
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 27 January 2022
Sec. Rheumatology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.785744
This article is part of the Research Topic Chronic Rheumatic Inflammatory Conditions and Cardiovascular Health View all 18 articles
 Boglárka Soós1†
Boglárka Soós1† Miklós Fagyas2†Ágnes Horváth1Edit Végh1Anita Pusztai1Monika Czókolyová1Alexandra Csongrádi2Attila Hamar1Zsófia Pethő1Nóra Bodnár1György Kerekes3Katalin Hodosi1Éva Szekanecz4Szilvia Szamosi1Sándor Szántó1,5Gabriella Szűcs1
Miklós Fagyas2†Ágnes Horváth1Edit Végh1Anita Pusztai1Monika Czókolyová1Alexandra Csongrádi2Attila Hamar1Zsófia Pethő1Nóra Bodnár1György Kerekes3Katalin Hodosi1Éva Szekanecz4Szilvia Szamosi1Sándor Szántó1,5Gabriella Szűcs1 Zoltán Papp2‡
Zoltán Papp2‡ Zoltán Szekanecz1*‡
Zoltán Szekanecz1*‡Introduction: Angiotensin-converting enzyme (ACE) and ACE2 have been implicated in the regulation of vascular physiology. Elevated synovial and decreased or normal ACE or ACE2 levels have been found in rheumatoid arthritis (RA). Very little is known about the effects of tumor necrosis factor α (TNF-α) inhibition on ACE or ACE2 homeostasis. In this study, we assessed the effects of one-year anti-TNF therapy on ACE and ACE2 production in RA and ankylosing spondylitis (AS) in association with other biomarkers.
Patients and Methods: Forty patients including 24 RA patients treated with either etanercept (ETN) or certolizumab pegol (CZP) and 16 AS patients treated with ETN were included in a 12-month follow-up study. Serum ACE levels were determined by commercial ELISA, while serum ACE2 activity was assessed using a specific quenched fluorescent substrate. Ultrasonography was performed to determine flow-mediated vasodilation (FMD), common carotid intima-media thickness (ccIMT) and arterial pulse-wave velocity (PWV) in all patients. In addition, CRP, rheumatoid factor (RF) and ACPA were also measured. All assessments were performed at baseline and 6 and 12 months after treatment initiation.
Results: Anti-TNF therapy increased ACE levels in the full cohort, as well as in the RA and AS subsets. ACE2 activity increased in the full cohort, while the ACE/ACE2 ratio increased in the full cohort and in the RA subset (p < 0.05). Uni- and multivariable regression analyses determined associations between ACE or ACE/ACE2 ratios at different time points and disease duration, CRP, RF, FMD and IMT (p < 0.05). ACE2 activity correlated with CRP. The changes of ACE or ACE2 over 12 months were determined by treatment together with either RF or FMD (p < 0.05).
Conclusions: Anti-TNF treatment may increase ACE and ACE2 in the sera of RA and AS patients. ACE and ACE2 may be associated with disease duration, markers of inflammation and vascular pathophysiology. The effects of TNF inhibition on ACE and ACE2 may reflect, in part, the effects of these biologics on the cardiovascular system.
Rheumatoid arthritis (RA) and ankylosing spondylitis (AS) have been associated with increased cardiovascular (CV) morbidity and mortality (1–5). Non-invasive ultrasound-based techniques are suitable to assess preclinical vascular pathophysiology in RA and AS (1, 6). Early endothelial dysfunction of the brachial artery, carotid atherosclerosis and increased arterial stiffness are indicated by abnormal endothelium-dependent, flow-mediated vasodilation (FMD) (4, 6, 7), common carotid intima-media thickness (IMT) and carotid plaques (4–6, 8), as well as arterial pulse-wave velocity (PWV) (5, 6, 9), respectively.
Systemic inflammation and pro-inflammatory cytokines including tumor necrosis factor α (TNF-α) are involved in the pathogenesis of arthritis-associated secondary atherosclerosis and CV disease (10, 11). Anti-TNF agents are effective and safe in the therapy of RA and AS (12–16). TNF inhibitors also suppress synovial angiogenesis and vascular endothelial growth factor (VEGF) production (17, 18). The control of inflammation by targeted therapies including TNF-α inhibitors, may decrease CV morbidity and mortality in arthritides (1, 10, 14, 19), especially in anti-TNF responder patients (14, 20). Anti-TNF biologics may improve or at least stabilize vascular physiology indicated by FMD, IMT and PWV (14, 19, 21–24).
Angiotensin-converting enzyme (ACE) is a member of the renin-angiotensin-aldosterone system (RAAS), which is an important regulator of blood pressure and salt-water homeostasis (25). ACE catalyzes the conversion of angiotensin I to angiotensin II, and the metabolism of bradykinin (25). ACE has been implicated in CV disease, myocardial infarction, hypertension, heart failure and diabetic nephropathy (25, 26). ACE inhibitors are among the most frequently prescribed drugs with antihypertensive and cardioprotective effects (25, 26). ACE2 is an ACE homolog with monocarboxypeptidase activity (27). ACE2 generates angiotensin peptides Ang1−9 and Ang1−7 from Ang-I and Ang-II, respectively (27, 28). ACE2, through Ang1−7, is capable of reducing myocardial oxidative stress and pathological remodeling (28, 29). TNF-α converting enzyme (TACE/ADAM17) is responsible for ACE2 shedding from cardiomyocytes and endothelial cells (30). Thus, opposite to the Ang-I-ACE-Ang-II pathway, ACE2 exerts vasculoprotective and antihypertensive mechanisms by the counter-regulation of the RAAS system (28, 31). Increased soluble ACE2 activity has been associated with advanced heart failure (32), ventricular arrhythmias (33) and hypertension (28).
ACE and ACE2 concentrations and activity are readily measurable in the serum. ACE activity is affected by the intake of ACE inhibitors while ACE2 activity is not (26, 28). Changes in soluble ACE and ACE2 may reflect redistribution and opposite changes of tissue activity of these enzymes (28, 34).
With respect to ACE and ACE2 in arthritides, ACE insertion-deletion (I/D) gene polymorphism has been associated with RA and AS in some populations, primarily in cohort studies carried out in Arabic countries (35–40). When studying DD, ID and II genotypes, the frequency of the D allele was higher in RA compared to healthy controls (35). Moreover, the DD genotype may confer increased susceptibility to RA (35, 38, 40). Results in AS are controversial. In one study, similarly to RA, the DD genotype has been associated with AS including sacroiliac and ocular involvement (39). Moreover, in AS, carrying the D allele was correlated with higher CRP levels (41). In contrast, another study reported association between the I allele and AS (42). The II genotype was also associated with juvenile idiopathic arthritis (37). ACE polymorphisms could not be correlated with psoriatic arthritis (PsA) (36).
Regarding serum or plasma ACE and ACE2 levels, the very first study on ACE serum and synovial fluid ACE levels in various arthritides was published as early as in 1986. In this study, serum ACE levels were similar in RA, AS, PsA, osteoarthritis (OA) patients and healthy controls. On the other hand, synovial fluid ACE levels were increased in RA vs. OA (43). More recently, 50 RA compared to 30 healthy women, RA patients had increased Ang-II, Ang1−7 and ACE plasma levels, as well as ACE/ACE2 ratios vs. controls. RA was associated with lower Ang-II/Ang1−7 ratios. ACE inhibitors did not significantly influence serum Ang-II, Ang1−7, ACE and ACE2 levels in RA patients. ACE2 levels inversely correlated with carotid IMT (44). Decreased ACE2 levels were also found in RA, systemic sclerosis (SSc) and systemic lupus erythematosus (SLE) vs. healthy controls (45). In contrast, other studies found similar serum ACE levels in RA, OA and healthy individuals (46). Increased synovial fluid ACE concentrations have been described in RA compared to OA (46, 47). Within the RA synovial tissue, ACE expression is localized to endothelial cells and synovial macrophages (48).
With respect to the regulation of ACE and ACE2 production in arthritides, in animal models of arthritis, Ang1−7 exerted significant anti-inflammatory effects as it attenuated oxidative stress, as well as TNF-α, interleukin 1 (IL-1) and IL-6 production (49). Moreover, IL-6 upregulated ACE2 in synovial tissues (50). Anti-ACE2 antibodies that inhibit the anti-inflammatory and anti-fibrotic effects of ACE2 have been described in connective tissue diseases with constrictive vasculopathies, such as SSc, SLE and mixed connective tissue disease (MCTD) (51, 52).
Therapeutically, both ACE inhibitors and angiotensin receptor blockers (ARB) may exert significant anti-inflammatory effects (53). However, in the multiple regression analysis of RA patients receiving ACE inhibitors or ARBs in comparison to those not taking such drugs, the use of either ACE inhibitors or ARBs was not associated with disease activity (54).
There have been very few studies on the possible effects of TNF inhibitors on ACE and ACE2. In a small study, ACE2 plasma levels were significantly lower in RA patients on anti-TNF treatment compared to healthy controls (55). In the study of RA, SSc and SLE patients, most antirheumatic treatments did not affect ACE2 levels (45). We have not found any studies where changes in ACE or ACE2 levels or activity were evaluated upon anti-TNF therapy.
Thus, ACE and ACE2 may be involved in the inflammatory processes underlying RA and AS, as well as in vascular pathology associated with these arthritides. Yet, very little information has become available on the effects of anti-TNF therapy on ACE and ACE2 production and on their correlation with disease activity, markers of inflammation, autoantibodies and vascular pathophysiology. We have recently set up a mixed cohort of RA and AS patients and reported multiple effects of anti-TNF treatment over one year on vascular pathophysiology (19) and various vascular biomarkers (56, 57). We have published data on vascular pathophysiology and bone in the very same cohort before (19, 56, 58–60). As a novelty in comparison to the previous publications of the same cohort, now we wished to study ACE and ACE2 production in context with inflammation, autoantibodies and vascular pathophysiology in the very same cohort in order to obtain more information on the possible effects of biologics on the RAAS.
Fifty one patients with inflammatory arthritis (35 RA and 16 axial radiographic AS) selected for the initiation of anti-TNF therapy but unselected for CV disease (any previous CV events) were enrolled in the study as described before (19, 56, 57). These patients were consecutively selected in one tertiary rheumatology center (University of Debrecen). Patient characteristics in the full, RA and AS cohorts are seen in Table 1. The full cohort included 33 women and 18 men with mean age of 51.4 ± 11.8 (range: 24–83) years, while mean age at diagnosis was 43.1 ± 10.8 (range: 11–71) years. The mean disease duration was 8.3 ± 7.6 (range: 1–44) years. Exclusion criteria included untreated, unstable hypertension (blood pressure >140/90 mmHg), current inflammatory disease other than RA or AS, infectious disease or renal failure (based on eGFR and hospital records). None of patients received antiplatelet (e.g., aspirin, clopidogrel) or anticoagulant therapy (e.g., heparin, warfarin) at the time of inclusion. As antihypertensive drugs may affect the vascular status, hypertension had been stabilized for at least 6 months before the onset of this study. Moreover, antihypertensive drugs remained unchanged throughout the study. Some patients received ACE inhibitors prior to the study. However, we have previously demonstrated that ACE inhibitor treatment does not have any effects on circulating ACE concentration or ACE2 activity (28).
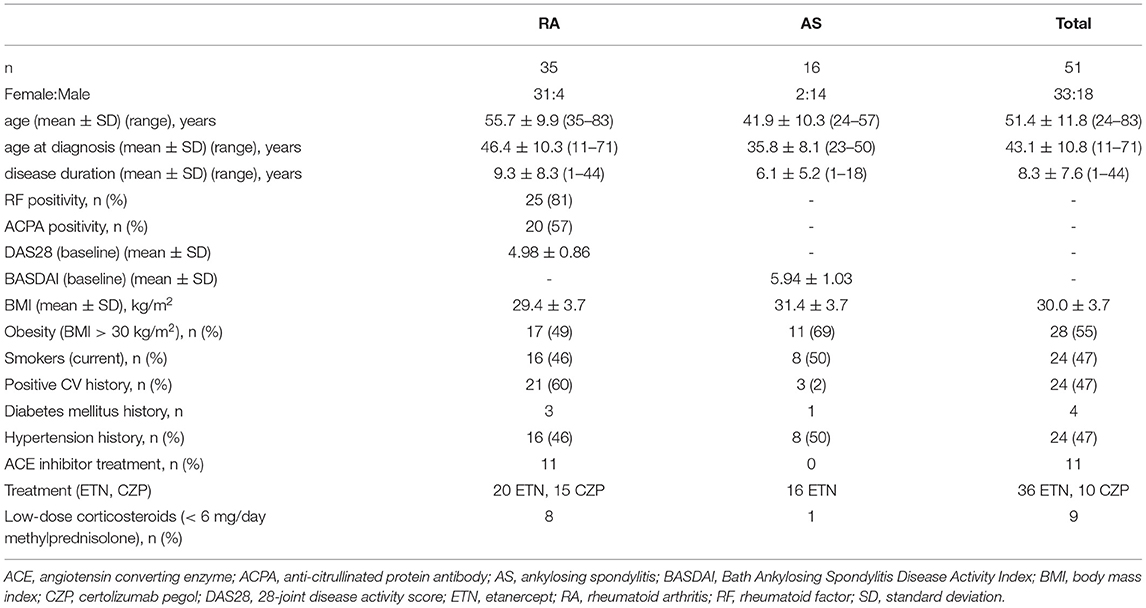
Table 1. Patient characteristics.
Patients with active disease were recruited prior to initiating a biological therapy. At baseline RA patients had a mean DAS28 of 4.98 ± 10.86, while AS patients exerted mean BASDAI of 5.94 ± 1.03. All patients started on an anti-TNF therapy at baseline and continued the same biological treatment during one year. Among the 35 RA patients, 20 received etanercept (ETN) 50 mg/week subcutaneous (SC) and 15 received certolizumab pegol (CZP) (400 mg at 0, 2 and 4 weeks, and thereafter 200 mg every two weeks SC). Altogether 12 RA patients were treated with ETN and eight with CZP in combination with methotrexate (MTX). The other patients received anti-TNF monotherapy. RA patients did not take DMARDs other than MTX. All 16 AS patients received ETN monotherapy 50 mg/week SC. Altogether eight RA and one AS patients took low-dose (<6 mg/day) methylprednisolone (Table 1).
The study was approved by the Hungarian Scientific Research Council Ethical Committee (approval No. 14804-2/2011/EKU). Written informed consent was obtained from each patient and assessments were carried out according to the Declaration of Helsinki.
First, detailed medical history was taken. We inquired for history of CVD, as well as current smoking, experience of chest pain resembling angina pectoris, hypertension and diabetes mellitus during the last 2 years prior to the start of this study by a questionnaire (Table 1). We also determined body mass index (BMI) and obesity (Table 1). Further clinical assessments including physical examination were performed at baseline (B), and after 6 (6 M) and 12 months (12 M) of therapy. At baseline RA patients had a mean DAS28 of 4.98±0.86, while AS patients exerted mean BASDAI of 5.94 ± 1.03 (Table 1).
Blood samples were collected from patients by using a standard aseptic technique. Native blood was incubated for 60 min at room temperature; serum fractions (separated by centrifugation at 1,500 g for 15 min) were stored at −20°C until further use.
Serum high sensitivity C reactive protein (hsCRP; normal: ≤ 5 mg/l) and IgM rheumatoid factor (RF; normal: ≤ 50 IU/ml) were measured by quantitative nephelometry (Cobas Mira Plus-Roche), using CRP and RF reagents (both Dialab). ACPA (anti-CCP; aCCP) autoantibodies were detected in serum samples using a second generation Immunoscan-RA CCP2 ELISA test (Euro Diagnostica; normal: ≤ 25 IU/ml). The assay was performed according to the manufacturer's instructions.
In order to exclude the possible interference effect of RF, we compared ACE concentration and ACE2 activity values in RF positive and negative patients at baseline. We did not find statistically significant differences between positive or negative patients, thus presence of RF in the sample may not interfere with the tests (data not shown in figure).
Serum ACE concentration was determined by a commercial human ACE ELISA development kit (R&D Systems) according to the manufacturer's instructions, with minor modifications, as described previously (61). Enzyme-linked immunosorbent plates (Greiner Bio-One) were coated with 80 ng/well capture antibody, and the remaining binding sites were then blocked with reagent diluent (10 mg/mL bovine serum albumin [Sigma-Aldrich] in Dulbecco's phosphate buffered saline solution [PBS, Gibco]). Diluted sera (in reagent diluent, 100-fold dilution) were added to the wells, and the antibody-antigen complexes were labeled with a biotinylated detection antibody (20 ng/well). Two hundred-fold-diluted streptavidin-conjugated horseradish-peroxidase (kit component) was added to the wells. Finally, the amounts of complexes were detected with a substrate solution containing 0.3 mg/mL tetramethylbenzidine, 0.1 mM H2O2 and 50 mM acetic acid. The reaction was terminated after 20 min by the addition of 0.5 M HCl, and the optical density was measured at 450 nm. Serum ACE concentration rather than activity was measured as 11 RA patients had been receiving ACE inhibitor treatment (Table 1). ACE levels are expressed in ng/mL units.
Serum ACE2 activity was determined using a specific quenched fluorescent substrate as previously described (28). The reaction mixture (200 μL) contained 20 μl serum, 80 μL buffer and 100 μl (100 μM) ACE2-specific fluorescent substrate (7-methoxycoumarin-4-yl)acetyl-Ala-Pro-Lys(2,4-dinitrophenyl)-OH [Mca-APK(Dnp)] (Peptide 2.0, USA). Serum ACE2 activity was measured by fluorometric assay of the enzymatic cleavage of K(Dnp) from the fluorogenic substrate Mca-APK(Dnp). The reaction mixture contained 500 mM NaCl, 10 μM ZnCl2 and 75 mM TRIS HCl, pH 6.5. All chemicals were from Sigma (St. Louis, MO, USA) if not stated otherwise. The reaction was performed in black 96-well microtiter plates (Greiner Bio-One, Frickenhauser, Germany). The assay was monitored continuously by measuring the increase in fluorescence (excitation wavelength = 340 nm, emission wavelength = 405 nm) upon substrate hydrolysis using a fluorescence microplate reader (NOVOstar; BMG Labtech GmbH, Offenburg, Germany). Initial enzyme activities were determined from the linear rate of fluorescence increase over the 0–120 min time course. The increase in fluorescence was plotted as a function of reaction time and fitted with a linear regression. Serum ACE2 activity was calculated by the equation:
ACE2 activity = (S/k) * D
S: rate of observed increase in fluorescence intensity;
k: change in fluorescence intensity upon the complete cleavage of 0.1 nmol of Mca-APK(Dnp);
D: dilution of the serum sample.
One unit of fluorescence (UF) corresponds to the quantity of enzyme which can degrade 0.1 nmol Mca-APK(Dnp) in one h at 37°C. The specificity of the serum ACE2 enzyme activity assay was tested using the specific human ACE2 inhibitor DX600 before (28). ACE2 activity is expressed in UF/mL units.
ACE inhibitors do not interfere with ACE2 activity.
The FMD, IMT and PWV assessments carried out in the very same cohort were performed and published previously (19).
Statistical analysis was performed using SPSS version 22.0 (IBM) software. Normally distributed data are expressed as the mean ± SD for continuous variables and percentages for categorical variables. Continuous variables were evaluated by paired two-tailed t-test and Wilcoxon test. Nominal variables were compared between groups using the chi-squared or Fisher's exact test, as appropriate. Matched data with not normal distribution are expressed in median [interquartile range] and compared with Wilcoxon matched-pairs signed rank tests. Correlations were determined by Pearson's and Spearman's analyses. Univariate and multiple regression analysis using the stepwise method was applied to investigate independent associations between angiogenic biomarkers (dependent variables) and other clinical, laboratory and imaging parameters (independent variables). The β standardized linear coefficients showing linear correlations between two parameters were determined. The B (+95% CI) regression coefficient indicated independent associations between dependent and independent variables during changes. Repeated measures analysis of variance (RM-ANOVA) was performed in order to determine the additional effects of multiple parameters on changes of vascular imaging markers between B and 12 M. The dependent variables were FMD, ccIMT and PWV. Partial η2 is given as indicator of effect size, with values of 0.01 suggesting small, 0.06 medium and 0.14 large effects. In all analyses, P values < 0.05 were considered significant.
Separate vascular imaging, as well as disease activity, inflammatory and vascular biomarker data obtained in the very same cohort have been published (19, 56, 57). Here we used those data to associate them with the ACE and ACE2 measurements. None of the data presented here have been published elsewhere.
In the mixed cohort of 51 arthritis (RA+AS) patients, serum ACE concentration significantly increased after 6 M (166.7 [124–232] ng/mL; p = 0.003) and 12 M of treatment (183.4 [121–222] ng/mL; p < 0.001) compared to B (142.7 [88–176] ng/mL). In the RA subset, ACE concentration also increased after 6 M (183.1 [140–291]; p = 0.006) and 12 M (186.6 [137–338] ng/mL; p = 0.001) vs. B (150.3 [131–198] ng/mL). Finally, in AS, ACE concentration did not change significantly after 6 M (118.7 [87–182] ng/mL; p = 0.245), however, it significantly increased after 12 M (140.5 [95–190] ng/mL; p = 0.043) compared to B (127.1 [76–154] ng/mL) (Figure 1A). ACE levels in RA and AS did not differ at B (p = 0.055). On the other hand, ACE concentration was significantly higher in RA compared to AS after 6 M (p = 0.004) and 12 M of treatment (p = 0.024) (Figure 1A).
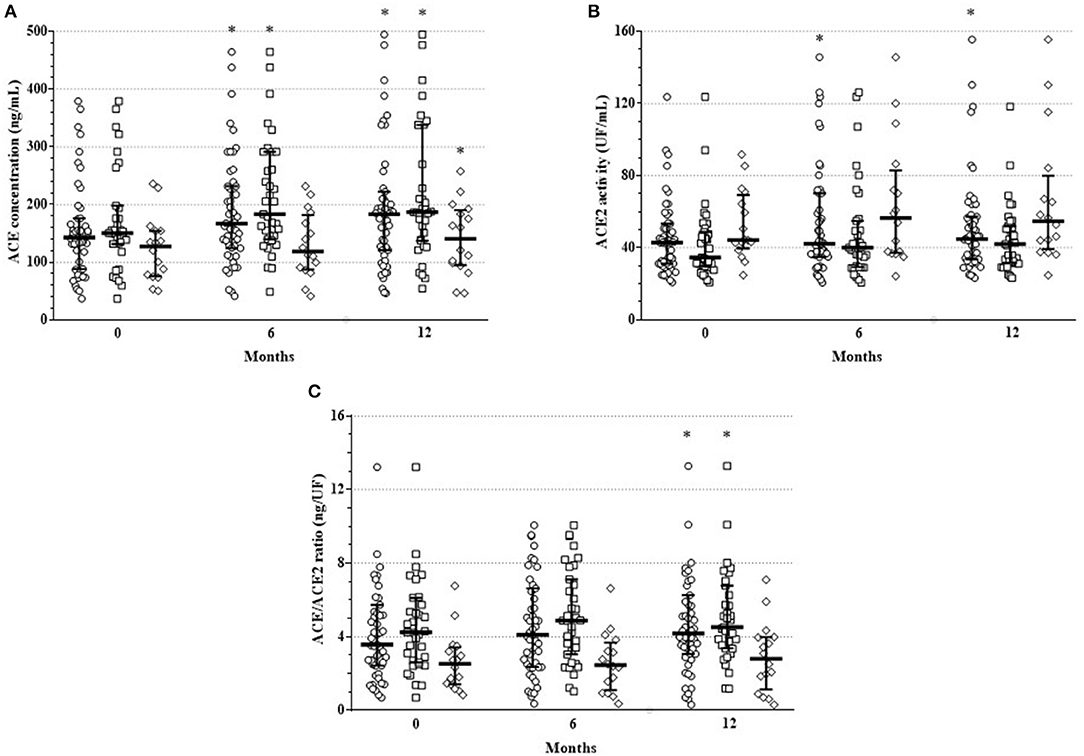
Figure 1. One-year changes of (A) ACE concentration, (B) ACE2 activity and (C) ACE/ACE2 ratio upon TNF inhibition in the full RA + AS cohort (cycle symbol), as well as in the RA (square symbol) and AS subsets (diamond symbol). Median and interquartile range are shown on the figure; each symbol corresponds to an individual value. *indicate significant differences compared to baseline using Wilcoxon matched-pairs signed rank tests (p < 0.05).
In the full RA+AS cohort, ACE2 activity significantly increased after 6 M (41.1 [35–70] UF/mL; p = 0.044) and 12 M (44.7 [34–57] UF/ml; p = 0.010) compared to B (42.8 [31–53] UF/mL). In RA, ACE2 activity did not change after 6 M (40.1 [29–55] UF/mL; p = 0.201) and after 12 M (42.0 [32–52] UF/mL; p = 0.080) vs. B (34.6 [30–49] UF/mL). Similarly, in AS, ACE2 activity remained unchanged after 6 M (56.6 [37–83] UF/mL; p = 0.088) and 12 M (54.7 [39–80] UF/mL; p = 0.063) compared to B (44.3 [40–69] UF/mL; p = 0.039) (Figure 1B). ACE2 activity was higher in AS than in RA at B (p = 0.037) and after 12 M (p = 0.019) (Figure 1B).
Finally, in order to reflect the ACE/ACE2 balance, we calculated ratios of ACE concentrations and ACE2 activity (ACE/ACE2 ratios) (Figure 1C). In the mixed cohort, ACE/ACE2 ratio did not change after 6 M (4.1 [2.4–6.6] ng/UF; p = 0.083) but significantly increased after 12 M (4.2 [3.1–6.3] ng/UF; p = 0.019) compared to B (3.6 [2.4–5.7] ng/UF). In RA, ACE/ACE2 ratio did not change after 6 M (4.87 [3.0–7.1] ng/UF; p = 0.069) but significantly increased after 12 M (4.52 [3.4–6.8] ng/UF; p = 0.035) vs. B (4.24 [2.6–6.1] ng/UF). In AS, ACE/ACE2 ratio remained unchanged after 6 M (2.46 [1.1–3.7] ng/UF; p = 0.990) and 12 M (2.8 [1.1–4.0] ng/UF; p = 0.501) compared to B (2.53 [1.4–3.4] ng/UF) (Figure 1C). ACE/ACE2 ratio was higher in RA than in AS at B (p = 0.004), as well as after 6 M (p = 0.001) and 12 M (p = 0.003) (Figure 1C).
All results of the simple correlation analysis are seen in Supplementary Table S1. This table indicates the full cohort, as well as the RA and AS subsets.
In the univariable regression analysis of the RA+AS cohort, ACE levels at various time points were independently and positively associated with RF and FMD and inversely with age, disease duration, CRP, RF, IMT and FMD (p < 0.05) (Table 2). ACE2 activity was independently determined by CRP (P < 0.05) (Table 2). The ACE/ACE2 ratio independently correlated with age, disease duration, CRP, RF, IMT and FMD (Table 2). In RA, ACE concentrations were only associated with CRP, RF and FMD, while ACE2 activity did not show any correlations. In RA, ACE/ACE2 ratio correlated with disease duration, CRP, RF and FMD (p < 0.05) (Table 2). In AS, ACE level was independently associated with BASDAI, while ACE2 activity did not show any correlations. ACE/ACE2 ratio positively associated only with disease duration (p < 0.05) (Table 2).
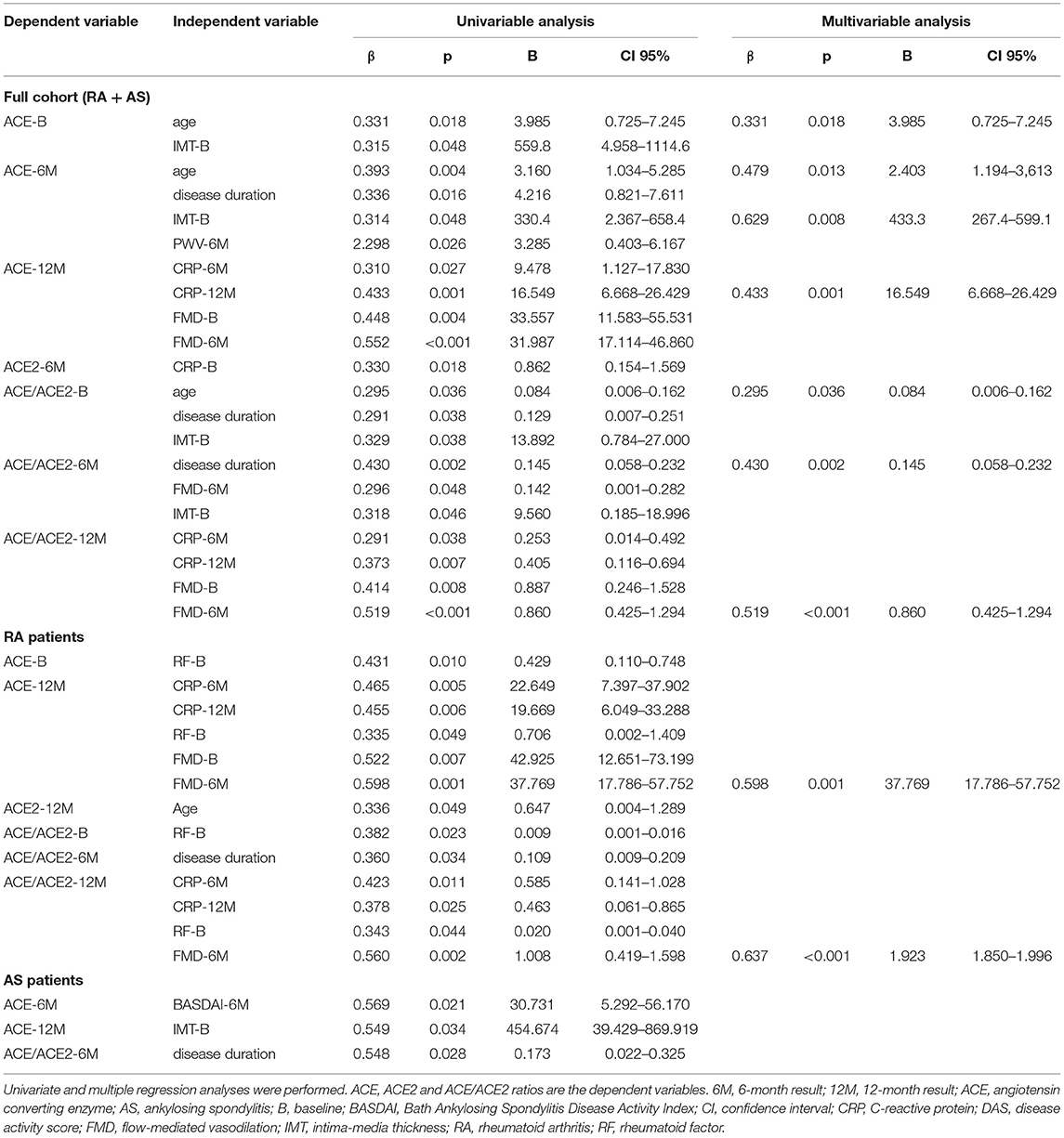
Table 2. Univariable and multivariable regression analysis of ACE and ACE2.
The multivariable regression analysis confirmed the positive correlation among ACE levels and age, CRP and IMT in the mixed cohort. Similarly, ACE/ACE2 ratios also correlated with age and disease duration (p < 0.05) (Table 2). In RA, the only correlation revealed was that between ACE levels and FMD (p < 0.05) (Table 2). No such correlations were observed in AS (Table 2).
Finally, RM-ANOVA analysis was performed in order to assess determinants of ACE or ACE2 changes over time. In the full cohort, one-year change in ACE concentration or in ACE/ACE2 ratio was determined by anti-TNF treatment together with higher RF or FMD at B (p < 0.05) (Table 3). In RA, ACE level or ACE/ACE2 ratio changes were associated with treatment along with higher RF (p < 0.005) (Table 3). No such associations were observed in AS (Table 3).
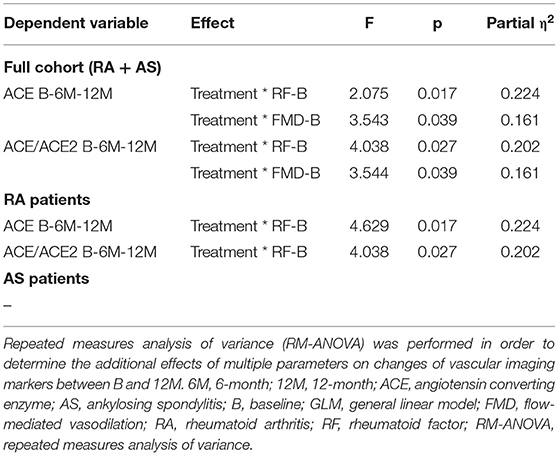
Table 3. Significant results of general linear model (GLM) repeated measures analysis of variance (RM-ANOVA) test determining the effects of treatment and other independent variables on ACE and ACE2 as dependent variables.
To our best knowledge, these may be the first data on the effects of one-year anti-TNF therapy on ACE level and ACE2 activity in arthritis patients. We found that one-year anti-TNF treatment significantly increased ACE concentration in the mixed cohort, as well as in the RA and AS subset. TNF inhibition also stimulated ACE2 activity in the RA+AS cohort but not in RA or AS. ACE/ACE2 ratios significantly increased in the mixed cohort and in RA, but not in AS. Interestingly, ACE levels and ACE/ACE2 ratios were higher in RA vs. AS, while ACE2 activity values were higher in AS vs. RA at most time points. Moreover, baseline, 6- and 12-month ACE levels, as well as ACE/ACE2 ratios variably correlated with disease duration, CRP, RF and various parameters of vascular pathophysiology. ACE2 activity only correlated with CRP.
We assessed ACE levels and ACE2 activity. ACE activity could not be included in this study as some patients received ACE inhibitors (Table 1), which could interfere with this parameter (26, 28). ACE2 activity is not affected by the use of ACE inhibitor treatment (28).
Upon TNF-α inhibition, ACE concentration increased in the mixed patient cohort throughout the treatment period. ACE levels also increased in RA and exerted a late, 12 M increase in AS. Similarly, anti-TNF therapy increased ACE2 activity in the mixed cohort, while ACE/ACE2 ratios showed late, 12 M increases in the full cohort and in RA. We could not compare our data from those published by others as, to our best knowledge, there have not been any studies evaluating the longitudinal effects of TNF-α inhibition on ACE or ACE2 levels or activity. In RA, increased synovial fluid ACE concentrations (46, 47) and decreased serum ACE2 (45) or unchanged serum ACE levels have been reported (46). Thus, in RA, there may be a redistribution of ACE and ACE2 from the serum to the synovium. This may be reversed by anti-TNF treatment, however, this was hypothetical as no prospective studies assessing the effects of TNF inhibition of ACE and ACE2 redistribution have been conducted. Similarly to RA, Potdar et al. (34) reported increased expression of colonic ACE2 in active ulcerative colitis (UC) and low expression of small bowel ACE in Crohn's disease (CD) vs. controls. Anti-TNF therapy restored ACE2 tissue expression by decreasing colonic ACE2 in UC and stimulating small bowel ACE2 in CD (34). The redistribution of ACE and ACE2 between tissue and serum was discussed above (28). Thus, although we did not conduct tissue expression studies, elevated serum ACE concentrations and ACE2 activity upon one-year anti-TNF treatment may reflect redistribution between tissue and serum of ACE and ACE2 (28, 45–47). The pattern of ACE2 changes in our cohort is similar to the UC results reported by Potdar et al., however, they only assessed tissue ACE2 levels and not serum ACE2 activity (34). In addition, increased synovial fluid RA ACE levels were reported in RA compared to controls (43, 46, 47). The synovial fluid compartment reflects the characteristics of the tissue better than the serum. Again, considering the redistribution patterns of ACE and ACE2 between serum and tissue (28), our results indicating increasing ACE levels and ACE2 activity upon treatment may reflect decreasing synovial ACE and ACE2 expression.
When comparing RA and AS, there was no difference in ACE level between the two groups at baseline. On the other hand, after 6 M and 12 M, ACE concentrations were higher in RA compared to AS suggesting that TNF inhibition might have a more pronounced effect on ACE in RA vs. AS. With respect to ACE2 activity, it was higher in AS compared to RA both at B and after 12 M. Yet, the difference was greater between AS and RA after one-year therapy again suggesting a stronger stimulating effect of anti-TNF agents on ACE2 activity in AS vs. RA. Moreover, one-year anti-TNF treatment resulted in the increase of ACE levels in both RA and AS. In contrast, ACE2 activity was only increased in the full cohort, but not in the RA and AS subsets. ACE/ACE2 ratios were higher in RA compared to AS at all time points. There have been no studies on the effects of biologics on ACE and ACE2 in RA or AS, therefore, we cannot compare our data to other reports.
The baseline time point represents a pre-treatment status when both RA and AS patients had higher inflammatory state and disease activity. After 12 M, as anti-TNF therapy was proven to be clinically effective, most RA and AS patients had remission or at least low disease activity (LDA). That is why it is important to compare correlations between ACE, ACE2 or ACE/ACE2 and other parameters at baseline and after 12 M. In the regression analyses, at baseline ACE concentration only correlated with age, RF and IMT. However, after one-year treatment it also correlated with CRP and FMD. The correlation between 6 and 12 M ACE and CRP or disease activity was observed in the full cohort (CRP), as well as in the RA (CRP) and AS subsets (BASDAI). Thus, in treated arthritis patients with lower degree of inflammation, ACE levels may become a marker of remaining systemic inflammation or disease activity. Moreover, high baseline CRP and RF levels may predict ACE levels after 12 M. Increased plasma ACE levels were found in some cohorts (44), while others reported similar ACE levels in RA and controls (46). Our study was an uncontrolled, longitudinal therapeutic study comparing post- and pre-treatment ACE levels. ACE2 activity after 6 M positively correlated with baseline CRP suggesting that more extensive baseline inflammation may predict higher ACE2 activity after 6 M, when inflammation is already dampened. In another study of Tang et al. (39), decreased ACE2 levels were found in RA vs. healthy controls. Yet, that study included RA patients with mixed characteristics and the study did not assess longitudinal treatment effects (39). ACE/ACE2 ratios at baseline exerted correlations with disease duration, RF and IMT. Again, after 6 and 12 M, ACE/ACE2 ratios also correlated with CRP and FMD, which pattern is similar to that observed with ACE discussed above.
In the RM-ANOVA analysis, changes of ACE levels or ACE/ACE2 ratios over 12 M were positively associated with baseline RF and FMD in the full cohort and with baseline RF in the RA subset. Thus, in RA, high baseline RF may be the most important denominator of ACE concentration changes among the disease-related parameters included in this study.
Our study certainly has strengths and limitations. The strength of this study is that, for the first time, we assessed the effects of biologics on ACE and ACE2 in arthritides in a prospective manner. In addition, ours is the first study to evaluate ACE and ACE2 in association with multiple disease-related laboratory markers and those of vascular pathophysiology. Possible limitations may include the relatively low patient number and the lack of control groups.
In conclusion, anti-TNF treatment may increase ACE and ACE2 in the sera of RA and AS patients, which may reflect the shedding and redistribution of ACE and ACE2 from the tissue to the blood. Baseline ACE and ACE2 may be associated with disease duration, markers of inflammation (CRP), autoimmunity (RF) and vascular pathophysiology (FMD, IMT). The effects of TNF inhibition on ACE and ACE2 release may reflect, in part, the beneficial effects of biologics on vascular pathology.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was approved by the Hungarian Scientific Research Council Ethical Committee (Approval No. 14804-2/2011/EKU). Written informed consent was obtained from each patient and assessments were carried out according to the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
BS: study conceptualization, patient recruitment, data collection, and draft writing. MF, ZPa, and AC: ACE and ACE2 measurements, data analysis, and manuscript drafting. ÁHo, EV, AHa, ZPe, NB, SSzam, and SSzán: patient recruitment and examination and data collection. AP and MC: laboratory assessments and data analysis. GK: vascular ultrasound examination and data collection. KH: statistical analysis and interpretation. ÉS: study concept, data interpretation, and draft writing. GS: study concept, draft writing, manuscript finalization, and senior coordinator. ZS: study concept, principal investigator, senior coordinator, and manuscript finalization. All authors contributed to the article and approved the submitted version.
This study received funding from the European Union and the State of Hungary and co-financed by the European Social Fund in the framework of TAMOP-4.2.4.A/2-11/1-2012-0001 National Excellence Program (ZS); by the European Union grant GINOP-2.3.2-15-2016-00050 (ZS); by the Pfizer Investigator Initiated Research Grants Nos. WS1695414 and WS1695450 (ZS) and the National Research, Development and Innovation Fund of Hungary Grant FK 128809 (MF). MF was supported by the ÚNKP-21-5-DE-458 New National Excellence Program of the Ministry for Innovation and Technology. This paper was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/00069/21/5). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.785744/full#supplementary-material
1. Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJ, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. (2017) 76:17–28. doi: 10.1136/annrheumdis-2016-209775
2. Nurmohamed MT, Heslinga M, Kitas GD. Cardiovascular comorbidity in rheumatic diseases. Nat Rev Rheumatol. (2015) 11:693–704. doi: 10.1038/nrrheum.2015.112
3. Castaneda S, Nurmohamed MT, Gonzalez-Gay MA. Cardiovascular disease in inflammatory rheumatic diseases. Best Pract Res Clin Rheumatol. (2016) 30:851–69. doi: 10.1016/j.berh.2016.10.006
4. Kerekes G, Szekanecz Z, Der H, Sandor Z, Lakos G, Muszbek L, et al. Endothelial dysfunction and atherosclerosis in rheumatoid arthritis: a multiparametric analysis using imaging techniques and laboratory markers of inflammation and autoimmunity. J Rheumatol. (2008) 35:398–406.
5. Bodnar N, Kerekes G, Seres I, Paragh G, Kappelmayer J, Nemethne ZG, et al. Assessment of subclinical vascular disease associated with ankylosing spondylitis. J Rheumatol. (2011) 38:723–9. doi: 10.3899/jrheum.100668
6. Kerekes G, Soltesz P, Nurmohamed MT, Gonzalez-Gay MA, Turiel M, Vegh E, et al. Validated methods for assessment of subclinical atherosclerosis in rheumatology. Nat Rev Rheumatol. (2012) 8:224–34. doi: 10.1038/nrrheum.2012.16
7. Gonzalez-Gay MA, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Martin J, Llorca J. Endothelial dysfunction, carotid intima-media thickness, and accelerated atherosclerosis in rheumatoid arthritis. Semin Arthritis Rheum. (2008) 38:67–70. doi: 10.1016/j.semarthrit.2008.02.001
8. Gonzalez-Juanatey C, Llorca J, Martin J, Gonzalez-Gay MA. Carotid intima-media thickness predicts the development of cardiovascular events in patients with rheumatoid arthritis. Semin Arthritis Rheum. (2009) 38:366–71. doi: 10.1016/j.semarthrit.2008.01.012
9. Soltesz P, Der H, Kerekes G, Szodoray P, Szucs G, Danko K, et al. A comparative study of arterial stiffness, flow-mediated vasodilation of the brachial artery, and the thickness of the carotid artery intima-media in patients with systemic autoimmune diseases. Clin Rheumatol. (2009) 28:655–62. doi: 10.1007/s10067-009-1118-y
10. Greenberg JD, Kremer JM, Curtis JR, Hochberg MC, Reed G, Tsao P, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. (2011) 70:576–82. doi: 10.1136/ard.2010.129916
12. Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 79:1–15. doi: 10.1136/annrheumdis-2019-216655
13. Damjanov N, Nurmohamed MT, Szekanecz Z. Biologics, cardiovascular effects and cancer. BMC Med. (2014) 12:48. doi: 10.1186/1741-7015-12-48
14. Szekanecz Z, Kerekes G, Soltesz P. Vascular effects of biologic agents in RA and spondyloarthropathies. Nat Rev Rheumatol. (2009) 5:677–84. doi: 10.1038/nrrheum.2009.219
15. Murdaca G, Colombo BM, Puppo F. Anti-TNF-alpha inhibitors: a new therapeutic approach for inflammatory immune-mediated diseases: an update upon efficacy and adverse events. Int J Immunopathol Pharmacol. (2009) 22:557–65. doi: 10.1177/039463200902200301
16. Murdaca G, Negrini S, Pellecchio M, Greco M, Schiavi C, Giusti F, et al. Update upon the infection risk in patients receiving TNF alpha inhibitors. Expert Opin Drug Saf. (2019) 18:219–29. doi: 10.1080/14740338.2019.1577817
17. Murdaca G, Spano F, Miglino M, Puppo F. Effects of TNF-alpha inhibitors upon the mechanisms of action of VEGF. Immunotherapy. (2013) 5:113–5. doi: 10.2217/imt.12.151
18. Szekanecz Z, Koch AE. Targeting angiogenesis in rheumatoid arthritis. Curr Rheumatol Rev. (2008) 4:298–303. doi: 10.2174/157339708786263942
19. Vegh E, Kerekes G, Pusztai A, Hamar A, Szamosi S, Vancsa A, et al. Effects of 1-year anti-TNF-alpha therapy on vascular function in rheumatoid arthritis and ankylosing spondylitis. Rheumatol Int. (2020) 40:427–36. doi: 10.1007/s00296-019-04497-0
20. Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. (2007) 56:2905–12. doi: 10.1002/art.22809
21. Angel K, Provan SA, Hammer HB, Mowinckel P, Kvien TK, Atar D. Changes in arterial stiffness during continued infliximab treatment in patients with inflammatory arthropathies. Fundam Clin Pharmacol. (2011) 25:511–7. doi: 10.1111/j.1472-8206.2010.00872.x
22. Del Porto F, Lagana B, Lai S, Nofroni I, Tinti F, Vitale M, et al. Response to anti-tumour necrosis factor alpha blockade is associated with reduction of carotid intima-media thickness in patients with active rheumatoid arthritis. Rheumatology (Oxford). (2007) 46:1111–5. doi: 10.1093/rheumatology/kem089
23. Kotani K, Miyamoto M, Ando H. The effect of treatments for rheumatoid arthritis on endothelial dysfunction evaluated by flow-mediated vasodilation in patients with rheumatoid arthritis. Curr Vasc Pharmacol. (2017) 15:10–8. doi: 10.2174/1570161114666161013113457
24. van Sijl AM, van Eijk IC, Peters MJ, Serne EH, van der Horst-Bruinsma IE, Smulders YM, et al. Tumour necrosis factor blocking agents and progression of subclinical atherosclerosis in patients with ankylosing spondylitis. Ann Rheum Dis. (2015) 74:119–23. doi: 10.1136/annrheumdis-2013-203934
25. Corvol P, Michaud A, Soubrier F, Williams TA. Recent advances in knowledge of the structure and function of the angiotensin I converting enzyme. J Hypertens Suppl. (1995) 13:S3–10. doi: 10.1097/00004872-199509003-00002
26. Fagyas M, Uri K, Siket IM, Darago A, Boczan J, Banyai E, et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) I: endogenous angiotensin converting enzyme (ACE) inhibition. PLoS ONE. (2014) 9:e87843. doi: 10.1371/journal.pone.0087843
27. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. (2000) 87:E1–9. doi: 10.1161/01.RES.87.5.e1
28. Uri K, Fagyas M, Manyine Siket I, Kertesz A, Csanadi Z, Sandorfi G, et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE. (2014) 9:e87845. doi: 10.1371/journal.pone.0087845
29. Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. (2010) 122:717–28. doi: 10.1161/CIRCULATIONAHA.110.955369
30. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
31. Wang W, Bodiga S, Das SK, Lo J, Patel V, Oudit GY. Role of ACE2 in diastolic and systolic heart failure. Heart Fail Rev. (2012) 17:683–91. doi: 10.1007/s10741-011-9259-x
32. Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, Klein AL, et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. (2009) 15:565–71. doi: 10.1016/j.cardfail.2009.01.014
33. Lehmann HI, Wolke C, Malenke W, Rohl FW, Hammwohner M, Bukowska A, et al. Enzymatic activity of DPIV and renin-angiotensin system (RAS) proteases in patients with left ventricular dysfunction and primary prevention implantable cardioverter/defibrillator (ICD). Int J Cardiol. (2013) 168:255–60. doi: 10.1016/j.ijcard.2012.09.083
34. Potdar AA, Dube S, Naito T, Li K, Botwin G, Haritunians T, et al. Altered intestinal ACE2 levels are associated with inflammation, severe disease, and response to anti-cytokine therapy in inflammatory bowel disease. Gastroenterology. (2021) 160:809–22.e7. doi: 10.1053/j.gastro.2020.10.041
35. Ahmed AZ, El-Shahaly HA, Omar AS, Ghattas MH. Patterns of angiotensin converting enzyme insertion/deletion gene polymorphism among an Egyptian cohort of patients with rheumatoid arthritis. Int J Rheum Dis. (2013) 16:284–90. doi: 10.1111/j.1756-185X.2012.01820.x
36. Al-Awadhi AM, Hasan EA, Sharma PN, Haider MZ, Al-Saeid K. Angiotensin-converting enzyme gene polymorphism in patients with psoriatic arthritis. Rheumatol Int. (2007) 27:1119–23. doi: 10.1007/s00296-007-0349-y
37. Alsaeid K, Haider MZ, Ayoub EM. Angiotensin converting enzyme gene insertion-deletion polymorphism is associated with juvenile rheumatoid arthritis. J Rheumatol. (2003) 30:2705–9.
38. Elshazli RM, Elsaid AM, Shawky DM, Barakat LAA. Genetic polymorphisms of ACE I/D, IL-1beta G > A and IL-4 VNTR among Egyptian subjects with rheumatoid arthritis. Arch Physiol Biochem. (2019) 13:1–10. doi: 10.1080/13813455.2019.1703005
39. Inanir A, Yigit S, Tural S, Ozturk SD, Akkanet S, Habiboglu A. Significant association between insertion/deletion polymorphism of the angiotensin-convertig enzyme gene and ankylosing spondylitis. Mol Vis. (2012) 18:2107–13.
40. Song GG, Bae SC, Kim JH, Lee YH. The angiotensin-converting enzyme insertion/deletion polymorphism and susceptibility to rheumatoid arthritis, vitiligo and psoriasis: a meta-analysis. J Renin Angiotensin Aldosterone Syst. (2015) 16:195–202. doi: 10.1177/1470320313478285
41. Inal EE, Gorukmez O, Eroglu S, Solak O, Topak A, Yakut T. Association of GSTM1, GSTT1, GSTP1-ILE105VAL and ACE I/D polymorphisms with ankylosing spondylitis. Rheumatol Int. (2016) 36:17–23. doi: 10.1007/s00296-015-3317-y
42. Shehab DK, Al-Jarallah KF, Al-Awadhi AM, Al-Herz A, Nahar I, Haider MZ. Association of angiotensin-converting enzyme (ACE) gene insertion-deletion polymorphism with spondylarthropathies. J Biomed Sci. (2008) 15:61–7. doi: 10.1007/s11373-007-9203-1
43. Lowe JR, Dixon JS, Guthrie JA, McWhinney P. Serum and synovial fluid levels of angiotensin converting enzyme in polyarthritis. Ann Rheum Dis. (1986) 45:921–4. doi: 10.1136/ard.45.11.921
44. Braz NFT, Pinto MRC, Vieira ELM, Souza AJ, Teixeira AL, Simoes ESAC, et al. Renin-angiotensin system molecules are associated with subclinical atherosclerosis and disease activity in rheumatoid arthritis. Mod Rheumatol. (2021) 31:119–26. doi: 10.1080/14397595.2020.1740418
45. Tang X, Geng L, Feng X, Sun L. Decreased serum ACE2 levels in patients with connective tissue diseases. Rheumatology (Oxford). (2020). doi: 10.1093/rheumatology/keaa898
46. Cobankara V, Ozturk MA, Kiraz S, Ertenli I, Haznedaroglu IC, Pay S, et al. Renin and angiotensin-converting enzyme (ACE) as active components of the local synovial renin-angiotensin system in rheumatoid arthritis. Rheumatol Int. (2005) 25:285–91. doi: 10.1007/s00296-004-0564-8
47. Wu Y, Li M, Zeng J, Feng Z, Yang J, Shen B, et al. Differential expression of renin-angiotensin system-related components in patients with rheumatoid arthritis and osteoarthritis. Am J Med Sci. (2020) 359:17–26. doi: 10.1016/j.amjms.2019.10.014
48. Veale D, Yanni G, Bresnihan B, FitzGerald O. Production of angiotensin converting enzyme by rheumatoid synovial membrane. Ann Rheum Dis. (1992) 51:476–80. doi: 10.1136/ard.51.4.476
49. Liu J, Liu Y, Pan W, Li Y. Angiotensin-(1-7) attenuates collagen-induced arthritis via inhibiting oxidative stress in rats. Amino Acids. (2021) 53:171–81. doi: 10.1007/s00726-020-02935-z
50. Mokuda S, Tokunaga T, Masumoto J, Sugiyama E. Angiotensin-converting enzyme 2, a SARS-CoV-2 receptor, is upregulated by interleukin 6 through STAT3 signaling in synovial tissues. J Rheumatol. (2020) 47:1593–5. doi: 10.3899/jrheum.200547
51. Chappell MC. Angiotensin-converting enzyme 2 autoantibodies: further evidence for a role of the renin-angiotensin system in inflammation. Arthritis Res Ther. (2010) 12:128. doi: 10.1186/ar3052
52. Takahashi Y, Haga S, Ishizaka Y, Mimori A. Autoantibodies to angiotensin-converting enzyme 2 in patients with connective tissue diseases. Arthritis Res Ther. (2010) 12:R85. doi: 10.1186/ar3012
53. Ranjbar R, Shafiee M, Hesari A, Ferns GA, Ghasemi F, Avan A. The potential therapeutic use of renin-angiotensin system inhibitors in the treatment of inflammatory diseases. J Cell Physiol. (2019) 234:2277–95. doi: 10.1002/jcp.27205
54. Sluijsmans D, Rohrich DC, Popa CD, van den Bemt BJF. Angiotensin-inhibiting drugs do not impact disease activity in patients with rheumatoid arthritis: a retrospective cross-sectional study. J Clin Med. (2021) 10:1985. doi: 10.3390/jcm10091985
55. Keewan E, Beg S, Naser SA. Anti-TNF-alpha agents modulate SARS-CoV-2 receptors and increase the risk of infection through notch-1 signaling. Front Immunol. (2021) 12:641295. doi: 10.3389/fimmu.2021.641295
56. Balogh E, Végh E, Kerekes G, Pusztai A, Hamar A, Hodosi K, et al. Effects of one-year anti-TNF-α therapy on biomarkers of angiogenesis and functional vascular parameters in arthritides. Rheumatol Orthop Med. (2019) 4:1–8. doi: 10.15761/ROM.1000169
57. Pusztai A, Hamar A, Horvath A, Gulyas K, Vegh E, Bodnar N, et al. Soluble vascular biomarkers in rheumatoid arthritis and ankylosing spondylitis: effects of one-year anti-TNF-alpha therapy. J Rheumatol. (2020) 48:821–8. doi: 10.3899/jrheum.200916
58. Juhasz B, Gulyas K, Horvath A, Vegh E, Pusztai A, Szentpetery A, et al. Peripheral quantitative computed tomography in the assessment of bone mineral density in anti-TNF-treated rheumatoid arthritis and ankylosing spondylitis patients. BMC Musculoskelet Disord. (2021) 22:817. doi: 10.1186/s12891-021-04708-5
59. Pusztai A, Hamar A, Czokolyova M, Gulyas K, Horvath A, Vegh E, et al. Associations of vascular and bone status in arthritis patients. Sci Rep. (2021) 11:19504. doi: 10.1038/s41598-021-99071-9
60. Gulyas K, Horvath A, Vegh E, Pusztai A, Szentpetery A, Petho Z, et al. Effects of 1-year anti-TNF-alpha therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin Rheumatol. (2020) 39:167–75. doi: 10.1007/s10067-019-04771-3
61. Fagyas M, Uri K, Siket IM, Darago A, Boczan J, Banyai E, et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) III: endogenous inhibition of angiotensin converting enzyme (ACE) provides protection against cardiovascular diseases. PLoS ONE. (2014) 9:e93719. doi: 10.1371/journal.pone.0093719
Keywords: rheumatoid arthritis, ankylosing spondylitis, angiotensin converting enzyme, vascular disease, biologics, anti-TNF therapy
Citation: Soós B, Fagyas M, Horváth Á, Végh E, Pusztai A, Czókolyová M, Csongrádi A, Hamar A, Pethő Z, Bodnár N, Kerekes G, Hodosi K, Szekanecz É, Szamosi S, Szántó S, Szűcs G, Papp Z and Szekanecz Z (2022) Angiotensin Converting Enzyme Activity in Anti-TNF-Treated Rheumatoid Arthritis and Ankylosing Spondylitis Patients. Front. Med. 8:785744. doi: 10.3389/fmed.2021.785744
Received: 29 September 2021; Accepted: 10 December 2021;
Published: 27 January 2022.
Edited by:
Giuseppe Mandraffino, University of Messina, ItalyReviewed by:
Giuseppe Murdaca, University of Genoa, ItalyCopyright © 2022 Soós, Fagyas, Horváth, Végh, Pusztai, Czókolyová, Csongrádi, Hamar, Pethő, Bodnár, Kerekes, Hodosi, Szekanecz, Szamosi, Szántó, Szűcs, Papp and Szekanecz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoltán Szekanecz, c3pla2FuZWN6LnpvbHRhbkBtZWQudW5pZGViLmh1
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.