal I, Lengana T, et al. (225)Ac-PSMA-617 in chemotherapy-naive patients with advanced prostate cancer: a pilot study. Eur J Nucl Med Mol Imaging. (2019) 46:129–38. doi: 10.1007/s00259-018-4167-0
86. Sathekge M, Bruchertseifer F, Vorster M, Lawal IO, Knoesen O, Mahapane J, et al. Predictors of Overall and Disease-Free Survival in Metastatic Castration-Resistant Prostate Cancer Patients Receiving 225Ac-PSMA-617 Radioligand Therapy. J Nucl Med. (2020) 61:62–9. doi: 10.2967/jnumed.119.229229
87. Kratochwil C, Bruchertseifer F, Giesel F, Apostolidis C, Haberkorn U, Morgenstern A. Ac-225-DOTATOC - an empiric dose finding for alpha particle emitter based radionuclide therapy of neuroendocrine tumors. J Nucl Med. (2015) 56(Suppl. 3):1232.
88. Morgenstern A, Bruchertseifer F, Apostolidis C. Targeted alpha therapy with 213Bi. Curr Radiopharm. (2011) 4:295–305. doi: 10.2174/1874471011104040295
89. Apostolidis C, Molinet R, Rasmussen G, Morgenstern A. Production of Ac-225 from Th-229 for targeted alpha therapy. Anal Chem. (2005) 77:6288–91. doi: 10.1021/ac0580114
90. Zielinska B, Apostolidis C, Bruchertseifer F, Morgenstern A. An Improved Method for the Production of Ac-225/Bi-213 from Th-229 for Targeted Alpha Therapy. Solv Extract Ion Exchang. (2007) 25:339–49. doi: 10.1080/07366290701285108
91. Boll RA, Malkemus D, Mirzadeh S. Production of actinium-225 for alpha particle mediated radioimmunotherapy. Appl Radiat Isot. (2005) 62:667–79. doi: 10.1016/j.apradiso.2004.12.003
92. Samsonov M, Nerozin N, Podsoblyaev D, Prokof’ev I, Tkachev S, Khamianov S, et al. Isolation of alpha-emitting radionuclides for nuclear medicine in JSC “SSC RF–IPPE”. Proceedings of the 10th International Symposium on Targeted Alpha Therapy. Kanazawa (2017).
93. Morgenstern A, Apostolidis C, Kratochwil C, Sathekge M, Krolicki L, Bruchertseifer F. An overview of targeted alpha therapy with 225Actinium and 213Bismuth. Curr Radiopharm. (2018) 11:200–8. doi: 10.2174/1874471011666180502104524
94. Ahenkorah S, Cassells I, Deroose CM, Cardinaels T, Burgoyne AR, Bormans G, et al. Bismuth-213 for targeted radionuclide therapy: from atom to bedside. Pharmaceutics. (2021) 13:599. doi: 10.3390/pharmaceutics13050599
95. Weidner JW, Mashnik SG, John KD, Hemez F, Ballard B, Bach H, et al. Proton-induced cross sections relevant to production of 225Ac and 223Ra in natural thorium targets below 200 MeV. Appl Radiat Isot. (2012) 70:2602–7. doi: 10.1016/j.apradiso.2012.07.006
96. Weidner JW, Mashnik SG, John KD, Ballard B, Birnbaum ER, Bitteker LJ, et al. 225Ac and 223Ra production via 800 MeV proton irradiation of natural thorium targets. Appl Radiat Isot. (2012) 70:2590–5. doi: 10.1016/j.apradiso.2012.07.003
97. Ermolaev SV, Zhuikov BL, Kokhanyuk VM, Matushko VL, Kalmykov SN, Aliev RA, et al. Production of actinium, thorium and radium isotopes from natural thorium irradiated with protons up to 141 MeV. Radiochim Acta. (2012) 100:223–9. doi: 10.1524/ract.2012.1909
98. Zhuikov BL, Kalmykov SN, Ermolaev SV, Aliev RA, Kokhanyuk VM, Matushko VL, et al. Production of 225Ac and 223Ra by irradiation of Th with accelerated protons. Radiochemistry. (2011) 53:73–80. doi: 10.1134/S1066362211010103
99. Abergel R, An D, Lakes A, Rees J, Gauny S. Actinium Biokinetics and Dosimetry: What is the Impact of Ac-227 in Accelerator-Produced Ac-225? J Med Imaging Radiat Sci. (2019) 50(Suppl. 1):S23. doi: 10.1016/j.jmir.2019.03.073
100. Apostolidis C, Molinet R, McGinley J, Abbas K, Möllenbeck J, Morgenstern A. Cyclotron production of Ac-225 for targeted alpha therapy. Appl Radiat Isot. (2005) 62:383–7. doi: 10.1016/j.apradiso.2004.06.013
101. Braccini S. Compact Medical Cyclotrons and their use for Radioisotope Production and Multi-disciplinary Research. Proceedings of Cyclotrons 2016. Zurich (2017).
102. Morgenstern A, Abbas K, Bruchertseifer F, Apostolidis C. Production of alpha emitters for targeted alpha therapy. Curr Radiopharm. (2008) 1:135–43. doi: 10.2174/1874471010801030135
103. Melville G, Meriarty H, Metcalfe P, Knittel T, Allen BJ. Production of Ac-225 for cancer therapy by photon-induced transmutation of Ra-226. Appl Radiat Isot. (2007) 65:1014–22. doi: 10.1016/j.apradiso.2007.03.018
104. Robertson KHA, Ramogida FC, Schaffer P, Radchenko V. Development of 225Ac Radiopharmaceuticals: TRIUMF perspectives and experiences. Curr Radiopharm. (2018) 11:156–72. doi: 10.2174/1874471011666180416161908
105. Sgouros G, He B, Ray N, Ludwig DL, Frey EC. Dosimetric impact of Ac-227 in accelerator-produced Ac-225 for alpha-emitter radiopharmaceutical therapy of patients with hematological malignancies: a pharmacokinetic modeling analysis. EJNMMI Phys. (2021) 8:60. doi: 10.1186/s40658-021-00410-6
106. Ackerman NL, Graves EE. The potential for Cerenkov luminescence imaging of alpha-emitting radionuclides. Phys Med Biol. (2012) 57:771–83. doi: 10.1088/0031-9155/57/3/771
107. Rasheed R, Usmani S, Naqvi SAR, Alkandari F, Marafi F. Alpha Therapy with 225Actinium Labeled Prostate Specific Membrane Antigen: Reporting New Photopeak of 78 Kilo-electron Volts for Better Image Statistics. Indian J Nucl Med. (2019) 34:76–7. doi: 10.4103/ijnm.IJNM_115_18
108. Usmani S, Rasheed R, Al Kandari F, Marafi F, Naqvi SAR. 225Ac Prostate-Specific Membrane Antigen Posttherapy α Imaging: Comparing 2 and 3 Photopeaks. Clin Nucl Med. (2019) 44:401–3. doi: 10.1097/RLU.0000000000002525
109. Vatsa R, Sood A, Vadi SK, Das CK, Kaur K, Parmar M, et al. 225Ac-PSMA-617 radioligand posttherapy imaging in metastatic castrate-resistant prostate cancer patient using 3 photopeaks. Clin Nucl Med. (2020) 45:437–8. doi: 10.1097/RLU.0000000000003031
110. McDevitt MR, Finn RD, Sgouros G, Ma D, Scheinberg DA. An 225Ac/213Bi generator system for therapeutic clinical applications: construction and operation. Appl Radiat Isot. (1999) 50:895–904. doi: 10.1016/S0969-8043(98)00151-1
111. Tafreshi NK, Tichacek CJ, Pandya DN, Doligalski ML, Budzevich MM, Kil H, et al. Melanocortin 1 Receptor-Targeted α-Particle Therapy for Metastatic Uveal Melanoma. J Nucl Med. (2019) 60:1124–33. doi: 10.2967/jnumed.118.217240
112. Kratochwil C, Giesel FL, Stefanova M, Benešová M, Bronzel M, Afshar-Oromieh A, et al. PSMA-targeted radionuclide therapy of metastatic castration-resistant prostate cancer with 177Lu-Labeled PSMA-617. J Nucl Med. (2016) 57:1170–6. doi: 10.2967/jnumed.115.171397
113. Kabasakal L, AbuQbeitah M, Aygün A, Yeyin N, Ocak M, Demirci E, et al. Pre-therapeutic dosimetry of normal organs and tissues of 177Lu-PSMA-617 prostate-specific membrane antigen (PSMA) inhibitor in patients with castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. (2015) 42:1976–83. doi: 10.1007/s00259-015-3125-3
114. Jaggi JS, Kappel BJ, McDevitt MR, Sgouros G, Flombaum CD, Cabassa C, et al. Efforts to control the errant products of a targeted in vivo generator. Cancer Res. (2005) 65:4888–95. doi: 10.1158/0008-5472.CAN-04-3096
115. Hooijman EL, Chalashkan Y, Ling SW, Kahyargil FF, Segbers M, Bruchertseifer F, et al. Development of [225Ac]Ac-PSMA-I&T for Targeted Alpha Therapy According to GMP Guidelines for Treatment of mCRPC. Pharmaceutics. (2021) 13:715. doi: 10.3390/pharmaceutics13050715
116. McDevitt MR, Ma D, Simon J, Frank RK, Scheinberg DA. Design and synthesis of 225Ac radioimmunopharmaceuticals. Appl Radiat Isot. (2002) 57:841–7. doi: 10.1016/S0969-8043(02)00167-7
117. Miederer M, Henriksen G, Alke A, Mossbrugger I, Quintanilla-Martinez L, Senekowitsch-Schmidtke R, et al. Preclinical evaluation of the alpha-particle generator nuclide 225Ac for somatostatin receptor radiotherapy of neuroendocrine tumors. Clin Cancer Res. (2008) 14:3555–61. doi: 10.1158/1078-0432.CCR-07-4647
118. Yang H, Wilson JJ, Orvig C, Li Y, Wilbur DS, Ramogida CF, et al. Harnessing α-emitting radionuclides for therapy: radiolabeling method review. J Nucl Med. (2022) 63:5–13. doi: 10.2967/jnumed.121.262687
119. Yang H, Zhang C, Yuan Z, Rodriguez-Rodriguez C, Robertson A, Radchenko V, et al. Synthesis and evaluation of a macrocyclic Actinium-225 Chelator, Quality Control and In Vivo Evaluation of (225) Ac-crown-αMSH Peptide. Chemistry. (2020) 26:11435–40. doi: 10.1002/chem.202002999
120. Thiele NA, Brown V, Kelly JM, Amor-Coarasa A, Jermilova U, MacMillan SN, et al. An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew Chem Int Ed Engl. (2017) 56:14712–7. doi: 10.1002/anie.201709532
121. Li L, Rousseau J, Jaraquemada-Peláez MG, Wang X, Robertson A, Radchenko V, et al. (225)Ac-H(4)py4pa for Targeted Alpha Therapy. Bioconjug Chem. (2021) 32:1348–63. doi: 10.1021/acs.bioconjchem.0c00171
122. Pretze M, Kunkel F, Runge R, Freudenberg R, Braune A, Hartmann H, et al. Ac-EAZY! Towards GMP-Compliant Module Syntheses of 225Ac-Labeled Peptides for Clinical Application. Pharmaceuticals. (2021) 14:652. doi: 10.3390/ph14070652
123. Kelly J, Amor-Coarasa A, Sweeney E, Wilson J, Causey P, Babich J. A Consensus Time for Performing Quality Control of 225Ac-Labeled Radiopharmaceuticals. Durham, NC: Research Square (2021). doi: 10.21203/rs.3.rs-39342/v2
124. Thakral P, Simecek J, Marx S, Kumari J, Pant V, Sen IB. In-House Preparation and Quality Control of Ac-225 Prostate-Specific Membrane Antigen-617 for the Targeted Alpha Therapy of Castration-Resistant Prostate Carcinoma. Ind J Nucl Med. (2021) 36:114–9. doi: 10.4103/ijnm.ijnm_200_20
125. Bass RT, Buckwalter BL, Patel BP, Pausch MH, Price LA, Strnad J, et al. Identification and characterization of novel somatostatin antagonists. Mol Pharmacol. (1996) 50:709–15.
126. Reubi JC, Schaer JC, Wenger S, Hoeger C, Erchegyi J, Waser B, et al. SST3-selective potent peptidic somatostatin receptor antagonists. Proc Natl Acad Sci U.S.A. (2000) 97:13973–8. doi: 10.1073/pnas.250483897
127. Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X, et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U.S.A. (2006) 103:16436–41. doi: 10.1073/pnas.0607761103
128. Wang X, Fani M, Schulz S, Rivier J, Reubi JC, Maecke HR. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur J Nucl Med Mol Imaging. (2012) 39:1876–85. doi: 10.1007/s00259-012-2231-8
129. Wadas TJ, Eiblmaier M, Zheleznyak A, Sherman CD, Ferdani R, Liang K, et al. Preparation and biological evaluation of 64Cu-CB-TE2A-sst2-ANT, a somatostatin antagonist for PET imaging of somatostatin receptor-positive tumors. J Nucl Med. (2008) 49:1819–27. doi: 10.2967/jnumed.108.054502
130. Baum RP, Zhang J, Schuchardt C, Müller D, Mäcke H. First-in-Humans Study of the SSTR Antagonist 177Lu-DOTA-LM3 for Peptide Receptor Radionuclide Therapy in Patients with Metastatic Neuroendocrine Neoplasms: Dosimetry, Safety, and Efficacy. J Nucl Med. (2021) 62:1571–81. doi: 10.2967/jnumed.120.258889
131. Cescato R, Erchegyi J, Waser B, Piccand V, Maecke HR, Rivier JE, et al. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J Med Chem. (2008) 51:4030–7. doi: 10.1021/jm701618q
132. Fani M, Braun F, Waser B, Beetschen K, Cescato R, Erchegyi J, et al. Unexpected sensitivity of sst2 antagonists to N-terminal radiometal modifications. J Nucl Med. (2012) 53:1481–9. doi: 10.2967/jnumed.112.102764
133. Fani M, Del Pozzo L, Abiraj K, Mansi R, Tamma ML, Cescato R, et al. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: the chelate makes the difference. J Nucl Med. (2011) 52:1110–8. doi: 10.2967/jnumed.111.087999
134. Nicolas GP, Mansi R, McDougall L, Kaufmann J, Bouterfa H, Wild D, et al. Biodistribution, Pharmacokinetics, and Dosimetry of 177 Lu-, 90 Y-, and 111In-Labeled Somatostatin Receptor Antagonist OPS201 in Comparison to the Agonist 177Lu-DOTATATE: The Mass Effect. J Nucl Med. (2017) 58:1435–41. doi: 10.2967/jnumed.117.191684
135. Dalm SU, Nonnekens J, Doeswijk GN, de Blois E, van Gent DC, Konijnenberg MW, et al. Comparison of the Therapeutic Response to Treatment with a 177Lu-labeled somatostatin receptor agonist and antagonist in preclinical models. J Nucl Med. (2016) 57:260–5. doi: 10.2967/jnumed.115.167007
136. Wild D, Fani M, Fischer R, Del Pozzo L, Kaul F, Krebs S, et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: a pilot study. J Nucl Med. (2014) 55:1248–52. doi: 10.2967/jnumed.114.138834
137. Fani M, Mansi R, Nicolas GP, Wild D. Radiolabeled somatostatin analogs-a continuously evolving class of radiopharmaceuticals. Cancers. (2022) 14:1172. doi: 10.3390/cancers14051172
138. Reidy-Lagunes D, Pandit-Taskar N, O’Donoghue JA, Krebs S, Staton KD, Lyashchenko SK, et al. Phase I Trial of Well-Differentiated Neuroendocrine Tumors (NETs) with Radiolabeled Somatostatin Antagonist 177Lu-Satoreotide Tetraxetan. Clin Cancer Res. (2019) 25:6939–47. doi: 10.1158/1078-0432.CCR-19-1026
139. Nicolas G, Ansquer C, Lenzo N, Grønbaek H, Haug A, Navalkissoor S, et al. 1160O An international open-label study on safety and efficacy of 177Lu-satoreotide tetraxetan in somatostatin receptor positive neuroendocrine tumours (NETs): An interim analysis. Ann Oncol. (2020) 31:S771. doi: 10.1016/j.annonc.2020.08.1373
140. Mansi R, Plas P, Vauquelin G, Fani M. Distinct In Vitro Binding Profile of the Somatostatin Receptor Subtype 2 Antagonist [177Lu]Lu-OPS201 Compared to the Agonist [177Lu]Lu-DOTA-TATE. Pharmaceuticals. (2021) 14:1265. doi: 10.3390/ph14121265
141. Fani M, Nicolas GP, Wild D. Somatostatin receptor antagonists for imaging and therapy. J Nucl Med. (2017) 58(Suppl. 2):61S–6S. doi: 10.2967/jnumed.116.186783
142. Klomp MJ, Dalm SU, de Jong M, Feelders RA, Hofland J, Hofland LJ. Epigenetic regulation of somatostatin and somatostatin receptors in neuroendocrine tumors and other types of cancer. Rev Endocr Metab Disord. (2021) 22:495–510. doi: 10.1007/s11154-020-09607-z
143. Papotti M, Bongiovanni M, Volante M, Allìa E, Landolfi S, Helboe L, et al. Expression of somatostatin receptor types 1-5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. a correlative immunohistochemical and reverse-transcriptase polymerase chain reaction analysis. Virchows Arch. (2002) 440:461–75. doi: 10.1007/s00428-002-0609-x
144. Kulaksiz H, Eissele R, Rössler D, Schulz S, Höllt V, Cetin Y, et al. Identification of somatostatin receptor subtypes 1, 2A, 3, and 5 in neuroendocrine tumours with subtype specific antibodies. Gut. (2002) 50:52–60. doi: 10.1136/gut.50.1.52
145. Jaïs P, Terris B, Ruszniewski P, LeRomancer M, Reyl-Desmars F, Vissuzaine C, et al. Somatostatin receptor subtype gene expression in human endocrine gastroentero-pancreatic tumours. Eur J Clin Invest. (1997) 27:639–44. doi: 10.1046/j.1365-2362.1997.1740719.x
146. Hu Y, Ye Z, Wang F, Qin Y, Xu X, Yu X, et al. Role of somatostatin receptor in pancreatic neuroendocrine tumor development, diagnosis, and therapy. Front Endocrinol. (2021) 12:679000. doi: 10.3389/fendo.2021.679000
147. Wängberg B, Nilsson O, Johanson VV, Kölby L, Forssell-Aronsson E, Andersson P, et al. Somatostatin receptors in the diagnosis and therapy of neuroendocrine tumor. Oncologist. (1997) 2:50–8. doi: 10.1634/theoncologist.2-1-50
148. Remes SM, Leijon HL, Vesterinen TJ, Arola JT, Haglund CH. Immunohistochemical expression of somatostatin receptor subtypes in a panel of neuroendocrine neoplasias. J Histochem Cytochem. (2019) 67:735–43. doi: 10.1369/0022155419856900
149. Jonas S, John M, Boese-Landgraf J, Häring R, Prevost G, Thomas F, et al. Somatostatin receptor subtypes in neuroendocrine tumor cell lines and tumor tissues. Langenbecks Arch Chir. (1995) 380:90–5. doi: 10.1007/BF00186414
150. Lambertini C, Barzaghi-Rinaudo P, D’Amato L, Schulz S, Nuciforo P, Schmid HA. Evaluation of somatostatin receptor subtype expression in human neuroendocrine tumors using two sets of new monoclonal antibodies. Regul Pept. (2013) 187:35–41. doi: 10.1016/j.regpep.2013.10.007
151. Lou E, Gandhi N, Farrell A, Xiu J, Seeber A, Beg SS, et al. Differential expression of somatostatin receptor (SSTR) subtypes across a spectrum of neuroendocrine neoplasms (NENs). J Clin Oncol. (2022) 40(16_suppl):3071. doi: 10.1200/JCO.2022.40.16_suppl.3071
152. Kaemmerer D, Athelogou M, Lupp A, Lenhardt I, Schulz S, Luisa P, et al. Somatostatin receptor immunohistochemistry in neuroendocrine tumors: comparison between manual and automated evaluation. Int J Clin Exp Pathol. (2014) 7:4971–80.
153. Kanakis G, Grimelius L, Spathis A, Tringidou R, Rassidakis GZ, Öberg K, et al. Expression of Somatostatin Receptors 1-5 and dopamine receptor 2 in lung carcinoids: implications for a therapeutic role. Neuroendocrinology. (2015) 101:211–22. doi: 10.1159/000381061
154. Reubi JC, Maecke HR. Approaches to multireceptor targeting: hybrid radioligands, radioligand cocktails, and sequential radioligand applications. J Nucl Med. (2017) 58(Suppl. 2):10S–6S. doi: 10.2967/jnumed.116.186882
156. Graf F, Fahrer J, Maus S, Morgenstern A, Bruchertseifer F, Venkatachalam S, et al. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS One. (2014) 9:e88239. doi: 10.1371/journal.pone.0088239
157. Tafreshi NK, Pandya DN, Tichacek CJ, Budzevich MM, Wang Z, Reff JN, et al. Preclinical evaluation of [225Ac]Ac-DOTA-TATE for treatment of lung neuroendocrine neoplasms. Eur J Nucl Med Mol Imaging. (2021) 48:3408–21. doi: 10.1007/s00259-021-05315-1
158. Zhang J, Singh A, Kulkarni HR, Schuchardt C, Müller D, Wester HJ, et al. From bench to bedside-the bad berka experience with first-in-human studies. Semin Nucl Med. (2019) 49:422–37. doi: 10.1053/j.semnuclmed.2019.06.002
159. Ballal S, Yadav MP, Bal C, Sahoo RK, Tripathi M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to (177)Lu-DOTATATE PRRT: first clinical experience on the efficacy and safety. Eur J Nucl Med Mol Imaging. (2020) 47:934–46. doi: 10.1007/s00259-019-04567-2
160. Ocak M, Toklu T, Demirci E, Selçuk N, Kabasakal L. Post-therapy imaging of 225Ac-DOTATATE treatment in a patient with recurrent neuroendocrine tumor. Eur J Nucl Med Mol Imaging. (2020) 47:2711–2. doi: 10.1007/s00259-020-04725-x
161. Kamaleshwaran KK, Suneelkumar M, Madhusairam R, Radhakrishnan EK, Arunpandiyan S, Arnold VJ. Whole-body and Single-Photon Emission Computed Tomography/Computed Tomography Postpeptide Receptor Alpha Radionuclide Therapy Images of Actinium 225-Tetraazacyclododecanetetraacetic Acid-Octreotide as a Primary Modality of Treatment in a Patient with Advanced Rectal Neuroendocrine Tumor with Metastases. Indian J Nucl Med. (2020) 35:226–8. doi: 10.4103/ijnm.IJNM_58_20
162. Zhang J, Kulkarni HR, Baum RP. Peptide Receptor Radionuclide Therapy Using 225Ac-DOTATOC Achieves Partial Remission in a Patient With Progressive Neuroendocrine Liver Metastases After Repeated β-emitter peptide receptor radionuclide therapy. Clin Nucl Med. (2020) 45:241–3. doi: 10.1097/RLU.0000000000002915
163. Zhang J, Kulkarni HR, Baum RP. 225Ac-DOTATOC-Targeted Somatostatin Receptor α-Therapy in a Patient With Metastatic Neuroendocrine Tumor of the Thymus, Refractory to β-Radiation. Clin Nucl Med. (2021) 46:1030–1. doi: 10.1097/RLU.0000000000003792
164. Satapathy S, Sood A, Das CK, Kavanal AJ, Mittal BR. Alpha Before Beta: Exceptional Response to First-Line 225Ac-DOTATATE in a Patient of Metastatic Neuroendocrine Tumor With Extensive Skeletal Involvement. Clin Nucl Med. (2022) 47:e156–7. doi: 10.1097/RLU.0000000000003823
165. Kavanal AJ, Satapathy S, Sood A, Khosla D, Mittal BR. Subclinical Hypothyroidism After 225Ac-DOTATATE Therapy in a Case of Metastatic Neuroendocrine Tumor: Unknown Adverse Effect of PRRT. Clin Nucl Med. (2022) 47:e184–6. doi: 10.1097/RLU.0000000000003893
166. Budlewski T, Król ZJ, Bruchertseifer F, Majkowska-Pilip A, Morgenstern A, Wierzba W. Innovative radioisotope therapy for patients with neuroendocrine tumors using an alpha (225Ac) emitter labeled somatostatin analog: octreotate. a promising new treatment for advanced, progressive neuroendocrine neoplasms. Pol Arch Intern Med. (2022) 132:16275. doi: 10.20452/pamw.16275
167. Alan Selçuk N, Demirci E, Ocak M, Toklu T, Ergen S, Kabasakal L. Almost complete response with a single administration 225Ac-DOTATATE in a patient with a metastatic neuroendocrine tumor of unknown primary. Mol Imaging Radionucl Ther. (2022) 31:139–41. doi: 10.4274/mirt.galenos.2022.64497
168. Ballal S, Yadav MP, Tripathi M, Sahoo RK, Bal C. Survival outcomes in metastatic gastroenteropancreatic neuroendocrine tumor patients receiving concomitant 225Ac-DOTATATE targeted alpha therapy and capecitabine: a real-world scenario management based long-term outcome study. J Nucl Med. (2022) [Epub ahead of print]. doi: 10.2967/jnumed.122.264043
Keywords: actinium-225, neuroendocrine tumor, peptide receptor radionuclide therapy (PRRT), targeted α-particle therapy, SSTR, SSTR antagonist
Citation: Shi M, Jakobsson V, Greifenstein L, Khong P-L, Chen X, Baum RP and Zhang J (2022) Alpha-peptide receptor radionuclide therapy using actinium-225 labeled somatostatin receptor agonists and antagonists. Front. Med. 9:1034315. doi: 10.3389/fmed.2022.1034315
Received: 01 September 2022; Accepted: 21 November 2022;
Published: 07 December 2022.
Edited by:
Maria Picchio, Vita-Salute San Raffaele University, ItalyReviewed by:
Marco Maccauro, Fondazione IRCCS Istituto Nazionale Tumori, ItalyCopyright © 2022 Shi, Jakobsson, Greifenstein, Khong, Chen, Baum and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingjing Zhang, ai56aGFuZ0BudXMuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.