 Xi Jiang
Xi Jiang Zhongxiu Jiang
Zhongxiu Jiang Qi Cheng
Qi Cheng Wei Sun
Wei Sun Min Jiang
Min Jiang Yan Sun
Yan Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Med., 23 September 2022
Sec. Gastroenterology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1000563
This article is part of the Research TopicReviews in GastroenterologyView all 10 articles
The incidence and mortality of colorectal cancer (CRC) have been markedly increasing worldwide, causing a tremendous burden to the healthcare system. Therefore, it is crucial to investigate the risk factors and pathogenesis of CRC. Cholecystectomy is a gold standard procedure for treating symptomatic cholelithiasis and gallstone diseases. The rhythm of bile acids entering the intestine is altered after cholecystectomy, which leads to metabolic disorders. Nonetheless, emerging evidence suggests that cholecystectomy might be associated with the development of CRC. It has been reported that alterations in bile acid metabolism and gut microbiota are the two main reasons. However, the potential mechanisms still need to be elucidated. In this review, we mainly discussed how bile acid metabolism, gut microbiota, and the interaction between the two factors influence the development of CRC. Subsequently, we summarized the underlying mechanisms of the alterations in bile acid metabolism after cholecystectomy including cellular level, molecular level, and signaling pathways. The potential mechanisms of the alterations on gut microbiota contain an imbalance of bile acid metabolism, cellular immune abnormality, acid-base imbalance, activation of cancer-related pathways, and induction of toxin, inflammation, and oxidative stress.
Colorectal cancer (CRC) is the third most malignancy worldwide for humans (1–3). The incidence and mortality of CRC are terrifyingly high. CRC accounts for over 9% of all cancers incidence (4). It has been estimated that approximately 53,200 deaths projected in 2020 (5) and 3.2 million new CRC cases projected in 2040 (6). CRC exerts a significant geographic difference, more common in the western developed countries (7–9). The incidence rate was 10-fold higher in the highest rate countries than that in the lowest rate countries (10). The incidence of CRC in China is 23.3 per 100,000 (11). In addition, the prevalence of CRC has been growing in the young individuals (12–14), contributing to substantial social and economic burden to the healthcare system (2, 6). Therefore, it is essential to explore the risk factors and pathogenesis of CRC.
Several etiologies have been implicated in the pathogenesis of CRC, including genetic susceptibility and environmental factors, such as consumption of tobacco and alcohol, inflammatory bowel disease (IBD), adenomatous polyps, family history, unhealthy diet, physical inactivity and obesity (8, 15–17). Cholecystectomy is a standard procedure for treatment of symptomatic cholelithiasis and gallstone diseases. The number of this procedure has been increasing. It has been reported that approximately 800,000 cases of cholecystectomy are performed in the United States per year, and the number is also growing in China (18, 19). In the past, cholecystectomy was deemed to be almost harmless. Nevertheless, an increasing body of evidence suggests that cholecystectomy might be associated with the development of CRC (20–25). Alterations of bile acid metabolism and gut microbiota have been demonstrated to play significant roles in CRC. However, the potential specific mechanisms are still unclear. Therefore, in the present study, we described the current knowledge on the association between cholecystectomy and CRC, and summarized the potential mechanisms.
CRC is the third most common cause of cancer-related mortality worldwide (2), and is also the second most common cause of cancer mortality in the United States (26). It has estimated that more than 1.8 million cases were diagnosed and 881,000 deaths occurred in 2018, accounting for 1 in 10 cancer cases and deaths (11). More recently, over 1.9 million new cases were reported in 2020 (27). Globally, CRC incidence and mortality vary widely across countries, according to GLOBOCAN 2020 data (28). The incidence of CRC is higher in males than in females, and the trend is younger in recent years (12–14). In general, the incidence of proximal colon tumors is the highest, while that of distal colon tumors is the lowest, which is more common in the elderly. The incidence rates increased by 1 and 2% each year among the 50–64 age group and under 50 years of age, respectively (26). The population of CRC patients as a whole is rapidly getting younger as the declining incidence in the older population coincides with the increasing incidence in the younger population, causing huge burden on the healthcare system. It has been well-acknowledged that CRC is associated with several risk factors such as smoking, unhealthy diet, alcohol abuse, physical inactivity and obesity (15, 16, 29). Nevertheless, the risk factors and pathogenesis of CRC still need to be further explored.
Cholecystectomy is the most common procedure performed in biliary surgery. In most cases, the procedure is relatively standardized and the long-term results after surgery are satisfactory. Cholecystectomy can be performed in two main ways: a laparoscopic or a classic open operation technique. Compared to the classic way, the laparoscopic cholecystectomy is a relatively minimally invasive surgical procedure and has basically replaced the open technique for cholecystectomies since the early 1990s (30). Several advantages have made it a popular procedure over the past few decades, including a short hospital stay, quick return to normal activities, and reduced pain after surgery, more acceptable cosmetic results, less morbidity and less mortality (31–33). The reported short-term complications include postoperative bleeding, biliary leakage biliary peritonitis, subhepatic effusion or subphrenic abscess, postoperative jaundice, postoperative pancreatitis, residual common bile duct stones, and gastrointestinal fistula, and the long-term complications include bile duct stricture, recurrent common bile duct stones, biliary bleeding, post-cholecystectomy syndrome, residual overgrown bile duct syndrome, and increased incidence of CRC.
A meta-analysis of 10 cohort studies described that there was an increased risk for colon cancer up to 30% higher than the non-cholecystectomized group (24). In addition, the study also observed a positive relationship between the female gender and CRC. Moreover, a previous study confirmed that patients who performed cholecystectomy presented a 108% higher risk of developing CRC than the common population. The male and female patients who underwent cholecystectomy were reported to have a 74 and 154% higher risk of CRC, respectively (23). Furthermore, age was also regarded as a risk factor for gastrointestinal cancers in patients with a history of cholecystectomy (23). The standardized incidence ratio (SIR) was highest in individuals between 40 and 49 years old, followed by those in their ages more than 80 years old (23). The reported median duration from cholecystectomy to the diagnosis of CRC was about 5–15 years or more (34, 35). Additionally, the right colon is more prone to be affected by cholecystectomy. Giovaimucci et al. (34) believed that the proximal and distal colon is related to the different sources of embryos, and they present different sensitivity to carcinogens. The right colon is more sensitive to bile acids, probably due to the high amount of stool fluid in the right colon. Thomas et al. (36) believed that the concentration of secondary bile acids and the activity of 7α-dehydroxylase were higher in the right colon than in the left colon, and there were obvious differences in bile acid metabolism, leading to the susceptibility of CRC in the right colon after cholecystectomy.
Bile acids are the main components of bile and are synthesized in the hepatocytes via cytochrome P450-mediated oxidation of cholesterol (37, 38). This process takes place through two biosynthetic pathways: the “classical” and an “alternative” pathway (39). During the “classical” pathway, three cholesterol hydroxylase enzymes cholesterol 7α-hydroxylase (CYP7A1), sterol 12α-hydroxylase (CYP8B1) and mitochondrial sterol 27-hydroxylase (CYP27A1) produce the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA) (40, 41). The “alternative” pathway produces CDCA via the hydroxylation of the cholesterol side chain by CYP27A1, and the oxysterol intermediates are then formed by 7α-hydroxylation by CYP7B141 (40). Bile acids can be divided into free bile acids and conjugated bile acids according to their structures. Free bile acids include CA, deoxycholic acid (DCA), CDCA, and lithocholic acid (LCA). The free bile acids are combined with glycine or taurine respectively to form various corresponding conjugated bile acids, including glycocholic acid, taurocholic acid, glycochenodeoxycholic acid and taurochenodeoxycholic acid. Conjugated bile acids are more water-soluble and generally exist in the body as sodium salts, which are more stable than free bile acids. In addition, bile acids can be divided into primary and secondary bile acids according to their sources. Primary bile acids are synthesized directly from cholesterol in hepatocytes including CA and CDCA, while secondary bile acids are formed when primary bile acids are secreted into the intestine and undergo 7-α-hydroxylation by intestinal bacteria including DCA, LCA, ursodeoxycholic acid (UDCA), and tauroursodeoxycholic acid (TUDC). Bile acids are secreted through the tubular membrane into the bile and stored in the gallbladder. After the animal eats, the duodenum secretes cholecystokinin, which stimulates gallbladder contraction, thereby releasing bile acids into the small intestine. In the small intestine, conjugated bile acids specifically activate pancreatic lipases and enhance fat-soluble vitamins solubilization by creating mixed micelles of dietary lipids, sterols, and fat-soluble vitamins. Finally, about 95% bile acids are reabsorbed in the ileum and returned to the liver via the portal vein. However, approximately 5% bile acids escaping from intestinal reabsorption enter the colon, where they are further converted to secondary, more hydrophilic bile acids by the intestinal flora (40, 42, 43).
Under normal conditions, the gallbladder controls the rate and flow of bile into the intestine and enterohepatic circulation of bile acids, which plays a key role in regulating physiological homeostasis (44). However, the rhythm of bile acids entering the intestine is altered after cholecystectomy, which leads to metabolic disorders. The normal bile acid pool is the total amount of bile acids in the enterohepatic circulation, which is about 3 g and consists of 50% CA, 30% CDCA, 20% DCA and very small amounts of other bile acids. Previous studies showed that the bile acid reabsorption and enterohepatic circulation increase due to the sphincter of Oddi disorders after cholecystectomy (45, 46). However, there is also small number of studies finding that the bile acid pool decreases or remains unchanged after cholecystectomy. For example, a previous study observed that the bile acid pool decreased by about 16% three months after cholecystectomy (47). Two animal experiments showed that the total bile acid pool decreased by about 40% two weeks after cholecystectomy, as well as the reduction of circadian rhythm (48, 49). In addition, the total amount of bile acids remained essentially unchanged after five to eight years of cholecystectomy (46). The above findings suggest that the size of bile acid pool decreases in the short-term outcome after cholecystectomy, but there is no significant effect on the long-term outcome. Moreover, it has been confirmed that cholecystectomy increases the bacterial uncoupling and dehydroxylation of bile acids, thereby leading to the high proportion of secondary bile acids (44). Zhang et al. (48) demonstrated that the contents of DCA, LCA and their binding products with taurine were significantly increased in the ileum of mice after cholecystectomy, together with the increasing of fecal bile acid.
Numerous experimental studies have confirmed the tumorigenic potential of bile acids, particularly the secondary bile acids DCA and lesser extent of the LCA (50–55). It is important to note that the bile acids are usually considered as tumor promoters rather than tumor inducers, because the changed bile acid concentrations depend on exposure to carcinogenic chemicals or genetic susceptibility (56). The carcinogenic effects of secondary bile acids on CRC are summarized in Table 1.
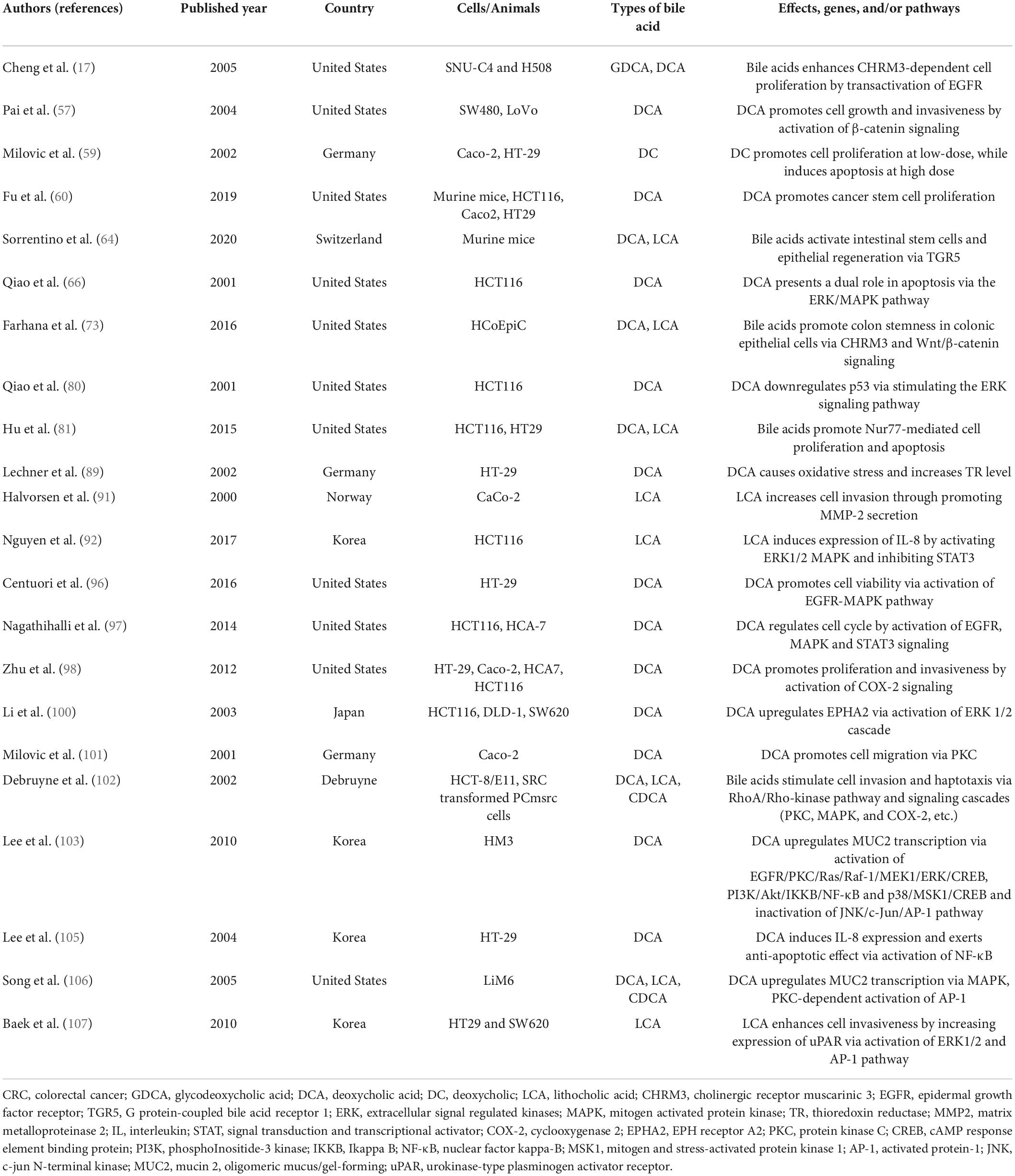
Table 1. The carcinogenic effects of secondary bile acids on CRC.
DCA has been reported to enhance colonic epithelial and colon cancer cell proliferation and/or invasiveness (57–60), promotes dysplasia (61), and disrupts the cell monolayer integrity of intestinal cancer and precancerous cells, increases the production of pro-inflammatory cytokines (51, 62), promotes cell cycle arrest (63), and activate intestinal stem cells and epithelial regeneration (64). In addition, DCA has been demonstrated to inhibit wound healing in wounded colonic epithelial monolayers by impairing cell migration ability (65). Interestingly, DCA exerts pro-apoptotic and anti-apoptotic effects on colon cells (66). It has been revealed that DCA could promote transition from adenoma to carcinoma and resist apoptosis (67), and also induce epithelial-mesenchymal transition (EMT) process, increase vasculogenic mimicry (VM) formation (68). Furthermore, DCA could help cancer cells to escape immune surveillance (69). Besides, DCA was found to cause a redistribution of cholesterol and decrease the fluidity of the membranes (70). As well, previous studies have confirmed that DCA could be converted into a powerful carcinogen 3-methylcholanthrene (3-MC) (71), and also regulate cell junction and increase intestinal permeability (72). Besides, DCA/LCA could increase drug resistance and induce colon carcinogenesis (73).
With respect to the molecular mechanism, DCA and/or LCA was reported to induce expressions of cyclooxygenase (COX)-2 promoter (74) by transactivation of the epidermal growth factor receptor (EGFR) in HCT116, H508 and SNU-C4 human colon cancer cell lines (17, 75), promote the stable and translocated pronucellin entering the nucleus and stimulate the expression of uPA, urokinase-type plasminogen activator receptor (uPAR) and cyclin D1 in SW480 and LoVo cells (57), activate muscarinic receptor (MR) in H508 human colon cancer cells (76), increase the expression of matrix metalloproteases (MMPs) in H508 cells (77), inhibit the effect of microRNA (miR)-199a-5p and/or promote the expression of CDK2 associated cullin domain 1 (CAC1) in HCT-8 cells (78). These above processes were associated with proliferation and invasion of DCA. For the anti-apoptotic characteristics, DCA was shown to upregulate the expression of X-linked inhibitor of apoptosis protein (XIAP) in normal intestinal epithelial cells (IEC-6), while downregulate the expression of p53 in HCT116 cells (79, 80). It is noted that Hu et al. suggested that DCA and/or LCA presented a dual role in modulating cell survival and death by regulating expression of Nur77 and intracellular location in HCT116 and HT29 colon cancer cells (81). Likewise, DCA was revealed to decrease the expression of human leukocyte antigen (HLA) class I antigens on the surface of HT29, SK-CO-l and SW1116 cells to help cancer cells to escape immune surveillance (69). As well, DCA was also described to prompt colonic epithelial cells HCoEpiC into becoming cancer stem cells (CSCs) (73), and form aberrant crypt foci (ACF) and high-grade dysplasia in AKR/J mice (82). Besides, DCA endorses the recruitment of tumor-associated macrophages (TAM) (83), decrease the levels of secretory antibodies of the type IgA (sIgA) and promotes polarization of M2 macrophages in APCmin/+ mice (51). Interestingly, DCA has also been reported to cause genomic instability including heteroploidy, intrachromosomal instability and gene point mutations (84). The genomic instability appears via several mechanisms, comprising DNA oxidative damage, mitochondria damage, endoplasmic reticulum damage, micronucleus rate increase, disruption of mitosis, and mutations of chromosome aneuploidy (85, 86). DCA-induced DNA oxidative damage is caused after long-term exposure to high concentrations of nitro DCA and oxidation, which can induce apoptosis or DNA damage. Long-term DNA damage leads to mutation and natural selection of mutant cells, and ultimately promotes the development of cancer cells (87). Moreover, DCA can cause abnormal functions of some DNA mismatch repair enzymes by inducing mutations, such as adenomatous polyposis coli (APC) and tumor protein p53 (TP53). Subsequently, the dysfunction of DNA mismatch repair causes genome microsatellite instability (88). Long-term exposure to a high concentration of secondary bile acids can generate reactive oxygen species (ROS), induce oxidative stress (89), active nitrogen species, and cause DNA damage in intestinal epithelial cells, leading to genomic instability and increase gene mutations (90). In contrast, LCA has been reported to promote CRC via promoting expression of MMP-2 in CaCo-2 cells (91), interleukin (IL)-8 in HCT116 cells (92), ATP binding cassette subfamily B member 1 (ABCB1), ATP binding cassette subfamily G member 2 (ABCG2) in HCoEpiC cells (73), and miR-21, and inhibition of PTEN in HCT116 cells (93). As well, LCA induces DNA single-strand breaks (94) and inhibits mammalian DNA polymerase β in rat colon epithelial cells (95).
Several signaling pathways have been reported to be involved in the tumor-promoting effect of DCA on CRC. For example, DCA promotes CRC by activation of EGFR-mitogen activated protein kinase (MAPK), and induction of calcium in HT-29 cells (96) and signal transduction and transcriptional activator (STAT) 3 signaling pathways in HCT116 and HCA-7 cells (97). DCR facilitates proliferation and invasiveness through COX-2 in HT-29, Caco-2, HCA7, and HCT116 cells (98) and/or COX-2/prostaglandin E2 (PGE2) signaling pathway in human colonic fibroblasts CCD-18Co cells (99). In addition, DCA and/or LCA has been demonstrated to promote CRC by regulating Wnt/β-catenin signaling in SW480, LoVo, or HCoEpiC cells (57, 73), activation of extracellular signal regulated kinases (ERK) 1/2 cascade in HCT116, DLD-1, and SW620 cells (100), protein kinase C (PKC) in Caco-2 cells (101), RhoA/Rho-kinase pathway in HCT-8/E11 and SRC transformed PCmsrc cells (102), EGFR/PKC/Ras/ERK/cAMP response element binding protein (CREB), phosphoInositide-3 kinase (PI3K)/Akt/IkappaB (IKKB)/nuclear factor kappa-B (NF-κB) and p38/mitogen and stress-activated protein kinase 1 (MSK1)/CREB pathways and inactivates c-jun N-terminal kinase (JNK)/c-Jun/activated protein-1 (AP-1) pathway (103) and p53 pathway (80). Moreover, DCA activates JNK 1/2, and AKT signaling pathways that result in selective resistance to apoptosis, angiogenesis, proliferation and oxidative stress (40, 104). Furthermore, DCA is also reported to activate anti-apoptotic effect of NF-κB and induces IL-8 (105) and to upregulate MUC2 transcription via MAPK, PKC-dependent activation of AP-1 pathway in LiM6 cells (106). Meanwhile, LCA induces expression of uPAR and increases cell invasiveness via activation of ERK1/2 MAPK and AP-1 pathway in HT29 and SW620 cells (107) and inactivation of STAT3 and Src/EGFR pathways in HCT116 cells (92, 108, 109).
The intestinal flora is a great deal number and diversity of microbial species, which is the most significant micro-ecosystem in the human body. It has been estimated that approximately more than 500 species of bacteria from 30 genera exist in healthy adult intestines (110). These bacteria are composed of aerobes, facultative anaerobes and anaerobes, and most of them are obligate anaerobes or facultative anaerobes. Among the bacteria, 90% of the intestinal flora is Bacteroidetes and Firmicutes (111). The intestinal flora is a significant contributor in several physiological activities, such as food residue metabolism, micronutrient synthesis, primary bile acid metabolism, secondary bile acid synthesis, and immune response regulation (112). In addition, these bacteria is able to establish a biological barrier in the gut via space-occupying effect, nutrient competition, and some secreted metabolites (113), which can decrease low-grade inflammatory response in the body and maintain the integrity of the intestinal wall. Motivating the intestine to create an effective immune defense system can modulate the absorption and conversion of sugar and fat in the intestinal tract, subsequently ameliorate glucose tolerance and oxidative stress, and lower blood glucose (114). Therefore, the homeostasis of gut microbiota plays significant roles in maintaining human health (115–118). However, dysbiosis of intestinal flora is involved in a wide range of human diseases. A large of human and animal experiments has confirmed that the dysbiosis of gut microbiota shows cancer-promoting effects on gastrointestinal carcinogenesis, especially CRC (119–126).
It has been well-acknowledged that cholecystectomy induces tremendous changes in the composition and function of the gut microbiota. For example, previous studies confirmed that after cholecystectomy, the number of Bifidobacteria and Lactobacillus was significantly decreased, while the number of Enterococcus, Oscillospira, Escherichia coli, Bacteroidaceae and Bacteroidetes was significantly increased (127–131). A previous study demonstrated that 1 mmol/L of DCA can effectively inhibit the growth of Clostridium perfringens, Bacteroides fragilis, Lactobacillus and Bifidobacterium in the intestinal tract (132). In addition, Cao et al. found that DCA significantly upregulated the populations of opportunistic pathogens, including Ruminococcus, Escherichia-Shigella, Desulfovibrio, and Dorea. Moreover, they also confirmed that DCA significantly increased the levels of Clostridium and Escherichia-Shigella, but markedly decreased the abundance of Lactobacillus_gasseri and mostly butyrate-producing bacteria, such as Clostridium leptum Lachnospiraceae bacterium and Eubaterium coprostanoligenes (83). On the contrary, an animal research showed that the population level of Bacteroides was increased in the ceca of rats fed with DCA (133). The main potential mechanisms include imbalance of bile acid metabolism, cellular immune abnormality, acid-base imbalance, and activation of cancer-related pathways and induction of toxin, inflammation and oxidative stress.
Fibroblast growth factors (FGF) are cellular factors that are synthesized by the terminal epithelial cells of the ileum and are involved in the regulation of bile acid metabolism (134). The FGF19 or FGF15 is transported to the liver through the portal vein system to inhibit bile acid synthesis. A previous study showed that the levels of FGF19 mRNA in the epithelial tissues of the gallbladder were 250 times higher than that in the terminal ileal epithelium (135). After cholecystectomy, the balance of bile acid metabolism is disturbed as the expression of FGF19 decreases and the primary bile acid production increases, altering the bidirectional interaction between bile acid and intestinal flora (128). The continuous drainage of bile into the intestinal lumen continuously stimulates intestinal motility, which increases peristalsis and shortens the total intestinal transit time. The enterohepatic circulation of bile acid is accelerated and the production of secondary bile acids is increased. The hydrophobic nature of secondary bile acids increases their affinity for the phospholipid bilayer of the intestinal bacterial cell membrane, leading to cell membrane damage and bacterial lysis and death (70).
The mucosal epithelium of the gallbladder could synthesize surfactant protein D (SP-D) (136). The SP-D is excreted into the intestinal lumen with bile and facilitates the synthesis of intestinal T cells (137). Intestinal T cells are involved in the regulation of inflammatory responses in the intestine. After cholecystectomy, the lack of SP-D in the gallbladder drastically reduces the number of intestinal T cells and predisposes the intestinal tract to bacterial infection and dysbiosis (137). Gallbladder surface protein D can also inhibit the growth of Lactobacilli in the intestinal tract by directly binding to Lactobacilli to induce lysis of Lactobacilli (137). Although Lactobacillus is beneficial to the human body, its excessive growth can affect the growth of other bacteria in the intestinal tract, thus causing dysbiosis of the intestinal flora.
Small intestinal fluid is weakly alkaline, with a pH value of 8.0–9.0. Normal bile is weakly acidic, and its pulsatile secretion helps to create a good intestinal microbiological environment and maintain a stable pH value in the intestine. After cholecystectomy, alkaline bile is continuously secreted, which affects the pH balance in the intestine. The optimal pH values for the growth of Lactobacilli and Bifidobacteria are 5.5–6.0 and 6.5–7.0, respectively (138). Therefore, the increase in pH in the intestine inhibits the growth of beneficial bacteria such as Lactobacillus and Bifidobacterium, leading to dysbiosis.
Activation of Wnt/β-catenin pathway has been implicated in the development and progression of CRC (139–141). E-cadherin is a well-known tumor suppressor, which can exert its function through β-catenin (142). The correlation between gut bacteria and E-cadherin/β-catenin has been reported. For example, Fusobacterium nucleatum has been demonstrated to attach E-cadherin on epithelial cells via its toxic factor FadA adhesin and stimulate β-catenin signaling pathway, and subsequently induce the gene expression of Wnt pathway (143). Meanwhile, a recent study also confirmed that, in addition to the effect on alteration of gut microbiota, DCA could downregulate the expression of E-cadherin, and increase nuclear β-catenin expression, as well as initiation of the downstream Wnt signaling molecules (83). Furthermore, Fusobacterium nucleatum has been reported to release RNA into the host cell cytoplasm, which could be detected by cytosolic retinoic acid-inducible gene 1 (RIG-1), and then activate NF-κB pathway, ultimately induce the expression of inflammatory genes and oncogenes (144, 145). Besides, FadA has been illustrated to bind to vascular endothelial cadherin (VE-cadherin), causing VE-cadherin to relocate and then increasing the permeability of endothelial cells, which enables Fusobacterium and other bacteria species to enter into the blood stream (146). Peptostreptococcus anaerobius has been identified as a novel microbial promoter of intestinal inflammation and tumor (147, 148). A recent research found that Peptostreptococcus anaerobius could interact with toll-like receptors (TLR)-2 and TLR-4 to motivate the generation of reactive oxidative species (ROS), which can stimulate the biosynthesis of cholesterol, leading to colon cell proliferation and dysplasia in mice (149). Additionally, alternation of bile acid has been revealed to induce the growth of pro-inflammatory bacteria, such as Mogibacterium and Sutterella, which may cause DNA damage and inflammatory response (150). Chronic inflammation could then indorse the event of IBD-associated dysplasia and development of adenoma-carcinoma sequence (151, 152). It has been revealed that Bacteroides fragilis could release bacteroides fragilis toxin (BFT), which activates a pro-carcinogenic multi-step inflammatory cascade through IL-17R, NF-κB and STAT3 pathways in colon epithelial cells (153) and contributes the development of polyp-adenoma-CRC (150). Escherichia coli, Bacteroides fragilis, Providencia ewing, Micromonospora, and Peptostreptococcus anaerobius have been displayed to induce CRC by production of a genotoxin colibactin that could induce DNA damage (154, 155).
Although DCA and LCA present tumor-promoting effects, UDCA is a therapeutic bile acid and has been reported to have a chemopreventive effect based in vitro and in vivo (156–160). Recently, UDCA was demonstrated to reduce the risk for advanced colorectal adenoma (161, 162) and CRC (156, 163). In addition, UDCA could modulate the gut microbiome (162). UDCA can inhibit DCA-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in HCT116 cells (164). Moreover, co-treatment with low-dose celecoxib and UDCA reveals to decrease cell growth in HT-29 colon tumor cells (165). Besides, UDCA inhibits Ras mutations, wild-type Ras activation, and expression of COX-2 in azoxymethane (AOM)-induced colon cancer in rats (166). However, a previous study found that long-term administration of high-dose UDCA was associated with an increased risk of colorectal neoplasia in patients with ulcerative colitis (UC) and primary sclerosing cholangitis (PSC) (167). Currently, there are inadequate data to support the routine application of UDCA for chemoprevention of CRC, either in the common population or among individuals who are at higher risk for CRC.
There are many studies on the pathogenesis of CRC. In this paper, we reviewed the recent studies on the effects of cholecystectomy on CRC (Figure 1). The results show that cholecystectomy might promote the development of CRC by alteration of bile acid metabolism and the gut microbiota. The occurrence of CRC is related to changes in bile acid metabolism, the composition and function of the gut microbiota, and/or the interaction between the two factors. General surgeons should strictly grasp the indications of cholecystectomy. Cholecystectomy is necessary for acute and chronic cholecystitis, symptomatic cholelithiasis, biliary tract movement disorders, non-calculous cholecystitis, gallbladder tumors or polyps, and biliary pancreatitis. Gallbladders with good contractile function should be preserved as much as possible, not blindly removed. However, whether other physiological changes after cholecystectomy are associated with intestinal flora, affecting the occurrence and development of CRC, and whether there is a direct correlation between the carcinogenic effect of secondary bile acids and intestinal microorganisms after cholecystectomy are still unclear and need to be further investigated. With the continuous research on the pathogenesis of secondary bile acids-induced CRC, targeted therapies, including targeted bile acid metabolism and intestinal microflora regulation, may be promising treatment strategies for CRC.
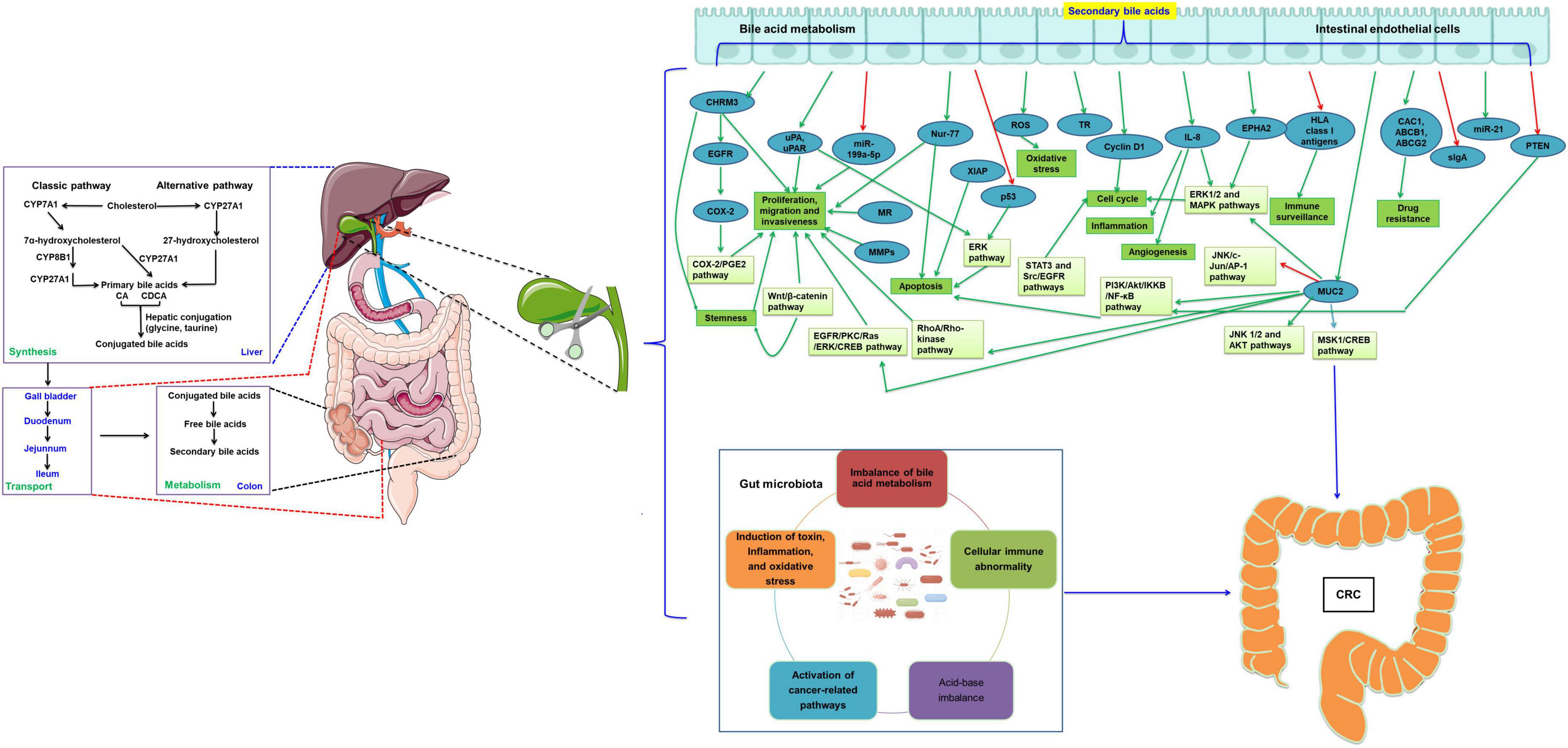
Figure 1. Cholecystectomy promotes the development of CRC by the alternation of bile acid metabolism and the gut microbiota. The green arrow indicates the levels are upregulated or the pathway is activated, while the red arrow indicates the levels are downregulated or the pathway is inactivated. CRC, colorectal cancer; CYP7A1, cholesterol 7α-hydroxylase; CYP8B1, sterol 12α-hydroxylase; CYP27A1, mitochondrial sterol 27-hydroxylase; CA, cholic acid; CDCA, chenodeoxycholic acid; COX-2, cyclooxygenase 2; EGFR, epidermal growth factor receptor; uPAR, urokinase-type plasminogen activator receptor; MR, muscarinic receptor; MMPs, matrix metalloproteinases; miR, microRNA; MUC2, mucin 2, oligomeric mucus/gel-forming; TR, thioredoxin reductase; IL, interleukin; EPHA2, EPH receptor A2; ABCB1, ATP binding cassette subfamily B member 1; ABCG2, ATP binding cassette subfamily G member 2; HLA, human leukocyte antigen; sIgA, secretory antibodies of the type IgA; XIAP, X-linked inhibitor of apoptosis protein; ROS, reactive oxygen species; PGE2, prostaglandin E2; ERK, extracellular signal regulated kinases; CREB, cAMP response element binding protein; PI3K, phosphoInositide-3 kinase; IKKB, Ikappa B; NF-κB, nuclear factor kappa-B; MAPK, mitogen activated protein kinase; STAT, signal transduction and transcriptional activator; PKC, protein kinase C; MSK1, mitogen and stress-activated protein kinase 1; AP-1, activated protein-1; JNK, c-jun N-terminal kinase.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
YS conceived the project. XJ and ZJ performed the research. QC and WS collected the background information. XJ, ZJ, QC, WS, and MJ drafted the manuscript. YS revised the manuscript. All authors approved the publication of the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81700216), the 345 Talent Project of Shengjing Hospital of China Medical University, and the Doctoral Start-up Foundation of Liaoning Province (2019-BS-274).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. (2019) 14:89–103. doi: 10.5114/pg.2018.81072
2. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. (2019) 16:713–32. doi: 10.1038/s41575-019-0189-8
3. Bien J, Lin AA. Review of the diagnosis and treatment of metastatic colorectal cancer. JAMA. (2021) 325:2404–5. doi: 10.1001/jama.2021.6021
4. Favoriti P, Carbone G, Greco M, Pirozzi F, Pirozzi RE, Corcione F. Worldwide burden of colorectal cancer: a review. Updates Surg. (2016) 68:7–11. doi: 10.1007/s13304-016-0359-y
5. Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. (2021) 325:669–85. doi: 10.1001/jama.2021.0106
6. Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Translational Oncol. (2021) 14:101174. doi: 10.1016/j.tranon.2021.101174
7. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. (2009) 22:191–7. doi: 10.1055/s-0029-1242458
8. Hull R, Francies FZ, Oyomno M. Colorectal cancer genetics, incidence and risk factors: in search for targeted therapies. Cancer Manag Res. (2020) 12:9869–82. doi: 10.2147/CMAR.S251223
9. Boyle P, Langman JS. ABC of colorectal cancer: epidemiology. BMJ. (2000) 321:805–8. doi: 10.1136/bmj.321.7264.805
10. Wilmink AB. Overview of the epidemiology of colorectal cancer. Dis Colon Rectum. (1997) 40:483–93. doi: 10.1007/BF02258397
11. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
12. Siegel RL, Jakubowski CD, Fedewa SA, Davis A, Azad NS. Colorectal cancer in the young: epidemiology, prevention, management. Am Soc Clin Oncol. (2020) 40:1–14. doi: 10.1200/EDBK_279901
13. Mauri G, Sartore-Bianchi A, Russo AG, Marsoni S, Bardelli A, Siena S. Early-onset colorectal cancer in young individuals. Mol Oncol. (2019) 13:109–31. doi: 10.1002/1878-0261.12417
14. Patel SG, Ahnen DJ. Colorectal cancer in the young. Curr Gastroenterol Rep. (2018) 20:15. doi: 10.1007/s11894-018-0618-9
15. Wong MCS, Huang J, Lok V, Wang J, Fung F, Ding H, et al. Differences in incidence and mortality trends of colorectal cancer worldwide based on sex, age, and anatomic location. Clin Gastroenterol Hepatol. (2021) 19:955–66.e61. doi: 10.1016/j.cgh.2020.02.026
16. Sawicki T, Ruszkowska M, Danielewicz A, Niedźwiedzka E, Arłukowicz T, Przybyłowicz KE. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers. (2021) 13:2025. doi: 10.3390/cancers13092025
17. Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol. (2005) 70:1035–47. doi: 10.1016/j.bcp.2005.07.023
18. Brescia A, Gasparrini M, Nigri G, Cosenza UM, Dall’Oglio A, Pancaldi A, et al. Laparoscopic cholecystectomy in day surgery: feasibility and outcomes of the first 400 patients. Surgeon. (2013) 11:S14–8. doi: 10.1016/j.surge.2012.09.006
19. Lamberts MP, Kievit W, Özdemir C, Westert GP, van Laarhoven CJ, Drenth JP. Value of EGD in patients referred for cholecystectomy: a systematic review and meta-analysis. Gastrointest Endosc. (2015) 82:24–31. doi: 10.1016/j.gie.2015.01.024
20. Aurif F, Kaur H, Chio JPG, Kittaneh M, Malik BH. The association between cholecystectomy and colorectal cancer in the female gender. Cureus. (2021) 13:e20113. doi: 10.7759/cureus.20113
21. Schernhammer ES, Leitzmann MF, Michaud DS, Speizer FE, Giovannucci E, Colditz GA, et al. Cholecystectomy and the risk for developing colorectal cancer and distal colorectal adenomas. Br J Cancer. (2003) 88:79–83. doi: 10.1038/sj.bjc.6600661
22. Altieri A, Pelucchi C, Talamini R, Bosetti C, Franceschi S, La Vecchia C. Cholecystectomy and the risk of colorectal cancer in Italy. Br J Cancer. (2004) 90:1753–5. doi: 10.1038/sj.bjc.6601721
23. Kim SB, Kim KO. Prevalence and risk factors of gastric and colorectal cancer after cholecystectomy. J Korean Med Sci. (2020) 35:e354. doi: 10.3346/jkms.2020.35.e354
24. Zhang Y, Liu H, Li L, Ai M, Gong Z, He Y, et al. Cholecystectomy can increase the risk of colorectal cancer: a meta-analysis of 10 cohort studies. PLoS One. (2017) 12:e0181852. doi: 10.1371/journal.pone.0181852
25. Shao T, Yang YX. Cholecystectomy and the risk of colorectal cancer. Am J Gastroenterol. (2005) 100:1813–20. doi: 10.1111/j.1572-0241.2005.41610.x
26. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. (2020) 70:145–64. doi: 10.3322/caac.21601
27. Bray F, Colombet M, Mery L, Piñeros M, Znaor A, Zanetti R, et al. Cancer Incidence in Five Continents Volume XI: Cancer Today. Lyon: IARC Scientific Publication (2021).
28. Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Piñeros M, et al. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer (2021).
29. Lewandowska A, Rudzki G. Title: risk factors for the diagnosis of colorectal cancer. Cancer Control. (2022) 29:10732748211056692. doi: 10.1177/10732748211056692
30. Begum S, Khan MR, Gill RC. Cost effectiveness of glove endobag in laparoscopic cholecystectomy: review of the available literature. J Pak Med Assoc. (2019) 69:S58–61.
31. Chattopadhyay K, Das R. Laparoscopic and open cholecystectomy: a comparative study. Int J Surg Sci. (2020) 4:427–30. doi: 10.33545/surgery.2020.v4.i1h.375
32. Islam MR, Ali MS, Azam SG, Islam MR. Comparative study between laparoscopic and open cholecystectomy: complications and management. Med Today. (2021) 33:19–21. doi: 10.3329/medtoday.v33i01.52152
33. Lombardo S, Rosenberg JS, Kim J, Erdene S, Sergelen O, Nellermoe J, et al. Cost and outcomes of open versus laparoscopic cholecystectomy in Mongolia. J Surg Res. (2018) 229:186–91. doi: 10.1016/j.jss.2018.03.036
34. Giovannucci E, Colditz GA, Stampfer MJ. A meta-analysis of cholecystectomy and risk of colorectal cancer. Gastroenterology. (1993) 105:130–41. doi: 10.1016/0016-5085(93)90018-8
35. Moorehead RJ, McKelvey ST. Cholecystectomy and colorectal cancer. Br J Surg. (1989) 76:250–3. doi: 10.1002/bjs.1800760312
36. Thomas LA, Veysey MJ, French G, Hylemon PB, Murphy GM, Dowling RH. Bile acid metabolism by fresh human colonic contents: a comparison of caecal versus faecal samples. Gut. (2001) 49:835–42. doi: 10.1136/gut.49.6.835
37. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
38. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. (2009) 50:1955–66. doi: 10.1194/jlr.R900010-JLR200
39. Axelson M, Ellis E, Mörk B, Garmark K, Abrahamsson A, Björkhem I, et al. Bile acid synthesis in cultured human hepatocytes: support for an alternative biosynthetic pathway to cholic acid. Hepatology. (2000) 31:1305–12. doi: 10.1053/jhep.2000.7877
40. Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. (2009) 50:1509–20. doi: 10.1194/jlr.R900007-JLR200
41. Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. (2004) 40:539–51. doi: 10.1016/j.jhep.2003.11.006
42. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
43. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. (2016) 7:22–39. doi: 10.1080/19490976.2015.1127483
44. Housset C, Chrétien Y, Debray D, Chignard N. Functions of the Gallbladder. Compr Physiol. (2016) 6:1549–77. doi: 10.1002/cphy.c150050
45. Cortes V, Amigo L, Zanlungo S, Galgani J, Robledo F, Arrese M, et al. Metabolic effects of cholecystectomy: gallbladder ablation increases basal metabolic rate through G-protein coupled bile acid receptor Gpbar1-dependent mechanisms in mice. PLoS One. (2015) 10:e0118478. doi: 10.1371/journal.pone.0118478
46. Kullak-Ublick GA, Paumgartner G, Berr F. Long-term effects of cholecystectomy on bile acid metabolism. Hepatology. (1995) 21:41–5. doi: 10.1002/hep.1840210109
47. Berr F, Stellaard F, Pratschke E, Paumgartner G. Effects of cholecystectomy on the kinetics of primary and secondary bile acids. J Clin Invest. (1989) 83:1541–50. doi: 10.1172/JCI114050
48. Zhang F, Qin H, Zhao Y, Wei Y, Xi L, Rao Z, et al. Effect of cholecystectomy on bile acids as well as relevant enzymes and transporters in mice: implication for pharmacokinetic changes of rifampicin. Eur J Pharm Sci. (2017) 96:141–53. doi: 10.1016/j.ejps.2016.09.006
49. Zhang F, Duan Y, Xi L, Wei M, Shi A, Zhou Y, et al. The influences of cholecystectomy on the circadian rhythms of bile acids as well as the enterohepatic transporters and enzymes systems in mice. Chronobiol Int. (2018) 35:673–90. doi: 10.1080/07420528.2018.1426596
50. Dong W, Liu L, Dou Y, Xu M, Liu T, Wang S, et al. Deoxycholic acid activates epidermal growth factor receptor and promotes intestinal carcinogenesis by ADAM 17-dependent ligand release. J Cell Mol Med. (2018) 22:4263–73. doi: 10.1111/jcmm.13709
51. Liu L, Dong W, Wang S, Zhang Y, Liu T, Xie R, et al. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. (2018) 9:5588–97. doi: 10.1039/C8FO01143E
52. Yao Y, Li X, Xu B, Luo L, Guo Q, Wang X, et al. Cholecystectomy promotes colon carcinogenesis by activating the Wnt signaling pathway by increasing the deoxycholic acid level. Cell Commun Signal. (2022) 20:85. doi: 10.1186/s12964-022-00890-8
53. Nagengast F, Grubben M, Van Munster I. Role of bile acids in colorectal carcinogenesis. Eur J Cancer. (1995) 31:1067–70. doi: 10.1016/0959-8049(95)00216-6
54. Nagengast FM. Bile acids and colonic carcinogenesis. Scand J Gastroenterol. (1988) 23:76–81. doi: 10.3109/00365528809095955
55. Kuhls S, Osswald A, Ocvirk S. Bile acids, bile pigments and colorectal cancer risk. Curr Opin Gastroenterol. (2022) 38:173–8. doi: 10.1097/MOG.0000000000000820
56. Ocvirk S, O’Keefe SJ. Dietary fat, bile acid metabolism and colorectal cancer. Semin Cancer Biol. (2021) 73:347–55. doi: 10.1016/j.semcancer.2020.10.003
57. Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates β-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell. (2004) 15:2156–63. doi: 10.1091/mbc.e03-12-0894
58. Peiffer L, Peters D, McGarrity T. Differential effects of deoxycholic acid on proliferation of neoplastic and differentiated colonocytes in vitro. Dig Dis Sci. (1997) 42:2234–40. doi: 10.1023/A:1018806431866
59. Milovic V, Teller IC, Faust D, Caspary WF, Stein J. Effects of deoxycholate on human colon cancer cells: apoptosis or proliferation. Eur J Clin Invest. (2002) 32:29–34. doi: 10.1046/j.0014-2972.2001.00938.x
60. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F, et al. FXR regulates intestinal cancer stem cell proliferation. Cell. (2019) 176:1098–112.e18. doi: 10.1016/j.cell.2019.01.036
61. Flynn CG. Understanding the Promotional Effect of Deoxycholic Acid During Colorectal Cancer Development. Doctoral dissertations. Mansfield, CT: University of Connecticut (2008).
62. Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. (2019) 12:851–61. doi: 10.1038/s41385-019-0162-4
63. Zeng H, Umar S, Rust B. Secondary bile acids and short chain fatty acids in the colon: a focus on colonic microbiome, cell proliferation, inflammation, and cancer. Int J Mol Sci. (2019) 20:1214. doi: 10.3390/ijms20051214
64. Sorrentino G, Perino A, Yildiz E, El Alam G, Bou Sleiman M, Gioiello A, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. (2020) 159:956–68.e8. doi: 10.1053/j.gastro.2020.05.067
65. Mroz MS, Lajczak NK. The bile acids, deoxycholic acid and ursodeoxycholic acid, regulate colonic epithelial wound healing. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G378–87. doi: 10.1152/ajpgi.00435.2016
66. Qiao D, Stratagouleas ED, Martinez JD. Activation and role of mitogen-activated protein kinases in deoxycholic acid-induced apoptosis. Carcinogenesis. (2001) 22:35–41. doi: 10.1093/carcin/22.1.35
67. Shah SA, Volkov Y, Arfin Q, Abdel-Latif MM, Kelleher D. Ursodeoxycholic acid inhibits interleukin 1 beta [corrected] and deoxycholic acid-induced activation of NF-kappaB and AP-1 in human colon cancer cells. Int J Cancer. (2006) 118:532–9. doi: 10.1002/ijc.21365
68. Song X, An Y, Chen D, Zhang W, Wu X, Li C, et al. Microbial metabolite deoxycholic acid promotes vasculogenic mimicry formation in intestinal carcinogenesis. Cancer Sci. (2022) 113:459–77. doi: 10.1111/cas.15208
69. Arvind P, Papavassiliou ED, Tsioulias GJ, Duceman BW, Lovelace CI, Geng W, et al. Lithocholic acid inhibits the expression of HLA class I genes in colon adenocarcinoma cells. Differential effect on HLA-A, -B and -C loci. Mol Immunol. (1994) 31:607–14. doi: 10.1016/0161-5890(94)90168-6
70. Jean-Louis S, Akare S, Ali MA, Mash EA Jr., Meuillet E, Martinez JD. Deoxycholic acid induces intracellular signaling through membrane perturbations. J Biol Chem. (2006) 281:14948–60. doi: 10.1074/jbc.M506710200
71. Bhattacharya S, Haldar PK. Chemopreventive property of Trichosanthes dioica root against 3-methylcholanthrene-induced carcinogenesis in albino mice. J Environ Pathol Toxicol Oncol. (2012) 31:109–19. doi: 10.1615/JEnvironPatholToxicolOncol.v31.i2.30
72. Zeng H, Safratowich BD, Cheng WH, Larson KJ, Briske-Anderson M. Deoxycholic acid modulates cell-junction gene expression and increases intestinal barrier dysfunction. Molecules. (2022) 27:723. doi: 10.3390/molecules27030723
73. Farhana L, Nangia-Makker P, Arbit E, Shango K, Sarkar S, Mahmud H, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. (2016) 7:181. doi: 10.1186/s13287-016-0439-4
74. Oshio H, Abe T, Onogawa T, Ohtsuka H, Sato T, Ii T, et al. Peroxisome proliferator-activated receptor alpha activates cyclooxygenase-2 gene transcription through bile acid transport in human colorectal cancer cell lines. J Gastroenterol. (2008) 43:538–49. doi: 10.1007/s00535-008-2188-3
75. Merchant NB, Rogers CM, Trivedi B, Morrow J, Coffey RJ. Ligand-dependent activation of the epidermal growth factor receptor by secondary bile acids in polarizing colon cancer cells. Surgery. (2005) 138:415–21. doi: 10.1016/j.surg.2005.06.030
76. Cheng K, Chen Y, Zimniak P, Raufman JP, Xiao Y, Frucht H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim Biophys Acta. (2002) 1588:48–55. doi: 10.1016/S0925-4439(02)00115-1
77. Cheng K, Xie G, Raufman JP. Matrix metalloproteinase-7-catalyzed release of HB-EGF mediates deoxycholyltaurine-induced proliferation of a human colon cancer cell line. Biochem Pharmacol. (2007) 73:1001–12. doi: 10.1016/j.bcp.2006.11.028
78. Kong Y, Bai PS, Sun H, Nan KJ, Chen NZ, Qi XG. The deoxycholic acid targets miRNA-dependent CAC1 gene expression in multidrug resistance of human colorectal cancer. Int J Biochem Cell Biol. (2012) 44:2321–32. doi: 10.1016/j.biocel.2012.08.006
79. Turner DJ, Alaish SM, Zou T, Rao JN, Wang JY, Strauch ED. Bile salts induce resistance to apoptosis through NF-kappaB-mediated XIAP expression. Ann Surg. (2007) 245:415–25. doi: 10.1097/01.sla.0000236631.72698.99
80. Qiao D, Gaitonde SV, Qi W, Martinez JD. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis. (2001) 22:957–64. doi: 10.1093/carcin/22.6.957
81. Hu Y, Chau T, Liu HX, Liao D, Keane R, Nie Y, et al. Bile acids regulate nuclear receptor (Nur77) expression and intracellular location to control proliferation and apoptosis. Mol Cancer Res. (2015) 13:281–92. doi: 10.1158/1541-7786.MCR-14-0230
82. Flynn C, Montrose DC, Swank DL, Nakanishi M, Ilsley JN, Rosenberg DW. Deoxycholic acid promotes the growth of colonic aberrant crypt foci. Mol Carcinog. (2007) 46:60–70. doi: 10.1002/mc.20253
83. Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int J Cancer. (2017) 140:2545–56. doi: 10.1002/ijc.30643
84. Payne CM, Crowley-Skillicorn C, Bernstein C, Holubec H, Moyer MP, Bernstein H. Hydrophobic bile acid-induced micronuclei formation, mitotic perturbations, and decreases in spindle checkpoint proteins: relevance to genomic instability in colon carcinogenesis. Nutr Cancer. (2010) 62:825–40. doi: 10.1080/01635581003695756
85. Payne CM, Bernstein C, Dvorak K, Bernstein H. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin Exp Gastroenterol. (2008) 1:19–47. doi: 10.2147/CEG.S4343
86. Degirolamo C, Modica S, Palasciano G, Moschetta A. Bile acids and colon cancer: solving the puzzle with nuclear receptors. Trends Mol Med. (2011) 17:564–72. doi: 10.1016/j.molmed.2011.05.010
87. Peng S, Huo X, Rezaei D, Zhang Q, Zhang X, Yu C, et al. In Barrett’s esophagus patients and Barrett’s cell lines, ursodeoxycholic acid increases antioxidant expression and prevents DNA damage by bile acids. Am J Physiol Gastrointest Liver Physiol. (2014) 307:G129–39. doi: 10.1152/ajpgi.00085.2014
88. Noffsinger AE. Serrated polyps and colorectal cancer: new pathway to malignancy. Annu Rev Pathol. (2009) 4:343–64. doi: 10.1146/annurev.pathol.4.110807.092317
89. Lechner S, Müller-Ladner U, Schlottmann K, Jung B, McClelland M, Rüschoff J, et al. Bile acids mimic oxidative stress induced upregulation of thioredoxin reductase in colon cancer cell lines. Carcinogenesis. (2002) 23:1281–8. doi: 10.1093/carcin/23.8.1281
90. Ignacio Barrasa J, Olmo N, Pérez-Ramos P, Santiago-Gómez A, Lecona E, Turnay J, et al. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis. (2011) 16:1054–67. doi: 10.1007/s10495-011-0633-x
91. Halvorsen B, Staff AC, Ligaarden S, Prydz K, Kolset SO. Lithocholic acid and sulphated lithocholic acid differ in the ability to promote matrix metalloproteinase secretion in the human colon cancer cell line CaCo-2. Biochem J. (2000) 349:189–93. doi: 10.1042/bj3490189
92. Nguyen TT, Lian S, Ung TT, Xia Y, Han JY, Jung YD. Lithocholic acid stimulates IL-8 expression in human colorectal cancer cells via activation of Erk1/2 MAPK and suppression of STAT3 activity. J Cell Biochem. (2017) 118:2958–67. doi: 10.1002/jcb.25955
93. Nguyen TT, Ung TT, Li S. Lithocholic acid induces miR21, promoting PTEN inhibition via STAT3 and ERK-1/2 signaling in colorectal cancer cells. Int J Mol Sci. (2021) 22:10209. doi: 10.3390/ijms221910209
94. Kulkarni MS, Cox BA, Yielding KL. Requirements for induction of DNA strand breaks by lithocholic acid. Cancer Res. (1982) 42:2792–5.
95. Ogawa A, Murate T, Suzuki M, Nimura Y, Yoshida S. Lithocholic acid, a putative tumor promoter, inhibits mammalian DNA polymerase beta. Jpn J Cancer Res. (1998) 89:1154–9. doi: 10.1111/j.1349-7006.1998.tb00510.x
96. Centuori SM, Gomes CJ, Trujillo J, Borg J, Brownlee J, Putnam CW, et al. Deoxycholic acid mediates non-canonical EGFR-MAPK activation through the induction of calcium signaling in colon cancer cells. Biochim Biophys Acta. (2016) 1861:663–70. doi: 10.1016/j.bbalip.2016.04.006
97. Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, et al. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-α: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. (2014) 74:2062–72. doi: 10.1158/0008-5472.CAN-13-2329
98. Zhu Y, Zhu M, Lance P. Stromal COX-2 signaling activated by deoxycholic acid mediates proliferation and invasiveness of colorectal epithelial cancer cells. Biochem Biophys Res Commun. (2012) 425:607–12. doi: 10.1016/j.bbrc.2012.07.137
99. Nguyen TT, Ung TT, Kim NH, Jung YD. Role of bile acids in colon carcinogenesis. World J Clin Cases. (2018) 6:577–88. doi: 10.12998/wjcc.v6.i13.577
100. Li Z, Tanaka M, Kataoka H, Nakamura R, Sanjar R, Shinmura K, et al. EphA2 up-regulation induced by deoxycholic acid in human colon carcinoma cells, an involvement of extracellular signal-regulated kinase and p53-independence. J Cancer Res Clin Oncol. (2003) 129:703–8. doi: 10.1007/s00432-003-0493-z
101. Milovic V, Teller IC, Murphy GM, Caspary WF, Stein J. Deoxycholic acid stimulates migration in colon cancer cells. Eur J Gastroenterol Hepatol. (2001) 13:945–9. doi: 10.1097/00042737-200108000-00012
102. Debruyne PR, Bruyneel EA, Karaguni IM, Li X, Flatau G, Müller O, et al. Bile acids stimulate invasion and haptotaxis in human colorectal cancer cells through activation of multiple oncogenic signaling pathways. Oncogene. (2002) 21:6740–50. doi: 10.1038/sj.onc.1205729
103. Lee HY, Crawley S, Hokari R, Kwon S, Kim YS. Bile acid regulates MUC2 transcription in colon cancer cells via positive EGFR/PKC/Ras/ERK/CREB, PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative JNK/c-Jun/AP-1 pathway. Int J Oncol. (2010) 36:941–53. doi: 10.3892/ijo_00000573
104. Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. (2005) 589:47–65. doi: 10.1016/j.mrrev.2004.08.001
105. Lee DK, Park SY, Baik SK, Kwon SO, Chung JM, Oh ES, et al. [Deoxycholic acid-induced signal transduction in HT-29 cells: role of NF-kappa B and interleukin-8]. Korean J Gastroenterol. (2004) 43:176–85.
106. Song S, Byrd JC, Koo JS, Bresalier RS. Bile acids induce MUC2 overexpression in human colon carcinoma cells. Cancer. (2005) 103:1606–14. doi: 10.1002/cncr.21015
107. Baek MK, Park JS, Park JH, Kim MH, Kim HD, Bae WK, et al. Lithocholic acid upregulates uPAR and cell invasiveness via MAPK and AP-1 signaling in colon cancer cells. Cancer Lett. (2010) 290:123–8. doi: 10.1016/j.canlet.2009.08.030
108. Nguyen TT, Ung TT, Li S, Lian S, Xia Y, Park SY, et al. Metformin inhibits lithocholic acid-induced interleukin 8 upregulation in colorectal cancer cells by suppressing ROS production and NF-kB activity. Sci Rep. (2019) 9:2003. doi: 10.1038/s41598-019-38778-2
109. Li S, Nguyen TT, Ung TT, Sah DK, Park SY, Lakshmanan VK. Piperine attenuates lithocholic acid-stimulated interleukin-8 by suppressing Src/EGFR and reactive oxygen species in human colorectal cancer cells. Antioxidants. (2022) 11:530. doi: 10.3390/antiox11030530
110. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. (2012) 489:220–30. doi: 10.1038/nature11550
111. Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. (2015) 26:26191. doi: 10.3402/mehd.v26.26191
112. Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, et al. A new genomic blueprint of the human gut microbiota. Nature. (2019) 568:499–504. doi: 10.1038/s41586-019-0965-1
113. Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, et al. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. (2014) 52:398–406. doi: 10.1128/JCM.01500-13
114. Liu Y, Zhang S, Zhou W, Hu D, Xu H, Ji G. Secondary bile acids and tumorigenesis in colorectal cancer. Front Oncol. (2022) 12:813745. doi: 10.3389/fonc.2022.813745
115. Greenhalgh K, Meyer KM, Aagaard KM, Wilmes P. The human gut microbiome in health: establishment and resilience of microbiota over a lifetime. Environ Microbiol. (2016) 18:2103–16. doi: 10.1111/1462-2920.13318
116. Hajiagha MN, Taghizadeh S, Asgharzadeh M, Dao S, Ganbarov K, Köse Ş. Gut microbiota and human body interactions; Its impact on health: a review. Curr Pharm Biotechnol. (2022) 23:4–14. doi: 10.2174/1389201022666210104115836
117. Ogunrinola GA, Oyewale JO, Oshamika OO, Olasehinde GI. The human microbiome and its impacts on health. Int J Microbiol. (2020) 2020:8045646. doi: 10.1155/2020/8045646
118. Althani AA, Marei HE, Hamdi WS, Nasrallah GK, El Zowalaty ME, Al Khodor S, et al. Human microbiome and its association with health and diseases. J Cell Physiol. (2016) 231:1688–94. doi: 10.1002/jcp.25284
119. Song M, Chan AT, Sun J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology. (2020) 158:322–40. doi: 10.1053/j.gastro.2019.06.048
120. Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. (2013) 105:1907–11. doi: 10.1093/jnci/djt300
121. Gao R, Gao Z, Huang L, Qin H. Gut microbiota and colorectal cancer. Eur J Clin Microbiol Infect Dis. (2017) 36:757–69. doi: 10.1007/s10096-016-2881-8
122. Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene. (2020) 39:4925–43. doi: 10.1038/s41388-020-1341-1
123. Wang G, Yu Y, Wang YZ, Wang JJ, Guan R, Sun Y, et al. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J Cell Physiol. (2019) 234:17023–49. doi: 10.1002/jcp.28436
124. Tilg H, Adolph TE, Gerner RR, Moschen AR. The intestinal microbiota in colorectal cancer. Cancer Cell. (2018) 33:954–64. doi: 10.1016/j.ccell.2018.03.004
125. Lucas C, Barnich N, Nguyen HTT. Microbiota, inflammation and colorectal cancer. Int J Mol Sci. (2017) 18:1310. doi: 10.3390/ijms18061310
126. Saus E, Iraola-Guzmán S, Willis JR, Brunet-Vega A, Gabaldón T. Microbiome and colorectal cancer: roles in carcinogenesis and clinical potential. Mol Aspects Med. (2019) 69:93–106. doi: 10.1016/j.mam.2019.05.001
127. Ren X, Xu J, Zhang Y, Chen G, Zhang Y, Huang Q, et al. Bacterial Alterations in Post-Cholecystectomy Patients Are Associated With Colorectal Cancer. Front Oncol. (2020) 10:1418. doi: 10.3389/fonc.2020.01418
128. Keren N, Konikoff FM, Paitan Y, Gabay G, Reshef L, Naftali T, et al. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ Microbiol Rep. (2015) 7:874–80. doi: 10.1111/1758-2229.12319
129. Wang W, Wang J, Li J, Yan P, Jin Y, Zhang R, et al. Cholecystectomy damages aging-associated intestinal microbiota construction. Front Microbiol. (2018) 9:1402. doi: 10.3389/fmicb.2018.01402
130. Yoon WJ, Kim HN, Park E, Ryu S, Chang Y, Shin H, et al. The impact of cholecystectomy on the gut microbiota: a case-control study. J Clin Med. (2019) 8:79. doi: 10.3390/jcm8010079
131. Frost F, Kacprowski T, Rühlemann M, Weiss S, Bang C, Franke A, et al. Carrying asymptomatic gallstones is not associated with changes in intestinal microbiota composition and diversity but cholecystectomy with significant dysbiosis. Sci Rep. (2021) 11:6677. doi: 10.1038/s41598-021-86247-6
132. Floch MH, Binder HJ, Filburn B, Gershengoren W. The effect of bile acids on intestinal microflora. Am J Clin Nutr. (1972) 25:1418–26. doi: 10.1093/ajcn/25.12.1418
133. Sakai K, Makino T, Kawai Y, Mutai M. Intestinal microflora and bile acids. Effect of bile acids on the distribution of microflora and bile acid in the digestive tract of the rat. Microbiol Immunol. (1980) 24:187–96. doi: 10.1111/j.1348-0421.1980.tb00578.x
134. Kim Y, Lee S, Kim S, Kim TY, Lee SH, Chang JH, et al. LKB1 in intestinal epithelial cells regulates bile acid metabolism by modulating FGF15/19 production. Cell Mol Gastroenterol Hepatol. (2022) 13:1121–39. doi: 10.1016/j.jcmgh.2021.12.017
135. Barrera F, Azócar L, Molina H, Schalper KA, Ocares M, Liberona J, et al. Effect of cholecystectomy on bile acid synthesis and circulating levels of fibroblast growth factor 19. Ann Hepatol. (2015) 14:710–21. doi: 10.1016/S1665-2681(19)30766-5
136. Adolph TE, Grander C, Moschen AR, Tilg H. Liver-microbiome axis in health and disease. Trends Immunol. (2018) 39:712–23. doi: 10.1016/j.it.2018.05.002
137. Sarashina-Kida H, Negishi H, Nishio J, Suda W, Nakajima Y, Yasui-Kato M, et al. Gallbladder-derived surfactant protein D regulates gut commensal bacteria for maintaining intestinal homeostasis. Proc Natl Acad Sci USA. (2017) 114:10178–83. doi: 10.1073/pnas.1712837114
138. Garro MS, Aguirre L, Savoy de Giori G. Biological activity of Bifidobacterium longum in response to environmental pH. Appl Microbiol Biotechnol. (2006) 70:612–7. doi: 10.1007/s00253-005-0102-y
139. Bian J, Dannappel M, Wan C, Firestein R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells. (2020) 9:2125. doi: 10.3390/cells9092125
140. Cheng X, Xu X, Chen D, Zhao F, Wang W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed Pharmacother. (2019) 110:473–81. doi: 10.1016/j.biopha.2018.11.082
141. Cho YH, Ro EJ. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun. (2020) 11:5321. doi: 10.1038/s41467-020-19173-2
142. Tian X, Liu Z, Niu B, Zhang J, Tan TK, Lee SR, et al. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol. (2011) 2011:567305. doi: 10.1155/2011/567305
143. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
144. Lee P, Tan KS. Fusobacterium nucleatum activates the immune response through retinoic acid-inducible gene I. J Dent Res. (2014) 93:162–8. doi: 10.1177/0022034513516346
145. Allen-Vercoe E, Jobin C. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunol Lett. (2014) 162:54–61. doi: 10.1016/j.imlet.2014.05.014
146. Fardini Y, Wang X, Témoin S, Nithianantham S, Lee D, Shoham M, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. (2011) 82:1468–80. doi: 10.1111/j.1365-2958.2011.07905.x
147. Yang Y, Jobin C. Novel insights into microbiome in colitis and colorectal cancer. Curr Opin Gastroenterol. (2017) 33:422–7. doi: 10.1097/MOG.0000000000000399
148. Gao ZY, Cui Z, Yan YQ, Ning LJ, Wang ZH, Hong J. Microbe-based management for colorectal cancer. Chin Med J. (2021) 134:2922–30. doi: 10.1097/CM9.0000000000001887
149. Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology. (2017) 152:1419–33.e5. doi: 10.1053/j.gastro.2017.01.009
150. Yu S, Shao X, Zhou Y, Yu Y, Kuai X, Zhou C. Bidirectional regulation of bile acid on colorectal cancer through bile acid-gut microbiota interaction. Am J Transl Res. (2021) 13:10994–1003.
151. Dhir M, Montgomery EA, Glöckner SC, Schuebel KE, Hooker CM, Herman JG, et al. Epigenetic regulation of WNT signaling pathway genes in inflammatory bowel disease (IBD) associated neoplasia. J Gastrointest Surg. (2008) 12:1745–53. doi: 10.1007/s11605-008-0633-5
152. Hale VL, Chen J, Johnson S, Harrington SC, Yab TC, Smyrk TC, et al. Shifts in the fecal microbiota associated with adenomatous polyps. Cancer Epidemiol Biomark Prevent. (2017) 26:85–94. doi: 10.1158/1055-9965.EPI-16-0337
153. Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. (2018) 23:203–14.e5. doi: 10.1016/j.chom.2018.01.007
154. Arthur JC. Microbiota and colorectal cancer: colibactin makes its mark. Nat Rev Gastroenterol Hepatol. (2020) 17:317–8. doi: 10.1038/s41575-020-0303-y
155. Dubinsky V, Dotan I, Gophna U. Carriage of colibactin-producing bacteria and colorectal cancer risk. Trends Microbiol. (2020) 28:874–6. doi: 10.1016/j.tim.2020.05.015
156. Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, et al. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer. (1998) 31:111–8. doi: 10.1080/01635589809514689
157. Narisawa T, Fukaura Y, Terada K, Sekiguchi H. Prevention of N-methylnitrosourea-induced colon tumorigenesis by ursodeoxycholic acid in F344 rats. Jpn J Cancer Res. (1998) 89:1009–13. doi: 10.1111/j.1349-7006.1998.tb00489.x
158. Narisawa T, Fukaura Y, Takeba N, Nakai K. Chemoprevention of N-methylnitrosourea-induced colon carcinogenesis by ursodeoxycholic acid-5-aminosalicylic acid conjugate in F344 rats. Jpn J Cancer Res. (2002) 93:143–50. doi: 10.1111/j.1349-7006.2002.tb01252.x
159. Narisawa T, Fukaura Y, Terada K, Sekiguchi H. Inhibitory effects of ursodeoxycholic acid on N-methylnitrosourea-induced colon carcinogenesis and colonic mucosal telomerase activity in F344 rats. J Exp Clin Cancer Res. (1999) 18:259–66.
160. Earnest DL, Holubec H, Wali RK, Jolley CS, Bissonette M, Bhattacharyya AK, et al. Chemoprevention of azoxymethane-induced colonic carcinogenesis by supplemental dietary ursodeoxycholic acid. Cancer Res. (1994) 54:5071–4.
161. Alberts DS, Martínez ME, Hess LM, Einspahr JG, Green SB, Bhattacharyya AK, et al. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J Natl Cancer Inst. (2005) 97:846–53. doi: 10.1093/jnci/dji144
162. Pearson T, Caporaso JG. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. (2019) 8:617–28. doi: 10.1002/cam4.1965
163. Zhang H, Xu H, Zhang C, Tang Q, Bi F. Ursodeoxycholic acid suppresses the malignant progression of colorectal cancer through TGR5-YAP axis. Cell Death Discov. (2021) 7:207. doi: 10.1038/s41420-021-00589-8
164. Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J Nutr. (2004) 134:483–6. doi: 10.1093/jn/134.2.483
165. van Heumen BW, Roelofs HM, Te Morsche RH, Marian B, Nagengast FM, Peters WH. Celecoxib and tauro-ursodeoxycholic acid co-treatment inhibits cell growth in familial adenomatous polyposis derived LT97 colon adenoma cells. Exp Cell Res. (2012) 318:819–27. doi: 10.1016/j.yexcr.2012.02.004
166. Khare S, Cerda S, Wali RK, von Lintig FC, Tretiakova M, Joseph L, et al. Ursodeoxycholic acid inhibits Ras mutations, wild-type Ras activation, and cyclooxygenase-2 expression in colon cancer. Cancer Res. (2003) 63:3517–23. doi: 10.1016/S0016-5085(03)83066-4
167. Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. (2011) 106:1638–45. doi: 10.1038/ajg.2011.156
Keywords: colorectal cancer, cholecystectomy, bile acid metabolism, gut microbiota, development
Citation: Jiang X, Jiang Z, Cheng Q, Sun W, Jiang M and Sun Y (2022) Cholecystectomy promotes the development of colorectal cancer by the alternation of bile acid metabolism and the gut microbiota. Front. Med. 9:1000563. doi: 10.3389/fmed.2022.1000563
Received: 22 July 2022; Accepted: 06 September 2022;
Published: 23 September 2022.
Edited by:
Enis Kostallari, Mayo Clinic, United StatesReviewed by:
Bei Tan, Peking Union Medical College Hospital (CAMS), ChinaCopyright © 2022 Jiang, Jiang, Cheng, Sun, Jiang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Sun, eWFuczE5ODZAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.