 Giovanna Gallo
Giovanna Gallo Massimo Volpe
Massimo Volpe Carmine Savoia
Carmine Savoia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 20 January 2022
Sec. Hematology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.798958
This article is part of the Research TopicEndotheliopathies: Current Concepts and Importance in Clinical PracticeView all 12 articles
Endothelium plays a fundamental role in the cardiovascular system, forming an interface between blood and adjacent tissues by regulating the vascular tone through the synthesis of nitric oxide, prostaglandins and other relaxing factors. Endothelial dysfunction is characterized by vasoconstriction, cell proliferation and shifting toward a proinflammatory and prothrombic state. In hypertension endothelial dysfunction may be involved in the initiation and development of vascular inflammation, vascular remodeling, and atherosclerosis and is independently associated with increased cardiovascular risk. Different conditions such as impaired vascular shear stress, inflammation and oxidative stress, activation of the renin angiotensin system have been described as important pathophysiological mechanisms involved in the development of endothelial dysfunction. The release of extracellular vesicles by neighboring cells in the vascular wall has emerged as an important regulator of endothelial function and with potential antihypertensive properties and beneficial effects by counteracting the hypertension mediated organ damage. Furthermore, macrovesicles are emerging as an innovative therapeutic approach for vascular protection, allowing the delivery of bioactive molecules, such as miRNA and drugs interacting with the renin angiotensin system. In this review we summarize the available evidence about the pathophysiological implications of endothelial dysfunction in cardiovascular diseases, focusing on hypertension and its sequelae, and the potential innovative therapeutic strategies targeting the endothelium with the aim to improve vascular function and remodeling.
Vascular endothelium plays an important role in cardiovascular (CV) physiology, forming an interface between blood and adjacent tissues and it is involved in nutrients and metabolites transport as well as in the interaction with circulating cells, hormones, and cytokines (1). Endothelial cells regulate the vascular tone through the synthesis of nitric oxide (NO), prostaglandins and other relaxing factors. Moreover, healthy endothelium provides antioxidant, anti-inflammatory, and antithrombotic functions and contributes to the maintenance of vascular tone, serving as a gatekeeper for organ/tissue homeostasis and blood pressure control (2).
Endothelial dysfunction is characterized by a shift of the actions of the endothelium toward reduced vasodilation, cell proliferation, platelet adhesion and activation and proinflammatory and prothrombic state. Endothelial dysfunction occurs in association with several CV risk factors, including hypertension, hypercholesterolemia and insulin resistance, contributing to inflammation in the vascular wall, of resistance arteries as well as to increased lipoprotein oxidation, smooth muscle cell proliferation, extracellular matrix deposition, cell adhesion, and thrombus formation in conducting arteries (3–5).
It should be noted that the manifestations of endothelial dysfunction may precede the development of hypertension (6). Essential hypertension is characterized by functional and structural alterations in resistance arteries which lead to increased peripheral vascular resistance (7). Endothelial dysfunction may contribute to the increased peripheral resistance by several mechanisms that leads to the enhanced constriction and vascular remodeling (i.e., structural, mechanical, and functional alterations) of resistance arteries, which is associated to the development and complications of hypertension (6, 8). In particular, endothelial dysfunction may participate to the increased myogenic tone of resistance arteries through the activation of the renin-angiotensin system (RAS), endothelin-1, catecholamines, and growth factors production, leading to vasoconstriction, vascular remodeling and then to increased resistance to blood flow and ultimately to increased peripheral blood pressure. The induction of inflammatory processes in the vascular wall may be associated to endothelial dysfunction and may contribute further to the remodeling of resistance arteries (6, 9), and conduit arteries which is associated with the increased risk of atherosclerosis and the development of CV disease (CVD) (6, 10–12).
In this review we will discuss the available evidence on the pathophysiological implications of endothelial dysfunction in hypertension, as well as the potential innovative therapeutic strategies targeting the endothelium.
Endothelial function is tightly regulated by the activation of several mediators and systems including NO, prostaglandins and other relaxing factors as well as by mechanical stimuli including vascular shear that stimulates numerous downstream signaling pathways to maintain and regulate endothelial function and vascular tone (13, 14). A fundamental distinction should be made between steady laminar and oscillatory flow (Figure 1). It has been shown that laminar flow enhances the production of vasodilator factors such as NO, prostacyclin, tissue-type plasminogen activator by the activation of mechanosensors and mechanosensitive channels which have been proposed to regulate a broad range of endothelial and vascular functions (15). Laminar shear activates the glycocalyx mechanosensing which is transferred by the cytoskeleton to integrins that distribute the force via actin microfilaments, microtubules, and intermediate filaments through the focal adhesion of c-Src kinases (15) leading to the maintenance of endothelial integrity. Increased laminar shear stress results also in elevated concentration of endothelial cytosolic calcium, leading to the activation of NO synthase (eNOS) and the increased production of NO. Elevated cytosolic calcium levels also trigger the opening of calcium-activated potassium channels, which is associated to endothelial cells hyperpolarization and thereby to vasorelaxation. Moreover, platelet endothelial cell adhesion molecule-1 (PECAM-1) along with caveolin, tyrosine-specific phospho-transferase Fyn, vascular endothelial growth factor (VEGF)-receptor 2 and the vascular endothelial cadherin (VE-cadherin) forms a mechanosensory complex which confers an adequate responsiveness to the beneficial effects of shear stress in endothelial cells (16). In particular, in laminar flow the force exerted on PECAM-1 triggers the activation of VEGF- receptor 2 in the absence of its ligand, which in turn induces integrin-mediated signaling and ultimately leads to the suppression of inflammatory pathways (17).
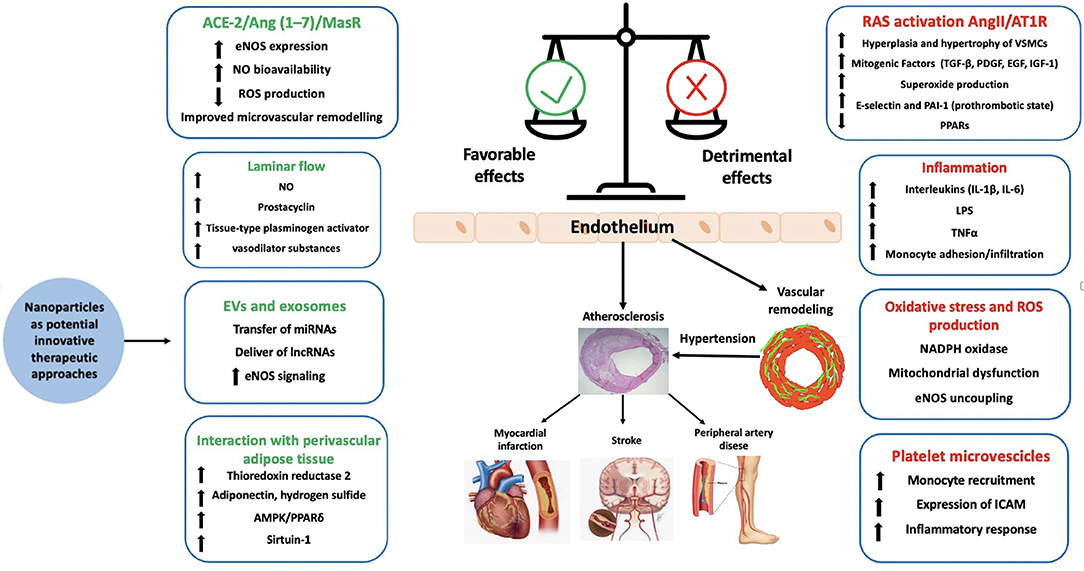
Figure 1. Factor contributing to endothelial function and dysfunction. ACE-2, angiotensin converting enzyme-2; AMPK, AMP-activated protein kinase; Ang, angiotensin; AT1R, type 1 angiotensin II receptor; EGF, epidermal growth factor eNOS, endothelial nitric oxide synthase; EVs, extracellular vesicles; ICAM, intercellular adhesion molecule 1; IGF-1, insulin-like growth factor-1; lncRNA, long-non-coding RNA; LPS, lipopolysaccharide; MCP-1, monocyte chemotactic protein 1; NAPDH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; PDGF, platelet derived growth factor; PPAR, peroxisome proliferator receptor; ROS, reactive oxygen species; TGF-β, tumor growth factor-β; TNFα, tumor necrosis factor.
In response to increased shear stress, AMPK-induced phosphorylation and sirtuin-1-mediated deacetylation promoted eNOS compartmentalization and activation with atheroprotective effects in an in vivo mouse model (18), thus contributing to vascular protection. Moreover, laminar flow increases JNK-mediated p53 phosphorylation, GADD45 and p21cip1, inhibiting endothelial cells growth and atherosclerotic plaque development (19, 20).
On the other hand, oscillatory flow reduces eNOS expression, promotes leukocyte infiltration, smooth muscle proliferation and the secretion of proinflammatory molecules, such as MCP-1 (monocyte chemotactic protein 1), PDGFs (platelet derived growth factor), and endothelin-1 leading to vasoconstriction, increased blood pressure (BP) and atherosclerosis development in larger arteries (21). These processes involve the activation of mechanosensitive genes in endothelial cells, inducing the increase of reactive oxygen species (ROS) and the activation of several transcription factors, such as Kruppel-like factor [KLF2/4], NF-κB, AP-1, early growth response-1, c-Jun, c-fos, and c-myc, as well as the activation of mitogen-activated protein kinases (MAPKs) and small ubiquitin-like modifier (SUMO) signaling (15, 22–24). Interestingly the SUMOylation process can downregulate the expression of the protective transcription factor p53 which in turn can be associated to the development of CV complications in hypertension. Furthermore, oscillatory flow induces the activation of the PI3Kinase-Akt pathway which leads to the assembly of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 and to the production of ROS (15), thus contributing to the vascular inflammation and remodeling. Other possible mechanisms involved in endothelial dysfunction triggered by oscillatory flow include the expression of the transcriptional factor Yes-associated protein (YAP) and its related coactivator PDZ binding motif (TAZ) that enhances cell cycle regulatory genes such as cyclin A1 (CNNA1) and E2F transcription factor 1 (E2F1) and increases inflammation and monocyte attachment, contributing in turn to endothelial dysfunction (15).
A large body of evidence over the past years has shown that ROS are involved in endothelium dysregulation. In the vascular system the major source of ROS production is NADPH oxidase whose expression is increased in hypertensive conditions by several stimuli including shear stress alterations, renin angiotensin system (RAS) and endothelin activation (25).
ROS are key signaling molecules through which vasoactive agents such as angiotensin II (Ang II), endothelin-1 and prostanoids mediate effects at cellular level, and may modify cell function through highly regulated redox-sensitive signal transduction. This may occur through the alteration of intracellular calcium homeostasis contributing to vasoconstriction, cell growth and inflammation which lead to hypertension development and hypertension mediated organ damage (HMOD) (26, 27). ROS stimulate multiple signaling pathways involved in inflammation, cell growth and vascular remodeling. These pathways include the activation of NF-κB, MAPK, JAK-2, STAT, p21Ras, Pyk-2 (Proline-rich Tyrosine Kinase 2) and AKT, receptor tyrosine kinases such as EGFR (Epidermal Growth Factor Receptor), IGFR (Insulin-like Growth Factor Receptor 1) and PDGFR (Platelet Derived Growth Factor Receptor), protein tyrosine phosphatases and redox-sensitive transcriptor factors such as Activator Protein 1 (AP)-1 and Hypoxia-inducible factor 1 (HIF-1) (15, 22–24, 28–33).
In hypertension, oxidative stress promotes aberrant cell signaling and post-translational modification (oxidation and phosphorylation) of proteins and in turn cell and tissue damage (34). In particular, protein phosphatases such as tyrosine phosphatases and protein serine/threonine phosphatases are inactive in the oxidized state, resulting in increased phosphorylation and activation of downstream protein targets involved in cell growth and inflammation which may contribute to vascular remodeling and hypertension development (34, 35) (Figure 1). ROS can also inhibit SIRT1 activity through oxidative modifications on its cysteine residues. Decreased activity of SIRT1 enhances the NF-κB signaling, which supports inflammatory responses (36). Moreover, reduced SIRT1 activity is associated with a decreased AMP-activated protein kinase (AMPK) activation, which results in a reduced expression of antioxidant enzymes such as manganese superoxide dismutase, catalase, γ glutamylcysteine synthase, and thioredoxin (37).
Increased ROS concentration induces the reduction of NO bioavailability by the increased quenching (34). Furthermore, the ROS-dependent phosphorylation of ERK5 by phosphokinase-C-ζ (PKCζ) and the activation of tumor necrosis factor α (TNFα)- mediated pathway induces the degradation of eNOS leading to the reduced production of NO concentration and in turn contributing further to endothelial dysfunction (34).
RAS and in particular its key effector Ang II play a fundamental role in the development of hypertension and its sequelae, contributing to endothelial dysfunction, cell growth, oxidative stress, vasoconstriction and inflammation. Ang II induces hyperplasia and hypertrophy of vascular smooth muscle cells (VSMC) in resistance arteries by modulating the endogenous production of mitogenic factors (including TGF-β (tumor growth factor-β), PDGF (platelet-derived growth factor), EGF (epidermal growth factor), IGF-1 (insulin-like growth factor 1) (38) and by enhancing basal superoxide production through the activation of cSrc, PKC, phospholipase A2 (PLA2) and phospholipase D (PLD) and increased NADPH oxidase and ROS generation (27, 39, 40). Moreover, Ang II stimulates the production of E-selectin and plasminogen activator inhibitor-1 (PAI-1), contributing to a prothrombotic state and to atherosclerotic plaque rupture (41). In addition, Ang II downregulates PPARs which have been largely demonstrated to reduce inflammatory response in experimental animals and to decrease serum markers of inflammation in humans (42). Through the stimulation of AT1 (Ang II type 1) receptors, Ang II also induces the synthesis of aldosterone which activates mineralocorticoid receptors enhancing inflammation, fibrosis, and endothelial damage (43).
As a matter of fact, RAS inhibitors and mineralocorticoid receptor antagonists have been demonstrated to reduce the proinflammatory and pro-fibrotic effects of Ang II and aldosterone, improving endothelial function and reducing oxidative stress (44).
Available evidence suggests that RAS blockade obtained by angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) is associated with improved function and structure of resistance arteries (6, 45, 46). The activation of complementary protective axes of the RAS may potentially contribute to the beneficial effects of RAS blockers. This includes the expression of angiotensin II type 2 receptor (AT2R) through the activation of a functional crosstalk between AT1R/AT2R during selective AT1R blockade (47–50). AT2R may contribute to improve endothelial dysfunction and arterial remodeling in hypertensive conditions, as its activation is linked to vasodilation, NO production and antiproliferative and anti-inflammatory effects (51). Thus, AT2R may participate to the mechanisms whereby therapeutic use of ARBs induces cardiovascular protection (52).
Experimental studies suggest that also the activation of ACE-2/Ang (1–7)/MasR axis may in part counteract the Ang II–induced actions in the cardiovascular system, including endothelial dysfunction, vasoconstriction and cell growth (53). In this regard, we have recently shown that ACE-2/Ang (1–7)/MasR axis plays an important role in arterial protection during selective AT1R blockade through the improvement of endothelial function and remodeling of resistance arteries via the reduction of ROS production and increased NO bioavailability (54) (Figure 1).
Other antihypertensive agents recommended in clinical practice have also shown vascular protective effects. Mineralocorticoid receptor antagonists have been demonstrated to reduce arterial stiffness and to improve endothelial function, measured by flow-mediated dilation (55). Calcium channel blockers have been demonstrated to have pleiotropic effects leading to the improvement of endothelial function and to the reduction of central aortic pressures (56). These effects are not directly linked to the antagonism of voltage-dependent calcium channels but rather are associated to the reduction of ET-1, monocyte chemoattractant protein-1 and C-reactive protein (57). Third generation beta-blockers with α1-adrenergic receptor antagonist activity have also been shown to improve endothelial function through antioxidant mechanisms and cause NO-dependent vasodilation (57) (Figure 2). Furthermore, endothelin receptor antagonists, might represent feasible future therapeutic agents to prevent endothelial dysfunction, vascular remodeling and organ damage in hypertension (58).
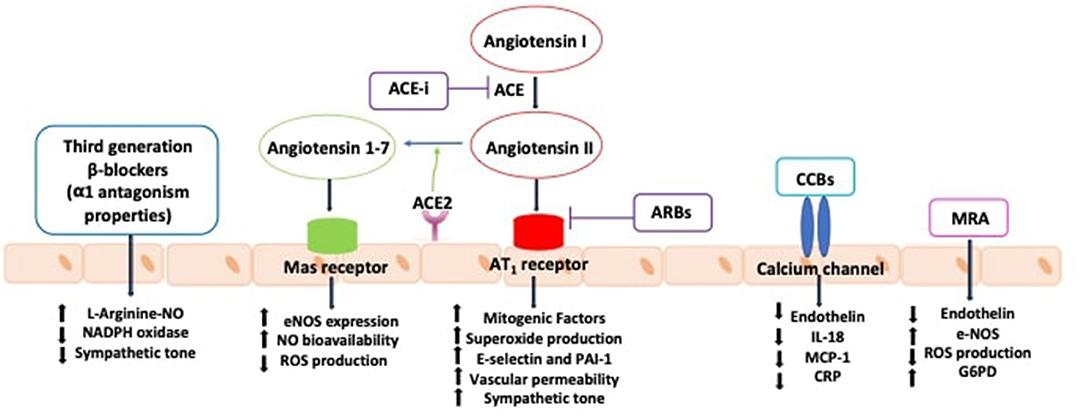
Figure 2. Effects of antihypertensive agents in improving endothelial function. ACE, angiotensin converting enzyme; ACE-i, angiotensin converting enzyme inhibitors; ARBs: angiotensin receptor blockers; AT1R, type 1 angiotensin II receptor; CCBs, calcium channel blockers; CRP, C-reactive protein; eNOS, nitric oxide synthase; G6PD, glucose-6-phosphate dehydrogenase; IL-18, interleukin-18; MCP-1, monocyte chemotactic protein 1; MRA, mineralocorticoid receptor antagonists; NAPDH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species.
Extracellular vesicles (EVs) are released in plasma from cells after the fusion of multivesicular bodies with the plasmic membrane and can deliver their cargo, including mRNA, microRNA (miRNA), small amounts of DNA, transcription factors, cytokines, and growth factors, to other cells in remote locations (59, 60). EVs can also be released into the extracellular space by neighboring cells through paracrine mechanisms along with the systemic release in plasma (61). Available findings have shown a correlation between endothelial dysfunction and circulating levels of EVs, particularly in patients with hypertension, coronary artery disease (CAD) and diabetes, although conflicting evidence exists with respect to their protective or harmful role (62–64). EVs have been identified as potential novel biomarkers and bioactivators in the development of hypertension affecting vascular tone in patients with endothelial dysfunction (65). It has been shown that EVs may reduce endothelial-dependent vasodilation and impair acetylcholine (ACh) induced vasorelaxation in a concentration-dependent manner. However, it has not been completely clarified how circulating EVs may affect resistance artery function during the basal state and when overt hypertension may occur (66). A recent study has demonstrated that an enriched EVs preparation from normotensive individuals (humans or rats) impair vasodilation in response to endothelial-dependent vasodilators, potentially through L-NAME inhibitory effects on eNOS. These findings support a paracrine/endocrine role of circulating EVs in the regulation of vascular tone in resistance arteries (67). Other animal studies showed that the dilatation of mouse mesenteric arteries induced by shear stress was impaired by the infusion of EVs isolated from diabetic patients (68) and that endothelial derived EVs decreased NO and increased ROS production, impairing ACh-mediated vasorelaxation, at aortic ring level (69).
On the other hand, EVs have shown beneficial effects on endothelial cells by inhibiting hyperproliferative pathways, through the activation of eNOS signaling mediated by miRNA (70, 71). Moreover, MiR-143/miR-145 contained in EVs has been shown to reduce atherosclerotic lesion formation in the aorta of ApoE–/– mice (72). MiR-19a72 and miR-23b70 mediate the atheroprotective laminar shear stress-induced cell cycle arrest via a decrease in E2F1 and hypophosphorylation of retinoblastoma or directly targeting cyclin D1 (73).
Interestingly, an increasing body of evidence have shown that long non-coding RNA (lncRNAs) can be selectively packaged into EVs and may act as regulators of endothelial function which may represent a promising therapeutic tool, although further studies are required to clarify the specific targets (74, 75).
Over the past decades several non-pharmacological (i.e., diet, antioxidants, and vitamin supplementation) and pharmacological approaches have been suggested in order to improve endothelial function as mentioned previously. Recently, selective, and more specific approaches such as nanoparticles have been proposed. Nanomedicine is emerging as an innovative approach with the aim to target specific endocytic pathways throughout the formulation of different nanoparticles, encapsulating therapeutic agents with enhanced bioavailability and ensuring treatment effectiveness. Indeed, the challenge to drug delivery in the endothelium consists in the selection of appropriate targets and in the design of nanoparticle-formulations with appropriate binding-properties to the vascular endothelium in micro, small, medium and in large vessels against continuous flowing blood (76, 77). In particular, the delivery through nanoparticles of many clinically used drugs such as antihypertensive agents, statins, antidiabetic drugs and interleukin 1β monoclonal antibodies may represent a potential target for treatment of endothelial dysfunction thus yielding potential new therapeutic approaches (78).
Future application may include the use of several types of small molecules that target complementary epigenetic pathways. More specifically, histone deacetylase inhibitors, DNA methyltransferase inhibitors, histone methyltransferases and demethylase inhibitors have been demonstrated to play an essential role in the regulation of endothelial stem/progenitor cell functions through modifying chromatin structure. In such a context, nanoparticles might be used to modulate the activities of epigenetic enzymes to enhance the vascular repair function of endothelial cells (79).
Several lines of evidence support the role of endothelium in physiology of peripheral arteries. Impairment of vascular flow, RAS activation, oxidative stress and inflammation have been demonstrated to play a fundamental role in the development of endothelial dysfunction in hypertensive patients, leading to vascular remodeling, atherosclerotic plaques progression and eventually increased risk of CV events (Figure 1).
The modulation of vascular inflammation through RAS blockers and other antihypertensive drugs is a well assessed therapeutic approach to improve vascular function and remodeling.
Recent evidence suggests that EVs have attracted increasing interest both as biomarkers or mediators of disease, as well as vehicles for delivering bioactive molecules, such as miRNA and drugs interacting with RAS, with potential beneficial effects on the endothelium. Hence EVs have emerged as important regulators of endothelial function and potentially as a promising novel therapeutic approach to improve endothelial dysfunction.
Moreover, a better molecular understanding of organ vasculature-bed heterogeneity and of organ/tissue microenvironment-governed endothelial cell phenotypic changes may represent the lead foundation for innovative tissue specific therapies (80).
GG, MV, and CS substantially contributed to the conception and design, drafted the article, and approved the final version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Alexander Y, Osto E, Schmidt-Trucksäss A, Shechter M, Trifunovic D, Duncker DJ, et al. Endothelial function in cardiovascular medicine: a consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc Res. (2021) 117:29–42. doi: 10.1093/cvr/cvaa085
2. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. (2015) 116:1231–44. doi: 10.1161/CIRCRESAHA.116.302855
3. Durier S, Fassot C, Laurent S, Boutouyrie P, Couetil JP, Fine E, et al. Physiological genomics of human arteries: quantitative relationship between gene expression and arterial stiffness. Circulation. (2003) 108:1845–51. doi: 10.1161/01.CIR.0000091407.86925.7A
4. Yu Y, Fukuda N, Yao EH, Matsumoto T, Kobayashi N, Suzuki R, et al. Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens. (2008) 21:72–7. doi: 10.1038/ajh.2007.5
5. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
6. Savoia C, D'Agostino M, Lauri F, Volpe M. Angiotensin type 2 receptor in hypertensive cardiovascular disease. Curr Opin Nephrol Hypertens. (2011) 20:125–32. doi: 10.1097/MNH.0b013e3283437fcd
7. Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci. (2007) 112:375–84. doi: 10.1042/CS20060247
8. Murray EC, Nosalski R, MacRitchie N, Tomaszewski M, Maffia P, Harrison DG, et al. Therapeutic targeting of inflammation in hypertension: from novel mechanisms to translational perspective. Cardiovasc Res. (2021) 117:2589–609. doi: 10.1093/cvr/cvab330
9. Park JB, Schiffrin EL. Small artery remodeling is the most prevalent (earliest?) form of target organ damage in mild essential hypertension. J Hypertens. (2001) 19:921–30. doi: 10.1097/00004872-200105000-00013
10. Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. (2004) 15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA
11. Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens. (2006) 15:152–8. doi: 10.1097/01.mnh.0000203189.57513.76
12. Savoia C, Schiffrin EL. Inhibition of the renin angiotensin system: implications for the endothelium. Curr Diab Rep. (2006) 6:274–8. doi: 10.1007/s11892-006-0060-5
13. Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, et al. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol. (2020) 17:52–63. doi: 10.1038/s41569-019-0239-5
14. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. (2014) 34:2191–8. doi: 10.1161/ATVBAHA.114.303422
15. Chatterjee S. Endothelial mechanotransduction, redox signaling and the regulation of vascular inflammatory pathways. Front Physiol. (2018) 9:524. doi: 10.3389/fphys.2018.00524
16. He M, Martin M, Marin T, Chen Z, Gongol B. Endothelial mechanobiology. APL Bioeng. (2020) 4:010904. doi: 10.1063/1.5129563
17. Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. (2015) 208:975–86. doi: 10.1083/jcb.201408103
18. Shentu TP, He M, Sun X, Zhang J, Zhang F, Gongol B, et al. AMP-activated protein kinase and sirtuin 1 coregulation of cortactin contributes to endothelial function. Arterioscler Thromb Vasc Biol. (2016) 36:2358–68. doi: 10.1161/ATVBAHA.116.307871
19. Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, et al. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. (2011) 193:867–84. doi: 10.1083/jcb.201010051
20. Davies PF, Civelek M, Fang Y, Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res. (2013) 99:315–27. doi: 10.1093/cvr/cvt101
21. Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol. (2014) 34:2378–86. doi: 10.1161/ATVBAHA.114.303428
22. Godo S, Shimokawa H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Rad Biol Med. (2017) 109:4–10. doi: 10.1016/j.freeradbiomed.2016.12.019
23. Godo S, Sawada A, Saito H, Ikeda S, Enkhjargal B, Suzuki K, et al. Disruption of physiological balance between nitric oxide and endothelium-dependent hyperpolarization impairs cardiovascular homeostasis in mice. Arterioscler Thromb Vasc Biol. (2016) 36:97–107. doi: 10.1161/ATVBAHA.115.306499
24. Gutterman DD, Chabowski DS, Kadlec AO, Durand MJ, Freed JK, Ait-Aissa K, et al. The human microcirculation: regulation of flow and beyond. Circ Res. (2016) 118:157–72. doi: 10.1161/CIRCRESAHA.115.305364
25. Poznyak AV, Grechko AV, Orekhova VA, Khotina V, Ivanova EA, Orekhov AN. NADPH oxidases and their role in atherosclerosis. Biomedicines. (2020) 8:206. doi: 10.3390/biomedicines8070206
26. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
27. Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. (2004) 122:339–52. doi: 10.1007/s00418-004-0696-7
28. Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, et al. Shear stress regulation of Krüppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun. (2006) 341:1244–51. doi: 10.1016/j.bbrc.2006.01.089
29. Wang X, Fang X, Zhou J, Chen Z, Zhao B, Xiao L, et al. Shear stress activation of nuclear receptor PXR in endothelial detoxification. Proc Natl Acad Sci USA. (2013) 110:13174–9. doi: 10.1073/pnas.1312065110
30. Guo D, Chien S, Shyy JY. Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ Res. (2007) 100:564–71. doi: 10.1161/01.RES.0000259561.23876.c5
31. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA. (2000) 97:9052–7. doi: 10.1073/pnas.97.16.9052
32. Wilson SH, Caplice NM, Simari RD, Holmes Jr DR, Carlson PJ, et al. Activated nuclear factor-kappaB is present in the coronary vasculature in experimental hypercholesterolemia. Atherosclerosis. (2000) 148:23–30. doi: 10.1016/S0021-9150(99)00211-7
33. Orr AW, Stockton R, Simmers MB, Sanders JM, Sarembock IJ, Blackman BR, et al. Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. (2007) 176:719–27. doi: 10.1083/jcb.200609008
34. Touyz RM, Rios FJ, Alves-Lopes R, Neves KB, Camargo LL, Montezano AC. Oxidative stress: a unifying paradigm in hypertension. Can J Cardiol. (2020) 36:659–70. doi: 10.1016/j.cjca.2020.02.081
35. Tabet F, Schiffrin EL, Callera GE, He Y, Yao G, Ostman A, et al. Redox-sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circ Res. (2008) 103:149–58. doi: 10.1161/CIRCRESAHA.108.178608
36. Zhang W, Huang Q, Zeng Z, Wu J, Zhang Y, Chen Z. Sirt1 inhibits oxidative stress in vascular endothelial cells. Oxidative Med Cell Longev. (2017) 2017:7543973. doi: 10.1155/2017/7543973
37. Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. (2009) 105:114–27. doi: 10.1161/CIRCRESAHA.109.201590
38. Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, et al. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol. (2006) 26:e133–7. doi: 10.1161/atvb.26.9.1976
39. Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol. (2003) 30:860–6. doi: 10.1046/j.1440-1681.2003.03930.x
40. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. (2000) 52:639–672.
41. Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2003) 23:981–7. doi: 10.1161/01.ATV.0000069236.27911.68
42. Diep QN, Amiri F, Touyz RM, Cohn JS, Endemann D, Neves MF, et al. PPARalpha activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension. (2002) 40:866–71. doi: 10.1161/01.HYP.0000037969.41360.CC
43. Joffe HV, Adler GK. Effect of aldosterone and mineralocorticoid receptor blockade on vascular inflammation. Heart Fail Rev. (2005) 10:31–7. doi: 10.1007/s10741-005-2346-0
44. Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. (2002) 40:504–10. doi: 10.1161/01.HYP.0000034738.79310.06
45. Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. (2014) 16:431. doi: 10.1007/s11906-014-0431-2
46. Ghiadoni L, Taddei S, Virdis A. Hypertension and endothelial dysfunction: therapeutic approach. Curr Vasc Pharmacol. (2012) 10:42–60. doi: 10.2174/157016112798829823
47. Savoia C, Sada L, Zezza L, Pucci L, Lauri FM, Befani A, et al. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens. (2011) 2011:281240. doi: 10.4061/2011/281240
48. Cosentino F, Savoia C, De Paolis P, Francia P, Russo A, Maffei A, et al. Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists. Am J Hypertens. (2005) 18:493–9. doi: 10.1016/j.amjhyper.2004.11.007
49. Savoia C, Touyz RM, Volpe M, Schiffrin EL. Angiotensin type 2 receptor in resistance arteries of type 2 diabetic hypertensive patients. Hypertension. (2007) 49:341–6. doi: 10.1161/01.HYP.0000253968.95136.b8
50. Kaschina E, Namsolleck P, Unger T. AT2 receptors in cardiovascular and renal diseases. Pharmacol Res. (2017) 125:39–47. doi: 10.1016/j.phrs.2017.07.008
51. Carey RM, Park J. Role of angiotensin type 2 receptors in vasodilation of resistance and capacitance vessels. Hypertension. (2006) 48:824–5. doi: 10.1161/01.HYP.0000244109.55948.bc
52. Siragy HM. The role of the AT2 receptor in hypertension. Am J Hypertens. (2000) 13:62S−7S. doi: 10.1016/S0895-7061(00)00274-0
53. Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. (2013) 216:R1–17. doi: 10.1530/JOE-12-0341
54. Savoia C, Arrabito E, Parente R, Nicoletti C, Madaro L, Battistoni A, et al. Mas receptor activation contributes to the improvement of nitric oxide bioavailability and vascular remodeling during chronic AT1R (angiotensin type-1 receptor) blockade in experimental hypertension. Hypertension. (2020) 76:1753–61. doi: 10.1161/HYPERTENSIONAHA.120.15527
55. Sakima A, Arima H, Matayoshi T, Ishida A, Ohya Y. Effect of mineralocorticoid receptor blockade on arterial stiffness and endothelial function: a meta-analysis of randomized trials. Hypertension. (2021) 77:929–37. doi: 10.1161/HYPERTENSIONAHA.120.16397
56. Williams B, Lacy PS, Thom SM, Cruickshank K, Stanton A, Collier D, et al. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. (2006) 113:1213–25. doi: 10.1161/CIRCULATIONAHA.105.595496
57. Silva I, de Figueiredo RC, Rios D. Effect of different classes of antihypertensive drugs on endothelial function and inflammation. Int J Mol Sci. (2019) 20:3458. doi: 10.3390/ijms20143458
58. Schiffrin EL. Vascular endothelin in hypertension. Vasc Pharmacol. (2005) 43:19–29. doi: 10.1016/j.vph.2005.03.004
59. Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. (2014) 3:23743. doi: 10.3402/jev.v3.23743
60. Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, et al. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res. (2015) 116:255–63. doi: 10.1161/CIRCRESAHA.116.304360
61. Jansen F, Li Q, Pfeifer A, Werner N. Endothelial- and immune cell-derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl Sci. (2017) 2:790–807. doi: 10.1016/j.jacbts.2017.08.004
62. Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Leftheriotis G, et al. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol. (2008) 173:1210–9. doi: 10.2353/ajpath.2008.080228
63. Amabile N, Cheng S, Renard JM, Larson MG, Ghorbani A, McCabe E, et al. Association of circulating endothelial microparticles with cardiometabolic risk factors in the Framingham Heart Study. Eur Heart J. (2014) 35:2972–9. doi: 10.1093/eurheartj/ehu153
64. Chen Y, Feng B, Li X, Ni Y, Luo Y. Plasma endothelial microparticles and their correlation with the presence of hypertension and arterial stiffness in patients with type 2 diabetes. J Clin Hypertens. (2012) 14:455–60. doi: 10.1111/j.1751-7176.2012.00631.x
65. Wang JM, Su C, Wang Y, Huang YJ, Yang Z, Chen L, et al. Elevated circulating endothelial microparticles and brachial-ankle pulse wave velocity in well-controlled hypertensive patients. J Hum Hypertens. (2009) 23:307–15. doi: 10.1038/jhh.2008.137
66. Burger D, Turner M, Munkonda MN, Touyz RM. Endothelial microparticle-derived reactive oxygen species: role in endothelial signaling and vascular function. Oxidative Med Cell Longev. (2016) 2016:5047954. doi: 10.1155/2016/5047954
67. Good ME, Musante L, La Salvia S, Howell NL, Carey RM, Le TH, et al. Circulating extracellular vesicles in normotension restrain vasodilation in resistance arteries. Hypertension. (2020) 75:218–28. doi: 10.1161/HYPERTENSIONAHA.119.13363
68. Martin S, Tesse A, Hugel B, Martínez MC, Morel O, Freyssinet JM, et al. Shed membrane particles from T lymphocytes impair endothelial function and regulate endothelial protein expression. Circulation. (2004) 109:1653–9. doi: 10.1161/01.CIR.0000124065.31211.6E
69. Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circul Physiol. (2004) 286:H1910–5. doi: 10.1152/ajpheart.01172.2003
70. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. (2007) 9:654–9. doi: 10.1038/ncb1596
71. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/ncb2441
72. Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, et al. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol. (2014) 74:139–50. doi: 10.1016/j.yjmcc.2014.05.001
73. Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, et al. Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci USA. (2010) 107:3234–9. doi: 10.1073/pnas.0914825107
74. Voellenkle C, Garcia-Manteiga JM, Pedrotti S, Perfetti A, De Toma I, Da Silva D, et al. Implication of long noncoding RNAs in the endothelial cell response to hypoxia revealed by RNA-sequencing. Sci Rep. (2016) 6:24141. doi: 10.1038/srep24141
75. Fiedler J, Breckwoldt K, Remmele CW, Hartmann D, Dittrich M, Pfanne A, et al. Development of long noncoding RNA-based strategies to modulate tissue vascularization. J Am Coll Cardiol. (2015) 66:2005–2015. doi: 10.1016/j.jacc.2015.07.081
76. Cicha I. Strategies to enhance nanoparticle-endothelialinteractions under flow. J Cell Biotehnol. (2016) 1:191–208. doi: 10.3233/JCB-15020
77. Sharma AK, Kumar A, Taneja G, Nagaich U, Deep A, Datusalia AK, et al. Combined and individual strategy of exercise generated preconditioning and low dose copper nanoparticles serve as superlative approach to ameliorate ISO-induced myocardial infarction in rats. Pharmacol Rep. (2018) 70:789–95. doi: 10.1016/j.pharep.2018.02.023
78. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. (2021) 73:924–67. doi: 10.1124/pharmrev.120.000096
79. Fraineau S, Palii CG, Allan DS, Brand M. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. (2015) 282:1605–29. doi: 10.1111/febs.13183
Keywords: endothelium, inflammation, angiotensin II, vascular function, reactive oxygen species (ROS)
Citation: Gallo G, Volpe M and Savoia C (2022) Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 8:798958. doi: 10.3389/fmed.2021.798958
Received: 20 October 2021; Accepted: 23 December 2021;
Published: 20 January 2022.
Edited by:
Eleni Gavriilaki, G. Papanikolaou General Hospital, GreeceReviewed by:
Sanda Predescu, Rush University Medical Center, United StatesCopyright © 2022 Gallo, Volpe and Savoia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmine Savoia, Y2FybWluZS5zYXZvaWFAdW5pcm9tYTEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.