Abstract
Genetic testing has uncovered rare variants in complement proteins associated with thrombotic microangiopathy (TMA) and C3 glomerulopathy (C3G). Approximately 50% are classified as variants of uncertain significance (VUS). Clinical risk assessment of patients carrying a VUS remains challenging primarily due to a lack of functional information, especially in the context of multiple confounding factors in the setting of kidney transplantation. Our objective was to evaluate the clinicopathologic significance of genetic variants in TMA and C3G in a kidney transplant cohort. We used whole exome next-generation sequencing to analyze complement genes in 76 patients, comprising 60 patients with a TMA and 16 with C3G. Ten variants in complement factor H (CFH) were identified; of these, four were known to be pathogenic, one was likely benign and five were classified as a VUS (I372V, I453L, G918E, T956M, L1207I). Each VUS was subjected to a structural analysis and was recombinantly produced; if expressed, its function was then characterized relative to the wild-type (WT) protein. Our data indicate that I372V, I453L, and G918E were deleterious while T956M and L1207I demonstrated normal functional activity. Four common polymorphisms in CFH (E936D, N1050Y, I1059T, Q1143E) were also characterized. We also assessed a family with a pathogenic variant in membrane cofactor protein (MCP) in addition to CFH with a unique clinical presentation featuring valvular dysfunction. Our analyses helped to determine disease etiology and defined the recurrence risk after kidney transplant, thereby facilitating clinical decision making for our patients. This work further illustrates the limitations of the prediction models and highlights the importance of conducting functional analysis of genetic variants particularly in a complex clinicopathologic scenario such as kidney transplantation.
Introduction
Two prototypical complement-mediated kidney diseases are atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy (C3G) (1). Atypical hemolytic uremic syndrome is a primary complement-mediated thrombotic microangiopathy (TMA) characterized by hemolytic anemia, thrombocytopenia, and acute kidney injury (AKI) (2). C3 glomerulopathy is a group of nephritides in which complement activation leads to a predominant C3 fragment glomerular deposition as seen by immunofluorescence (3). It is subclassified as dense deposit disease or C3 glomerulonephritis (C3GN) based on the type and location of deposits as visualized by electron microscopy.
Recent progress in our understanding of the role of complement dysregulation and efficacy of complement blockade has revolutionized the clinical course and outcome of these diseases (4). Therapeutic advances render kidney transplantation as a viable option for patients with aHUS and C3G who previously were considered non-transplantable.
However, many challenges remain. First, aHUS may be mimicked by other disease processes, currently classified under secondary TMAs, which include infections, pregnancy, autoimmune conditions, and graft rejection (3). Kidney transplantation poses an especially problematic setting since there are multiple potential triggers (transplant surgery, drugs, rejection, and infections) for TMA development. Consequently, it can be “tricky” for transplant clinicians to distinguish aHUS from these secondary TMAs. However, the distinction is critical since complement-mediated TMAs are often non-responsive to conservative measures (removal of the offending drug or treating the underlying rejection/infection) (5). Second, both aHUS and C3G have a high risk of recurrence after kidney transplantation and the outcome can vary depending on the underlying complement abnormality (6, 7). Genetic variants in complement proteins are identified in approximately 60% of patients with aHUS, 10–60% of patients with secondary causes, and 30–40% of patients with C3G (8). However, <50% of the identified variants have a known functional consequence and are classified by the American College of Medical Genetics (ACMG) as variants of uncertain significance (VUS). Thus, the inability to ascertain the functional impact of a VUS poses another major barrier for clinical management after kidney transplantation. Finally, decisions about accepting living related donors for these patients can be complicated since there may be risk of recurrent disease in the recipient and de novo disease in the donor if the underlying etiology of disease is unclear.
We have previously published functional studies on VUS in complement factor I (CFI) in patients with a TMA that facilitated establishing their clinicopathologic relevance (9, 10). We have also characterized complement factor H (CFH) and CFI variants identified in patients with age-related macular degeneration (AMD), a leading cause of blindness in the developed world. These findings helped explain the early onset of advanced AMD in the patients as well as the high burden of disease in their families (10, 11). We now present studies on the functional characterization of rare genetic variants and single nucleotide polymorphisms (SNPs) in CFH in our kidney transplant cohort (Figure 1). This study highlights how a systematic and comprehensive analysis can help to address the challenges described above, fill the knowledge gap relative to underlying mechanism of disease, and facilitate therapeutic decisions in a kidney transplant population. Our work also emphasizes several additional key concepts relative to limitations of the computational predictive models, the role of multiple CFH variants (common and rare), the impact of concomitant variants in CFI and membrane cofactor protein (MCP, CD46), and modifications due to underlying diseases including especially lupus.
Figure 1
Materials and Methods
Study Population
Patients diagnosed with a TMA or with biopsy-proven C3G between 2015 and 2020 at Washington University School of Medicine (WUSM) were evaluated. These patients were identified during the pretransplant evaluation at our center or were referred to us from other centers. Patients with a TMA included those with a biopsy-proven diagnosis in their native kidney or those who developed a de novo TMA after transplantation in the renal allograft. The diagnosis of C3G was based on the 2013 consensus report guidelines which required intensity of C3 staining at least two orders of magnitude greater than any other immunoreactant (i.e., IgG, IgM, IgA, and C1q) on a scale of 0–3 (including 0, trace, 1+, 2+, 3+) in native kidney or allograft (11). This research followed the tenets of the Declaration of Helsinki. The study was approved by the institutional review board at Washington University School of Medicine in St. Louis, MO. Informed consent was obtained from each subject.
Sequencing
Next generation sequencing was performed by the Genomic and Pathology Services (GPS) at WUSM (12). The clinically validated aHUS/C3G next generation sequencing-based assay is derived from the Agilent Clinical Research Exome capture reagent (Agilent Technologies, Inc., Santa Clara, CA), with analysis of all exonic sequences for 13 genes (ADAMTS13, C3, CD46, CFB, CFH, CFHR1, CFHR2, CFHR3, CFHR4, CFHR5, CFI, DGKE, THBD, MMACHC, and PLG). CFHR3-CFHR1 copy number was assessed using multiplex ligation-dependent probe amplification (MLPA). All variants are reported according to Human Genome Variation Society (HGVS) nomenclature and classified based on the guidelines established by the joint consensus of the ACMG and the Association of Molecular Pathology (AMP). Based on these standards, variants are classified as: Level 1: pathogenic; Level 2: likely pathogenic; Level 3: variants of uncertain clinical significance (VUS); Level 4: likely benign; and Level 5: benign. The CFH reference wild-type (WT) sequence NM_000186.4 was employed for variant calling.
Molecular Engineering of Factor H CCP 1–8 and CCP 15–20 Fragments
Preparation of the FH CCP 18–20 vector has been described (14). A two-step DNA synthesis method was used to construct the FH CCP 1–8 and CCP 15–20 vectors (15). The CCP 1–8 and CCP 15–20 fragments were generated by PCR with a C-terminal 6× His tag. These products were used as templates for a second PCR reaction in which the outer primer contained an N-terminal signal peptide and an EcoRI restriction site. The PCR products were subsequently digested with EcoRI and BamHI and then ligated with the pSG5 vector. The fidelity of FH CCP 1–8, CCP 15–20, and CCP 18–20 pSG5 vectors was verified by DNA sequencing. Primers used for PCR amplification are listed in Supplementary Table 1.
Preparation and Expression of FH Variants
The FH variants (Figures 1B–D) were produced using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Each CFH cDNA clone was sequenced. The variants were transfected into human embryonic kidney 293T cells and DMEM was replaced with serum-free OptiMEM® (Invitrogen, NY). Transient transfections were performed using the Xfect reagent (Takara Bio USA, Mountain View, CA). Supernatants were collected after 72 h, concentrated 80×, and stored in aliquots at −80°C (9).
Quantifying FH Variants by ELISA and Western Blotting
The quantity of each recombinant FH variant protein was determined by ELISA (9, 16). The microtiter plates were coated with capture antibodies (Abs) (A254 for CCP 1–8; A229 for CCP 15–20, and CCP 18–20, Quidel) at 2 μg/ml overnight at 4°C and blocked for 1 h at 37°C (in 10 mM Tris, 150 mM NaCl pH 7.4, 1% BSA, and 0.05% Tween 20). The WT, variant fragments and purified human FH (Complement Technologies, TX) were diluted, incubated for 1 h at 37°C, and then washed with PBS pH 7.4 containing 0.05% Tween 20 (PBS-T). A polyclonal goat antiserum to human FH (A312, San Diego, CA) was employed to detect bound FH using a standard ELISA protocol (9, 16). For Western blots, supernatants were electrophoresed under reducing conditions using 4–20% SDS-PAGE, transferred to nitrocellulose, and then probed with 1:5,000 mouse Monoclonal TetraHis Ab (Qiagen, USA), followed by a 1:10,000 HRP goat anti-mouse IgG (Abcam, Cambridge, MA) (Figure 2).
Figure 2
Functional Studies
The variants were produced as recombinant protein fragments of FH and their function was tested against the corresponding WT FH fragments and also the WT full-length protein. Patient serum was also used for fluid-phase and cell-surface assays. A genetic variant was considered to be deleterious if the protein was (a) not synthesized and/or not secreted in vitro (as measured by Western blot and ELISA) or (b) secreted in normal amounts but was dysfunctional based on any one of the assays outlined below.
Binding Assays
Variant and WT FH binding to C3b and heparin was assessed by ELISAs. Microtiter plates were coated with human C3b (37.5 nM for CCP 1–8; 15 nM for CCP 15–20 and CCP 18–20) or unfractionated heparin (2 mg/ml; Sigma-Aldrich) overnight at 4°C in NaHCO3 buffer [50 mM for C3b binding (17); 100 mM for heparin binding, pH 9.6) (18)], followed by blocking with 4% BSA in PBS-T (15, 19). Samples were diluted in ELISA buffer and incubated for 1 h at 37°C. Bound FH was detected as described previously (9, 16). On at least three occasions, binding assays were performed employing serially diluted samples.
Cofactor Activity
Factor H proteins were diluted in physiologic salt buffer (10 mM Tris pH 7.4, 150 mM NaCl). C3b (15 ng) was incubated at 37°C with purified FH (200 ng), equivalent moles of WT CCP 1–8 or variant FH protein, and FI (20 ng) in a total reaction volume of 20 μl. Kinetic analyses were performed at 0, 5, 10, and 20 min. The samples were electrophoresed on a 4–20% gradient Tris-glycine gel and then transferred to nitrocellulose for Western blotting (9).
Serum-Based Assays
These included the sheep red blood cell hemolytic assay, the C3b deposition assay and the C3G biomarker panel. These tests were conducted at the Molecular Otolaryngology and Renal Research Laboratories (MORL), Iowa (https://morl.lab.uiowa.edu/). The hemolytic assay measures complement-mediated lysis of sheep erythrocytes secondary to activation of the alternative pathway. The C3b deposition measures complement-mediated C3b deposition on the surface of cultured MES-13 cells as visualized using Alexa-488 labeled anti-C3 antibody.
Statistical Analysis
Statistical analyses were performed using Prism software version 8 (GraphPad). Comparisons between two groups were assessed using paired t-test (non-parametric). Comparisons among groups were performed using a one-way ANOVA with Dunnett's multiple comparison test (P < 0.05 was considered as significant).
Results
A cohort of 76 kidney transplant patients (comprising 60 patients with a TMA and 16 with C3G) were identified and underwent genetic sequencing. Thirty-three of the 76 patients (~ 44%) carried a variant in a complement protein (Supplementary Table 2). Twenty-one of the 33 variants (~64%) were classified as a VUS. We have previously reported functional studies on VUS in CFI in this cohort that helped to establish their clinical significance (9). Variants in CFH were the focus of the current investigation. Ten rare genetic variants and four SNPs (E936D, N1050Y, I1059T, Q1143E) in CFH were identified (Supplementary Tables 2, 3). Of the 10 CFH variants, four were classified as pathogenic (C611Stop, C853R, c.619 + 1G>A, c.3493 + 1G>A), five as VUS (I372V, I453L, G918E, T956M, and L1207I) and one as likely benign (T1017I). We investigated the effects of the five VUS in CFH in eight patients. We also functionally characterized the SNPs (alone and in combination with a rare variant). Two patients in one family also carried concomitant variants in CFI (K441R, classified as likely benign) and MCP (c.287-2A>G, classified as pathogenic).
We hereby present “real-life” cases of eight patients with a detailed clinical history for each. Comprehensive antigenic, functional, and structural analyses were conducted to determine the underlying etiology of disease which was key for clinical decision-making regarding treatment and risk of recurrence.
Complement-Mediated TMA Vs. Secondary TMA After Kidney Transplantation
Clinical History
Patient 1
A 45-year-old Asian female developed end-stage renal disease (ESRD) secondary to biopsy-proven lupus nephritis. She underwent a deceased donor kidney transplant (DDKT) in 2008, which was complicated on post-operative day 5 by a TMA in the allograft leading to a transplant nephrectomy (Supplementary Figure 1). The etiology of TMA was presumed to be the calcineurin inhibitor (CNI). Therefore, during the second DDKT in 2012, belatacept was used instead of a CNI. Two years later, the patient developed AKI requiring hemodialysis (HD). Allograft biopsy (not shown) revealed acute cellular rejection and endothelialitis which did not respond to conventional treatment with steroids and she remained dialysis dependent. Given the loss of two allografts due to TMA and pathologic evidence of endothelial cell injury, genetic testing for complement variants was performed during the evaluation for a third transplant (Table 1).
Table 1
| Patients | Variants | MAF (%) | Allele count (gnomAD)/Number of homozygotes | C3 level (mg/dl) | C4 level (mg/dl) | Factor H (mg/dl) | FH autoantibody (≤22 unit/ml) | C3Nef (0.0–0.26) | Transplant history | |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Population | |||||||||
| 1 | I372V | 0.0008 | 0.011 (East Asian) | 2/0 | 72 (83–185) | 19.2 (12–54) | 66.2 (37–68) | Negative | Negative | 2 failed |
| 2 | I453L* | 0.0032 | 0.012 (African American) | 1/0 | 57 (90–180) | 11 (10–40) | 38.8 (37–68) | Negative | Negative | 1 failed |
| 3 - 5 | G918E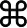 | 0.0008 | 0.003 (Latino) | 2/0 | 72.2 (79–152) | NT | 47.1 (16–41.2) | Negative | Negative | None |
| 6 | T956M | 0.13 | 0.17 (Caucasian) | 366/2 | 68 (90–180) | NT | 56.8 (37–68) | 38 | 1.02 | 1 failed |
| 7 | T956M | 0.13 | 0.17 (Caucasian) | 366/2 | 105 (75–175) | 25 (14–40) | 79.9 (37–68) | Negative | 0.3 | Allograft functioning well |
| 8 | L1207I | Novel | NT | Not reported/NA | NT | NT | NT | Negative | NT | Donor candidacy declined |
Genetic, immunologic, and clinical data from the TMA/C3G cohort at Washington University School of Medicine (WUSM).
Minor allele frequency (MAF) values were based on total and specific population frequency from Genome aggregation database (gnomAD).
Laboratory tests showed characteristics of a TMA—anemia [hemoglobin, 7.7 g/dl (normal range, 12.0–15.0 g/dl); elevated LDH, 292 U/L (normal range, 100–250 U/L); low haptoglobin, 17 mg/dl (normal range, 30–200 mg/dl)]; thrombocytopenia [platelet count, 66 k/μl (normal range, 150–400 k/μl)], and AKI (creatinine, 8.94 mg/dl with baseline 1.5 mg/dl); schistocytes on peripheral smear (3–7/HPF).
Laboratory studies were consistent with a TMA—anemia [hemoglobin, 3.9 g/dl (normal range, 12–15.5 g/dl); elevated LDH, 1,723 U/L (normal range, 140–280 U/L); undetectable haptoglobin, <30 mg/dl (normal range, 50–220 mg/dl)]; thrombocytopenia [platelet count, 71 k/μl (normal range, 150–450 k/μl)], and AKI (creatinine, 7.0 mg/dl with baseline 0.7 mg/dl); and schistocytes on peripheral smear (3–4/HPF). NT, not tested. I, isoleucine; V, valine; L, leucine; G, glycine; E, glutamic acid; T, threonine; M, methionine.
Patient 2
A 36-year-old African American female developed ESRD secondary to biopsy-proven lupus nephritis. She received a DDKT which failed within 1 year and she returned to dialysis. Allograft biopsy demonstrated arterioles with marked mucoid intimal edema and fragmented red cells. Laboratory results suggested a TMA (Table 1). Therefore, she underwent genetic testing during the evaluation for a second transplant.
For both patients described above, there was no family history of kidney disease. ADAMTS13 activity was normal and elevated levels of anti-phospholipid antibodies (APL Abs) or FH autoantibodies were not detected. Testing for Shiga toxin was also negative.
Genetic Analysis
We identified rare heterozygous CFH variants, I372V in patient 1 and I453L in patient 2 (Table 1). I372V has been reported in gnomAD with a MAF of 0.0008% in East Asians only and I453L with a MAF of 0.0032% in an individual of African ancestry. They were classified as VUS due to their rarity and lack of functional data or other evidence to support pathogenicity.
Structural Analysis
The I372 residue is a moderately conserved amino acid in CCP 6 (Figure 3) that is spatially adjacent to a putative GAG binding site in CCP 6–8 of the FH crystal structure (PDBID:2UWN and 2W80) (Figures 4A,B) (20, 21). The I372V substitution involves two hydrophobic amino acids. Both crystal structures (PDBID 2UWN and 2W80) demonstrate that the hydrophobic side chain of I372 is buried inside the CCP 6 protein core, away from the interacting surface. However, the alteration to a smaller hydrophobic residue V may influence the external surface of CCP 6 and its binding interactions through an internal structural rearrangement.
Figure 3
Figure 4
Sequence alignments of mammalian CCP 8 revealed that I453 is moderately conserved in 10 mammalian sequences (Figure 3) (22). Although I453 is spatially away from R444 (a putative heparin binding site) (19) (Figure 4A), the substitution of I to L may influence the external surface of CCP 8 and its binding interactions.
Antigenic and Functional Analyses
Factor H antigenic levels in both patients were in the normal range (Table 1). Secretion of the recombinantly produced variants (I372V and I453L) and their binding affinities to C3b were comparable to those of WT and full-length FH (Figures 2, 4C; Table 2). Cofactor activity in our standard assay with the recombinant proteins was also normal (Figure 4D). However, the variants showed a modest increase in binding affinity for heparin compared to WT. Given the aggressive nature of the TMA, loss of multiple allografts, rarity of the variants, and the structural data supporting a possible deleterious effect, we conducted further investigation using serum-based assays. The samples were sent to MORL, Iowa for the C3b deposition and sheep red blood cell hemolytic assays. The C3b deposition assay was abnormal for I372V (40–60% C3b deposition detected; reference range, negative, MORL, Iowa) and the sheep hemolytic assay was abnormal for I453L (20–40% hemolysis detected; normal <3%, MORL, Iowa).
Table 2
| Patient (s) | Variant | Location | Recombinant secretion (μg/ml) | C3b binding | Heparin binding | Cofactor activity | Cell-surface regulation using patient serum | ACMG interpretation | Modified interpretation (based on functional and structural analysis) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | I372V | CCP 6 | Comparable to WT | N | N | N | Defective (C3b deposition assay) | VUS | Deleterious |
| 2 | I453L | CCP 8 | Comparable to WT | N | N | N | Defective (sheep red cell hemolytic assay) | VUS | Deleterious |
| 3, 4 | G918E | CCP 15 | Decreased | ND | ND | __ | Not done | VUS | Deleterious |
| 3, 4 | G918E/N1050Y | CCP 15/18 | Decreased | ND | ND | __ | Not done | VUS | Deleterious |
| 6 | T956M/E936D | CCP 16 | Comparable to WT | N | N | __ | Not done | VUS | Normal function |
| 7 | T956M | CCP 16 | Comparable to WT | MD | N | __ | Not done | VUS | Likely benign |
| 8 | L1207I | CCP20 | Comparable to WT | N | N | __ | Not done | VUS | Normal function |
Summary of the functional analyses for CFH variants.
Secretion of I372V (5.9 μg/ml) and I453L (6.2 μg/ml) was comparable to WT CCP1–8 (4.0 μg/ml) (P > 0.05, SEM 1.15) as measured by ELISA. C3b binding and cofactor activity were normal but cell surface regulation was defective for both I372V and I453L. Variant G918E was not secreted. Secretion of SNP N1050Y (4.1 μg/ml) was comparable to WT CCP 15–20 (3.6 μg/ml) (P < 0.05, SEM 0.1). Secretion of G918E/N1050Y was markedly reduced compared to WT. Secretion of T956M (2.6 μg/ml) and T956M/E936D (3.5 μg/ml) were comparable with WT CCP 15–20 (3.6 μg/ml) (P = 0.7855, SEM 0.1). Secretion of L1207I was comparable to WT CCP 18–20 (2 μg/ml) (P > 0.05, SEM 0.16). ACMG, American College of Medical Genetics; VUS, variant of uncertain significance. Patient 5 did not undergo genetic testing due to financial/insurance issues. N, normal; ND, not done; MD, marginally decreased. I, isoleucine; V, valine; L, leucine; G, glycine; E, glutamic acid; T, threonine; M, methionine.
Implications
Patients 1 and 2 represent examples of post-transplant TMA in patients with SLE in which a rare complement genetic variant was identified. This clinical scenario is challenging for clinicians to determine if such patients have a primary complement-mediated TMA or a secondary SLE-associated or a transplant-related TMA or a combination of the above. Our analyses demonstrate that the variants I372V and I453L are produced normally and thus have no quantitative defect but are deleterious due to a qualitative defect based on the distinctly abnormal serum-based assays. The structural analysis also supports a likely deleterious effect.
However, given that the recombinant FH protein demonstrated normal results but serum-based assay revealed impaired function in the C3b deposition or the sheep red blood cell hemolytic assays led us to consider other possibilities. The question was raised that elevated FHR levels in the serum might be the cause of impaired FH function of complement regulation. To address this issue, we re-examined the next generation sequencing data which included CFHR1, CFHR2, CFHR3, CFHR4, and CFHR5 genes. Additionally, CFHR3-CFHR1 copy number was assessed. Neither patient carried a variant in the CFHR genes. A heterozygous CFHR3-1 deletion was identified in patient carrying the variant I453L but not in patient carrying the variant I372V. Since altered FHR levels have not been associated with a heterozygous CFHR3-1 deletion, we think that it is less likely that the patients have elevated FHR levels but this possibility cannot entirely be ruled out.
To summarize, these two patients did not develop TMA in the native kidney but developed an early and aggressive disease in the allograft in the presence of a trigger(s) that included transplant surgery and/or rejection. Overall, these data help to clarify that the TMA in these two patients is likely complement-mediated. Therefore, in both cases it was recommended that eculizumab be administered at the time of the next kidney transplant.
Familial Postpartum TMA Syndrome With Multiple Genetic Variants
Clinical History
Patient 3 (Sister 1, Proband)
In 2015, a 25-year-old Hispanic female presented 10 days after an uncomplicated vaginal delivery with laboratory results consistent with a TMA (Table 1). Treatment with prednisone and plasma exchange (PE) was unsuccessful and HD was initiated. ADAMTS13 activity and APL Abs were normal. Eculizumab was started after kidney biopsy confirmed a TMA (not shown). Hemodialysis was discontinued after 8 weeks. Two months later, she developed flash pulmonary edema due to acute mitral valve regurgitation secondary to an anterior leaflet perforation, which necessitated a mitral valve replacement. The patient is currently doing well with stable kidney function. In retrospect, the proband's two sisters had similar clinical presentations.
Patient 4 (Sister 2)
In 2014, this 24-year-old presented 7 days postpartum after an uncomplicated vaginal delivery with anemia, thrombocytopenia, and AKI. She was unsuccessfully treated with PE and HD had to be initiated. The proband diagnosis was established 1 year later, leading to aHUS being considered in sister 2 and eculizumab was initiated. Her creatinine (Cr) initially stabilized at 2–2.5 mg/dl and was 0.9 mg/dl in December 2020. However, she did not continue eculizumab due to health insurance issues. Since discontinuation of eculizumab, the renal function has remained stable, but she has developed hematuria and proteinuria (1.2 g).
Patient 5 (Sister 3)
In 2006, this 18-year-old presented 7 days postpartum after an uncomplicated vaginal delivery with anemia, thrombocytopenia, and AKI. Similar to her sisters, she did not respond to PE and HD was initiated. The course was complicated by heart failure due to severe mitral and tricuspid regurgitation. She was not treated with eculizumab as aHUS was not considered until after the diagnosis had been made in her sisters (8 years later). She currently remains on dialysis.
Family history in this kindred is significant for a paternal grandmother who lost 10 of 12 pregnancies due to miscarriages, and a paternal aunt who is currently on dialysis (Supplementary Figure 2). No medical history is available for the father. The mother delivered five healthy children (the three sisters mentioned above and two brothers) with no postpartum complications.
Genetic Analysis
The proband was a compound heterozygote for two non-synonymous variants in CFH, G918E, and N1050Y (Table 1; Supplementary Table 3). The G918E variant is rare being identified in two individuals in gnomAD, one each of European and Latino ancestry, and has also been reported in a patient with a TMA (23). The G918 residue is highly conserved across species and in silico predictions point to a deleterious effect of the G918E variant. Due primarily to a lack of in vitro studies, it was classified as a VUS.
The N1050Y SNP occurs at a position that is not highly conserved and in silico analyses yield inconsistent predictions. This variant has been associated with reduced AMD risk (24) and is not enriched in aHUS (25, 26). Based on these criteria, the N1050Y variant was classified as benign.
A heterozygous splice-site variant (c.287-2A>G) was identified in the MCP (CD46) (27). In vitro studies are consistent with an exon skipping effect predicted to result in protein truncation (27, 28). This variant has been identified in aHUS, and is classified as a pathogenic variant based on functional, computational and population evidence (2, 29, 30).
A heterozygous missense variant, K441R, in CFI was also identified and previously described as likely benign (31). Sister 2 carries the same genetic variants in CFH and MCP as the proband but does not carry the variant in CFI. Unfortunately sister 3 did not undergo genetic testing.
Structural Analysis
G918 is located in CCP 15 between the highly conserved C915 and W920 residues (Figure 3). Substitution of G with E introduces a negatively charged side chain at the main surface loop between β-strands 6 and 7 (Figure 5A). This alteration could generate side chain clashes between E918 and I868 that perturb the conformation of the adjacent C870–C915 disulfide bond and thereby disrupt FH protein folding.
Figure 5
N1050 is located in CCP 18 (Figure 3), close to the interdomain linker between CCP 17 and CCP 18, which could alter the inter-CCP domain arrangements. Because Y has a larger relative size than N, its presence in the variant may modulate the overall folded-back CCP domain structure but should not affect FH stability.
Antigenic and Functional Analyses
Factor H and FI antigenic levels in the proband were normal, but MCP expression was low (Table 1). Secretion of the recombinantly produced variant protein G918E was undetectable (Figure 2B; Table 2). By contrast, the recombinant N1050Y SNP displayed higher secretion than WT (Table 2). We produced the combined variant G918E/N1050Y and showed that the secretion was markedly reduced (Figure 2B). We also recombinantly produced the CFI variant K441R which displayed normal secretion and no defect in cofactor activity (not shown).
Implications
These cases represent pregnancy-associated TMA in a family that carries genetic variants in multiple complement proteins (FH, MCP, and FI). The MCP mutation is pathogenic given the low expression in our patient and having been reported previously as having low expression on granulocytes (27). Our structural and functional data establish that the CFH variant G918E is also deleterious because it abolishes FH protein expression. The proband though had a normal FH level. We speculate that in our patient this may be due to enhanced protein production by the N1050Y SNP, likely consistent with N1050Y location on the other allele. This hypothesis is supported by the fact that another patient carrying G918E had a reduced FH level of 168 mg/l (normal range 180–420 mg/l; personal communication, Richard Smith, Iowa).
These results indicate that TMA etiology in these patients is complement-mediated based on defective CFH and MCP variants. The risk of recurrent TMA after a kidney transplant in patients carrying an MCP mutation is low since the allograft comes with a normal MCP. However, since the two sisters also harbor the dysfunctional CFH mutation, they are at higher risk for recurrent disease.
We propose that complement dysregulation may have caused multiple miscarriages in the paternal grandmother and ESRD in the paternal aunt. The presentation of acute heart failure (AHF) due to valvular disease in two sisters is unusual and has not been reported previously. We propose that the mechanism underlying AHF is TMA-associated vasculitis with thrombus formation leading to valvular dysfunction.
Our work highlights a unique postpartum aHUS presentation with a remarkable phenotype involving the renal and cardiac systems and illustrates the putative role of multiple variants in disease etiology (33).
C3 Glomerulopathy Associated With and Without a Monoclonal Gammopathy
Clinical History
Patient 6
A 67-year-old Caucasian female with biopsy-proven C3GN in the setting of a monoclonal gammopathy (elevated IgG kappa on serum protein electrophoresis and immunofixation) underwent a DDKT in 2016. She had an elevated FH autoantibody and a C3 nephritic factor (C3Nef; 1.02, normal range 0.0–0.26). Prior to transplant, she was treated with bortezemib, cyclophosphamide, and dexamethasone, which reduced the M-protein (≥90%) and led to negative tests for both autoantibodies. She developed recurrent C3GN in 2018 (Supplementary Figure 3). A bone marrow biopsy revealed involvement by the previously diagnosed plasma cell neoplasm. A year later the patient died secondary to refractory congestive heart failure.
Patient 7
A 28-year-old Caucasian male with biopsy-proven C3GN underwent a living unrelated kidney transplant from his wife in Feb 2020. He had an elevated C3Nef (0.3, normal range 0–0.26, Blood Center of Wisconsin) associated with a high Bb level (2.7 mg/l, normal <1.2 mg/l) and an elevated soluble C5b-9 (1.51 mg/l, normal <0.3 mg/l). The alternative pathway complement functional assay (AH50) was low (72%, normal 75–170) as well as the properdin level (7.5 mg/l, normal 10–33 mg/l). A monoclonal gammopathy was not detected (Table 1). There was no family history of kidney disease. At last follow-up in March 2021, Cr was stable at 1.2 mg/dl with no proteinuria.
Genetic Analysis
A rare heterozygous variant T956M was identified in patients 6 and 7 (Table 1). This variant is located at a position that is not highly conserved but has been reported previously in patients with aHUS (34–36). Most in silico tools predict that this variant is tolerated; however, MutationAssessor and Polphen2-Hdiv predict a damaging effect. It was classified as a VUS due to a lack of functional studies. Patient 6 also carried a SNP, E936D, which is common across multiple populations and is associated with a predisposition to aHUS (Supplementary Table 3) (37). In silico tools however predict that E936D is benign.
Structural Analysis
The T956 residue in CCP 16 is less conserved and adjacent to the highly conserved Y957 residue (Figure 3). The hydrophobic Y957 side chain is packed in the CCP 16 interior, whereas the hydrophilic T956 side chain is exposed on the protein surface (Figure 5B). The substitution of hydrophilic T with the long hydrophobic side chain M in the T956M variant may perturb the beta sheet orientation, and therefore alter CCP 16 folding.
The E936 residue in CCP 16 is less conserved in multiple CCP sequence alignments (Figure 3). E and D are both negatively charged hydrophilic amino acids in the E936D variant, and the substitution is highly tolerated without interfering with protein folding (Figure 5B).
Antigenic and Functional Analysis
Factor H antigenic levels in both patients were normal (Table 1). Secretion of the recombinantly produced T956M variant was comparable to that of WT (Figure 2; Table 2). The variant had slightly reduced affinity for C3b binding compared to WT, but unchanged affinity for heparin binding (Figure 5C). Defective C3b binding was corrected with the double mutant E936D/T956M.
Implications
These two patients with C3GN carry a genetic variant (T956M) and acquired defects in the form of autoantibodies (C3Nef and/or FH). Our analyses demonstrate that T956M may have a modest functional defect due to ~30% decrease in C3b binding. However, the function improved when combined as variant E936D/T956M (Figure 5C). The T956M variant was also previously reported in an aHUS patient and functional data showed no defect (36). Thus, while the structural analysis proposes a possible deleterious effect, functional analysis by us and others establish that T956M is likely benign. Therefore, the development of autoantibodies (FH and C3Nef) in patient 6 is most likely related to the underlying monoclonal gammopathy, a well-known association of C3G (38). Patient 7 persistently harbors an elevated C3Nef and an abnormal complement biomarker profile (due to an unclear etiology) despite being on immunosuppression and thus is at risk for recurrent C3GN.
Living Donor Kidney Transplantation in aHUS
Clinical History
Patient 8
A 63-year-old Caucasian male was evaluated as a kidney donor for his 36-year-old son. The son had developed ESRD at age 33 and was given a diagnosis of possible aHUS. He was initially treated with plasmapheresis and subsequently switched to eculizumab. The son underwent genetic testing which identified a heterozygous CFHR3-1 deletion. The father underwent genetic testing as part of donor evaluation work-up. This case was referred to us from an outside hospital to assist with genetic interpretation of the VUS identified in the father as described below.
Genetic Analysis
A novel heterozygous missense variant L1207I was identified. This variant is located within a poorly conserved nucleotide position in CCP 20 (Figure 3) (39). In silico functional prediction indicated a tolerated effect of this alteration. This variant though was categorized as a VUS due to the paucity of functional studies and lack of other pathogenicity supporting evidence. Two SNPs (I1059T and Q1143E) and a homozygous CFHR3-1 deletion were also identified. The father or son did not carry FH-autoantibodies.
Structural Analysis
CCP 20 interacts with the C3d cleavage fragment of C3b and GAGs (14). C3d binds electrostatically with CCP 20. The interacting surface consists of a positively charged pole formed by R1203, K1230, and R1231 from CCP 20, and a negatively charged cleft formed by E160, D163, and D292 from the C3d moiety of C3b (Figure 6A) (40). CCP 20 has a GAG binding site that is formed by a hydrophobic pocket. The key amino acids identified in this region are L1181, R1182, W1183, S1191, E1195, S1196, E1198, and R1215 (Figure 6B) (14). L1207 does not appear to directly coordinate either C3d or GAG binding. L and I are two hydrophobic and highly interchangeable amino acids, and the substitution of L to I likely does not alter the overall conformation of the C3b and heparin binding sites.
Figure 6
Antigenic and Functional Analysis
Secretion of the recombinantly produced variant protein in the supernatant was normal compared to that of WT (WT, 1.1 μg/ml; L1207I, 1.07 μg/ml) (Figure 2C). Functional studies confirmed that the variant had no significant effect on either C3b or heparin binding compared to that of WT (P > 0.05) (Figure 6C). Functional analysis of the SNPs (I1059T and Q1143E) was also performed and demonstrated no defect (Supplementary Figure 4).
Implications
This case is challenging because it involves a living donor transplant for a father-son pair. The son (potential recipient) has a presumptive diagnosis of aHUS with no identified genetic variants, whereas the healthy father (potential donor) has a rare genetic variant (L1207I) and SNPs in CFH. Our analyses showed that L1207I is not functionally or structurally deleterious and probably explains why the father did not develop kidney disease. In this case, donor candidacy was declined since functional analysis of the VUS was not available at the time of transplantation. More importantly, a definitive diagnosis of aHUS and its etiology in the son remains unknown.
Living-related donor kidney transplant is generally contraindicated for patients with aHUS (43) because a nephrectomy may trigger TMA in the genetically susceptible donor. Although donor genetic analysis may be performed, some recipients have more than one mutation and genetic testing does not reveal a variant in the complement gene in approximately one-third of patients with aHUS (such as in this case). We may consider living-related donor transplants in patients who carry a known pathogenic mutation in a complement protein that causes disease in the patient if the donor tests negative for that mutation. The recipient and living donor should participate in the decision-making process after they understand the risks and benefits.
Discussion
We describe clinical cases of eight patients with a TMA or C3G from our kidney transplant cohort. In each case, we were faced with the challenge of determining the disease etiology and recurrence risk because of the presence of confounding risk factors in their clinical history. Moreover, these patients carried genetic variants in CFH that were classified as a VUS. Since there was a lack of information in the literature about the functional impact of these variants, we employed an integrative approach by utilizing the clinical, genetic, antigenic and structural data in addition to the functional analyses, to further define the implications for each variant (5).
Our work highlights a multi-modality approach that substantially facilitated: (i) Establishing the diagnosis of a complement-mediated TMA in a complex clinical setting (lupus) with multiple triggers (Patients 1 and 2) (ii) Defining the role of multiple deleterious variants in a unique postpartum TMA presentation featuring a three-sister kindred with multiple organ system involvement (Patients 3, 4, and 5). (iii) Illustrating the prominent role of acquired factors (monoclonal gammopathy, C3 nephritic factor, and FH autoantibody) in C3G even though a rare variant was identified but demonstrated normal function (Patients 6 and 7) (iv) Formulating an informed therapeutic decision about a potential living donor transplantation in the setting of a TMA (Patient 8).
Our results point out potential problems in using the ACMG guidelines to interpret variants. These guidelines are designed for Mendelian diseases and heavily weigh population frequency. Consequently, despite functional data demonstrating no defect, the interpretation for many of the genetic variants described in our cases remained a VUS primarily due to the rarity of the variant. Based on these results, we suggest that modifications to the ACMG framework for diseases with variable penetrance, such as aHUS, be considered. For combined variants, we speculated but could not confirm if they were present on the same or different alleles. One way to further address this issue is to test family members to determine whether variants are shared by other affected and unaffected relatives. In our cases, the family samples were not available for sequencing.
There are two quite informative studies on CFH variants in aHUS and C3G by Merinero et al. that are particularly worth noting here. In the first one, the authors report functional characterization of CFH variants using FH purified from plasma samples of FH Y402H heterozygous carriers (36). This was an elegant approach to obtaining purified full-length FH which was available for 7 of 12 variants. While our manuscript was being prepared for submission, a second comprehensive study reported a functional assessment for 105 CFH variants associated with aHUS (44). In this study, full length FH was prepared recombinantly by ThermoFisher Scientific (Regensburg, Germany). Of note, four of the five VUS we describe here have not been reported previously including in this recent publication. The one overlapping variant, T956M, demonstrated similar results to our data. Also, the SNPs (E936D, N1050Y, I1059T, Q1143E) have been reported and results of our functional data are comparable (Supplementary Figure 4) (44). Additionally, we demonstrate that the SNP E936D may have a potentially protective role since it helped improve the C3b binding for rare variant, T956M. We used FH fragments which have been shown by us (16) and others (45–47) to be highly predictive of the results obtained with full-length protein (36, 44). Nevertheless, the availability of commercially available full-length recombinant FH and the serum-based assays is an advance that should be taken advantage of.
In summary, there are challenges in reconciling genetic data with clinical management. The strategy of recombinant protein production followed by systematic antigenic, functional, and structural assessment remains the gold standard (Figure 7). We predict that such functional assessments will become more widely available in the future and will be critical to address knowledge gaps and identify the etiology and pathogenic mechanisms in patients with complement-mediated kidney diseases, especially in a complex clinical setting of kidney transplantation. The ongoing development of complement biomarkers and novel assays will provide clinicians with the tools to stratify patients for targeted therapy and adapt precision medicine for genetic kidney diseases.
Figure 7
Funding
This study was supported by the Barnes-Jewish Hospital Foundation Fund (AJ). Research reported in this publication was also supported by NIGMS, NEI, and NIAMS of the National Institutes of Health under award numbers R35 GM136352, R01 EY028602, and R21 AR076534 (JA). The manuscript was edited by the Scientific Editing Service of the Institute of Clinical and Translational Sciences at Washington University, which is supported by an NIH Clinical and Translational Science Award (UL1 TR002345).
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The original contributions presented in the study are publicly available. These data can be found here: ncbi.nlm.nih.gov/clinvar/, SCV001905506–SCV001905512.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board Washington University School of Medicine in St. Louis, MO. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZR, AJ, and JA conceived and designed the experiments, analyzed the data, and interpreted results. ZR performed the experiments. ZR and SP conducted the structural analysis. LL-G classified variants in accordance with ACMG 2015 criteria. ZR prepared the figures and drafted the manuscript. ZR, SP, LL-G, JA, and AJ edited the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors thank Dr. Aubrey Morrison (Washington University School of Medicine) for his helpful suggestions during manuscript preparation; Drs. Appalanaidu Sasapu (University of Arkansas for Medical Sciences), Yuzhou Zhang, and Richard Smith (Molecular Otolaryngology and Renal Research Laboratories, University of Iowa) for their assistance in conducting serum-based assays for patients 1 and 2 and for providing details for variant G918E (relevant for patient 3, 4, and 5); Dr. Daniel Brennan (Johns Hopkins University School of Medicine) for providing patient details on patient 8; Dr. Joseph Gaut (Washington University School of Medicine in St. Louis, MO) for providing the kidney biopsy image for patient 1; and Dr. Hongjie Gu (Division of Biostatistics, Washington University School of Medicine) for support in reviewing statistical methods and performing the statistical analyses.
Conflict of interest
AJ reports serving on the scientific advisory boards of Alexion Pharmaceuticals and Novartis Pharmaceuticals and being a consultant for Gemini Therapeutics and Chinook Therapeutics. AJ is a principal investigator on a trial by Apellis pharmaceuticals. JA reports serving as a consultant for Celldex Therapeutics, Clinical Pharmacy Services, Kypha Inc., Achillion Pharmaceuticals Inc., and BioMarin Pharmaceutical Inc. and having stock or equity options in Compliment Corporation, Kypha Inc., Gemini Therapeutics, and Q32 BIO INC. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.775280/full#supplementary-material
References
1.
GoodshipTHCookHTFakhouriFFervenzaFCFremeaux-BacchiVKavanaghDet al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. (2017) 91:539–51. 10.1016/j.kint.2016.10.005
2.
KavanaghDGoodshipTHRichardsA. Atypical hemolytic uremic syndrome. Semin Nephrol. (2013) 33:508–30. 10.1016/j.semnephrol.2013.08.003
3.
Caravaca-FontanFLucientesLCaveroTPragaM. Update on C3 glomerulopathy: a complement-mediated disease. Nephron. (2020) 144:272–80. 10.1159/000507254
4.
Gonzalez SuarezMLThongprayoonCMaoMALeeaphornNBathiniTCheungpasitpornW. Outcomes of kidney transplant patients with atypical hemolytic uremic syndrome treated with eculizumab: a systematic review and meta-analysis. J Clin Med. (2019) 8:919. 10.3390/jcm8070919
5.
GavriilakiEAnagnostopoulosAMastellosDC. Complement in thrombotic microangiopathies: unraveling ariadne's thread into the labyrinth of complement therapeutics. Front Immunol. (2019) 10:337. 10.3389/fimmu.2019.00337
6.
NorisMRemuzziG. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. (2010) 10:1517–23. 10.1111/j.1600-6143.2010.03156.x
7.
Regunathan-ShenkRAvasareRSAhnWCanettaPACohenDJAppelGBet al. Kidney transplantation in C3 glomerulopathy: a case series. Am J Kidney Dis. (2019) 73:316–23. 10.1053/j.ajkd.2018.09.002
8.
PalmaLMPSridharanMSethiS. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. (2021) 6:11–23. 10.1016/j.ekir.2020.10.009
9.
JavaAPozziNLove-GregoryLDHeuselJWSungYJHuZet al. A multimodality approach to assessing factor I genetic variants in atypical hemolytic uremic syndrome. Kidney Int Rep. (2019) 4:1007–17. 10.1016/j.ekir.2019.04.003
10.
JavaABaciuPWidjajahakimRSungYJYangJKavanaghDet al. Functional analysis of rare genetic variants in complement factor I (CFI) using a serum-based assay in advanced age-related macular degeneration. Transl Vis Sci Technol. (2020) 9:37. 10.1167/tvst.9.9.37
11.
PickeringMCD'agatiVDNesterCMSmithRJHaasMAppelGBet al. C3 glomerulopathy: consensus report. Kidney Int. (2013) 84:1079–89. 10.1038/ki.2013.377
12.
WilsonPCLove-GregoryLCorlissMMcNultySHeuselJWGautJP. Beyond panel-based testing: exome analysis increases sensitivity for diagnosis of genetic kidney disease. Kidney360. (2020) 1:772–80. 10.34067/KID.0001342020
13.
Sanchez-CorralPPouwRBLopez-TrascasaMJozsiM. Self-damage caused by dysregulation of the complement alternative pathway: relevance of the factor H protein family. Front Immunol. (2018) 9:1607. 10.3389/fimmu.2018.01607
14.
MorganHPSchmidtCQGuarientoMBlaumBSGillespieDHerbertAPet al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. (2011) 18:463–70. 10.1038/nsmb.2018
15.
LiszewskiMKLeungMKHauhartRBullerRMBertramPWangXet al. Structure and regulatory profile of the monkeypox inhibitor of complement: comparison to homologs in vaccinia and variola and evidence for dimer formation. J Immunol. (2006) 176:3725–34. 10.4049/jimmunol.176.6.3725
16.
WagnerEKRaychaudhuriSVillalongaMBJavaATriebwasserMPDalyMJet al. Mapping rare, deleterious mutations in Factor H: association with early onset, drusen burden, and lower antigenic levels in familial AMD. Sci Rep. (2016) 6:31531. 10.1038/srep31531
17.
BuhlmannDEberhardtHUMedyukhinaAProdingerWMFiggeMTZipfelPFet al. FHR3 blocks C3d-mediated coactivation of human B cells. J Immunol. (2016) 197:620–9. 10.4049/jimmunol.1600053
18.
WangXSappMChristensenNDDillnerJ. Heparin-based ELISA reduces background reactivity in virus-like particle-based papillomavirus serology. J Gen Virol. (2005) 86:65–73. 10.1099/vir.0.80472-0
19.
WangZFlaxLAKempMMLinhardtRJBaronMJ. Host and pathogen glycosaminoglycan-binding proteins modulate antimicrobial peptide responses in Drosophila melanogaster. Infect Immun. (2011) 79:606–16. 10.1128/IAI.00254-10
20.
ProsserBEJohnsonSRoversiPHerbertAPBlaumBSTyrrellJet al. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med. (2007) 204:2277–83. 10.1084/jem.20071069
21.
SchneiderMCProsserBECaesarJJKugelbergELiSZhangQet al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature. (2009) 458:890–3. 10.1038/nature07769
22.
OsborneAJBrenoMBorsaNGBuFFremeaux-BacchiVGaleDPet al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol. (2018) 200:2464–78. 10.4049/jimmunol.1701695
23.
BuFBorsaNGJonesMBTakanamiENishimuraCHauerJJet al. High-throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. J Am Soc Nephrol. (2016) 27:1245–53. 10.1681/ASN.2015040385
24.
YuYWagnerEKSouiedEHSeitsonenSImmonenIJHappolaPet al. Protective coding variants in CFH and PELI3 and a variant near CTRB1 are associated with age-related macular degeneration dagger. Hum Mol Genet. (2016) 25:5276–85. 10.1093/hmg/ddw336
25.
NeumannHPSalzmannMBohnert-IwanBMannuelianTSkerkaCLenkDet al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. (2003) 40:676–81. 10.1136/jmg.40.9.676
26.
RichardsABuddlesMRDonneRLKaplanBSKirkEVenningMCet al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet. (2001) 68:485–90. 10.1086/318203
27.
Fremeaux-BacchiVMoultonEAKavanaghDDragon-DureyMABlouinJCaudyAet al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2006) 17:2017–25. 10.1681/ASN.2005101051
28.
CaprioliJNorisMBrioschiSPianettiGCastellettiFBettinaglioPet al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. (2006) 108:1267–79. 10.1182/blood-2005-10-007252
29.
GautJPJainSPfeiferJDVigh-ConradKACorlissMSharmaMKet al. Routine use of clinical exome-based next-generation sequencing for evaluation of patients with thrombotic microangiopathies. Mod Pathol. (2017) 30:1739–47. 10.1038/modpathol.2017.90
30.
MariniSCGomesMGuilhermeRCardaJPPintoCSFidalgoTet al. Atypical hemolytic-uremic syndrome: recurrent phenotypic expression of a patient with MCP gene mutation combined with risk haplotypes. Blood Coagul Fibrinolysis. (2019) 30:68–70. 10.1097/MBC.0000000000000793
31.
KavanaghDYuYSchrammECTriebwasserMWagnerEKRaychaudhuriSet al. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet. (2015) 24:3861–70. 10.1093/hmg/ddv091
32.
BarlowPNSteinkassererANormanDGKiefferBWilesAPSimRBet al. Solution structure of a pair of complement modules by nuclear magnetic resonance. J Mol Biol. (1993) 232:268–84. 10.1006/jmbi.1993.1381
33.
BresinERuraliECaprioliJSanchez-CorralPFremeaux-BacchiVRodriguez De CordobaSet al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. (2013) 24:475–86. 10.1681/ASN.2012090884
34.
MagaTKNishimuraCJWeaverAEFreesKLSmithRJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. (2010) 31:E1445–60. 10.1002/humu.21256
35.
Perez-CaballeroDGonzalez-RubioCGallardoMEVeraMLopez-TrascasaMRodriguez De CordobaSet al. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. (2001) 68:478–84. 10.1086/318201
36.
MerineroHMGarciaSPGarcia-FernandezJArjonaETortajadaARodriguez De CordobaS. Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int. (2018) 93:470–81. 10.1016/j.kint.2017.07.015
37.
NorisMCaprioliJBresinEMossaliCPianettiGGambaSet al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. (2010) 5:1844–59. 10.2215/CJN.02210310
38.
RavindranAFervenzaFCSmithRJHSethiS. C3 glomerulopathy associated with monoclonal Ig is a distinct subtype. Kidney Int. (2018) 94:178–86. 10.1016/j.kint.2018.01.037
39.
ClarkSJRidgeLAHerbertAPHakobyanSMulloyBLennonRet al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol. (2013) 190:2049–57. 10.4049/jimmunol.1201751
40.
KajanderTLehtinenMJHyvarinenSBhattacharjeeALeungEIsenmanDEet al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci USA. (2011) 108:2897–902. 10.1073/pnas.1017087108
41.
BlaumBSHannanJPHerbertAPKavanaghDUhrinDStehleT. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol. (2015) 11:77–82. 10.1038/nchembio.1696
42.
Sanchez-CorralPPerez-CaballeroDHuarteOSimckesAMGoicoecheaELopez-TrascasaMet al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. (2002) 71:1285–95. 10.1086/344515
43.
JavaA. Peri- and post-operative evaluation and management of atypical hemolytic uremic syndrome (aHUS) in kidney transplantation. Adv Chronic Kidney Dis. (2020) 27:128–37. 10.1053/j.ackd.2019.11.003
44.
Martin MerineroHZhangYArjonaEDel AngelGGoodfellowRGomez-RubioEet al. Functional characterization of 105 factor H variants associated with atypical HUS: lessons for variant classification. Blood. (2021) 2021:blood.2021012037. 10.1182/blood.2021012037
45.
FerreiraVPHerbertAPHockingHGBarlowPNPangburnMK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol. (2006) 177:6308–16. 10.4049/jimmunol.177.9.6308
46.
WongEKSHallamTMBrocklebankVWalshPRSmith-JacksonKShuttleworthVGet al. Functional characterization of rare genetic variants in the n-terminus of complement factor H in aHUS, C3G, and AMD. Front Immunol. (2020) 11:602284. 10.3389/fimmu.2020.602284
47.
PechtlICKavanaghDMcintoshNHarrisCLBarlowPN. Disease-associated N-terminal complement factor H mutations perturb cofactor and decay-accelerating activities. J Biol Chem. (2011) 286:11082–90. 10.1074/jbc.M110.211839
Summary
Keywords
kidney transplantation, complement, complement regulators, atypical hemolytic uremic syndrome, variants of uncertain significance, thrombotic microangiopathy, C3 glomerulopathy
Citation
Ren Z, Perkins SJ, Love-Gregory L, Atkinson JP and Java A (2021) Clinicopathologic Implications of Complement Genetic Variants in Kidney Transplantation. Front. Med. 8:775280. doi: 10.3389/fmed.2021.775280
Received
13 September 2021
Accepted
08 November 2021
Published
29 November 2021
Volume
8 - 2021
Edited by
Sophia Lionaki, National and Kapodistrian University of Athens, Greece
Reviewed by
Jia Rao, Fudan University, China; Li Zhu, Peking University First Hospital, China
Updates
Copyright
© 2021 Ren, Perkins, Love-Gregory, Atkinson and Java.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anuja Java ajava@wustl.edu
This article was submitted to Nephrology, a section of the journal Frontiers in Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.