 Chih-Ru Lin
Chih-Ru Lin Karim Bahmed2,3
Karim Bahmed2,3 Beata Kosmider
Beata Kosmider
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 03 January 2022
Sec. Pulmonary Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.762878
This article is part of the Research TopicManaging Chronic Obstruction Pulmonary Disease: From Translational Research to Public Health PracticeView all 15 articles
Pulmonary emphysema is characterized by the destruction of alveolar septa and irreversible airflow limitation. Cigarette smoking is the primary cause of this disease development. It induces oxidative stress and disturbs lung physiology and tissue homeostasis. Alveolar type II (ATII) cells have stem cell potential and can repair the denuded epithelium after injury; however, their dysfunction is evident in emphysema. There is no effective treatment available for this disease. Challenges in this field involve the large complexity of lung pathophysiological processes and gaps in our knowledge on the mechanisms of emphysema progression. It implicates dysregulation of various signaling pathways, including aberrant inflammatory and oxidative responses, defective antioxidant defense system, surfactant dysfunction, altered proteostasis, disrupted circadian rhythms, mitochondrial damage, increased cell senescence, apoptosis, and abnormal proliferation and differentiation. Also, genetic predispositions are involved in this disease development. Here, we comprehensively review studies regarding dysregulated cell signaling, especially in ATII cells, and their contribution to alveolar wall destruction in emphysema. Relevant preclinical and clinical interventions are also described.
Over 300 million people suffer from chronic obstructive pulmonary disease (COPD) worldwide (1). It is the third leading cause of death, resulting in ~3 million deaths every year, according to World Health Organization (WHO) (2). COPD includes lung parenchymal destruction (emphysema) and airway disease (chronic bronchitis). The extent of lung tissue damage in emphysema is measured by chest computed tomography (CT) density. Emphysema is a progressive and irreversible disease with limited therapeutic strategies. Lung volume reduction surgery and lung transplantation represent promising options for end-stage disease (3). At the cellular level, emphysema is characterized by alveolar epithelial cell death and impaired re-epithelialization (Figure 1), which causes alveolar wall destruction and decreased surface area in the lung parenchyma for gas exchange (4). Pulmonary vasculature is linked to alveolar architectures and function, whereas endothelial dysfunction and vascular abnormalities were observed in emphysema (5, 6). Furthermore, extracellular matrix (ECM), including elastin, collagen, and proteoglycans, tethers the parenchymal compartments to the airway and affects the tissue mechanics and airway smooth muscle contraction. The process of mechanotransduction provides mechanical cues of the microenvironment to control many cellular events such as proliferation and differentiation and maintain tissue integrity (7). Changes in ECM composition in emphysema may impact airway smooth muscle cells function, including hypertrophy and hyperplasia, resulting in airway remodeling and obstruction. Also, the loss of elastic recoil leads to airspace enlargements and irreversibly weakens respiratory airflow (8). Increased lung volume, gas trapping, and reduced alveolar units are observed in emphysema patients compared to controls (4).
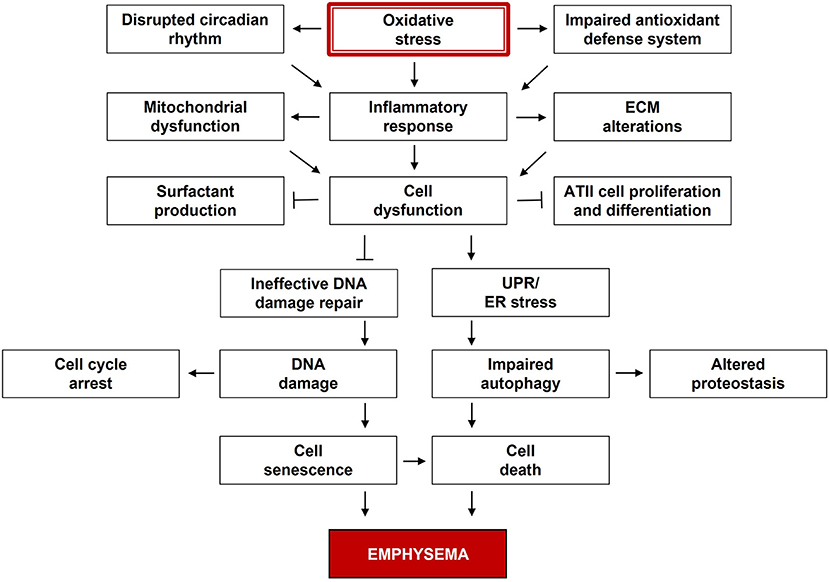
Figure 1. Multiple dysregulated signaling pathways in the pathogenesis of emphysema. Oxidative stress is the major contributor to emphysema. Reactive oxygen species (ROS) and related species disturb cell signaling and impair cell functions, while the antioxidant defense system is overwhelmed. These components are dynamically and progressively interactive over time, and their alterations can lead to emphysema development.
Environmental irritants, especially cigarette smoke, are the main risk factors of emphysema development. Cigarette smoke contains thousands of chemicals and oxidants that can induce oxidative stress and modify biomolecules, including proteins, lipids, nucleic acids, or carbohydrates, thus affecting lung physiology and tissue homeostasis (9). Surfactant is essential for alveoli and smoking has adverse effects on its function and composition through oxidation and altered proteostasis (10). For instance, Takamiya et al. showed that cigarette smoke extract and its component acrolein modified the surfactant protein (SP)-A in the alveolar epithelium (11). The ability of SP-A to inhibit bacterial growth and macrophage phagocytosis in vitro was attenuated due to its conformational changes. The antioxidant defense systems can protect from damage, whereas they are overwhelmed by persisting oxidative stress (9). Cigarette smoke-induced reactive oxygen species (ROS) disturb cell signaling and impair cell functions, including inflammatory responses and protease-antiprotease balance, resulting in ECM destruction and alveolar epithelial cell injury and death (12). Furthermore, emerging senescent cells and vascular dysfunction in the lungs fail the microenvironment for alveolar re-epithelialization.
Besides environmental factors, genetic susceptibility has a significant impact on the morbidity of COPD, such as alpha-1 antitrypsin (AAT) deficiency (13). Studies have identified multiple genetic loci related to this disease susceptibility, e.g., HHIP, FAM13A, IREB2, MMP12, MMP1, RAGE, SFTPD, FBLN5 in humans and mice (14). These genes are associated with surfactant function, tissue growth, remodeling, and homeostasis. The large complexity of lung pathophysiological processes implicated in emphysema contributes to its heterogeneity. Genetic susceptibility, age, sex, and race-dependent differences may also contribute to this disease development.
Cigarette smoke-induced oxidative stress and redox imbalance are major contributors to emphysema (15). ROS and related species act as second messengers in cell signal transduction and participate in cellular physiological responses, including the inflammatory immune system and mitochondrial respiration. Oxidative stress refers to high ROS levels that overwhelm the antioxidant defense system. It can alter the activities and functions of redox-sensitive molecules and metabolic enzymes such as p53, NRF2, NF-κB, JNK, MAPK, protein tyrosine phosphatases, glutathione, thioredoxin, peroxiredoxins, and histone deacetylase (15, 16). High oxidative stress and the defective antioxidant defense system were detected in the lungs and alveolar type II (ATII) cells in emphysema patients (15, 17). Especially reduced NRF2 (18) and FOXO3 (19) levels were observed, which are important transcription factors regulating multiple antioxidant genes such as HO-1, NQO1, and GPX1. Also, NF-E2 was downregulated in ATII cells in this disease (20). Several NRF2 activators and different classes of antioxidants have been developed and tested in clinical trials, however, without promising results (15). Increasing evidence shows that the small redox protein thioredoxin relieved animal emphysema and pulmonary inflammation through multiple mechanisms (21). Its therapeutic potential was recently proposed.
We have demonstrated that DJ-1 protein modulates the NRF2-mediated antioxidant defense system in human primary ATII cells after exposure to cigarette smoke (22). Altered DJ-1 function, its overoxidation to sulfonate form (-) at the cysteine-106 and ubiquitination, were reported in the pathophysiological response to oxidative damage and emphysema patients (17). Of note, DJ-1 oxidation to sulfinate form (-) was detected in smokers, which reflected its cytoprotective activity. Oxidative stress-induced DJ-1 overoxidation and cleavage in mitochondria were observed in A549 cells treated with hydrogen peroxide. DJ-1 ablation resulted in mitochondrial dysfunction. We also showed impaired S100A8 function in ATII cells in emphysema (23). S100A8 belongs to the S100 protein family and responds to oxidative stress. ATII cell death in this disease correlated with decreased S100A8 sulfination (24). Furthermore, the cytoprotective function of S100A8 compensated for the absence of DJ-1 in vitro and in vivo. Targeting redox regulation appears to be an ideal approach to this disease yet challenging, possibly due to delicate redox balance, disparities between animal models and human diseases, species differences, compound bioavailability, and other variables.
Environmental irritants can promote the recruitment of inflammatory cells into the lungs. The pro-inflammatory and immune responses are evident in emphysema pathophysiology. Glucocorticoids and bronchodilators are commonly used to treat COPD, although both have many side effects, including immunosuppression (21). The PDE4 inhibitor roflumilast is an approved drug that selectively inhibits PDE4 and increases cAMP levels, leading to an anti-inflammatory response. Numerous PDE inhibitors are in clinical testing. Dual PDE3/4 inhibitors such as RPL554 have gained interest in enhanced efficacy as PDE3 is expressed in vascular smooth muscle cells, and its inhibitor induces bronchodilation (25). Similarly, drug combinations may have a synergistic effect over mono-components. For example, combination of PDE4 and PI3Kδ inhibitors significantly increased protection against cigarette smoke extract-induced apoptosis of lung epithelial cells and reduced inflammatory responses of neutrophils and macrophages in vitro.
Under normal conditions, the immune cells balance lung defense, tolerance, or tissue repair. Particularly, alveolar macrophages (AM) interact with pulmonary surfactants in the innate immune responses. AM are potent phagocytes, regulate adaptive immunity and recruit neutrophils and monocytes into the lungs. They secrete inflammatory mediators and proteases such as ECM degrading enzymes and cathepsins upon activation, resulting in elastolysis, alveolar tissue damage, and remodeling of alveoli (8, 12). AM show plasticity based on the microenvironment. Lechner et al. indicated that M2-like macrophages and bone marrow-derived monocytes constitute a regenerative ATII cell niche component, modulating their proliferation and differentiation (26). Using knockout mice and adoptive transfer studies, they found that macrophages accumulated in the lung via the CCL2-CCR2 chemokine recruitment axis during lung regeneration after pneumonectomy. F4/80+ M2-like macrophage and ATII cell co-culture supported the formation of pneumospheres.
Nevertheless, high inflammation in smokers correlated with emphysema development (8, 27). Inflammatory responses downregulated SFTPB, SFTPD, SCNN1A, and SCNN1B gene expression, related to surfactant production and alveolar fluid clearance in ATII cells (28). Decreased SP-D expression was detected in the lungs of emphysema patients (29), indicating the defects in ATII cell function and innate immunity. Especially the number of AM is negatively related to alveolar parenchymal tissue density (27), and impaired phagocytosis is positively associated with the severity of emphysema (30). Increased microRNA (miR)-155 expression was recently identified in the smokers' lungs and in AM of mice exposed to cigarette smoke (31), contributing to inflammation and this disease development. ATII cells isolated from subjects with this disease showed high oxidative stress (17, 32) and ECM degradation with the elevation of matrix metalloproteinase (MMP) 9, CD147, and cathepsin B (20). Other proteases elevated in patients with emphysema include neutrophil elastase, dipeptidyl peptidase-4, proteinase-3, MMPs 1, 8, 12, and 13, cathepsins C, D, E, G, K, and S, and caspases 1, 3, 7, 8, 9, and 11 (33). Several studies have shown the efficacy of antiprotease therapy in emphysema; however, without promising results in clinical trials (21). On the other hand, ADAM8 belongs to a disintegrin and metalloproteinase family. It was reported as a protective proteinase negatively related to emphysema development (34). ADAM8 levels were decreased in macrophages and alveolar epithelial cells in patients with this disease and mice exposed to cigarette smoke. Moreover, ADAM8 knockout mice displayed higher inflammatory and oxidative stress levels, mucus cell metaplasia, alveolar septal cell death, and reduced ATII cell proliferation and repair, contributing to the lung destruction caused by cigarette smoke exposure. Accordingly, pro-inflammatory mediators, oxidative stress, protease-antiprotease imbalance, and impaired pathogen phagocytosis contribute to the destruction of lung tissue and this disease progression (12). Many anti-inflammatory drugs have been developed but have limited efficacy (21). Further studies of immune responses and homeostasis can provide a clue for therapeutic interventions and lung regeneration.
Increasing evidence indicates that the nuclear hormone receptor PPARγ, a regulator of lipid metabolism, adipogenesis, and inflammation, represents a potential therapeutic target for emphysema. It was downregulated in antigen-presenting cells (APCs) isolated from the lungs of patients with this disease and mice exposed to cigarette smoke, thus directing Th1 and Th17 cell differentiation (35, 36). Also, overexpression of dominant-negative PPARγ in murine ATII cells induced emphysema with increased inflammatory cytokines, MMPs, and accumulation of myeloid-derived suppressor cells (MDSCs) in the lungs and circulation system (37). As expected, treatment with a PPARγ agonist ciglitazone inhibited pathogenic lung APCs in vitro in humans and mice. Further, it attenuated cigarette smoke-induced emphysema in mice (35, 36). Thiazolidinediones (TZDs), including ciglitazone, pioglitazone, rosiglitazone, and troglitazone, are valuable drugs for type 2 diabetes and have shown anti-inflammatory effects through selective stimulation of PPARγ. Their application to emphysema needs to be further evaluated.
Chronic inflammation can increase susceptibility to lung infection. Cigarette smoke-induced emphysema in animals was challenged with bacterial or viral infections to study the mechanism of this disease exacerbation (38). H3N1 influenza A virus infection caused higher virus titers in the lung, inflammation around bronchi and in the parenchyma, and mucus exudates in the airways in cigarette smoke-exposed mice compared to the influenza virus alone (39). Also, Pseudomonas aeruginosa infection resulted in impaired autophagy with an increased number of aggresomes, cell senescence, and alveolar space enlargement in mice exposed to cigarette smoke compared to controls (40). Cysteamine, an antioxidant drug with autophagy-inducing properties, alleviated these changes. Nippostrongylus brasiliensis, a nematode in rodents, was used to develop a mouse model of progressive emphysema (38). This infection induced a long-lasting Th2 immune response and alternatively activated M2 macrophages accompanied by substantially upregulated MMP12 expression, resulting in alveolar wall destruction and airspace enlargement. Increasing evidence shows the formation of autoimmune components, including autoantibodies and key mediators (such as BAFF and IL-17A), increased B cell counts, and B cell-rich lymphoid follicles in emphysema patients, which were associated with this disease severity (41, 42). Zhou et al. exposed mice to 150–180 mg/m3 cigarette smoke for 2 weeks followed by 2 weeks of rest before the elastin challenge to study the autoimmunity in emphysema development (43). MMP12-generated elastin fragments are self-antigens that induce monocyte chemotactic activity and contribute to this disease pathology. Exposure to cigarette smoke sensitized mice to elastin and elicited IL17A-predominant autoimmune processes leading to neutrophilic airway inflammation, mucus hyperproduction, airspace enlargement, and lung function decline. Of note, elastin-specific T cell response was also observed in COPD patients. The correlation of B cell adaptive immune responses with late-stage emphysema introduces opportunities for new therapeutic interventions. However, the crucial role of B cells in the defense against pathogenic infections and immune-regulatory activities limits anti-B cell therapies. Further characterization of B cell subsets and their contribution to disease progression are needed.
Cigarette smoke-induced oxidative stress increases protein misfolding (44), thus altering lung homeostasis. It induces endoplasmic reticulum (ER) stress and unfolded protein response (UPR), leading to inflammation and cell apoptosis. ER stress was observed in emphysema patients and animals (45). Activation of UPR with the upregulation of GRP78 was detected in human small airway epithelial cells and ATII cells obtained from smokers (46). The aggresome formation, involved in the cellular response to misfolded proteins and their clearance, was also observed in the lungs of smokers, emphysema patients, mice exposed to cigarette smoke, and aged mice (40). Specifically, the levels of aggresome correlated with smoking history and emphysema severity in humans. Cigarette smoke increased aggresome formation by accumulating ubiquitinated proteins and autophagy proteins p62 and valosin-containing protein (VCP) in murine lungs, indicating aberrant autophagy (40). Intraperitoneal administration with the autophagy-inducing drug cysteamine reduced cigarette smoke-induced aggresome formation in the lungs, inflammatory responses in bronchoalveolar lavage fluid (BALF), and emphysematous changes in mice. Similarly, inhibition of ER stress by treatment with 4-phenylbutyric acid reduced cigarette smoke-induced inflammatory response, alveolar cell apoptosis, and airspace enlargement in mice (47). Therefore, the ER stress-autophagy pathway represents a potential therapeutic intervention.
Several studies have pointed out the deleterious impact of ceramides or lactosylceramides accumulation in the human lung in emphysema progression (48), likely due to impaired autophagy resulting in sphingolipids imbalance. They were accumulated in different cell types, including alveolar epithelial and endothelial cells and macrophages in patients with this disease (49). Ceramides are components of sphingolipids and serve as second messenger lipids. Their upregulation leads to inflammatory responses, which can cause alveolar epithelial cell apoptosis and emphysema development in animal models (49). Cigarette smoke promoted the activation of membrane-localized acid sphingomyelinase, which catalyzes the hydrolysis of sphingomyelin to ceramide. This thereupon increased membrane and intracellular ceramide accumulation in aggresomes (48). Cysteamine reduced cigarette smoke-induced ceramide accumulation in Beas2b cells (48). It improved efferocytosis, cell viability, and decreased cell senescence. Also, intraperitoneal injection of autophagy-inducer gemfibrozil significantly reduced ceramide accumulation in murine lungs after cigarette smoke exposure.
The human respiratory system functions with a daily circadian rhythm, affecting airway resistance, ventilatory controls, and immune function, yet it changes with age and smoking (50, 51). Smokers have marked diurnal changes in pulmonary function tests compared to non-smokers (51). The lungs' molecular clock is also altered in response to environmental factors, including allergens, pollutions, pathogens, infections, oxidative stress, hypoxia/hyperoxia, jet lag, and shift work (52). Importantly, circadian gene expression positively connects to cell cycle and immune regulation in mice and humans (53). Increasing evidence shows a link between circadian rhythm disruption and susceptibility to lung infection (54) and inflammatory diseases (55), including asthma and COPD with severe symptoms in the early morning, which correlated with disease exacerbations.
Pekovic-Vaughan et al. have demonstrated the circadian regulation of the NRF2 pathway in the murine lung, mainly in the bronchial and alveolar epithelium (56). The loss of NRF2-dependent antioxidant defense system and reduced-glutathione levels increased oxidative damage in the lungs of ClockΔ19 mutant mice compared to wild-type mice. SIRT-1 functions as NAD+-dependent protein deacetylases and regulates many pathophysiological processes. It controls the circadian clock through binding with CLOCK-BMAL1 complexes and promoting the deacetylation and degradation of PER2 proteins (57). In the lungs of mice exposed to cigarette smoke, altered rhythms of SIRT-1 protein and core clock genes including Bmal1, Clock, Per1, Per2, Cry1, Cry2, Nr1d1, Nr1d2, and Rora were observed in a circadian manner, increasing the susceptibility to inflammation and emphysema development (58). Similarly, BMAL1 protein was down-regulated in the lungs of smokers and individuals with this disease (58). BMAL1 deficiency accelerated senescence and caused age-associated shrinkage of major organs, including lungs, in 40-week-old mice, which correlated with increased ROS levels and inflammation (59). Melatonin, a natural hormone that regulates sleep-wake cycles, has shown beneficial effects on lung diseases through anti-oxidation and anti-inflammaton (60). It increased SIRT-1 expression in vitro and in vivo (61, 62). Intraperitoneal injection with melatonin decreased inflammatory response in BALF, and the lungs of mice challenged with cigarette smoke and lipopolysaccharide (61). It also reduced ER stress and bronchial and alveolar epithelial cell apoptosis, and protected alveolar architecture in a rat model of emphysema (62). The daily rhythm of SP-A, SP-B, and SP-C gene expression in the rat lungs was reported (63). This indicates the critical role of circadian rhythm in ATII cell function; however, its alterations in lung pathophysiology are still poorly understood.
Cigarette smoke can cause DNA damage, while the impairment of double-strand DNA break repair in ATII cells was shown in emphysema patients (32). Oxidative stress and persistent DNA damage are associated with stress-induced premature senescence (SIPS) (64). SIPS induces irreversible cell cycle arrest, chromatin changes, and resistance to apoptosis. It also drives a senescence-associated secretory phenotype (SASP) in cells, including secretion of inflammatory cytokines IL-6 and IL-8 and remodeling factors, thereby affecting tissue microenvironment and promoting senescence in an autocrine and paracrine manner (65). DNA damage response (DDR) is required for SASP, including cell cycle regulatory protein NBS1 and checkpoint kinases ATM and CHK2 (66). Transient exposure to SASP promoted stem cell function and tissue regeneration, whereas prolonged exposure had an opposite effect in mice (67). Mice with GFP-expressing ATII cells (CBA/Ca × C57BL6J) exposed to cigarette smoke for over 3 months displayed airspace enlargement and alveolar epithelial cell apoptosis (68). The surviving ATII cells showed higher resistance to apoptosis, which was likely related to circadian rhythm. This suggests the contribution of ATII cell senescence and the circadian clock's regulation to emphysema development. Reduced anti-aging protein SIRT-1 levels were found in the lungs of smokers and patients with emphysema, including AM, airway, and alveolar epithelium (69, 70). Particularly, oxidative stress-induced miR-34a expression was detected in lung tissue in these patients leading to suppression of SIRT-1 and SIRT-6 expression via the PI3K pathway (70). Furthermore, increased lipofuscin levels, a marker of senescence, and cell cycle inhibitors p16 and p21 were mainly detected in ATII cells and endothelial cells in emphysema patients compared to controls (71). Telomere signal intensity in these cells was lower in smokers and emphysema than in controls. A negative correlation between p16 and proliferation cell nuclear antigen (PCNA) was also reported. Similarly, other cells displayed characteristics of cellular senescence, including airway epithelial cells, fibroblasts, and immune cells (72). Their accumulation indicates the loss of tissue homeostasis and an altered environment for alveolar re-epithelialization in emphysema. Senotherapies are effective in animal models, including SIRT-1 activators and inhibitors of mTOR, JAK, FOXO4, and anti-apoptotic proteins (72). Drugs targeting anti-apoptotic proteins in senescent cells such as dasatinib and quercetin were well-tolerated in patients with age-related diseases. Senotherapy seems promising since the mechanism of cellular senescence is likely shared between different diseases.
Furthermore, mitochondria generate ATP for cell metabolism, and their dysfunction has been linked to age-related lung diseases. Several mitochondria-targeted antioxidants, including mitoQ, mito-TEMPO, pyrroloquinoline quinone, and SkQl are in clinical trials for other age-related diseases (72). Increased mitochondrial (mt)DNA copy number has been detected in the blood of patients with COPD along with a positive correlation to the number of pack-years smoking (73). It was also found in urine and was associated with respiratory symptoms and emphysema severity (74). Importantly, elevated mitochondrial ROS levels, low mitochondrial amount, accumulation of mtDNA damage, and mitochondrial dysfunction were detected in human ATII cells in this disease progression (75). It has been shown that electronic (e)-cigarette aerosols containing nicotine caused airway hyperreactivity, alveolar destruction, reduced lung function, and emphysematous changes in mice (76). Nicotine altered calcium levels and mitochondrial membrane potential in human primary ATII cells with DJ-1 knockdown (77). Dysregulation of mitochondrial oxidative phosphorylation (OXPHOS) complexes was observed in the lungs of DJ-1 knockout mice exposed to aerosolized nicotine, which disrupted the nuclear/mitochondrial stoichiometry resulting in mitochondrial dysfunction. This was associated with increased AM number and ATII cell apoptosis. Both ER stress and mitochondrial dysfunction were detected in emphysema, however, with unknown integration. It was recently demonstrated that ER stress led to mitochondrial UPR in an ATF4-dependent manner in MLE-12 cells (78). Mitochondrial UPR has contributed to different disease pathogenesis (79), although its activation is also known to lifespan extension in both Caenorhabditis elegans and mice (80). Improving our understanding of the mitochondrial role in cellular senescence can greatly benefit treating emphysema.
The correlation of alveolar cell apoptosis and decreased lung surface area have been shown in emphysema (81). Intratracheal instillation of active caspase-3 protein led to alveolar epithelial cell apoptosis and this disease development in mice (82). Increased active caspase 3 levels were detected in ATII cells in smokers and individuals with emphysema (24). Histone deacetylase inhibition altered chromatin remodeling leading to ATII cell apoptosis and alveolar structure destruction in trichostatin A-treated rats through increased miR34a, p53, cleaved caspase 3, and microtubule-associated protein-1 light chain 3 (LC3) levels (83). Decreased hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF), lysyl oxidase, and collagen expression in this animal model pointed out the involvement of angiogenic factors in the alveolar structure. Subcutaneous injection of VEGF receptor (VEGFR) blocker led to emphysema without the infiltration of inflammatory cells in rats (84). Treatments targeting VEGF signaling have beneficial effects against emphysema development in animal models (85–87). Alveolar and endothelial cells can sense microenvironment changes; therefore, they serve as niches for regulating lung repair and integrity. Angiogenesis impairment by subcutaneous injection of sodium glutamate enhanced lung inflammation and emphysema in mice induced by intratracheal instillation of cigarette smoke extract, which was related to insufficient migration of pericytes and smooth muscle cells in lung tissue (88). Surprisingly, intravenous administration of healthy lung endothelial cells ameliorated emphysematous phenotype and lung function in an elastase-induced mouse model (89). It was recently reported that endothelial cells released angiocrine sphingosine-1-phosphate (S1P) after Pseudomonas aeruginosa infection in mice which promoted alveolar re-epithelialization via S1PR2-YAP signaling (90). Supplementation of S1P prevented alveolar cell apoptosis in VEGFR blockade-induced emphysema in mice (91). This suggests that sphingolipid balance is important for the maintenance of alveolar septal integrity.
In addition to endothelial dysfunction, pulmonary vascular remodeling characterized by increased smooth muscle cell proliferation and narrowing of the vascular lumen has been shown in smokers and to precede airspace enlargement in animal models of emphysema (92). It can subsequently lead to vascular wall thickening, pulmonary hypertension, and right heart failure. Bronchodilators that target the smooth muscles were beneficial in reducing COPD exacerbation risk vs. placebo in large-scale randomized trials (93). Significant improvements in forced expiratory volume (FVC) were reported after bronchodilator administration in subjects with emphysema (95, 96). Treatment with complementary bronchodilators or a combination of inhaled corticosteroids with bronchodilators was more effective than monotherapy (94). Pharmacological lung volume reduction through bronchodilator therapy is an important goal in these patients.
Smooth muscle cells are the mechanical sculptor of the epithelium (97). Nitric oxide (NO) production by nitric oxide synthase (NOS) regulates the degree of contraction of vascular smooth muscle cells and their proliferation. ATII cells express constitutive NOS as well as inducible NOS (iNOS) during inflammatory states, suggesting their interaction with vascular smooth muscle cells. The absence of SP-D in mice displayed iNOS-related chronic inflammation, alterations of surfactant homeostasis, and emphysematous changes. This elucidates the contribution of immune responses to NO/iNOS regulation (98). iNOS inactivation by genetic deficiency and pharmacological inhibition prevented cigarette smoke-induced pulmonary hypertension and emphysema, including structural and functional alterations of the lung vasculature and alveoli in mice (99). In clinical studies, elevated alveolar NO has been associated with COPD severity (100). These patients have higher numbers of iNOS+ ATII cells related to increased protein nitration and decreased lung function (101). Especially, the lung tissue from patients with emphysema has a higher ratio of the number of alveoli/vessels and an increased degree of vessel muscularization. Nitrated proteins in vasculature and alveoli were increased in these patients (99). NO can be toxic through combination with superoxide to generate peroxynitrites leading to nitration of biomolecules, altering their structure and function. Further, peroxynitrites induced alveolar epithelial and endothelial cell apoptosis (99). However, chronic exposure to the NOS inhibitor Nω-nitro-L-arginine methyl ester resulted in vascular senescence, hypertension, and emphysema development in mice (102). This emphasizes the importance of NO/NOS balance in vascular health and alveolar structure.
Chronic hypoxia in emphysema induces pulmonary artery smooth muscle cells proliferation and JAK2/STAT3 activation. JAK2 deficiency attenuated pulmonary vascular remodeling and smooth muscle hyperplasia in mice (103). Furthermore, HIF-1α promotes vascular smooth muscle cell proliferation under hypoxic conditions. Its increased expression was detected by immunohistochemistry in the lungs of emphysema patients, indicating vascular remodeling in this disease. Especially, HIF-1α was positively associated with disease severity (104). With oxygen treatment, the HIF-1α and erythropoietin decreased in COPD (105). It has been demonstrated that HIF-1α signaling in ATII cells promoted their proliferation after acute lung injury in mice induced by lipopolysaccharide or hydrochloric acid (106). ATII cell-specific HIF-1α deletion caused higher mortality in these mice. Also, HIF-1α is activated in the ATII to alveolar type I (ATI) cell transitional state (107). Together, these data show the intimate relationship between alveolar epithelium and smooth muscle cells for alveolar architecture and function.
ATII cells are progenitors in alveoli. Various ATII niches, mediators, and signaling support their functions for alveolar epithelial cell homeostasis and re-epithelialization. The interactions between signaling molecules arising from and acting on the alveolar epithelium, vasculature, mesenchyme, ECM, and immune cells institute ATII cell niches. Dynamic organization of WNT/β-catenin, TGF-β, YAP/TAZ, NOTCH, and TP53 signaling pathways participate in ATII cell growth and differentiation to ATI cells (108). The importance of WNT/β-catenin, YAP/TAZ, and TGF-β signaling in emphysema development has been demonstrated in animal models. Specifically, TGF-β2 was identified as a significant emphysema-associated genetic variant by human genome-wide association studies (GWAS) (109). TGF-β regulates multiple context-dependent cellular processes associated with tissue remodeling and is crucial for epithelial-mesenchymal interactions. It includes the regulation of cell polarity, ECM turnover, ATII to ATI cell transdifferentiation, and differentiation of lung fibroblasts to myofibroblasts positive for α-smooth muscle actin (α-SMA). Leucine-rich α-2-glycoprotein-1 (LRG1), known to regulate TGF-β signaling, was increased in endothelial cells in patients with emphysema and correlated to its severity (89). Elevated YAP protein levels were detected in the lungs of patients with this disease compared to healthy donors (110). In contrast, reduced WNT/β-catenin pathway in ATII cells was observed in emphysema patients (111, 112) while the non-canonical WNT pathway was upregulated (113, 114). These data suggest signaling imbalance and ATII cell dysfunction in the emphysematous lung. It is known that WNT/β-catenin and anti-inflammatory PPARγ signaling pathways are integrated, and they work in opposition, highlighting the relationship between immune response and ATII cell fate. Feller et al. demonstrated that a non-canonical WNT ligand WNT5a and pro-inflammatory cytokines can be transported through extracellular vesicles, leading to systemic inflammation in COPD patients (115). This further points out the importance of the interactive signaling and microenvironment in the pathogenesis of this disease.
HHIP is a component of hedgehog signaling, which is important in many developmental processes (116). It is a genetic locus associated with emphysema susceptibility in humans and mice. Sonic hedgehog signaling is required for myofibroblast differentiation and mesenchymal proliferation during alveologenesis, while it maintains quiescence in the adult murine lung (117). A broad population of hedgehog-receptive mesenchymal cells surrounding airways and alveoli was identified in the murine lung (118). The transgenic activation of the hedgehog in the mesenchyme disrupted the alveolar niche. This impaired ATII cell proliferation, increased airspace, and emphysematous changes. Single-cell transcriptome analysis of the human lung shows that mesenchymal subsets are conserved across species. The intermediate mesenchymal subset was involved in cholesterol metabolism, suggesting its role in surfactant biosynthesis of the alveolar epithelium. In addition, Kato et al. demonstrated that the paracrine signaling capabilities of pericytes, specialized mesenchymal cells surrounding the capillary, are crucial for alveologenesis. Loss of pericyte-specific YAP/TAZ reduced endothelial and ATII cell proliferation through paracrine regulation, resulting in defective alveolarization and a severe emphysema-like morphology in mice (119). It changed the growth factor expression profiles of these cells in the lungs, including reduced Angpt1, Tgfb2, Wnt11, Bmp4, and Hgf (119). Treatments targeting ATII cell proliferation and differentiation to ATI cells have not been developed owing to plenty of unknowns regarding the mechanism of alveolar re-epithelialization.
Oxidative stress, ER stress response, and inflammation are key contributors to emphysema development. Their interactive relationships have been reported (120). ROS provide an oxidizing environment and affect molecular chaperones and enzymes in the ER. In response to ER stress, the UPR helps cells adapt to and survive the stress condition by transcriptional and translational reprogramming. In contrast, programmed cell death signaling is activated when protein homeostasis cannot be achieved. UPR signaling is initiated by ER membrane-bound transducers: IRE1, ATF6, and PERK. Particularly, IRE1α regulates many cell functions, including metabolism, immunity, inflammatory cytokine production, cell differentiation, and apoptosis, through the RIDD mechanism, which induces the degradation of certain mRNAs or miRNAs. Of note, the IRE1α-mediated XBP1 pathway is essential for optimal expression of inflammatory cytokines and proangiogenic factors in human aortic endothelial cells (121, 122). Under chronic or severe ER stress, PERK-mediated phosphorylation of eIF2α induces expression of ATF4 and a proapoptotic factor CHOP/GADD153, resulting in ER stress-induced apoptosis. ATF4 can also induce growth arrest and DNA damage-inducible protein GADD34. Furthermore, ER and mitochondria are physically and functionally connected through mitochondria-associated ER membranes, which contain major calcium channels IP3R and VDAC. ROS can target ER calcium channels, leading to ER calcium release. It subsequently stimulates mitochondrial metabolism and ROS production. The delicate balance between different cell signaling can be the key to therapeutic benefit.
Given that ROS is involved in various physiological processes, the interplay between signaling potentially causes a cascade of cell dysfunction and imbalance under pathological stimuli, leading to tissue damage. Sirtuins (SIRT1-7) maintain cellular redox balance and are an important defense mechanism against oxidative stress according to the different subcellular localization of each sirtuin. Specifically, the ability of SIRT1 to intersect with different signaling, including the circadian clock, inflammation, cell cycle, and senescence, makes it a lucrative target for therapeutics. SIRT1 activators include baicalin, exendin-4, ginkgolide-B, pitavastatin, quercetin, SRT2104, and vitamin D. These medications are in clinical trials for other diseases. Of note, exendin-4 is a licensed medicine for patients with type 2 diabetes. Exendin-4 treatment improved lung function, mortality, and clinical appearance in mice with lipopolysaccharides-induced emphysema, possibly through bronchodilatory effects (123). The mechanistic links between diabetes and COPD imply the applicability of this medicine (124).
Despite numerous investigations, therapeutic strategies for pulmonary emphysema remain limited mainly due to the complexity and heterogeneity of disease manifestation. Multiple dysregulated signaling pathways affect physiological processes in emphysema development (Figure 1). Its pathological features include high inflammatory and immune response, oxidative stress, defective antioxidant defense system, altered protein homeostasis, cellular senescence, and apoptosis. Circadian rhythm disruption leads to abnormal cell cycle and immune dysregulation, which points out a poor lung repair in this disease. These components are dynamically and progressively interactive over time. Individual differences and genetic variations in surfactant function, tissue growth, remodeling, and homeostasis can also contribute to this disease development. Targeting the cellular processes to decrease alveolar injury and increase alveolar re-epithelialization may restore lung function. Various factors and signaling are interdependent in complex pathophysiology. Therefore, desired outcomes could be achieved by targeting multiple pathways. More systematic and comprehensive studies regarding interactions between different cell types, organelles, and signaling pathways are warranted to uncover new therapeutic strategies.
C-RL performed the literature search, prepared the figures, and wrote the manuscript. KB and BK provided significant works of literature, interpretations, and revision. All authors contributed to the article and approved the submitted version.
This work was supported by the American Heart Association Postdoctoral Fellowship 20POST35210318 (C-RL), R21 ES030808, Department of Defense W81XWH2110400, and the Catalyst Award from American Lung Association (KB), R01 ES032081, R01 HL150587, and the Department of Defense W81XWH2110414 (BK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ruvuna L, Sood A. Epidemiology of chronic obstructive pulmonary disease. Clin Chest Med. (2020) 41:315–27. doi: 10.1016/j.ccm.2020.05.002
3. Marchetti N, Criner GJ. Surgical approaches to treating emphysema: lung volume reduction surgery, bullectomy, and lung transplantation. Semin Respir Crit Care Med. (2015) 36:592–608. doi: 10.1055/s-0035-1556064
4. Tuder RM. Bringing light to chronic obstructive pulmonary disease pathogenesis and resilience. Ann Am Thorac Soc. (2018) 15:S227–33. doi: 10.1513/AnnalsATS.201808-583MG
5. Peinado VI, Barbera JA, Ramirez J, Gomez FP, Roca J, Jover L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. (1998) 274:L908–13. doi: 10.1152/ajplung.1998.274.6.L908
6. Estepar RSJ, Kinney GL, Black-Shinn JL, Bowler RP, Kindlmann GL, Ross JC, et al. Computed tomographic measures of pulmonary vascular morphology in smokers and their clinical implications. Am J Resp Crit Care. (2013) 188:231–9. doi: 10.1164/rccm.201301-0162OC
7. Bidan CM, Veldsink AC, Meurs H, Gosens R. Airway and extracellular matrix mechanics in COPD. Front Physiol. (2015) 6:346. doi: 10.3389/fphys.2015.00346
8. Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2001) 163:1304–9. doi: 10.1164/ajrccm.163.6.2009116
9. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. (2012) 5:9–19. doi: 10.1097/WOX.0b013e3182439613
10. Stenger PC, Alonso C, Zasadzinski JA, Waring AJ, Jung CL, Pinkerton KE. Environmental tobacco smoke effects on lung surfactant film organization. Biochim Biophys Acta. (2009) 1788:358–70. doi: 10.1016/j.bbamem.2008.11.021
11. Takamiya R, Uchida K, Shibata T, Maeno T, Kato M, Yamaguchi Y, et al. Disruption of the structural and functional features of surfactant protein A by acrolein in cigarette smoke. Sci Rep. (2017) 7:8304. doi: 10.1038/s41598-017-08588-5
12. Kapellos TS, Bassler K, Aschenbrenner AC, Fujii W, Schultze JL. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. (2018) 2018:2349045. doi: 10.1155/2018/2349045
13. Lowe KE, Regan EA, Anzueto A, Austin E, Austin JHM, Beaty TH, et al. COPDGene((R)) 2019: redefining the diagnosis of chronic obstructive pulmonary disease. Chronic Obstr Pulm Dis. (2019) 6:384–99. doi: 10.15326/jcopdf.6.5.2019.0149
14. Silverman EK. Genetics of COPD. Annu Rev Physiol. (2020) 82:413–31. doi: 10.1146/annurev-physiol-021317-121224
15. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. (2020) 33:101544. doi: 10.1016/j.redox.2020.101544
16. Pennington JD, Wang TJ, Nguyen P, Sun L, Bisht K, Smart D, et al. Redox-sensitive signaling factors as a novel molecular targets for cancer therapy. Drug Resist Updat. (2005) 8:322–30. doi: 10.1016/j.drup.2005.09.002
17. Bahmed K, Boukhenouna S, Karim L, Andrews T, Lin J, Powers R, et al. The effect of cysteine oxidation on DJ-1 cytoprotective function in human alveolar type II cells. Cell Death Dis. (2019) 10:638. doi: 10.1038/s41419-019-1833-5
18. Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. (2008) 178:592–604. doi: 10.1164/rccm.200803-380OC
19. Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, Huyck HL, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol. (2011) 187:987–98. doi: 10.4049/jimmunol.1001861
20. Tan LH, Bahmed K, Lin CR, Marchetti N, Bolla S, Criner GJ, et al. The cytoprotective role of DJ-1 and p45 NFE2 against human primary alveolar type II cell injury and emphysema. Sci Rep. (2018) 8:3555. doi: 10.1038/s41598-018-21790-3
21. Wang C, Zhou J, Wang J, Li S, Fukunaga A, Yodoi J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. (2020) 5:248. doi: 10.1038/s41392-020-00345-x
22. Bahmed K, Messier EM, Zhou W, Tuder RM, Freed CR, Chu HW, et al. DJ-1 modulates nuclear erythroid 2-related factor-2-mediated protection in human primary alveolar type II cells in smokers. Am J Respir Cell Mol Biol. (2016) 55:439–49. doi: 10.1165/rcmb.2015-0304OC
23. Lin CR, Bahmed K, Criner GJ, Marchetti N, Tuder RM, Kelsen S, et al. S100A8 protects human primary alveolar type II cells against injury and emphysema. Am J Respir Cell Mol Biol. (2019) 60:299–307. doi: 10.1165/rcmb.2018-0144OC
24. Lin CR, Bahmed K, Tomar D, Marchetti N, Criner GJ, Bolla S, et al. The relationship between DJ-1 and S100A8 in human primary alveolar type II cells in emphysema. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L791–804. doi: 10.1152/ajplung.00494.2018
25. Lakshmi SP, Reddy AT, Reddy RC. Emerging pharmaceutical therapies for COPD. Int J Chron Obstruct Pulmon Dis. (2017) 12:2141–56. doi: 10.2147/COPD.S121416
26. Lechner AJ, Driver IH, Lee J, Conroy CM, Nagle A, Locksley RM, et al. Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell. (2017) 21:120–34 e7. doi: 10.1016/j.stem.2017.03.024
27. Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. (1995) 152:1666–72. doi: 10.1164/ajrccm.152.5.7582312
28. Schwede M, Wilfong EM, Zemans RL, Lee PJ, Dos Santos C, Fang X, et al. Effects of bone marrow-derived mesenchymal stromal cells on gene expression in human alveolar type II cells exposed to TNF-alpha, IL-1beta, and IFN-gamma. Physiol Rep. (2018) 6:e13831. doi: 10.14814/phy2.13831
29. Cornwell WD, Kim C, Lastra AC, Dass C, Bolla S, Wang H, et al. Inflammatory signature in lung tissues in patients with combined pulmonary fibrosis and emphysema. Biomarkers. (2019) 24:232–9. doi: 10.1080/1354750X.2018.1542458
30. Berenson CS, Kruzel RL, Eberhardt E, Sethi S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis. (2013) 208:2036–45. doi: 10.1093/infdis/jit400
31. De Smet EG, Van Eeckhoutte HP, Avila Cobos F, Blomme E, Verhamme FM, Provoost S, et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. (2020) 13:423–36. doi: 10.1038/s41385-019-0241-6
32. Kosmider B, Lin CR, Vlasenko L, Marchetti N, Bolla S, Criner GJ, et al. Impaired non-homologous end joining in human primary alveolar type II cells in emphysema. Sci Rep. (2019) 9:920. doi: 10.1038/s41598-018-37000-z
33. Dey T, Kalita J, Weldon S, Taggart CC. Proteases and their inhibitors in chronic obstructive pulmonary disease. J Clin Med. (2018) 7:90244. doi: 10.3390/jcm7090244
34. Polverino F, Rojas-Quintero J, Wang X, Petersen H, Zhang L, Gai X, et al. A disintegrin and metalloproteinase domain-8: a novel protective proteinase in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2018) 198:1254–67. doi: 10.1164/rccm.201707-1331OC
35. Shan M, You R, Yuan XY, Frazier MV, Porter P, Seryshev A, et al. Agonistic induction of PPAR gamma reverses cigarette smoke-induced emphysema. J Clin Investig. (2014) 124:1371–81. doi: 10.1172/JCI70587
36. Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. (2009) 1:4ra10. doi: 10.1126/scitranlsmed.3000154
37. Wu LY, Wang GX, Qu P, Yan C, Du H. Overexpression of dominant negative peroxisome proliferator-activated receptor-gamma (PPAR gamma) in alveolar type II epithelial cells causes inflammation and T-cell suppression in the lung. Am J Pathol. (2011) 178:2191–204. doi: 10.1016/j.ajpath.2011.01.046
38. Craig JM, Scott AL, Mitzner W. Immune-mediated inflammation in the pathogenesis of emphysema: insights from mouse models. Cell Tissue Res. (2017) 367:591–605. doi: 10.1007/s00441-016-2567-7
39. Gualano RC, Hansen MJ, Vlahos R, Jones JE, Park-Jones RA, Deliyannis G, et al. Cigarette smoke worsens lung inflammation and impairs resolution of influenza infection in mice. Respir Res. (2008) 9:53. doi: 10.1186/1465-9921-9-53
40. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. (2018) 314:C73–87. doi: 10.1152/ajpcell.00110.2016
41. Sullivan JL, Bagevalu B, Glass C, Sholl L, Kraft M, Martinez FD, et al. B cell-adaptive immune profile in emphysema-predominant chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2019) 200:1434–9. doi: 10.1164/rccm.201903-0632LE
42. Polverino F, Seys LJ, Bracke KR, Owen CA. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L687–95. doi: 10.1152/ajplung.00304.2016
43. Zhou JS, Li ZY, Xu XC, Zhao Y, Wang Y, Chen HP, et al. Cigarette smoke-initiated autoimmunity facilitates sensitisation to elastin-induced COPD-like pathologies in mice. Eur Respir J. (2020) 56:2020. doi: 10.1183/13993003.00404-2020
44. Kelsen SG. The unfolded protein response in chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2016) 13:S138–45. doi: 10.1513/AnnalsATS.201506-320KV
45. Aggarwal S, Ahmad I, Lam A, Carlisle MA, Li C, Wells JM, et al. Heme scavenging reduces pulmonary endoplasmic reticulum stress, fibrosis, and emphysema. JCI Insight. (2018) 3:120694. doi: 10.1172/jci.insight.120694
46. Kelsen SG. Respiratory epithelial cell responses to cigarette smoke: the unfolded protein response. Pulm Pharmacol Ther. (2012) 25:447–52. doi: 10.1016/j.pupt.2012.07.005
47. Wang Y, Wu ZZ, Wang W. Inhibition of endoplasmic reticulum stress alleviates cigarette smoke-induced airway inflammation and emphysema. Oncotarget. (2017) 8:77685–95. doi: 10.18632/oncotarget.20768
48. Bodas M, Pehote G, Silverberg D, Gulbins E, Vij N. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis. Free Radic Biol Med. (2019) 131:81–97. doi: 10.1016/j.freeradbiomed.2018.11.023
49. Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. (2005) 11:491–8. doi: 10.1038/nm1238
50. Nosal C, Ehlers A, Haspel JA. Why lungs keep time: circadian rhythms and lung immunity. Annu Rev Physiol. (2020) 82:391–412. doi: 10.1146/annurev-physiol-021119-034602
51. Borsboom GJ, van Pelt W, van Houwelingen HC, van Vianen BG, Schouten JP, Quanjer PH. Diurnal variation in lung function in subgroups from two Dutch populations: consequences for longitudinal analysis. Am J Respir Crit Care Med. (1999) 159:1163–71. doi: 10.1164/ajrccm.159.4.9703106
52. Sundar IK, Yao H, Sellix MT, Rahman I. Circadian molecular clock in lung pathophysiology. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1056–75. doi: 10.1152/ajplung.00152.2015
53. Anafi RC, Francey LJ, Hogenesch JB, Kim J. CYCLOPS reveals human transcriptional rhythms in health and disease. Proc Natl Acad Sci USA. (2017) 114:5312–7. doi: 10.1073/pnas.1619320114
54. Sengupta S, Tang SY, Devine JC, Anderson ST, Nayak S, Zhang SL, et al. Circadian control of lung inflammation in influenza infection. Nat Commun. (2019) 10:4107. doi: 10.1038/s41467-019-11400-9
55. Krakowiak K, Durrington HJ. The role of the body clock in asthma and COPD: implication for treatment. Pulm Ther. (2018) 4:29–43. doi: 10.1007/s41030-018-0058-6
56. Pekovic-Vaughan V, Gibbs J, Yoshitane H, Yang N, Pathiranage D, Guo B, et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. (2014) 28:548–60. doi: 10.1101/gad.237081.113
57. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. (2008) 134:317–28. doi: 10.1016/j.cell.2008.06.050
58. Hwang JW, Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. (2014) 28:176–94. doi: 10.1096/fj.13-232629
59. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. (2006) 20:1868–73. doi: 10.1101/gad.1432206
60. Wang W, Gao J. Effects of melatonin on protecting against lung injury. Exp Ther Med. (2021) 21:228. doi: 10.3892/etm.2021.9659
61. Shin NR, Ko JW, Kim JC, Park G, Kim SH, Kim MS, et al. Role of melatonin as an SIRT1 enhancer in chronic obstructive pulmonary disease induced by cigarette smoke. J Cell Mol Med. (2020) 24:1151–6. doi: 10.1111/jcmm.14816
62. He BM, Zhang WX, Qiao JF, Peng ZY, Chai XP. Melatonin protects against COPD by attenuating apoptosis and endoplasmic reticulum stress via upregulating SIRT1 expression in rats. Can J Physiol Pharm. (2019) 97:386–91. doi: 10.1139/cjpp-2018-0529
63. Kim CM, Sohn JW, Yoon HJ, Shin DH, Park SS. Circadian rhythm of surfactant protein A, B and C mRNA in rats. Korean J Intern Med. (2003) 18:76–82. doi: 10.3904/kjim.2003.18.2.76
64. Park HY, Sin DD. Stress-induced premature senescence: another mechanism involved in the process of accelerated aging in chronic obstructive pulmonary disease. Inflam Adv Age Nutr Res Clin Interv. (2014) 2:193–202. doi: 10.1016/B978-0-12-397803-5.00016-2
65. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
66. Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
67. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. (2017) 31:172–83. doi: 10.1101/gad.290635.116
68. Tsutsumi A, Ozaki M, Chubachi S, Irie H, Sato M, Kameyama N, et al. Exposure to cigarette smoke enhances the stemness of alveolar type 2 cells. Am J Respir Cell Mol Biol. (2020) 63:293–305. doi: 10.1165/rcmb.2019-0188OC
69. Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2008) 177:861–70. doi: 10.1164/rccm.200708-1269OC
70. Baker JR, Vuppusetty C, Colley T, Papaioannou AI, Fenwick P, Donnelly L, et al. Oxidative stress dependent microRNA-34a activation via PI3Kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep. (2016) 6:35871. doi: 10.1038/srep35871
71. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. (2006) 174:886–93. doi: 10.1164/rccm.200509-1374OC
72. Baker JR, Donnelly LE, Barnes PJ. Senotherapy: a new horizon for COPD therapy. Chest. (2020) 158:562–70. doi: 10.1016/j.chest.2020.01.027
73. Carpagnano GE, Lacedonia D, Malerba M, Palmiotti GA, Cotugno G, Carone M, et al. Analysis of mitochondrial DNA alteration in new phenotype ACOS. BMC Pulm Med. (2016) 16:31. doi: 10.1186/s12890-016-0192-6
74. Zhang WZ, Rice MC, Hoffman KL, Oromendia C, Barjaktarevic IZ, Wells JM, et al. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight. (2020) 5:133984. doi: 10.1172/jci.insight.133984
75. Kosmider B, Lin CR, Karim L, Tomar D, Vlasenko L, Marchetti N, et al. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. EBioMedicine. (2019) 46:305–16. doi: 10.1016/j.ebiom.2019.07.063
76. Garcia-Arcos I, Geraghty P, Baumlin N, Campos M, Dabo AJ, Jundi B, et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax. (2016) 71:1119–29. doi: 10.1136/thoraxjnl-2015-208039
77. Bahmed K, Lin CR, Simborio H, Karim L, Aksoy M, Kelsen S, et al. The role of DJ-1 in human primary alveolar type II cell injury induced by e-cigarette aerosol. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L475–85. doi: 10.1152/ajplung.00567.2018
78. Jiang D, Cui H, Xie N, Banerjee S, Liu RM Dai H, Thannickal VJ, et al. ATF4 mediates mitochondrial unfolded protein response in alveolar epithelial cells. Am J Respir Cell Mol Biol. (2020) 63:478–89. doi: 10.1165/rcmb.2020-0107OC
79. Sorrentino V, Menzies KJ, Auwerx J. Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol Toxicol. (2018) 58:353–89. doi: 10.1146/annurev-pharmtox-010716-104908
80. Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. (2013) 497:451. doi: 10.1038/nature12188
81. Imai K, Mercer BA, Schulman LL, Sonett JR, D'Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. (2005) 25:250–8. doi: 10.1183/09031936.05.00023704
82. Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. (2003) 28:555–62. doi: 10.1165/rcmb.2002-0090OC
83. Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Natarajan R, Voelkel NF. Inhibition of histone deacetylase causes emphysema. Am J Physiol Lung Cell Mol Physiol. (2011) 300:L402–13. doi: 10.1152/ajplung.00207.2010
84. Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. (2003) 29:88–97. doi: 10.1165/rcmb.2002-0228OC
85. Adini A, Wu H, Dao DT, Ko VH, Yu LJ, Pan A, et al. PR1P stabilizes VEGF and upregulates its signaling to reduce elastase-induced murine emphysema. Am J Resp Cell Mol. (2020) 63:452–63. doi: 10.1165/rcmb.2019-0434OC
86. Dhapare S, Sakagami M. Salvianolic acid B as an anti-emphysema agent I: in vitro stimulation of lung cell proliferation and migration, and protection against lung cell death, and in vivo lung STAT3 activation and VEGF elevation. Pulm Pharmacol Ther. (2018) 53:107–15. doi: 10.1016/j.pupt.2018.10.001
87. Truong TM Li H, Dhapare S, Desai UR, Voelkel NF, Sakagami M. Sulfated dehydropolymer of caffeic acid: in vitro anti-lung cell death activity and in vivo intervention in emphysema induced by VEGF receptor blockade. Pulm Pharmacol Ther. (2017) 45:181–90. doi: 10.1016/j.pupt.2017.06.007
88. Pakhomova AV, Pershina OV, Ermakova NN, Krupin VA, Pan ES, Putrova OD, et al. Pericytes and smooth muscle cells circulating in the blood as markers of impaired angiogenesis during combined metabolic impairments and lung emphysema. B Exp Biol Med. (2020) 168:334–40. doi: 10.1007/s10517-020-04703-1
89. Hisata S, Racanelli AC, Kermani P, Schreiner R, Houghton S, Palikuqi B, et al. Reversal of emphysema by restoration of pulmonary endothelial cells. J Exp Med. (2021) 2021:218. doi: 10.1084/jem.20200938
90. Chen Q, Rehman J, Chan MW, Fu PF, Dudek SM, Natarajan V, et al. Angiocrine sphingosine-1-phosphate activation of S1PR2-YAP signaling axis in alveolar type II cells is essential for lung repair. Cell Rep. (2020) 31:107828. doi: 10.1016/j.celrep.2020.107828
91. Diab KJ, Adamowicz JJ, Kamocki K, Rush NI, Garrison J, Gu Y, et al. Stimulation of sphingosine 1-phosphate signaling as an alveolar cell survival strategy in emphysema. Am J Resp Crit Care. (2010) 181:344–52. doi: 10.1164/rccm.200906-0826OC
92. Weissmann N. Chronic obstructive pulmonary disease and pulmonary vascular disease. A comorbidity? Ann Am Thorac Soc. (2018) 15:S278–81. doi: 10.1513/AnnalsATS.201808-532MG
93. Halpin DM, Miravitlles M, Metzdorf N, Celli B. Impact and prevention of severe exacerbations of COPD: a review of the evidence. Int J Chron Obstruct Pulmon Dis. (2017) 12:2891–908. doi: 10.2147/COPD.S139470
94. Beeh KM. The role of bronchodilators in preventing exacerbations of chronic obstructive pulmonary disease. Tuberc Respir Dis. (2016) 79:241–7. doi: 10.4046/trd.2016.79.4.241
95. O'Donnell DE, Forkert L, Webb KA. Evaluation of bronchodilator responses in patients with “irreversible” emphysema. Eu Respirat J. (2001) 18:914–20. doi: 10.1183/09031936.01.00216501
96. Han MK, Wise R, Mumford J, Sciurba F, Criner GJ, Curtis JL, et al. Prevalence and clinical correlates of bronchoreversibility in severe emphysema. Eur Respir J. (2010) 35:1048–56. doi: 10.1183/09031936.00052509
97. Oczypok EA, Perkins TN, Oury TD. Alveolar epithelial cell-derived mediators: potential direct regulators of large airway and vascular responses. Am J Resp Cell Mol. (2017) 56:694–9. doi: 10.1165/rcmb.2016-0151PS
98. Knudsen L, Atochina-Vasserman EN, Guo CJ, Scott PA, Haenni B, Beers MF, et al. NOS2 is critical to the development of emphysema in Sftpd deficient mice but does not affect surfactant homeostasis. PLoS ONE. (2014) 9:85722. doi: 10.1371/journal.pone.0085722
99. Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. (2011) 147:293–305. doi: 10.1016/j.cell.2011.08.035
100. Brindicci C, Ito K, Resta O, Pride NB, Barnes PJ, Kharitonov SA. Exhaled nitric oxide from lung periphery is increased in COPD. Eur Respirat J. (2005) 26:52–9. doi: 10.1183/09031936.04.00125304
101. Maestrelli P, Paska C, Saetta M, Turato G, Nowicki Y, Monti S, et al. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur Respir J. (2003) 21:971–6. doi: 10.1183/09031936.03.00098203
102. Boe AE, Eren M, Morales-Nebreda L, Murphy SB, Budinger GR, Mutlu GM, et al. Nitric oxide prevents alveolar senescence and emphysema in a mouse model. PLoS ONE. (2015) 10:e0116504. doi: 10.1371/journal.pone.0116504
103. Zhang L, Wang Y, Wu GR, Rao LZ, Wei YQ, Yue HH, et al. Blockade of JAK2 protects mice against hypoxia-induced pulmonary arterial hypertension by repressing pulmonary arterial smooth muscle cell proliferation. Cell Proliferat. (2020) 53:12742. doi: 10.1111/cpr.12742
104. Fu X, Zhang F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med. (2018) 16:4553–61. doi: 10.3892/etm.2018.6785
105. Fernandez-Plata R, Thirion-Romero I, Nava-Quiroz KJ, Perez-Rubio G, Rodriguez-Llamazares S, Perez-Kawabe M, et al. Clinical markers of chronic hypoxemia in respiratory patients residing at moderate altitude. Life-Basel. (2021) 11:50428. doi: 10.3390/life11050428
106. McClendon J, Jansing NL, Redente EF, Gandjeva A, Ito Y, Colgan SP, et al. Hypoxia-inducible factor 1alpha signaling promotes repair of the alveolar epithelium after acute lung injury. Am J Pathol. (2017) 187:1772–86. doi: 10.1016/j.ajpath.2017.04.012
107. Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou JH, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol. (2020) 22:934. doi: 10.1038/s41556-020-0542-8
108. Aspal M, Zemans RL. Mechanisms of ATII-to-ATI cell differentiation during lung regeneration. Int J Mol Sci. (2020) 21:93188. doi: 10.3390/ijms21093188
109. Parker MM, Hao Y, Guo F, Pham B, Chase R, Platig J, et al. Identification of an emphysema-associated genetic variant near TGFB2 with regulatory effects in lung fibroblasts. Elife. (2019) 8:42720. doi: 10.7554/eLife.42720
110. Gokey JJ, Sridharan A, Xu Y, Green J, Carraro G, Stripp BR, et al. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight. (2018) 3:98738. doi: 10.1172/jci.insight.98738
111. Kneidinger N, Yildirim AO, Callegari J, Takenaka S, Stein MM, Dumitrascu R, et al. Activation of the WNT/beta-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med. (2011) 183:723–33. doi: 10.1164/rccm.200910-1560OC
112. Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, et al. Reduced frizzled receptor 4 expression prevents WNT/beta-catenin-driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2017) 196:172–85. doi: 10.1164/rccm.201605-0904OC
113. Heijink IH, de Bruin HG, Dennebos R, Jonker MR, Noordhoek JA, Brandsma CA, et al. Cigarette smoke-induced epithelial expression of WNT-5B: implications for COPD. Eur Respir J. (2016) 48:504–15. doi: 10.1183/13993003.01541-2015
114. Baarsma HA, Skronska-Wasek W, Mutze K, Ciolek F, Wagner DE, John-Schuster G, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. (2017) 214:143–63. doi: 10.1084/jem.20160675
115. Feller D, Kun J, Ruzsics I, Rapp J, Sarosi V, Kvell K, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. (2018) 9:1724. doi: 10.3389/fimmu.2018.01724
116. Zheng Y, Pan D. The Hippo signaling pathway in development and disease. Dev Cell. (2019) 50:264–82. doi: 10.1016/j.devcel.2019.06.003
117. Kugler MC, Loomis CA, Zhao Z, Cushman JC, Liu L, Munger JS. Sonic hedgehog signaling regulates myofibroblast function during alveolar septum formation in murine postnatal lung. Am J Respir Cell Mol Biol. (2017) 57:280–93. doi: 10.1165/rcmb.2016-0268OC
118. Wang C, de Mochel NSR, Christenson SA, Cassandras M, Moon R, Brumwell AN, et al. Expansion of hedgehog disrupts mesenchymal identity and induces emphysema phenotype. J Clin Invest. (2018) 128:4343–58. doi: 10.1172/JCI99435
119. Kato K, Dieguez-Hurtado R, Park DY, Hong SP, Kato-Azuma S, Adams S, et al. Pulmonary pericytes regulate lung morphogenesis. Nat Commun. (2018) 9:2448. doi: 10.1038/s41467-018-04913-2
120. Dandekar A, Mendez R, Zhang KZ. Cross talk between er stress, oxidative stress, and inflammation in health and disease. Stress Responses. (2015) 1292:205–14. doi: 10.1007/978-1-4939-2522-3_15
121. Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He AQ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscl Throm Vas. (2006) 26:2490–6. doi: 10.1161/01.ATV.0000242903.41158.a1
122. Pereira ER, Liao N, Neale GA, Hendershot LM. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE. (2010) 5:12521. doi: 10.1371/journal.pone.0012521
123. Viby NE, Isidor MS, Buggeskov KB, Poulsen SS, Hansen JB, Kissow H. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology. (2013) 154:4503–11. doi: 10.1210/en.2013-1666
Keywords: lung, alveolar epithelium, alveolar type II cells, emphysema, oxidative stress, tissue homeostasis
Citation: Lin C-R, Bahmed K and Kosmider B (2022) Dysregulated Cell Signaling in Pulmonary Emphysema. Front. Med. 8:762878. doi: 10.3389/fmed.2021.762878
Received: 23 August 2021; Accepted: 06 December 2021;
Published: 03 January 2022.
Edited by:
Shu-Chuan Ho, Taipei Medical University, TaiwanReviewed by:
Manish Bodas, University of Oklahoma Health Sciences Center, United StatesCopyright © 2022 Lin, Bahmed and Kosmider. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beata Kosmider, YmVhdGEua29zbWlkZXJAdGVtcGxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.