 Laura M. Glenn
Laura M. Glenn Lauren K. Troy1,2,3
Lauren K. Troy1,2,3- 1Department of Respiratory and Sleep Medicine, Royal Prince Alfred Hospital, Camperdown, NSW, Australia
- 2The University of Sydney School of Medicine (Central Clinical School), Sydney, NSW, Australia
- 3National Health and Medical Research Council (NHMRC) Centre of Research Excellence in Pulmonary Fibrosis, Camperdown, NSW, Australia
The multidisciplinary meeting (MDM) has been endorsed in current international consensus guidelines as the gold standard method for diagnosis of interstitial lung disease (ILD). In the absence of an accurate and reliable diagnostic test, the agreement between multidisciplinary meetings has been used as a surrogate marker for diagnostic accuracy. Although the ILD MDM has been shown to improve inter-clinician agreement on ILD diagnosis, result in a change in diagnosis in a significant proportion of patients and reduce unclassifiable diagnoses, the ideal form for an ILD MDM remains unclear, with constitution and processes of ILD MDMs varying greatly around the world. It is likely that this variation of practice contributes to the lack of agreement seen between MDMs, as well as suboptimal diagnostic accuracy. A recent Delphi study has confirmed the essential components required for the operation of an ILD MDM. The ILD MDM is a changing entity, as it incorporates new diagnostic tests and genetic markers, while also adapting in its form in response to the obstacles of the COVID-19 pandemic. The aim of this review was to evaluate the current evidence regarding ILD MDM and their role in the diagnosis of ILD, the practice of ILD MDM around the world, approaches to ILD MDM standardization and future directions to improve diagnostic accuracy in ILD.
Introduction
Interstitial lung disease (ILD) refers to a diverse group of disorders characterized by varying degrees of inflammation and/or fibrosis of the lung parenchyma (1). Due to multiple factors including atypical or overlapping patterns on radiology or histopathology, disease course heterogeneity and rarity of some diseases, the diagnosis of ILD is frequently challenging for clinicians. Despite extensive evaluation, the diagnosis remains unclassifiable in up to 10–20% of cases (2, 3).
Multidisciplinary discussion has been considered the gold standard method for diagnosis of ILDs for the past two decades. The ILD multidisciplinary meeting (MDM) involves dynamic discussion between different subspecialists whereby all available case details are carefully reviewed, and a consensus diagnosis reached. Previously, the findings obtained from surgical lung biopsy were considered the gold standard for ILD diagnosis. The MDM was first recommended to replace histopathology as the gold standard in the 2002 American Thoracic Society (ATS)/European Respiratory Society (ERS) Joint Statement on the Classification of Idiopathic Interstitial Pneumonias (4). This recommendation was re-emphasized in the 2013 update (1) as well as in more recent position statements (5, 6) and clinical practice guidelines for the diagnosis of idiopathic pulmonary fibrosis (7, 8), hypersensitivity pneumonitis (9) and progressive pulmonary fibrosis (8).
Despite strong support for the MDM from expert bodies, there is a paucity of evidence regarding ideal ILD MDM composition. Additionally, reduced access to specialist ILD centers impacts patients and clinicians in many parts of the world. Despite these challenges, it has been demonstrated that a multidisciplinary approach to ILD diagnosis has been widely adopted in routine care settings globally (10). The absence of defined “optimal” features of the ILD MDM has led to a lack of standardization between MDMs, with potential impact on diagnostic integrity and patient outcomes. An evidence-based approach to MDM constituency and processes is clearly a priority, particularly in view of the constantly evolving diagnostic and treatment landscape and the need to integrate new tools and technologies into the paradigm. In this review, we provide an overview of ILD MDMs worldwide, discussing recent developments and highlighting unmet research needs.
Role of the ILD MDM
Interstitial lung diseases place enormous encumbrance on patients, carers, and health care systems. Many patients with ILD are at risk of progressive fibrotic disease, associated with reduced quality of life and premature mortality. Idiopathic pulmonary fibrosis (IPF), the most common fibrotic ILD is almost universally progressive and up to 40% of non-IPF ILD are also observed to progress (11). Both early recognition of the condition and timely initiation of disease-specific therapy are important determinants of outcomes. Antifibrotic therapy to slow disease progression is now standard of care in IPF (12–15), whereas many non-IPF ILDs respond to treatment with immunosuppressive therapy (16). Recent landmark clinical trials such as INBUILD and SENSCIS have also demonstrated efficacy of antifibrotic therapies in non-IPF progressive fibrosing ILDs (17, 18), but there is much still to learn about disease-specific pathogenesis and targetable pathways.
For these reasons, early and accurate diagnosis is critical. ILD MDMs, involving case discussion between health professionals from different specialties to generate a consensus diagnosis for the patient, aim to maximize available clinical data, expertise and therefore, diagnostic accuracy. Traditionally, the ILD MDM is chaired by a respiratory physician with expertise in ILD. Other important contributors include other respiratory physicians, radiologists and histopathologists with expertise in ILD, rheumatologists and immunologists, ILD nurses, trainees, and other health professionals. Availability of resources will dictate the constitution of an MDM at each site. In addition to generation of a consensus ILD diagnosis, the ILD MDM also functions as a forum to consider patient prognosis and management.
Evidence for the practice of ILD MDMs
Since its implementation, a multidisciplinary approach has consistently been associated with higher levels of diagnostic confidence, better inter-observer agreement and lower rates of unclassifiable diagnoses—considered surrogate markers for diagnostic accuracy (19, 20). For example, Flaherty et al. (21) demonstrated significant improvements in inter-observer agreement and diagnostic confidence following multidisciplinary discussion between expert clinicians, radiologists and pathologists reviewing 58 cases of suspected idiopathic interstitial pneumonia, compared with each individual MDM participant working separately. These findings were replicated in another European study showing far superior levels of diagnostic agreement between local ILD MDMs and expert radiology and tissue pathology panels compared to individual specialties working apart (22).
An international study of IPF diagnosis by 34 expert ILD physicians and 370 non-expert respiratory physicians showed inter-observer agreement to be higher among expert physicians (Cohen's weighted kappa coefficient [κw] = 0.65, IQR 0.53–0.72) than non-expert physicians (κw = 0.53, IQR 0.41–0.63) and physicians without access to an ILD MDM demonstrated the lowest rate of inter-observer agreement (κw = 0.46, IQR 0.33–0.58). Importantly, association with an academic hospital or university, longer duration of ILD experience and access to an ILD MDM were all independently associated with greater prognostic accuracy of IPF diagnosis, thus demonstrating the clinical benefit of having experienced physicians present at the ILD MDM and training of non-experienced clinicians (23).
An Australian analysis of the clinical impact of the ILD MDM showed that multidisciplinary discussion resulted in a change in diagnosis in 53% of ninety consecutive patients with suspected ILD presenting to two specialist ILD centers (24). Importantly, there was a significant reduction in unclassifiable ILD diagnoses; with 42% of patients initially diagnosed with unclassifiable ILD by their referring physicians, and only 12% remaining unclassifiable following MDM discussion. These findings have been replicated in subsequent larger studies, which have also shown a trend toward greater prognostic separation for an MDM ILD diagnosis compared with pre-MDM individual clinician or radiologist diagnosis (25, 26).
Data that MDMs change therapeutic practice is limited. However, ILD MDMs have been shown to increase recommendations for antifibrotic therapy, steroid-sparing immunosuppressive agents, pulmonary vasodilators, clinical trial participation and supplemental oxygen prescription (24, 27).
Variability of multidisciplinary meetings around the world
Inter-MDM heterogeneity
Although this multidisciplinary approach to ILD diagnosis has been widely adopted, significant heterogeneity exists with regards to the structure and processes of ILD MDMs. The inconsistent diagnostic concordance between different MDMs may in-part reflect varying approaches to clinical decision-making or other MDM factors.
The largest multicenter evaluation of diagnostic concordance between different ILD MDMs to date was conducted by Walsh et al. in 2015 (26). Diagnostic concordance between seven ILD MDMs (each consisting of at least one clinician, radiologist, and pathologist) in Europe and the United Kingdom, was evaluated; based on sequential review of 70 patients presenting to an ILD expert center. Inter-MDM for first choice diagnoses overall was only moderate (κ = 0.50). Inter-MDM agreement was better for IPF [κw = 0.71 (IQR 0.64–0.77)] and connective tissue disease-associated ILD (CTD-ILD) [κw = 0.72 (0.68–0.78)]; however, only moderate for non-specific interstitial pneumonia (NSIP) [κw = 0.42 (0.37–0.49)], and fair for hypersensitivity pneumonitis (HP) [κw = 0.29 (0.24–0.40)].
Various reasons have been proposed as to why such discordance exists (19, 20, 28–33). Firstly, the intrinsic heterogeneity of individual ILDs and/or overlapping clinical, radiological, or histopathological features naturally results in difficulty distinguishing specific diagnoses. For example, chronic HP may also be associated with a UIP pattern on high-resolution computed tomography (HRCT), often making it difficult to distinguish from IPF. Secondly, differing approaches to interpretation of international clinical practice guidelines have been observed; with some ILD MDMs adhering more strictly to guidelines and others more likely to assign pragmatic clinical diagnoses with a view to facilitating treatment. Differences between individual physicians, including with regards to training and exposure to ILD logically impact on their diagnostic processes.
Importantly, factors relating to MDM structure, organization and administration, governance and clinical decision-making processes are likely to differ between MDMs. Jo et al. (34) surveyed ten expert ILD centers in Europe, North America and Australia; and identified significant differences in MDM constitution. Specifically, attendance by various specialists such as rheumatologists, quantity and method of data presentation and approach to formulation of diagnosis varied considerably between centers.
A global perspective of the ILD MDM
An evaluation of 457 centers across 64 countries in Europe, the Asia-Pacific region, North America, South and Latin America, Middle East and Africa performed between 2016 and 2017 via electronic questionnaires showed 79.6% held formal ILD MDMs to discuss patient diagnosis and management (10). However, the composition of the MDMs was heterogeneous. For example, centers in lower-income healthcare settings including Brazil, Russia, India, and China were more likely to discuss new cases at an ILD MDM than other countries, however held fewer formal meetings (median 50% compared with 80%). Responses from these countries were more likely to be from academic ILD centers than non-specialist centers, except for India which had a higher proportion of non-academic centers (52.6%).
Centers in high-income countries were more likely to hold meetings at least every 2 weeks (66.8 vs. 53.3%), hold solely face-to-face meetings (83.6 vs. 73.3%), have meetings of between 31 and 60 min duration (52.4 vs. 33.3%) and have at least four disciplines in attendance (60.8 vs. 33.3%) compared with participating centers in lower-middle income countries (10). Although low-income countries were under-represented in this study, it is fair to conclude that variability in MDM processes across the globe is likely at least in part related to reduced access to resources and multidisciplinary expertise in remote and poorer settings (35). A survey of 455 physicians managing IPF patients in Latin America published in 2018 showed that only 27.8% reported access to pathologists, 39.4% to radiologists and a mere 26.9% to multidisciplinary teams (36).
Diagnosing connective tissue disease-associated ILD (CTD-ILD) at ILD-MDM
Although up to 20% of ILDs are associated with underlying CTDs such as systemic sclerosis, rheumatoid arthritis and idiopathic inflammatory myopathies, rheumatologists and immunologists are not routinely involved in ILD MDMs (37, 38). In fact, an evaluation of global MDM practices showed only 37.1% of survey centers routinely involved a rheumatologist in ILD MDM discussions (10), and rheumatology opinion was otherwise only sought if the referring clinician suspected a systemic autoimmune disease based on their own assessment. The implications of this approach include the potential to miss CTD diagnoses, resulting in delayed institution of immunosuppressive treatments that might reverse disease and prevent irreversible lung fibrosis. Since rheumatologists or immunologists are not always accessible in every setting, it is somewhat reassuring the CTD-ILD is more reliably diagnosed at MDM than some other ILDs such as chronic hypersensitivity pneumonitis (HP) (26).
A recent observational study from a large Italian ILD expert center analyzed consecutive ILD MDM cases with suspected new diagnosis or progression of CTD-ILD, concluding that involvement of a rheumatologist in the MDD resulted in availability of more comprehensive clinical information and improved diagnostic accuracy (37).
Another recent Italian study involving a Delphi survey and additional questionnaires distributed to pulmonologists, rheumatologists and radiologists demonstrated high levels of agreement regarding the importance of a collaborative approach to diagnosis of CTD-ILD (38). Results were used to generate checklists of important “red flag” signs and symptoms suggestive of ILD in CTD patients, as well important “red flag” signs and symptoms suggestive of CTD in undifferentiated ILD patients. Importantly, the Delphi survey also addressed potential methods to improve recognition of CTD-ILD where rheumatologists are not present at the ILD MDM. These included creation of networks and collaborative research efforts between different centers across the country; and development of opportunities for clinicians to participate in multidisciplinary clinics or locoregional ILD MDMs discussing archetypal cases; with the aim of improving evidence-based approach to diagnostic formulation in challenging ILDs (38).
Expert consensus regarding essential features of the ILD MDM
Despite scarcity of supporting evidence, expert panels have suggested approaches to ILD MDM organization in attempts to provide a standard framework for universal implementation (5). As a minimum standard, these have recommended attendance by at least two respiratory physicians in addition to a radiologist and tissue pathologist, recognizing that this is clearly not feasible in every setting. Other key components, including core data inputs and outputs for each case are outlined in Table 1. Importantly, consensus should be achieved on whether there is a need for invasive testing with lung biopsy for each case (7, 8). Additionally, international consensus guidelines on standardized diagnostic ontology for fibrotic ILDs have also recommended classification of expected disease behavior to help inform patient prognosis, treatment goals and future monitoring strategy (1, 5, 28).

Table 1. Key data inputs and outputs for each case discussed at the ILD MDM, based on international expert consensus guidelines.
An example of a standardized framework for the ILD MDM is the 2017 Thoracic Society of Australia and New Zealand and the Lung Foundation Australia Position Statement on the ILD MDM and accompanying ILD toolkit, consisting of practical material designed to aid in the presentation and discussion of cases at ILD MDMs (5, 41)1 This included suggested template formats for presentation of case data, suggested sequence for case discussion and recommended standard nomenclature. The clinical impact of this toolkit is currently unknown.
It is also worth noting the proliferation of interstitial lung disease registries worldwide in the last 20 years, such as the Australasian ILD Registry (AILDR) (42), the Pulmonary Fibrosis Foundation Patient Registry (PFF-PR) (43), Canadian Registry for Pulmonary Fibrosis (CARE-PF) (44), and many others, including in developing countries (45–47). These important research repositories have provided a standardized platform for documentation of ILD MDM outcomes by participating centers, which will help to facilitate future collaborative research into ILD diagnostic and treatment pathways.
Worldwide standardization of the ILD MDM will require an understanding of factors contributing to diagnostic heterogeneity as well as consensus regarding the purpose and desired outcomes of multidisciplinary discussion. Recently, Teoh et al. (48) conducted informative research into the key components of an ILD MDM, through semi-structured interviews and subsequent Delphi surveys among ILD physicians across nine countries.
Experts strongly agreed upon five essential features for an ILD MDM, including: 1) the need for at least one radiologist, 2) high-quality HRCT images for each case, 3) technological infrastructure enabling real-time viewing of CT scans, 4) a quiet environment enabling uninterrupted, free-flowing discussion and 5) a standardized template for documentation of outputs from each case discussion. Additionally, experts agreed upon several other desirable components as outlined in Table 2. The need for validation of MDM processes following the genesis of robust evidence was identified as a priority.

Table 2. Essential and desirable features of the ideal ILD MDM based on expert consensusa.
Many questions remain. Notably, there was no consensus agreement upon the need for attendees from other specialty types such as rheumatology or immunology; and although a histopathologist was felt to be “highly desirable”, their presence was not considered essential. This likely reflects limited access, particularly in smaller and more remote centers. The authors also noted that viability is a concern, with ongoing increases in the workload of ILD MDM clinicians, radiologists, and pathologists, limiting time and resources available to dedicate to consideration of each available case. Of note, the pathologist is a key contributor in only a small fraction of cases where a biopsy has been performed. Additionally, in this modern era, lack of onsite pathology does not preclude involvement in the MDM as tissue pathologists can participate virtually even if a center does not have an onsite pathologist.
Interestingly, ILD experts agreed that research terminologies such as IPAF could also be documented as present by consensus at MDM. Although this term has not been validated as a distinct diagnostic entity, its use may be appropriate where the ILD MDM's favored diagnosis is suspected CTD-ILD, rather than idiopathic interstitial pneumonia, but the patient doesn't satisfy CTD diagnostic criteria. However, further studies using refined IPAF criteria are likely required before IPAF can be considered a distinct diagnosis and to inform evidence-based management of affected patients.
The ideal format of an ILD MDM also remains uncertain—whether meeting entirely face-to-face, virtually via videoconferencing or web-based platforms, or a hybrid model, is best. Nonetheless, these consensus agreements on the ideal features of an ILD MDM are fundamental in informing future research into MDM standardization.
Recent developments
Recent scientific and technological developments have had significant impact on both the inputs and outputs of the ILD MDM. Expanded therapeutic options and an increasing array of clinical trials have added to the complexities of management. Furthermore, the global pandemic has necessarily transformed the essence of human interaction, particularly in the healthcare setting. Some key recent influences on the ILD MDM are considered below.
Availability of antifibrotic therapies for IPF and progressive pulmonary fibrosis (PPF)
Evolution of clinical practice guidelines for diagnosis of ILDs and the availability of the antifibrotic drugs nintedanib and pirfenidone have been associated with increased frequency and complexity of referrals to ILD specialist centers for MDM consideration (49). Changes in patterns of MDM behavior have also been observed, particularly in parts of the world where an MDM diagnosis is required by regulatory prescribing bodies to access antifibrotic therapy.
In these settings, it can be tempting to label patients as having IPF or PPF despite an alternative leading differential diagnosis in order to overcome the limitations of regulatory prescribing. Although this is a pragmatic strategy, it is worth acknowledging the risks associated with such practice. With respect to the “PPF” (or progressive fibrosing ILD “PF-ILD”) label in centers where antifibrotics are available for this indication, “lumping” a heterogeneous group of patients together, rather than “splitting” into specific diagnostic groups risks limiting opportunities to identify inflammation-driven disease activity or other causes of deterioration. International guidelines emphasize the treatment for PPF must be agnostic to the underlying condition and all efforts must be made to procure and treat a leading diagnosis before labeling it as progressive (8).
Emergence of transbronchial lung cryobiopsy (TBLC)
TBLC has recently been demonstrated in both clinical trials and meta-analyses to be a reliable method for lung tissue sampling for histopathologic assessment and MDM diagnosis in ILD; a less invasive procedure with lower rates of complications such as pneumothorax and airway bleeding compared with traditional surgical lung biopsy although the quality of evidence is low (50–52). The landmark COLDICE trial demonstrated high levels of diagnostic concordance between TBLC and surgical lung biopsy ILD multidisciplinary discussions; and TBLC MDM diagnoses made with high confidence were even more reliable, showing excellent (95%) concordance with surgical lung biopsy MDM diagnosis (52).
The recently updated ATS/ERS/JRS/ALAT clinical practice guideline for IPF and PPF (8) includes a conditional recommendation to regard TBLC as an acceptable alternative to surgical lung biopsy in centers with appropriate expertise. The 2022 ERS Guidelines on TBLC in the diagnosis of interstitial lung diseases recommend TBLC as either a replacement first test in patients considered eligible for surgical lung biopsy (SLB); or as an option for patients considered unsuitable for SLB (53). As such, consideration of TBLC is likely to become more widely adopted into the ILD MDM diagnostic paradigm.
Novel diagnostic tools
There has been significant recent research interest into novel tools to increase diagnostic yield and accuracy in ILD diagnosis, including the use of genomic classifier testing (54).
Importantly, there is a growing understanding of pathogenic mutations linked to an inherited risk of pulmonary fibrosis. Mutations in telomere-related genes such as telomerase reverse transcriptase (TERT) have been identified in up to one-quarter of familial pulmonary fibrosis patients and have been associated with the “short telomere syndrome”, predisposing affected individuals to progressive ILD, premature hair graying, cryptogenic liver cirrhosis, hematological abnormalities, and reduced survival (55, 56). Despite variable pulmonary fibrosis phenotypes amongst patients with telomere-related gene mutations, affected individuals have been shown to experience consistently progressive disease, more rapid lung function decline and worse transplant-free survival than unaffected individuals (56). Rare variants in genes affecting surfactant metabolism in familial pulmonary fibrosis patients are also associated with an increased risk of lung adenocarcinoma (56). In addition to the prognostic implications, short telomere length has been associated with worse outcomes in patients treated with immunosuppressive medications and similar adverse outcomes have also been shown in patients with sporadic pathogenic mutations in telomere-related genes without a family history (56).
Integration of genetic data into the ILD MDM is yet to be validated, yet research is underway. A recent international survey including 352 respiratory physicians demonstrated support for the use of genetic testing in ILD, predominantly in view of its impact on diagnostic investigation approach and patient treatment, however 88% identified a need for more information on its role and interpretation of results (57).
Currently, genomic testing might be considered within the MDM for patients with a family history or atypical disease presentation, to help predict expected disease behavior and guide management discussions. For example, patients with inherited or sporadic ILD-associated genetic mutations might be recommended for earlier consideration of lung transplant referral, and avoidance or caution with immunosuppressive therapy. It could also be recommended with a view to potential clinical trial participation, such as current phase II studies into the use of the androgen danazol for treatment of familial pulmonary fibrosis associated with short telomere length (58). Additionally, genetic testing results have important implications for screening of family members of ILD patients. It is likely that genetic data is close to being implemented as an important consideration within the ILD MDM and will be used more widely in the future as access to testing and knowledge improves, and evidence-based guidelines become available.
The EnvisiaTM genomic classifier, which employs a machine learning algorithm developed to classify usual interstitial pneumonia (UIP) vs. non-UIP ILD patterns, uses bulk RNA-sequencing data obtained from high throughput sequencing of exome-enriched RNA extracted from transbronchial lung biopsies or TBLC (59). Numerous studies have demonstrated high specificity performance of the classifier to predict UIP in fibrotic ILD (60–63), however its sensitivity for UIP is only 68% and so many cases will still require lung tissue sampling (54). Kheir et al. (64) assessed the impact of sequentially presented data from TBLC and genomic classifier results on the diagnostic confidence of ILD MDMs. The classifier increased diagnostic confidence when added to TBLC for patients with a probable UIP pattern; although did not impact as significantly on the proportion of high-confidence diagnoses as did the addition of the TBLC result. The quality of available evidence was rated as low (54). As such, recent guidelines have made no recommendations either for or against the use of genomic classifier testing in clinical practice (8).
Other novel diagnostic methods with significant potential include the use of artificial intelligence and computer-based deep learning algorithms for the assessment of HRCT images and digital histology slides (65–67). As with genomic biomarkers, future controlled studies are required before these techniques are ready for widespread adoption into clinical practice and integration into the ILD MDM process.
Use of video and web-based technologies during the COVID-19 pandemic
The COVID-19 pandemic has dramatically impacted ILD services globally, with substantial disruption to usual clinician-patient interactions, access to diagnostic procedures and testing and face-to-face ILD MDMs. Due to social distancing requirements and widespread illness, many centers have adopted either entirely virtual MDMs or hybrid in-person and virtual MDMs, with some participants present via teleconferencing (68). Some limitations of this format of course exist, including potential technical issues and reduced participation by some attendees due to inherent differences between individuals and unfamiliarity or discomfort with the virtual format. However, it has generally been useful to allow continuation of ILD MDM service provision at both ILD expert centers and remote centers during the pandemic. Future studies are underway to inform the utility of virtual MDMs compared with conventional MDMs.
An innovative approach involving the use of a national cloud-based database integrating clinical, radiological and pathological data for ILD patients along with a web-based ILD MDM system was explored in Japan (69). Web-based MDMs were conducted with attendant pulmonologists, radiologists and pathologists for 465 cases of biopsy-proven IIPs. Web-based multidisciplinary discussion resulted in a change in diagnosis for 47% of patients, and improved prognostic discrimination between the pre-MDD and MDD diagnoses. Importantly, 5% of patients were diagnosed with non-idiopathic ILDs by MDD; and a substantial proportion of patients were identified as fulfilling IPAF criteria; with meaningful implications for patient management (69). Although the apparent feasibility of this system is promising, particularly in settings with limited access to ILD expertise, non-real time discussion of retrospective data by clinicians who have not physically reviewed the ILD patient likely limits the availability of important clinical data necessary for a precise diagnosis.
Suggested approach
Until additional evidence exists regarding the optimal ILD MDM, we suggest a multidisciplinary approach to ILD diagnosis as outlined in Figure 1; an approach adapted from the TSANZ/LFA 2017 position statement (5) and supported by preliminary data. Structure and coordination of the MDM should be based on current international expert consensus, as outlined in Table 2. Although the presence of at least one radiologist in addition to respiratory physicians is considered essential, other specialists' attendance will be dictated by availability. Ultimately, the ideal ILD MDM is comprised of a cohort of interested, enthusiastic individuals, since it should also be a learning environment in addition to its other functions. Uptake of the ILD MDM is essential to address current needs as well as train future ILD clinicians by propagating knowledge and expertise.
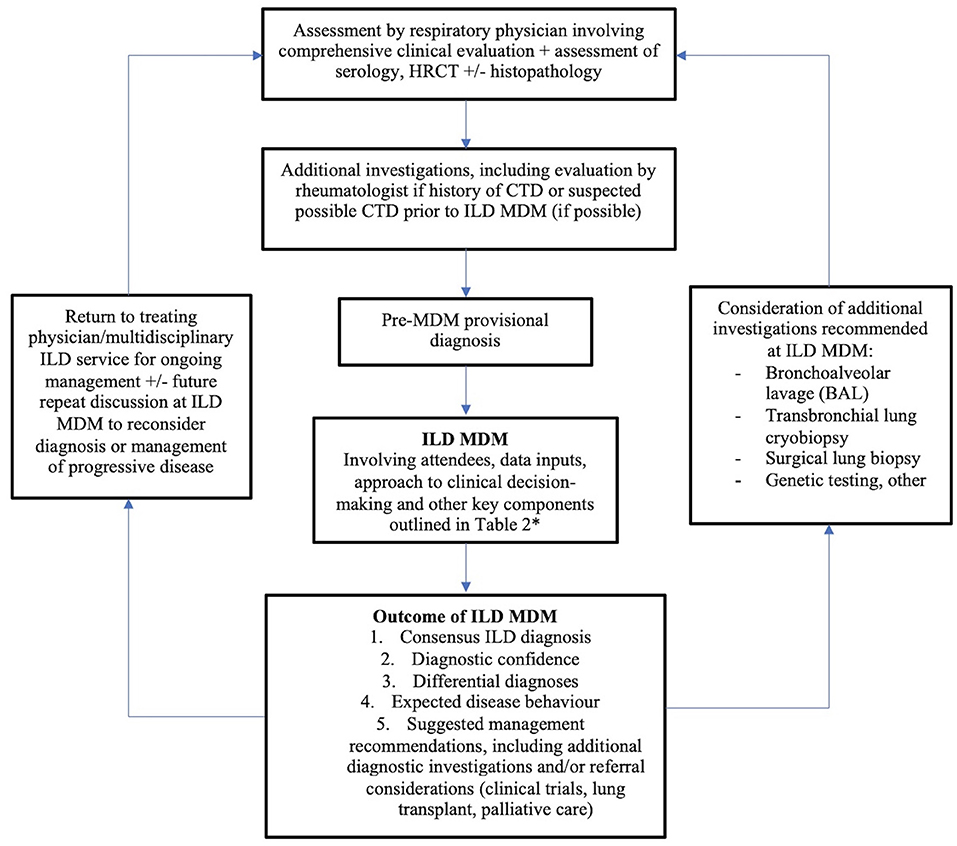
Figure 1. The role of the ILD MDM in diagnosis and ongoing clinical carea. aAdapted from Prasad et al. (5). *Plus attendance by rheumatologist and/or immunologist where available.
The treating physician should consider re-presentation of patient cases at the ILD where disease evolution or results of additional investigations are likely to result in a change in diagnosis; or to obtain consensus agreement upon management of progressive disease.
Conclusions and future directions
The MDM has an integral role in the diagnosis of ILD, with considerable implications for patient management and future outcomes. Therefore, it is critical that the optimal structure, processes, and governance of the ILD MDM are optimized and validated; with a view to standardization of the ILD MDM worldwide. Additional future research priorities will include the integration of novel diagnostic techniques such as genetic and molecular biomarker data for use within the ILD MDM, including consideration of their implication for personalized treatment approaches for ILD patients. The role of regional, national, or transnational ILD MDMs in order to standardize and improve ILD expertise and enhance access to multidisciplinary discussion should also be considered.
Author contributions
LG wrote the manuscript, with input from LT and TC. All authors reviewed the manuscript and agree with regard to the contents.
Funding
This research is supported by a Lung Foundation Australia scholarship, with matched funding provided by the University of Sydney (Lung Foundation Australia/Diana Cox Idiopathic Pulmonary Fibrosis Ph.D. Scholarship 2019).
Conflict of interest
LG has received travel and conference support from Boehringer Ingelheim. LT has provided paid consultancy for Erbe Elektromedezin and Boehringer Ingelheim. TC has received grant support, consultancy fees, and speaking honoraria from Boehringer Ingelheim and Hoffman-La Roche, consultancy fees from Bristol Myers Squibb; grant support from Biogen, and provides consultancy for DevPro and Ad Alta.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American thoracic society/European society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2013) 188:733–48. doi: 10.1164/rccm.201308-1483ST
2. Troy L, Glaspole I, Goh N, Zappala C, Hopkins P, Wilsher M, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. (2014) 43:1529–30. doi: 10.1183/09031936.00003414
3. Ryerson CJ, Corte TJ, Myers JL, Walsh SLF, Guler SA. A contemporary practical approach to the multidisciplinary management of unclassifiable interstitial lung disease. Eur Respir J. (2021) 58:2100276. doi: 10.1183/13993003.00276-2021
4. American Thoracic Society, European Respiratory Society. American thoracic society/European respiratory society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2002) 165:277–304. doi: 10.1164/ajrccm.165.2.ats01
5. Prasad JD, Mahar A, Bleasel J, Ellis SJ, Chambers DC, Lake F, et al. The interstitial lung disease multidisciplinary meeting: a position statement from the thoracic society of Australia and New Zealand and the Lung Foundation Australia. Respirology. (2017) 22:1459–72. doi: 10.1111/resp.13163
6. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a fleischner society white paper. Lancet Respir Med. (2018) 6:138–53. doi: 10.1016/S2213-2600(17)30433-2
7. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
8. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
9. Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vaskova M, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2020) 202:e36–69. doi: 10.1164/rccm.202005-2032ST
10. Richeldi L, Launders N, Martinez F, Walsh SLF, Myers J, Wang B, et al. The characterization of interstitial lung disease multidisciplinary team meetings: a global study. ERJ Open Res. (2019) 5:00209–2018. doi: 10.1183/23120541.00209-2018
11. Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med. (2020) 383:958–68. doi: 10.1056/NEJMra2005230
12. Anstrom KJ, King TE, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 366:1968–77. doi: 10.1056/NEJMoa1113354
13. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
14. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. (2011) 377:1760–9. doi: 10.1016/S0140-6736(11)60405-4
15. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosi. N Engl J Med. (2014) 370:2083–92. doi: 10.1056/NEJMoa1402582
16. Jee AS, Corte TJ. Current and emerging drug therapies for connective tissue disease-interstitial lung disease (CTD-ILD). Drugs. (2019) 79:1511–28. doi: 10.1007/s40265-019-01178-x
17. Flaherty K, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
18. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. (2019) 380:2518–28. doi: 10.1056/NEJMoa1903076
19. Walsh SLF. Multidisciplinary evaluation of interstitial lung diseases: current insights. Eur Respir Rev. (2017) 26:17002. doi: 10.1183/16000617.0002-2017
20. Furini F, Carnevale A, Casoni GL, Guerrini G, Cavagna L, Govoni M, et al. The role of the multidisciplinary evaluation of interstitial lung diseases: systematic literature review of the current evidence and future perspectives. Front Med. (2019) 6:246. doi: 10.3389/fmed.2019.00246
21. Flaherty KR, King TE, Raghu G, Lynch JP, Colby TV, Travis WD, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. (2004) 170:904–10. doi: 10.1164/rccm.200402-147OC
22. Thomeer M, Demedts M, Behr J, Buhl R, Costabel U, Flower CDR, et al. Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. (2008) 31:585–91. doi: 10.1183/09031936.00063706
23. Walsh SLF, Maher TM, Kolb M, Poletti V, Nusser R, Richeldi L, et al. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J. (2017) 50:1700396. doi: 10.1183/13993003.00936-2017
24. Jo HE, Glaspole IN, Levin KC, McCormack SR, Mahar AM, Cooper WA, et al. Clinical impact of the interstitial lung disease multidisciplinary service. Respirology. (2016) 21:1438–44. doi: 10.1111/resp.12850
25. De Sadeleer LJ, Meert C, Yserbyt J, Slabbynck H, Verschakelen JA, Verbeken EK, et al. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases: a retrospective observational study of 938 cases. Chest. (2018) 153:1416–23. doi: 10.1016/j.chest.2018.03.026
26. Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, Dubini A, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis of parenchymal lung disease: a case-cohort study. Lancet Respir Med. (2016) 4:557–65. doi: 10.1016/S2213-2600(16)30033-9
27. Grewal JS, Morisset J, Fisher JH, Churg AM, Bilawich A, Ellis J, et al. Role of a regional multidisciplinary conference in the diagnosis of interstitial lung disease. Ann Am Thorac Soc. (2019) 16:455–62. doi: 10.1513/AnnalsATS.201811-794OC
28. Ryerson CJ, Corte TJ, Lee JS, Richeldi L, Walsh SLF, Myers JL, et al. A standardized diagnostic ontology for fibrotic interstitial lung disease. An international working group perspective. Am J Respir Crit Care Med. (2017) 196:1249–54. doi: 10.1164/rccm.201702-0400PP
29. Moodley Y. A standardized diagnostic ontology for fibrotic interstitial lung disease. Am J Respir Crit Care Med. (2018) 197:1365–6. doi: 10.1164/rccm.201711-2299LE
30. Ryerson CJ, Walsh SLF, Collard HR. Reply to moodley: a standardized diagnostic ontology for fibrotic interstitial lung disease. Am J Respir Crit Care Med. (2018) 197:1366–7. doi: 10.1164/rccm.201712-2515LE
31. George PM, Spagnolo P, Kreuter M, Altinisik G, Bonifazi M, Martinez FJ, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. (2020) 8:925–34. doi: 10.1016/S2213-2600(20)30355-6
32. Meyer KC. Multidisciplinary discussions and interstitial lung disease diagnosis: how useful is a meeting of the minds? Lancet Respir Med. (2016) 4:529–31. doi: 10.1016/S2213-2600(16)30065-0
33. Brownell R, Moua T, Henry TS, Elicker BM, White D, Vittinghoff E, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax. (2017) 72:424–9. doi: 10.1136/thoraxjnl-2016-209671
34. Jo HE, Corte TJ, Moodley Y, Levin K, Westall G, Hopkins P, et al. Evaluating the interstitial lung disease multidisciplinary meeting: a survey of expert centres. BMC Pulm Med. (2016) 16:22. doi: 10.1186/s12890-016-0179-3
35. Rivera-Ortega P, Molina-Molina M. Interstitial lung diseases in developing countries. Ann Glob Health. (2019) 85:4. doi: 10.5334/aogh.2414
36. Cherrez-Ojeda I, Cottin V, Caldéron JC, Delgado C, Calero E, Simanca-Racines D, et al. Management and attitudes about IPF (Idiopathic Pulmonary Fibrosis) among physicians from Latin America. BMC Pulm Med. (2018) 18:5. doi: 10.1186/s12890-017-0569-1
37. De Lorenzis E, Bosello SL, Varone F, Sgalla G, Calandriello L, Natalello G, et al. Multidisciplinary evaluation of interstitial lung diseases: new opportunities linked to rheumatologist involvement. Diagnostics. (2020) 10:664. doi: 10.3390/diagnostics10090664
38. Bosello SL, Beretta L, Del Papa N, Harari S, Palmucci S, Pesci A, et al. Interstitial lung disease associated with autoimmune rheumatic diseases: checklists for clinical practice. Front Med. (2021) 8:732761. doi: 10.3389/fmed.2021.732761
39. Barnes H, Troy L, Lee CT, Sperling A, Strek M, Glaspole I. Hypersensitivity pneumonitis: current concepts in pathogenesis, diagnosis and treatment. Allergy. (2022) 77:442–53. doi: 10.1111/all.15017
40. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European respiratory society/American thoracic society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. (2015) 46:976–87. doi: 10.1183/13993003.00150-2015
41. Lung Foundation Australia. Interstitial Lung Disease Toolkit. (2017). Available online at: https://lungfoundation.com.au/resources/ild-mdm-toolkit-guide/ (accessed August 8, 2022).
42. Moore I, Wrobel J, Rhodes J, Lin Q, Webster S, Jo H, et al. Australasian interstitial lung disease registry (AILDR): objectives, design and rationale of a bi-national prospective database. BMC Pulm Med. (2020) 20:257. doi: 10.1186/s12890-020-01297-2
43. Wang BR, Edwards R, Freiheit EA, Ma Y, Burg C, de Andrade J, et al. The pulmonary fibrosis foundation patient registry. Rationale, design, and methods. Ann Am Thorac Soc. (2020) 17:1620–8. doi: 10.1513/AnnalsATS.202001-035SD
44. Ryerson CJ, Tan B, Fell C, Manganas H, Shapera S, Mittoo S, et al. The Canadian registry for pulmonary fibrosis: design and rationale of a national pulmonary fibrosis registry. Can Respir J. (2016) 2016:3562923. doi: 10.1155/2016/3562923
45. Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, et al. Interstitial lung disease in India: results of a prospective registry. Am J Respir Crit Care Med. (2017) 195:801–13. doi: 10.1164/rccm.201607-1484OC
46. Mohan BVM, Tousheed SZ, Manjunath PH, Ravichandra MR, Ranganatha R, Annapandian VM, et al. Multidisciplinary team obviates biopsy in most patients with diffuse parenchymal lung diseases: a retrospective study from India. Clin Respir J. (2021) 15:761–9. doi: 10.1111/crj.13358
47. Madan K, Hadda V, Mohan A, Guleria R. The ILD-India registry: look before you leap. Am J Respir Crit Care Med. (2017) 195:836–7. doi: 10.1164/rccm.201610-2099LE
48. Teoh AKY, Holland AE, Morisset J, Flaherty KR, Wells AU, Walsh SLF, et al. Essential features of an interstitial lung disease multidisciplinary meeting: an international Delphi survey. Ann Am Thorac Soc. (2022) 19:66–73. doi: 10.1513/AnnalsATS.202011-1421OC
49. Chaudhuri N, Leonard C. Beware weakening the ivory tower of MDT diagnosis in interstitial lung disease. J Clin Med. (2019) 8:1964. doi: 10.3390/jcm8111964
50. Kheir F, Becerra JPU, Bissell B, Ghazipura M, Herman D, Hon SM, et al. Transbronchial lung cryobiopsy in patients with interstitial lung disease: a systematic review. Ann Am Thorac Soc. (2022) 19:1193–202. doi: 10.1513/AnnalsATS.202102-198OC
51. Zayed Y, Alzghoul BN, Hyde R, Wadood Z, Banifadel M, Khasawneh M, et al. Role of transbronchial lung cyrobiopsy in the diagnosis of interstitial lung disease: a meta-analysis of 68 studies and 6300 patients. J Bronchol Interv Pulmonol. (2022) 10:1097. doi: 10.1097/LBR.0000000000000865
52. Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA, et al. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir Med. (2020) 8:171–81. doi: 10.1016/S2213-2600(19)30342-X
53. Korevaar DA, Colella S, Fally M, Camuset J, Colby TV, Hagmeyer L, et al. European respiratory society guidelines on transbronchial lung cryobiopsy in the diagnosis of interstitial lung diseases. Eur Respir J. (2022) 22:425. doi: 10.1183/13993003.00425-2022
54. Kheir F, Becerra JPU, Bissell B, Ghazipura M, Herman D, Hon SM, et al. Use of a genomic classifier in patients with interstitial lung disease: a systematic review and meta-analysis. Ann Am Thorac Soc. (2022) 19:827–32. doi: 10.1513/AnnalsATS.202102-197OC
55. Garcia CK. Insights from human genetic studies of lung and organ fibrosis. J Clin Invest. (2018) 128:36–44. doi: 10.1172/JCI93556
56. Zhang D, Newton CA. Familial pulmonary fibrosis: genetic features and clinical implications. Chest. (2021) 160:1764–73. doi: 10.1016/j.chest.2021.06.037
57. Teriwel M, Borie R, Crestani B, Galvin L, Bonella F, Fabre A, et al. Genetic testing in interstitial lung disease: an international survey. Respirology. (2022) 27:747–57. doi: 10.1111/resp.14303
58. Mackintosh JA, Pietsch M, Lutzky V, Enever D, Bancroft S, Apte SH, et al. TELO-SCOPE study: a randomised, double-blind, placebo-controlled, phase 2 trial of danazol for short telomere related pulmonary fibrosis. BMJ Open Respir Res. (2021) 8:e001127. doi: 10.1136/bmjresp-2021-001127
59. Kim SY, Diggans J, Pankratz D, Huang J, Pagan M, Sindy N, et al. Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high-dimensional transcriptional data. Lancet Respir Med. (2015) 3:473–82. doi: 10.1016/S2213-2600(15)00140-X
60. Pankratz DG, Choi Y, Imtiaz U, Fedorowicz GM, Anderson JD, Colby TV, et al. Usual interstitial pneumonia can be detected in transbronchial biopsies using machine learning. Ann Am Thorac Soc. (2017) 14:1646–54. doi: 10.1513/AnnalsATS.201612-947OC
61. Choi Y, Liu TT, Pankratz DG, Colby TV, Barth NM, Lynch DA, et al. Identification of usual interstitial pneumonia using RNA-Seq and machine learning: challenges and solutions. BMC Genom. (2018) 19:101. doi: 10.1186/s12864-018-4467-6
62. Raghu G, Flaherty KR, Lederer DJ, Lynch DA, Colby TV, Myers JL, et al. Use of a molecular classifer to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med. (2019) 7:487–96. doi: 10.1016/S2213-2600(19)30059-1
63. Richeldi L, Scholand MB, Lynch DA, Colby TV, Myers JL, Groshong SD, et al. Utility of a molecular classifier as a complement to high-resolution computed tomography to identify usual interstitial pneumonia. Am J Respir Crit Care Med. (2021) 23:211–20. doi: 10.1164/rccm.202003-0877OC
64. Kheir F, Alkhatib A, Berry GJ, Daroca P, Diethelm L, Rampolla R, et al. Using bronchoscopic lung cryobiopsy and a genomic classifier in the multidisciplinary diagnosis of diffuse interstitial lung diseases. Chest. (2020) 158:2015–25. doi: 10.1016/j.chest.2020.05.532
65. Soffer S, Morgenthau AS, Shimon O, Barash Y, Konen E, Glicksberg BS, et al. Artificial intelligence for interstitial lung disease analysis on chest computed tomography: a systematic review. Acad Radiol. (2022) 29:S226–35. doi: 10.1016/j.acra.2021.05.014
66. Humphries SM, Mackintosh JA, Jo HE, Walsh SLF, Silva M, Calandriello L, et al. Quantitative computed tomography predicts outcomes in idiopathic pulmonary fibrosis. Respirology. (2022). doi: 10.1111/resp.14333. [Epub ahead of print].
67. Testa LC, Jule Y, Lundh L, Bertotti K, Merideth MA, O'Brien KJ, et al. Automated digital quantification of pulmonary fibrosis in human histopathology specimens. Front Med. (2021) 8:607720. doi: 10.3389/fmed.2021.607720
68. Mackintosh JA, Glenn L, Barnes H, Dunn E, Bancroft S, Reddy T, et al. Benefits of a virtual interstitial lung disease multidisciplinary meeting in the face of COVID-19. Respirology. (2021) 26:612–5. doi: 10.1111/resp.14062
Keywords: interstitial lung disease (ILD), multidisciplinary meeting (MDM), diagnosis, connective tissue disease (CTD), progressive pulmonary fibrosis
Citation: Glenn LM, Troy LK and Corte TJ (2022) Diagnosing interstitial lung disease by multidisciplinary discussion: A review. Front. Med. 9:1017501. doi: 10.3389/fmed.2022.1017501
Received: 12 August 2022; Accepted: 05 September 2022;
Published: 21 September 2022.
Edited by:
Stefano Palmucci, University of Catania, ItalyCopyright © 2022 Glenn, Troy and Corte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura M. Glenn, bGdsZTkwNDFAdW5pLnN5ZG5leS5lZHUuYXU=