
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 22 October 2021
Sec. Gastroenterology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.761538
This article is part of the Research Topic Liver Fibrosis and MAFLD: from Molecular Aspects to Novel Pharmacological Strategies View all 15 articles
 Weiyi Qu1,2†
Weiyi Qu1,2† Tengfei Ma2,3,4†
Tengfei Ma2,3,4† Jingjing Cai2,5
Jingjing Cai2,5 Xiaojing Zhang2,6
Xiaojing Zhang2,6 Peng Zhang2,6
Peng Zhang2,6 Zhigang She1,2*Feng Wan3*
Zhigang She1,2*Feng Wan3* Hongliang Li1,2,4*
Hongliang Li1,2,4*Metabolic-associated fatty liver disease (MAFLD) is a new disease definition, and this nomenclature MAFLD was proposed to renovate its former name, non-alcoholic fatty liver disease (NAFLD). MAFLD/NAFLD have shared and predominate causes from nutrition overload to persistent liver damage and eventually lead to the development of liver fibrosis and cirrhosis. Unfortunately, there is an absence of effective treatments to reverse MAFLD/NAFLD-associated fibrosis. Due to the significant burden of MAFLD/NAFLD and its complications, there are active investigations on the development of novel targets and pharmacotherapeutics for treating this disease. In this review, we cover recent discoveries in new targets and molecules for antifibrotic treatment, which target pathways intertwined with the fibrogenesis process, including lipid metabolism, inflammation, cell apoptosis, oxidative stress, and extracellular matrix formation. Although marked advances have been made in the development of antifibrotic therapeutics, none of the treatments have achieved the endpoints evaluated by liver biopsy or without significant side effects in a large-scale trial. In addition to the discovery of new druggable targets and pharmacotherapeutics, personalized medication, and combinatorial therapies targeting multiple profibrotic pathways could be promising in achieving successful antifibrotic interventions in patients with MAFLD/NAFLD.
Due to the close association with metabolic disorders and the previous exclusionary diagnostic strategy facing many challenges, a new disease nomenclature, metabolic-associated fatty liver disease (MAFLD), was proposed by expert panels to renovate its former name, non-alcoholic fatty liver disease (NAFLD) (1, 2). However, there are committees and experts who believe that the molecular basis of the disease behind this new definition lacks sufficient understanding, which may lead to uncertainty and negative effects in this field (3). Although many aspects of MAFLD are not well-understood, the similarities of the prevalence, risk factors, and pathological and metabolic traits between MAFLD and NAFLD suggest that evidence from NAFLD over the past decades would provide valuable clues for the discovery of druggable targets for the treatment of MAFLD and its subsequent fibrosis (4–7).
Non-alcoholic fatty liver disease is the most common chronic liver disease globally and affects approximately a quarter of the world population (5, 8, 9). It progresses from simple liver steatosis to nonalcoholic steatohepatitis (NASH) and, in more severe cases, to liver fibrosis and cirrhosis (10). By 2030, the overall number of cases of this disease is projected to increase by 18.3%, and the number of cases of its related advanced liver disease and liver-related mortality will be doubled (11). Facing such a severe public health burden, MAFLD as a new concept still lacks direct and strong evidence from pharmaceutical investigations. Even for NAFLD with sufficient research data to endorse, there are no specific drugs approved by the United States Food and Drug Administration (US-FDA) or the European Medicines Agency (12, 13).
Fibrosis is a consequence of advanced liver injury that is closely associated with cirrhosis and liver carcinoma (14). Therefore, the improvement of liver fibrosis has become an important indicator for evaluating the efficacy of drugs for the treatment of NAFLD. However, the effectiveness of current drugs for hepatic fibrosis is limited (15). The identification of druggable targets and the development of novel reagents for the prevention and reversal of fibrosis will be an important mission of NAFLD/MAFLD research (16). This review summarizes the key endogenous molecules involved in the pathogenesis of NAFLD/MAFLD fibrosis and discusses the compounds or antibodies derived from these druggable targets that could potentially lead to successful treatments for NAFLD/MAFLD (Supplementary Table 1).
Fibrosis is the primary histological feature of the advanced form of NAFLD/MAFLD (17). Therefore, it is critical to elucidate the mechanism mediating liver fibrosis in NAFLD/MAFLD. Although the majority of the mechanistic discoveries were based on NAFLD, the new terminology MAFLD shares similar driving factors as NAFLD and knowledge from NAFLD provides important implication in the understanding of the pathogenesis of MAFLD. The pathophysiology of NAFLD progression is summarized as the “multiple hits” theory. That is, the “first hit” begins with hepatic triglyceride accumulation, and the responses of insulin resistance (IR)-related lipotoxic substances, and the increased de novo hepatic lipogenesis, thereafter oxidative stress, metabolic inflammation, endoplasmic reticulum stress, and autophagy together with the intestinal microbial signals and other links all involved, finally facilitating “parallel, multiple hits” to the liver (18). In the liver injury-repair process, dysregulated hepatocytes or inflammatory cells elicit paracrine signaling that promotes the hepatic stellate cells (HSCs) activation. Meanwhile, circulating factors (e.g., adipokine and fatty acids) from extrahepatic tissues (e.g., visceral adipose tissue or intestine) could activate HSCs directly or mediately (17). In addition, gene polymorphisms, such as PNPLA3, TM6SF2, and HSD17B13, may increase an individual's susceptibility to liver fibrosis during metabolic dysregulation (17, 19, 20). Upon the stimulation of the abovementioned profibrotic factors, HSCs turn into an active form and accelerate the production of fibroblasts, portal vein fibroblasts, and myofibroblasts, which ultimately result in exacerbated extracellular matrix formation (21). Overall, HSCs activation is a dominant manifestation during fibrosis in NAFLD (17, 21) (Figure 1).
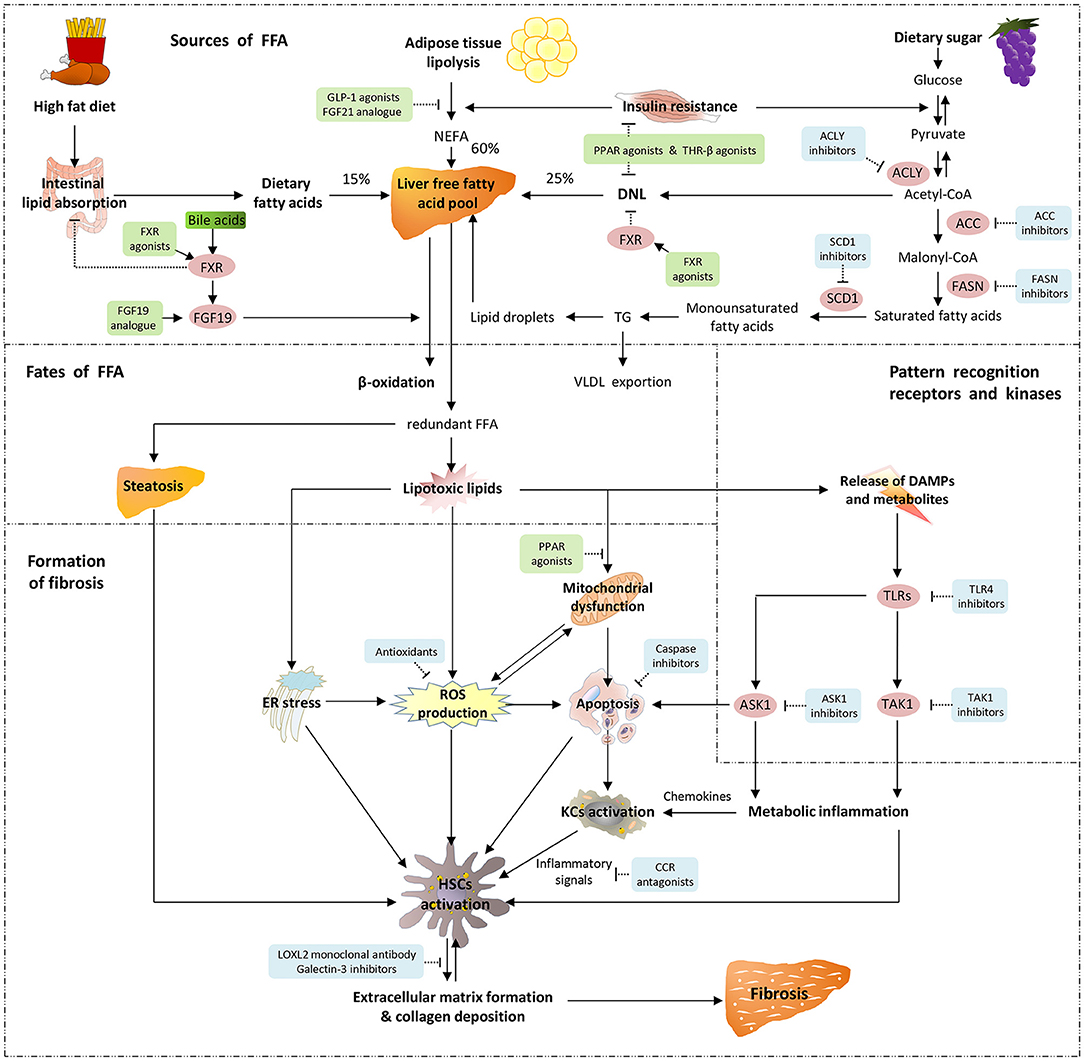
Figure 1. Pathogenetic mechanisms underlying fibrosis in MAFLD/NAFLD and molecular target of drug therapy. There are three sources of hepatic free fatty acids (FFA): 60% from the adipose tissue lipolysis or non-esterified fatty acid (NEFA) pool, 25% from de novo lipogenesis (DNL), and the remaining 15% from the intestinal absorption of diet. The two main metabolic pathways of hepatic FFA are mitochondria-mediated β-oxidation and esterification to form triglyceride (TG). Triglyceride is able to be exported into the circulation in the form of very-low-density lipoprotein (VLDL), and the excessive TG are stored in lipid droplets. When FFA are overaccumulated or their disposal is not timely, the redundant FFA act as substrates to produce lipotoxic lipids, which lead to endoplasmic reticulum (ER) stress, production of reactive oxygen species (ROS), impaired mitochondrial function and release of danger-associated molecular patterns (DAMPs). Pattern recognition receptors such as toll-like receptors (TLRs) sense the continuous production of DAMPs and metabolites, thereby triggering downstream signaling pathways. Apoptosis signal-regulating kinase 1 (ASK1) and TGF-β-activated kinase 1 (TAK1) are crucial intracellular signal transduction components that are activated by post-transcriptional modification, and further activate their downstream pivotal kinases and transcription factors, leading to the occurrence of metabolic inflammation. As metabolic stress leads to the expression and release of inflammatory chemokines, Kupffer cells (KCs) polarize into pro-inflammatory phenotypes and participate in the activation of hepatic stellate cells (HSCs) in coordination with ROS, apoptotic signals and ER stress. These exacerbate extracellular matrix formation and collagen deposition, and result in liver fibrosis. Emerging therapeutic agents and their molecular targets for fibrosis in MAFLD/NAFLD are also indicated. Agonists and analogs are marked in green, while antagonists, inhibitors and antibodies are marked in blue.
As the main target of liver injury, hepatocytes first face the imbalance of fatty acid and carbohydrate metabolism caused by metabolic overload in the early stage of NAFLD (22). With the further development of hepatic steatosis, excess, or not timely disposed fatty acids could be metabolized into toxic lipids (such as oxidized phospholipids), causing hepatocyte metabolic stress and damage or death (22). This lipotoxic response leads to hepatocytes apoptosis, which liberates reactive oxygen species (ROS) and free cholesterol (17). Damaged hepatocytes serve as a major driver for HSCs activation via paracrine signaling. For example, lipotoxic associated ROS production from mitochondria, endoplasmic reticulum, and NADPH oxidase (NOX) in hepatocytes have profound and direct impacts on HSCs activation (17, 23, 24). In addition, the receptors for advanced glycation end-products (RAGEs), which is pattern recognition receptor, are highly expressed in HSCs. ROSs are also generated in AGE formation, and oxidized RAGE stimulates NOX1, which contributes to ROS production in HSCs (24). Other signals from hepatocytes, such as leptin and osteopontin, are also involved in mediating the transformation of HSCs into a profibrotic and inflammatory phenotype (17). It should be noted that the innate immune response mainly regulates aseptic inflammation triggered by metabolic stress (25, 26). The innate immune system activates the release of inflammatory cytokines and chemokines by sensing metabolic stress and many of them have shown to be important in the pathogenesis of fibrosis, such as IL-1β and IL-18, C-C chemokine ligand types 2 and types 5 (CCL2 and CCL5), together with C-C chemokine receptor type 2 and types 5 (CCR2 and CCR5), etc. (27–29).
Hepatic macrophages can also polarize toward a proinflammatory phenotype, and their TLR4 signaling facilitates the production of transforming growth factor-beta 1 (TGF-β1) in response to metabolic insults. Transforming growth factor-beta 1 coordinates with HSCs to accelerates liver fibrosis (30). Hepatic T cell population is also essential in NASH-associated inflammation or stellate cell activation. Maintaining a good amount of CD8+ tissue-resident memory T cells protected mice from fibrosis progression by predisposing activated HSCs to FasL-Fas-mediated apoptosis in a CCR5-dependent manner (31, 32). There is a large number of B cells in the liver, immune regulatory properties of HSCs promote the profibrogenic activity of B cells (33). Platelets are an essential cellular source of PDGFβ and TGFβ that activate HSCs and promote fibrosis in NASH. Extracellular signals from resident and inflammatory cells collectively modulate HSCs activation by stimulating autophagy, oxidative stress, endoplasmic reticulum stress, and retinol metabolism, thereby further modulating liver fibrosis (17). Since NAFLD is a component of metabolic syndrome that affects multiple organs, circulating factors, and signals from extrahepatic tissues and organs, such as the intestinal microbiome (34), adipose tissue, and skeletal muscle, it also influences liver fibrosis (17, 35). Especially, intestinal flora-derived pathogen-associated molecular patterns and danger-associated molecular patterns, as well as endotoxins, could directly promote fibrosis by signaling through innate immune receptors like TLR4 on HSCs (17). Different types of epigenetic modifications, including DNA methylation, histone covalent modifications, and the expression of some non-coding RNAs (such as miR-29a and miR-590-5p), were found to play essential roles in regulating HSCs activation during the progression of NAFLD to NASH (36, 37).
Given the development of liver fibrosis as a result of interactions between various hepatic cell types under metabolic stress and the mediation of intercellular communication by secreted mediators or circulating mediators, which regulate lipid toxicity, inflammation, apoptosis, extracellular matrix formation, and fibrosis, the discovery of molecular targets and the development of novel pharmacological strategies covers all these aspects.
Glucagon-like peptide-1 (GLP-1) is a pleiotropic peptide hormone excreted by intestinal L cells that enhances insulin secretion and improves glucose homeostasis, thereby reducing liver non-esterified fatty acid (NEFA) overload caused by triglyceride decomposition. In addition to its metabolic benefits, GLP-1 has been shown to delay gastric emptying and limit body weight, as well as inhibit inflammation and cell apoptosis (38). These features make GLP-1 receptor agonists well-suited for the treatment of MAFLD, which is characterized by metabolic disorders, and allow them to help reduce multiple upstream links of HSCs activation, such as IR, lipid toxicity, and metabolic inflammation. Animal studies have shown that GLP-1 receptor agonists alleviate hepatic steatosis and inflammation, and play an antifibrotic role by improving HSCs phenotypes (12).
Liraglutide is a long-acting GLP-1 receptor agonist that reduces body weight in NAFLD patients. In a phase 2 study (NCT01237119), NAFLD patients who received subcutaneous injections of Liraglutide achieved histological improvement with attenuated fibrosis progression (39). Adverse effects of Liraglutide mainly consist of mild to moderate gastrointestinal reactions. Another GLP-1 receptor agonist in clinical application is Exenatide, which yields better improvement in the noninvasive Fibrosis 4 index and greater benefit in terms of body weight and liver enzymes than insulin Glargine in NAFLD patients with type 2 diabetes (NCT02303730) (40). A new-generation GLP-1 receptor agonist, Semaglutide, also improved steatohepatitis in a phase 2 trial (NCT02970942). It should be noted that although the administration of Semaglutide improved the fibrosis stage in nearly half of the patients, the excessive fibrosis regression rate in the placebo group made the difference between the groups non-significant (43 vs. 33%, P = 0.48) (41). Cotadutide is a dual receptor agonist of GLP-1 and Glucagon. Preclinical evidence has demonstrated that it is more effective than Liraglutide in improving liver fibrosis in NASH models (42). The current phase 2 study of NAFLD with compensatory fibrosis in patients with obesity (NCT04019561) completed the data collection of primary outcomes. Tirzepatide, a dual receptor agonist of GLP-1 and glucose-dependent insulinotropic polypeptide, also reported an improvement in fibrosis biomarkers in NAFLD patients with type 2 diabetes (NCT03131687) (43). Although it has only been approved by the US-FDA for the treatment of type 2 diabetes at present, a wide variety of GLP-1 receptor agonists have become attractive candidate drugs for the treatment of MAFLD due to their metabolic benefits.
De Novo lipogenesis (DNL) is a pivotal step in liver fatty acid metabolism and plays a major role in triglyceride accumulation in hepatocytes (44). Acetyl-CoA carboxylase (ACC) is a crucial enzyme in DNL regulation that catalyzes the rate-limiting step of acetyl-CoA to malonyl-CoA conversion and modulates mitochondrial fatty acid oxidation. Therefore, ACC is an attractive therapeutic target for restoring the balance of hepatic fatty acid metabolism (44). An animal study confirmed that inhibiting ACC reduced lipid toxicity in hepatocytes by lessening DNL. This mechanism resulted in the direct suppression of HSCs activation and impairment of HSCs profibrogenic activity, thereby reducing liver fibrosis in a rat model (45).
Firsocostat (also known as GS-0976 and NDI-010976) is a small-molecule allosteric inhibitor of ACC in the liver that significantly inhibited hepatic DNL in a metabolically overburdened population in a dose-dependent manner (46). Treatment with 20 mg of Firsocostat daily for 12 weeks reduced hepatic steatosis and fibrosis marker levels in patients with NASH (NCT02856555) (47). In order to explore more suitable treatment regimens for NAFLD patients with advanced liver fibrosis, the ATLAS phase 2b study (NCT03449446) focused on the improvement of F3–F4 fibrosis after 48 weeks of treatment with three agents alone or in combination (48). The combination of Firsocostat and either of the other two drugs allowed more patients to achieve the primary endpoint of fibrosis improvement ≥ 1 stage than the monotherapy regimen, although this benefit did not have a significant advantage over placebo. The combination of 20 mg of Firsocostat and 30 mg of Cilofexor transformed the fibrosis pattern in the biopsy area into ≤F2 and led to significant improvements in NASH activity, the enhanced liver fibrosis score and the liver stiffness as determined by transient elastography, revealing a potential antifibrotic effect (48). Most patients treated with Firsocostat experienced a remarkable increase in serum triglyceride levels. This asymptomatic hypertriglyceridemia subsided spontaneously in some patients and could partially be resolved by treatment with fish oil or fibrates (47, 48). Hypertriglyceridemia may be the result of decreased polyunsaturated fatty acids produced by malonyl-CoA, which makes the expression of sterol regulatory element-binding protein 1 increase in a compensatory manner, resulting in increased very-low-density lipoprotein secretion in the liver and peripheral triglyceride accumulation (44). In addition, Firsocostat in combination with Cilofexor increased the total cholesterol level and decreased the high-density lipoprotein cholesterol level in the ATLAS study (48). In consequence, further research on the long-term effects of Firsocostat on the cardiovascular system is needed.
PF-05221304 is another oral liver-directed ACC inhibitor that significantly improved the hepatic steatosis, fibrosis, and inflammation induced by metabolic stress in both human-derived in vitro systems and rodent models (49). An ongoing phase 2 study (NCT04321031) is evaluating whether it has a beneficial effect on fibrosis as assessed by liver biopsy in NASH patients.
ATP-citrate lyase (ACLY) is a pivotal lipogenic enzyme positioned at the coupling between glycocatabolism and lipid anabolism, and serves as a metabolic checkpoint for detecting excessive nutrients (12, 50). ATP-citrate lyase catalyzes mitochondrial-derived citrate into cytoplasmic oxaloacetate and acetyl-CoA, which in turn promotes the synthesis of fatty acids and cholesterol, and the acetylation of proteins (50). Studies have shown that the expression of ACLY is increased in liver samples from NAFLD patients (51). The increase of ACLY in hepatocytes may be due to the impairment of an E3 ligase-drived ubiquitination-dependent degradation of ACLY during metabolic stress (52). ATP-citrate lyase is involved in inflammatory and IL-4-induced macrophage polarization and activates the transcription factor Stat6 to induce coordinated fibrosis and tissue remodeling (50, 53). ATP-citrate lyase activity inhibition and gene silencing help prevent hepatic steatosis, reduce oxidative stress and prostaglandin E2 inflammatory mediators, thereby indirectly contribute to the improvement of fibrosis in metabolism-induced liver disease (12, 54, 55). Bempedoic acid (ETC-1002) is a first-class, prodrug-based direct competitive inhibitor of ACLY, which reduces ACLY activity and hepatic lipids in rodents (55, 56). Analysis of its phase 2 and 3 clinical studies demonstrated that bempedoic acid has good safety and favorable effects on lipid profiles and inflammation represented by high-sensitivity C-reactive protein (57). This evidence suggests that ACLY may serve as a new therapeutic target for regulating metabolism and MAFLD-related fibrosis. Further clinical trials are needed to evaluate the safety and efficacy of bempedoic acid in improving the primary outcomes of metabolic overload-induced liver disease and its fibrosis.
As a member of the lipogenic enzymatic cascade, fatty acid synthase (FASN) is a core modulator of hepatic DNL that catalyzes the synthesis of palmitate from acetyl-CoA and malonyl-CoA (58, 59). The powerful rate-limiting capacity of FASN for lipogenesis makes it a promising target for the treatment of liver disease caused by metabolic stress. The inhibition of FASN reduced hepatic DNL and improved liver steatosis in animal models and in patients with NAFLD (58–60). TVB-2640 is a novel FASN inhibitor that significantly reduces liver DNL and hepatic fat in obese subjects after 10 days' treatment (NCT02948569) (61). Given that the inhibition of hepatic DNL decreases intrahepatic fat accumulation, inflammation and fibrosis, a FASCINATE-1 (NCT03938246) phase 2a study focused on the efficacy of 25 mg or 50 mg of TVB-2640 in NASH patients (62). After 12 weeks of treatment, TVB-2640 reduced the liver fat and inflammatory biomarkers levels in a dose-dependent manner. More encouragingly, TVB-2640 produced significant improvements for several serum markers of fibrosis in NASH patients after such a short-term treatment (62). The benefit of TVB-2640 on fibrosis may be a result of the reduction in the DNL-triggered activation of HSCs or from inhibiting the indirect effect of lipotoxicity-mediated fibrosis (62). Encouraged by the success of the pilot study, a 52-week long-term study FASCINATE-2 (NCT04906421) was initiated with a higher drug dose (50 or 75 mg) and is recruiting NASH patients with F2–F3 liver fibrosis.
Stearoyl-CoA desaturase 1 (SCD1) is also a key enzyme for hepatic lipid anabolism that catalyzes the rate-limiting step of converting saturated fatty acids into monounsaturated fatty acids (63). Studies have indicated that SCD1 is overexpressed in activated HSCs, and is involved in diet-induced steatohepatitis and fibrosis by regulating Wnt signaling (64, 65). Decreasing SCD1 expression through genetic disruption or pharmacological inhibition reduced HSCs activation and alleviated liver fibrosis and steatohepatitis in murine models (66, 67). Arachidyl-amido cholanoic acid (Aramchol) downregulated SCD1 to inhibit DNL in the liver, reduce steatosis and inflammation, and reverse fibrosis in mice (66). In a phase 2a clinical trial (NCT01094158), 300 mg of Aramchol daily for 3 months dramatically reduced the liver fat content in NAFLD patients without significant adverse effects (67). Because the parameters in the above study, such as liver enzymes and insulin sensitivity did not improve, and the efficacy of the 300 mg was better than that of 100 mg, the ARREST phase 2b trial (NCT02279524) carried out a further study with doses of 400 and 600 mg. At the end of the 1-year treatment, Aramchol demonstrated dose-dependent benefits in reducing hepatic steatosis and fibrosis with good tolerability (63). Therefore, the ARMOR phase 3 trial (NCT04104321) was launched to evaluate the efficacy and safety of 300 mg of Aramchol twice a day in NASH patients with stage 2–3 fibrosis and metabolic disorders. The primary endpoint is fibrosis improvement for stage 1 or above and NASH regression, and the trial is currently in the recruitment stage (68).
Fanitol X receptor (FXR) is a nuclear receptor widely expressed in the liver and small intestinal mucosa that plays an essential role in the sensation of bile acid signals. Fanitol X receptor senses bile acid signals and regulates their secretion by negative feedback, resulting in decreased intestinal lipid absorption, downregulation of the expression of key lipogenic genes in the liver, and reduced hepatic lipid levels in the end (69). The beneficial effects of the ligation of FXR with bile acids on metabolism also include promoting fatty acid oxidation in the liver, regulating glycogenolysis and gluconeogenesis, as well as restoring insulin sensitivity in muscle and adipose tissue (12). These processes help to reduce toxic lipid production and suppress HSCs activation. The activation of FXR in HSCs has been confirmed to reduce extracellular matrix production and weaken the response of HSCs to profibrotic signals such as TGF-β, thus playing a protective role in fibrosis (70). In patients with fatty liver disease, the expression of hepatic FXR was negatively correlated with disease severity. In a rodent model, the deletion of FXR in the liver acerbates metabolic stress-induced liver steatosis, inflammation and fibrosis (12). Therefore, agonists of FXR are emerging as promising therapeutic agents for treating metabolic stress-induced liver fibrosis.
Obeticholic acid (OCA) is a semisynthetic FXR agonist that is more than 100-fold more potent than the endogenous ligand chenodeoxycholic acid (12). There is evidence from a number of clinical trials and animal studies showing that OCA has a promising effect on improving fibrosis due to NASH (71–73). Animal studies have indicated that OCA reduces metabolic stress-induced liver fibrosis and lipid infiltration and effectively improves systemic IR in obese and diabetic mice (12, 74). In the FLINT phase 2 clinical trial (NCT01265498), liver fibrosis and disease histological features were significantly improved in NASH patients who received 25 mg of OCA daily compared to those who received placebo (72). The benefits of OCA on NASH-induced liver fibrosis were also identified in interim analysis results published in the ongoing REGENERATE phase 3 trial (NCT02548351). Taking 25 mg of OCA daily improved liver fibrosis in nearly a quarter of NASH patients with stage 2–3 fibrosis, which is approximately twice the proportion observed in the control group (73). Twenty-five milligrams of OCA also outperformed placebo in reducing the NAFLD activity score (NAS). Regrettably, the expected endpoint of NASH regression was not achieved after 18 months of OCA treatment. The limitations of OCA treatment include not only mild to moderate dose-dependent pruritus but also more worrisome dyslipidemia, characterized by elevated low-density lipoprotein cholesterol (LDL-c) levels during treatment, which may pose an additional risk of atherosclerosis in NASH patients who are already overweight or suffering from type 2 diabetes (73). Overall, the treatment benefits on mid-term histological endpoint of the REGENERATE study are still uncertain, and the benefits do not significantly outweigh the potential risks. Therefore, in June 2020, the US-FDA rejected the pharmaceutical company Intercept's application for approval of OCA in treating fibrosis due to NASH, recommending that Intercept provide more interim data from REGENERATE and maintain long-term studies on the benefit-risk ratio of OCA. Currently, the efficacy and safety of OCA therapy in patients with compensated cirrhosis due to NASH are being evaluated in a phase 3 clinical trial (NCT03439254). Considering that FXR activation influences plasma lipoprotein concentrations, the combination of OCA and statins was considered and tested in the CONTROL phase 2 study (NCT02633956). After 16 weeks of treatment, OCA-induced increases in LDL-c in patients with NASH were mitigated with atorvastatin. This combination was generally safe and well-tolerated (75).
In addition to OCA, several FXR agonists have been examined in clinical trials or proven to be effective in animal studies. Cilofexor (formerly GS-9674), a small-molecule non-steroidal agonist of FXR, significantly reduced hepatic steatosis, liver biochemical marker, and serum bile acid levels, but did not improve liver fibrosis or stiffness after 24 weeks of treatment (NCT02854605) (76). However, as previously mentioned, it has preferable antifibrotic potential in combination with the ACC inhibitor Firsocostat (NCT03449446). A novel FXR agonist, EDP-305, also demonstrated a potent effect in terms of reducing liver fibrosis triggered by metabolic stress, bile duct ligation, and methionine-choline deficient (MCD) diet in rodent models (77, 78). Similar compounds, e.g., Tropifexor (LJN452) (NCT03517540 and NCT04065841), Nidufexor (LMB763), PX-104, EYP001, and TERN-101, are under investigation (79). In summary, FXR signaling decreases in patients with metabolic overload-induced fatty liver disease, especially in the fibrotic stages. Restoring FXR expression using a FXR agonist demonstrates promising therapeutic potential for treating fatty liver disease and fibrosis. Currently, there are side effects that limit the application of this strategy in clinical practice. Thus, the development of more precise drug targets or combination therapies is warranted.
Fibroblast growth factor 19 (FGF19), secreted by the intestines during feeding, is a signaling hormone downstream of FXR activation and is currently under clinical investigation for the potential treatment of NASH and its associated fibrosis (80). Fibroblast growth factor 19 (mouse ortholog FGF15) promotes NEFA oxidation in mitochondrial and glycogen synthesis via its interaction with FGF receptors (FGFR) in hepatocytes and thus suppresses metabolic stress-induced signaling activation in HSCs and liver fibrosis (80, 81). In addition, FGF19 plays a crucial role in the regulation of systemic glucose and lipid metabolism, as well as maintaining energy homeostasis (81). However, efforts involving the FGF19-FGFR pathway have encountered setbacks. Carcinogenicity were observed in mice treated with FGF19 (81, 82). To overcome these barriers, alternative approaches and molecules need to be developed. Aldafermin (alias NGM282) is an engineered non-carcinogenic FGF19 analog that maintains a key region of the protein involved in receptor interactions and signaling modulation but does not activate the FGF19-STAT3 carcinogenic pathway (82, 83). A 5-aminoacid deletion (P24–S28) coupled with the substitution of three amino acids at crucial positions (Ala30Ser, Gly31Ser, and His33Leu) within the amino terminus of Aldafermin enables biased FGFR4 signaling; thus, Aldafermin retains the ability to potently repress CYP7A1 expression (82). The first phase 2 trial (NCT02443116) assessing the safety and efficacy of Aldafermin for the treatment of NASH revealed that 12 weeks of Aldafermin treatment rapidly decreased liver fat content measured by MRI-proton density fat fraction (MRI-PDFF), and non-invasive markers of inflammation and fibrosis (84). Because the histological features of NASH were not assessed at the end of this study, an open-label trial of Aldafermin with histological endpoints in patients with NASH was conducted (NCT02443116). Consistently, this study confirmed that Aldafermin improved the NAS and fibrosis score of NASH patients after 12 weeks of treatment (85). Due to the encouraging success in trials of 12 weeks of Aldafermin treatment, the efficacy and safety of 24 weeks of treatment were further evaluated in patients with biopsy-proven NASH (NCT02443116). In this trial, Aldafermin reduced liver fat and produced a trend toward fibrosis improvement, and few adverse events were reported (86). To further evaluate the benefits of Aldafermin on liver fibrosis, two multicenter randomized controlled trials (RCTs) of Aldafermin in subjects with F2/F3 fibrosis (NCT03912532) or compensated cirrhosis (NCT04210245) are underway. It is worth mentioning that Aldafermin modulates CYP7A1-mediated bile acid homeostasis and may lead to an increase in serum cholesterol. An appropriate combination with statins may counteract Aldafermin-induced side effects on the lipid profile (NCT02443116) (87). Although no liver tumors were observed in multiple animal models after prolonged exposure to Aldafermin (82), trials of longer duration are still warranted for further safety evaluation.
Fibroblast growth factor 21 (FGF21) demonstrates large functional overlap with FGF19 in terms of regulating energy homeostasis and metabolism (80, 81). Unlike FGF19, FGF21 is produced in the liver during fasting and in response to elevated NEFA levels. The increased level of FGF21 in plasma has a negative-feedback effect on lipolysis in peripheral tissue (81). Moreover, FGF21 facilitates glucose and lipid uptake and adipogenesis in adipose and muscle tissue, which prevent ectopic lipid accumulation in the liver (80). Fibroblast growth factor 21 has shown great therapeutic potential as a treatment for NASH, but it has poor pharmacokinetic and biophysical properties (88). Numerous FGF21 analogs have been synthesized and developed for the treatment of metabolic diseases. Pegbelfermin (BMS-986036), a PEGylated human FGF21 analog, was tested in patients with NASH in a phase 2a clinical trial (NCT02413372). The administration of Pegbelfermin for 16 weeks significantly reduced the hepatic fat fraction as measured by MRI-PDFF by over 10%, and deceased liver fibrosis biomarkers, e.g., N-terminal type III collagen propeptide (pro-C3) (89). Further research using liver biopsies to assess the effects of 24 weeks of Pegbelfermin treatment on patients with histologically confirmed NASH with stage 3 liver fibrosis (FALCON 1; NCT03486899) or compensated cirrhosis (FALCON 2; NCT03486912) was initiated in 2018 (90). Efruxifermin is an engineered fusion protein formed by linking human IgG1 Fc to modified FGF21, and it has a balanced and long-lasting agonistic effect on FGFR1c, 2c, and 3c (91, 92). In the 16-week phase 2a BALANCED study (NCT03976401), Efruxifermin resulted in a significant reduction in the liver fat fraction as measured by MRI (92). This improvement in hepatic steatosis was accompanied by a reduction in biomarkers of fibrosis and the enhanced liver fibrosis scores. More importantly, 55% of patients achieved stage 1 or greater fibrosis improvement, and half of these patients even met all the exploratory endpoints (92). The major adverse effects of FGF21 analogs is mild gastrointestinal reactions. The promising efficacy and mild side effects of FGF21 analogs demonstrate their potential as treatments for NASH.
Peroxisome proliferator-activated receptors (PPAR), a superfamily of nuclear hormone receptors, are extensively involved in the regulation of metabolic homeostasis and inflammatory response in the liver (63). The distribution and functions of the three PPAR hypotypes, α, δ, and γ, are not identical. PPARα is mainly located on liver cells. After activation, it promotes the oxidation of fatty acids in the liver and enhances the expression of superoxide dismutase and catalase to protect liver cells from oxidative stress-induced damage (12). The expression of PPARδ is more extensive and serves various functions, such as inhibiting inflammation, enhancing NEFA oxidation, suppressing adipogenesis, and regulating the immune system (12). PPARα and PPARδ play an important role in suppressing liver fibrosis by inhibiting liver steatosis and inflammation (93). PPARγ is mainly expressed in adipocytes and pancreatic β cells and is able to accelerate the differentiation and storage capacity of adipocytes and regulate glucose metabolism (12, 94). Importantly, PPARγ can be activated by various ligands, such as fatty acids and thiazolidinedione, to inhibit HSCs proliferation and improve liver fibrosis (93). Research-based evidence suggests that PPARγ mediates the effect of liver-protective docosahexaenoic acid in ameliorating liver fibrosis by inducing cell cycle arrest and apoptosis in HSCs (95). In summary, some agonists targeting PPAR may have promise for the treatment of liver fibrosis.
Pirfenidone is an oral PPARα agonist with antisteatogenic and antifibrotic effects that is currently approved for the treatment of idiopathic pulmonary fibrosis (96). To evaluate its value for application in treating advanced liver fibrosis, the PROMETEO phase 2 study (NCT04099407) applied a sustained-release formulation with less potential toxicity to liver metabolism and a longer-lasting plasma concentration (97). The ratio of stage 3 to stage 4 fibrosis in the study population was approximately 1:3, and nearly half of the patients had advanced fibrosis due to NAFLD. Taking 600 mg of Pirfenidone twice a day improved fibrosis in 35% of patients after 12 months and reduced liver enzyme levels in nearly half of the patients. Moreover, the serum TGF-β1 level was lower, and the quality of life appraised by the Euro-QoL scale was better after Pirfenidone treatment. As the serum Pirfenidone concentration was higher in patients with fibrosis regression than in patients with fibrosis progression, Pirfenidone was associated with a better antifibrotic effect (97). The PPARγ agonist Pioglitazone, a first-generation thiazolidinedione agent, has been shown to improve the fibrosis score in NASH patients without diabetes (NCT00994682) (98). However, for patients with diabetes, the combination of Pioglitazone and vitamin E did not improve liver fibrosis, although this regimen was superior to vitamin E alone or placebo in terms of steatohepatitis resolution (NCT01002547) (99). Due to side effects, such as fluid retention, osteoporotic fracture or hypoglycemia, Pioglitazone may increase the overall risk of patients with metabolic disorders, and second-generation PPARγ agonists have been developed and tested in clinical trials. Novel agents, such as MSDC-0602K, limited the common side effects of Pioglitazone. However, MSDC-0602K failed to improve liver histological features in NASH patients with stage 1–3 fibrosis in a 52-week phase 2b trial (NCT02784444) (100).
Agonists that act on multiple PPAR hypotypes at the same time seem to be more effective than those that only act on one PPAR hypotype. Elafibranor (formerly GFT505) is a dual-pathway agonist that acts on both PPARα and PPARδ. It has been researched in a phase 2b study of NAFLD patients without cirrhosis (NCT01694849). In a post-hoc analysis aimed at the degree of steatohepatitis resolution, the researchers found that 120 mg of Elafibranor daily had a better effect on disease activity in the population with a NAS ≥4. More importantly, patients who achieved the primary outcome are often accompanied by a reduction in the degree of liver fibrosis (101). Another dual-path agonist, Saroglitazar, targets PPARα and PPARγ and has shown beneficial effects on serum lipid levels and liver biochemical parameters in patients with NAFLD (NCT03061721) (102, 103). It leads to improvement in non-invasive-assessed liver fibrosis parameters in NAFLD patients with diabetic dyslipidemia (104). It may produce antifibrotic effects by reducing oxidative stress and the production of lipotoxic substances, as well as inhibiting leptin signaling. The efficacy and safety of this drug in NAFLD patients with advanced fibrosis are still under evaluation (NCT04469920). Lanifibranor (IVA337), a pan-PPAR agonist that acts on all three receptor subtypes, causes fibrosis regression and ameliorates HSCs-related phenotypes in preclinical models of advanced chronic liver disease (105). In the phase 2b study NCT03008070 completed just a few months ago, Lanifibranor reached the steatosis active fibrosis score endpoint and demonstrated histological fibrosis improvement, with good tolerability. In the forthcoming multicenter phase 3 study NCT04849728, Lanifibranor will be further evaluated in adult patients with non-cirrhotic NAFLD and stage 2/3 liver fibrosis. In general, the benefits of poly/pan-PPAR agonists for liver fibrosis appear to be better than those of single-subtype agonists. We still need a lot of clinical evidence to highlight the direction for the application of such drugs.
As with the PPAR family, thyroid hormones widely participate in the regulation of lipid and glucose metabolism. Thyroid hormone receptor (THR)-β is mainly expressed in the liver and specifically enhances the oxidative utilization of hepatic fat and cholesterol metabolism (12). Preclinical studies revealed that the specific activation of THR-β reduces hepatic steatosis and fibrosis and improves insulin sensitivity and hepatocyte injury (106). There are two selective THR-β agonists presently under clinical development that can optimize the liver benefits while avoiding the adverse cardiac and skeletal effects of activating THR-α.
Resmetirom (MGL-3196) is a liver-directed THR-β agonist. In a phase 2 study (NCT02912260), the oral administration of 80 mg of Resmetirom daily for 36 weeks improved fibrosis activity markers in NAFLD patients with stage 1–3 fibrosis and caused the resolution of steatohepatitis in nearly 30% of patients, who also showed an improvement in the liver fibrosis stage compared with those treated with placebo (107). To obtain more data on the safety and efficacy of Resmetirom in non-invasive assessments, an open-label extension study (NCT02912260) was conducted in 31 patients from the aforementioned study with sustained mild to significantly increased liver enzyme levels. A reduction in fibrosis markers such as pro-C3 and liver stiffness assessed by transient elastography was observed after 36 weeks of Resmetirom treatment (108). Unlike the aforementioned OCA or Aldafermin, Resmetirom resulted in reduced levels of multiple lipids that carry the risk of atherosclerosis, such as LDL-c and triglycerides (108). In addition, Resmetirom was well-tolerated, and the main adverse effects were transient mild diarrhea or nausea (107, 108). The considerable efficacy and safety support the ongoing phase 3 clinical study (NCT03900429) to explore the efficacy for NAFLD patients with F2–F3 fibrosis. Another selective THR-β agonist is VK2809 (formerly called MB07344), which significantly improved liver fat content in NAFLD patients with hyperlipidemia (NCT02927184); the publication of the study result is expected. A 52-week phase 2b study, VOYAGE, is currently being conducted in NAFLD patients with stage F1–F3 fibrosis to investigate the benefits of 1.0, 2.5, 5.0, and 10 mg of VK2809 compared with placebo on liver fat content, fibrosis, and histopathology (NCT04173065). Compared with VK2809, Resmetirom has more clinical evidence to support its therapeutic potential in NASH.
Excessive fatty acids and subsequent mitochondrial dysfunction lead to ROS production, which plays a crucial role in NASH and advanced fibrosis (17, 24, 109, 110). Vitamin E, as a major fat-soluble chain-scission antioxidant, prevents plasma lipid and low-density lipoprotein peroxidation and protects the structural integrity of cells from damage caused by lipid peroxidation and oxygen-free radicals (110, 111). In addition to its powerful antioxidant effects, vitamin E also induces adiponectin expression, reduces inflammatory signaling, and regulates macrophage polarization, making it a potential treatment option for suppressing oxidative stress and metabolism-related liver diseases (111, 112). In the metabolic stress-induced NASH model in mice, vitamin E reduces oxidative stress, improves hepatic fibrosis and HSCs activation, and alleviates hyperinsulinemia. The antifibrotic properties may be due to the inhibition of TGF-β expression after the downregulation of ROS production, thereby reducing the activation of HSCs (112). A clinical study indicated that using 800 IU of vitamin E per day significantly improved transplant-free survival and liver decompensation in NASH patients with stage F3–F4 fibrosis (113). However, there is still a lack of direct and conclusive evidence of the beneficial effect of vitamin E on NASH-induced liver fibrosis. Vitamin E did not cause significant regression of liver biopsy-proven fibrosis in RCTs (NCT00063622, NCT00063635, and NCT01002547), although it induced varying degrees of improvement in NASH histology (99, 114, 115). In general, the liver benefits of vitamin E alone were more embodied in the reduction of oxidative stress marker levels and improvement of liver function and the NAS (99, 111, 116). It must be emphasized that there is evidence suggesting that dietary vitamin E supplementation might increase cancer risk (NCT00006392) and mortality in the healthy population (117, 118). However, meta-analyses from recent years have shown that the adverse effects of vitamin E supplementation on all-cause mortality or cancer risk are not significant, supporting dietary intake of this natural antioxidant (119, 120). Therefore, hepatologists may be inclined to use vitamin E in combination with other reagents in the study of NAFLD/MAFLD.
Chronic liver injury induced by excessive toxic lipid leads to increased hepatocyte apoptosis, which is an important feature of NASH (121, 122). Apoptotic hepatocytes activate immune cells and HSCs, thereby promoting liver fibrosis and cirrhosis (122, 123). Caspases, a family of cysteine proteases, play a central role in the progression of NAFLD/NASH due to their role in the regulation of liver apoptosis and inflammation (124). Caspase inhibitors have been studied and tested as therapeutic agents for NASH (122). Emricasan (IDN-6556) is an irreversible pan-caspase inhibitor that ameliorates apoptosis and liver fibrosis in NASH mouse models (125). In a short-term clinical study (NCT02077374), Emricasan suppressed caspase activation and liver enzyme levels in patients with NASH after 4 weeks of treatment, with decent safety and tolerance (126). Moreover, 3 months of treatment with Emricasan improved liver function more in patients with cirrhosis caused by NASH than in those with cirrhosis caused by viral hepatitis or alcoholic liver disease and showed a potential beneficial effect on portal hypertension (NCT02230670) (127). However, Emricasan did not show a significant beneficial effect on fibrosis regression in RCTs with a larger sample of NASH patients. In NASH patients with stage F1–F3 fibrosis (NCT02686762), usage of Emricasan at 10 or 100 mg daily for 72 weeks failed to improve liver fibrosis or lead to NASH resolution. Moreover, fibrosis and hepatocyte ballooning were aggravated in the Emricasan group (128). In another study conducted in NASH patients with compensatory cirrhosis (NCT02960204), Emricasan at 10, 50, or 100 mg daily did not cause an improvement in clinical outcomes (129). In NASH patients with decompensated cirrhosis (NCT03205345), administering 10 or 50 mg of Emricasan daily also did not reduce the amount of decompensation events or improve liver function after 48 weeks (130). These three studies indicated a robust effect of Emricasan on caspase activity inhibition and a good safety, but none demonstrated a significant therapeutic benefit. This may be due to excessive inhibition of apoptosis activating alternative forms of cell death, such as necroptosis and pyroptosis (124, 128). It is also possible that cirrhosis leads to many pathological changes, such as reduced number of functional hepatocytes, decreased hepatic blood flow and transporter protein expression, resulting in unsatisfactory drug bioavailability. Therefore, a daily dose of 50–100 mg may be insufficient for patients with cirrhosis (129, 130). Although none of these studies achieved the primary endpoint, they provided valuable reference data and ideas for design optimization in future clinical research on NASH-associated fibrosis. In a recent study in an HCV patient treated with liver transplantation, 24 months of Emricasan therapy showed a beneficial effect on moderate liver fibrosis. Although the pathogenesis of HCV-related fibrosis differs from that in NASH, this positive result inspires the initiation of treatment in NASH patients with moderate fibrosis (131).
The liver consists of a network of innate immune cells, which collectively form the first line of defense against invading organisms and toxins (132). Under excessive metabolic stress, the hepatic innate immune system is over-activated to further trigger hepatic cell injury and liver fibrosis (22, 25, 133, 134). There are a number of molecular targets that function as hubs controlling the inflammatory signaling flow in the progression of fatty liver disease, and they have been emerging as targets in the development of drugs for the treatment of MAFLD/NAFLD and fibrosis (48, 135–140).
Studies have revealed that apoptosis signal-regulating kinase 1 (ASK1), a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family, is hyperactivated in the liver of NASH patients. Upon receiving metabolic stress signals, ASK1 activates the downstream c-Jun N-terminal kinase (JNK) 1/2-mitogen-activated protein kinase 14 (p38) signaling cascade to trigger hepatic inflammation and fibrosis during the development of MAFLD (135, 136). Apoptosis signal-regulating kinase 1 functions as a molecular hub controlling cellular signal transduction in NASH. It has been considered an essential target for the development of drugs for NASH.
A number of clinical trials evaluating the efficacy of ASK1 inhibitors against NASH have been performed. There have been two large phase 3 studies in patients with NASH and advanced fibrosis. They compared the effect of the ASK-1 inhibitor Selonsertib (GS-4997) with that of placebo in ~1,700 patients with NASH and bridging fibrosis (F3, STELLAR-3) or compensated cirrhosis (F4, STELLAR-4) (NCT03053050 and NCT03053063). Although Selonsertib successfully suppressed the expression of hepatic phospho-p38, it did not significantly improve liver fibrosis on liver biopsy (137, 138). There are several explanations for the failure of these large trials. First, there are a number of signaling pathways involved in the pathogenesis of NASH, particularly in the advanced stages, which suggests that combination therapy may be required in the treatment of NASH. In a phase 2 clinical trial, 72 patients with NASH and stage F2–F3 fibrosis were treated with either 6 or 18 mg of GS-4997 orally once daily alone or in combination with a once-weekly injection of 125 mg of Simtuzumab (a humanized monoclonal antibody directed against lysyl oxidase-like molecule 2) for 24 weeks (NCT02466516). Reduced liver hardness on MRI elastography, reduced collagen content and lobular inflammation on liver biopsy, and improved serum markers of apoptosis and necrosis all suggested improvement in liver fibrosis. The assessed results showed that the proportion of patients with a reduction of fibrosis of at least one stage at week 24 was 20% in the Simtuzumab -alone group (2 of 10; 95% confidence interval (CI), 3–56), 30% in the 6-mg Selonsertib group (8 of 27; 95% CI, 14–50), and 43% in the 18-mg Selonsertib group (13 of 30; 95% CI, 26–63). The changes in fibrosis stage were correlated with the changes in hepatic collagen content (r = 0.54, P < 0.001). The median percent change in the morphometric collagen content of patients who were treated with Simtuzumab alone was 2.1%, while that of patients treated with 6 and 18 mg of Selonsertib was −8.2 and −8.7%, respectively. In summary, compared with patients treated with Simtuzumab alone, patients treated with Selonsertib showed a higher rate of fibrosis improvement and a lower rate of fibrosis progression. These findings suggest that Selonsertib combined with Simtuzumab may reduce liver fibrosis in patients with NASH and stage 2–3 fibrosis (139).
Another study was performed to evaluate the safety and efficacy of Selonsertib, Firsocostat, Cilofexor, and combinations in participants with bridging fibrosis or compensated cirrhosis due to NASH (NCT03449446). In this study, 392 patients with bridging fibrosis or compensated cirrhosis due to NASH were randomized to receive 18 mg of Selonsertib, 20 mg of Firsocostat, or 30 mg of Cilofexor, alone or in two-drug combinations, once daily for 48 weeks. Histological parameter analysis showed that for the primary endpoint of an improvement in fibrosis of ≥ 1 stage without the worsening of NASH, the proportion of patients was 12% (4 of 33, P = 0.94) in the Firsocostat group, 12% (4 of 34, P = 0.96) in the Cilofexor group, 15% (11 of 71, P = 0.62) in the Firsocostat/Selonsertib group, 19% (13 of 68, P = 0.26) in the Cilofexor/Selonsertib group, and 21% (14 of 67, P = 0.17) in the Cilofexor/Firsocostat group. A higher response rate was observed in the combination groups than in the monotherapy groups, but the differences between the treatment and placebo arms did not reach statistical significance. However, patients treated with Cilofexor/Selonsertib (8%; P = 0.018 vs. placebo) were significantly less likely to progress to cirrhosis than those treated with placebo (41%). These results suggest that combination therapy with Selonsertib offers the possibility of fibrosis reversal in the long-term treatment of patients with advanced NASH and fibrosis (48). Additional studies are warranted to confirm the potential therapeutic effects of ASK1 inhibitors on liver fibrosis in NASH.
Since ASK1 plays essential roles in physiological function, modulating its activity via posttranslational modification could be a more appropriate strategy in the treatment of disease. Many ASK1-negative regulators have been reported to significantly inhibit the development of NASH-associated fibrosis in rodents and preclinical models (140–142). For instance, the disassociation of milk fat globule-epidermal growth factor-factor 8 from ASK1 accelerates its dimerization and phosphorylation in hepatocytes under metabolic stress, thus leading to liver steatosis and fibrosis (140, 141). The deubiquitinating enzyme tumor necrosis factor-alpha-induced protein 3 (TNFAIP3) directly interacts with and deubiquitinates ASK1 in hepatocytes and ameliorates metabolic stress-induced hepatic inflammation and fibrosis (144). Tumor necrosis factor receptor-associated factor 6 promotes the polyubiquitination of Lys6 connections and the activation of ASK1, in turn exacerbating inflammatory and fibrotic responses in the liver (143). A high-fat diet also induces the overexpression of hepatic E3 ligase Skp1-Cul1-F-box protein F-box/WD repeat-containing protein 5 (FBXW5), which is a key endogenous activator of ASK1 ubiquitination and activation, and small molecules that mimic FBXW5 (S1) and FBXW5 (S3) can block the ubiquitination of ASK1 in MAFLD (144). Future clinical trials could aim to these molecules that regulate the activity of ASK1 in the posttranslational modification process, which may lead to better therapeutic effects in the treatment of NASH.
TGF-β-activated kinase 1 (TAK1) is a member of the MAP3K family and is known as a central signalosome in the regulation of the inflammatory response (145). Conventionally, TAK1 is activated by proinflammatory cytokines and agonists of toll-like receptors to activate MAPK and NF-κB signaling pathways (146). There is accumulating evidence showed that metabolic stress also promotes TAK1 signalosome formation and activity in hepatocytes, which leads to the development of NAFLD and NASH (147). However, previous studies showed that the complete deletion of TAK1 expression also accelerates NASH progression, suggesting that maintenance of the normal enzymatic activity of TAK1 is also critical for sustaining homeostasis in metabolism and inflammation (148). Therefore, posttranslational modifications are essential in fine-tuning the activity of the TAK1 signalosome under such conditions. Recent studies have revealed endogenous molecules that are important in the regulation of TAK1 ubiquitination or phosphorylation without suppressing its physiological activity, which may serve as potential targets in the development of treatments for NASH. Evidences from mouse or preclinical non-human primate models showed that the deubiquitinating enzyme cylindromatosis, TNFAIP3-interacting protein 3, ubiquitin-specific protease (USP) 4, and USP18 mitigate liver steatosis, inflammation, and fibrosis by deubiquitinating metabolic stress-induced TAK1 ubiquitination and activation (149–153), while dual-specificity phosphatase 14 and regulator of G protein signaling 5 dephosphorylate TAK1, resulting in the reduced activation of TAK1 and its downstream signaling pathways (154, 155). Although these molecules show strong potency, their safety and efficacy required to be tested in prospective studies.
Due to the unique anatomical association of the liver with the intestine, the blood supply of the liver is enriched in microbial-associated molecular patterns (PAMPs) and nutrients. Thus, Toll-like receptors (TLRs) play an essential role in liver physiology and pathophysiology (156). Previous evidence has shown that TLRs are involved in the pathogenesis of NASH and liver fibrosis (157–159). Among these TLRs, the role of TLR4 has been the most extensively studied due to its importance in recognizing gut-derived endotoxin (160). The genetic deletion or pharmaceutical inhibition of TLR4 improved liver steatosis, inflammation, and fibrosis in response to a high-fat diet in mice and nonhuman primates (158). A small long-acting molecule, JKB-121, inhibits TLR4, which inhibits liver fibrosis by repressing the redox status and stellate cell activation in the liver (NCT02442687). Recently, a novel TLR4 antagonist, JKB-122, was developed and shown to be effective in reducing autoimmune hepatitis-associated liver necrosis and inflammation in animal models (161). A phase 2 study testing the efficacy of JKB-122 for 52 weeks in subjects with NASH with fibrosis was initiated in 2020 (NCT04255069).
Vascular adhesion protein 1 (VAP-1) is continuously expressed as a membrane-bound amine oxidase along the sinusoidal endothelium, which facilitates the accumulation of inflammatory cells into the inflamed environment in concert with other leukocyte adhesion molecules (162). The soluble form of VAP-1 (sVAP-1) is also found in the serum of healthy adults, and its expression is increased under inflammatory conditions and in metabolic disorders (163). Vascular adhesion protein 1 can modulate leukocyte migration in both its transmembranous and soluble forms. Studies have shown that hepatic VAP-1 and serum sVAP-1 expression is increased in patients with NAFLD compared with control individuals (164). In addition, VAP-1 plays an essential role in hepatic fibrosis due to a number of etiologies, such as NAFLD, HBV, and HCV (163–165). Mechanistically, VAP-1 directly affects stellate cells by enhancing the expression of profibrotic genes and promoting liver fibrosis (163). The VAP-1 mutant strain showed significant attenuations of liver inflammation and fibrosis in the MCD diet model (163). PXS-4728A is a selective and orally active VAP-1 inhibitor with potent efficacy observed in animal trials (166). In 2015, the data from a phase 1 clinical trial showed that PXS-4728A administered for 14 days at doses between 3 and 10 mg was safe and well-tolerated. The data suggest that low doses are effective in inducing persistent enzyme inhibition, but further clinical trials are needed to verify the effectiveness of the drug in treating NASH.
During NASH progression, C-C chemokine receptor type 2 (CCR2) and C-C chemokine receptor type 5 (CCR5), together with their respective ligands, C-C chemokine ligand types 2 (CCL2) and C-C chemokine ligand types 5 (CCL5), promote liver fibrosis by increasing immune cell aggregation and infiltration and amplifying the inflammatory response (167–170). Cenicriviroc is a dual CCR2 and CCR5 antagonist with significant antifibrotic and anti-inflammatory activity in models of fibrosis, such as the mouse peritonitis model, mouse diet-induced NASH model, and rat thioacetamide-induced liver fibrosis model (171). Recently, it has been explored in the treatment of liver fibrosis associated with NASH (172). The 2-year phase 2b CENTAUR study showed that Cenicriviroc treatment resulted in liver fibrosis improvement compared to placebo, with a greater effect on advanced fibrosis (172). Due to the success in the phase 2 trial, a phase 3 study examining the efficacy and safety of Cenicriviroc in the treatment of liver fibrosis in adults with NASH was initiated in 2017. Unfortunately, there was a lack of efficacy at the 12-month follow-up in terms of achieving an improvement in fibrosis of at least 1 stage with no worsening of steatohepatitis (NCT03028740). There are emerging studies investigating whether combination therapy provides superior clinical effectiveness in the treatment of NASH and fibrosis. A phase 2 clinical trial in NASH patients exploring the efficacy of a combination of Tropifexor (LJN452, an FXR agonist) and Cenicriviroc has been completed, and the results are pending publication (NCT03517540).
Lysyl oxidase-like 2 (LOXL2) is an extracellular copper-dependent enzyme that catalyzes the cross-linking of structural extracellular matrix components in fibrous organs, including the liver (173). The serum LOXL2 level was associated with the severity of liver fibrosis (174). In a preclinical model characterized by advanced fibrosis and portal hypertension, an anti-LOXL2 antibody decreased the portal pressure in Mdr2-knockout mice (175). However, the LOXL2 monoclonal antibody Simtuzumab failed to reduce the liver collagen content and fibrosis in NASH patients with advanced fibrosis and cirrhosis (NCT01672866 and NCT01672879) (176). These findings were also observed in a phase 2 trial of the combination of the ASK 1 inhibitor Selonsertib and Simtuzumab. The coadministration of Selonsertib and Simtuzumab did not provide additional benefits over Selonsertib therapy alone in patients with NASH and moderate to severe fibrosis (139). There are several potential factors explaining the failure of the Simtuzumab trial. First, LOXL2 may be the driver for NASH, and fibrosis or redundancy in other pathways may mediate collagen formation. Second, although Simtuzumab effectively binds LOXL2, the dose and frequency at which it was applied in the study might be insufficient to neutralize its activity.
Galectin-3 is a β-galactoside-binding animal lectin in the nucleus and cytoplasm and on the cell surface that has been implicated in a variety of biological processes, including cell proliferation, survival and inflammation. Galectin-3 expression is upregulated in human fibrotic liver disease, and the level is associated with the induction and resolution of hepatic fibrosis in animal models (177). A mechanistic study showed that galectin-3 in HSC is required for TGF-β-mediated myofibroblast activation and matrix production during disease progression (177). Preclinical results showed that the galectin-3 inhibitor Belapectin (GR-MD-02) was effective in mouse models of NASH with advanced fibrosis or cirrhosis (178). Although Belapectin was safe and well-tolerated in a phase 1 trial (179), in this 16-week phase 2 clinical study, Belapectin treatment failed to alleviate liver fibrosis in patients with NASH with advanced fibrosis, as measured by multiparametric MRI corrected T1 mapping (NCT02421094). Similarly, another phase 2b trial of the safety and efficacy of Belapectin in patients with NASH, cirrhosis, and portal hypertension further showed that Belapectin was not associated with a significant reduction in the hepatic venous pressure gradient or fibrosis (NCT02462967) (180). However, treatment with Belapectin reduced the venous pressure gradient in a subset of patients without esophageal varices (180). To confirm this discovery, a phase 2b/3 trial evaluating the efficacy of Belapectin for the prevention of esophageal varices in NASH-associated cirrhosis was initiated and is expected to be completed in 2023 (NCT04365868).
TGF-β is an important pleiotropic cytokine involved in many biological processes such as cell survival, proliferation, differentiation, angiogenesis, and wound healing (181). In advanced MAFLD, TGF-β is activated by HSCs, triggering a series of responses including tissue repair, extracellular matrix production, growth regulation, and apoptosis, ultimately leading to liver fibrosis (182). Studies in NASH model of wild-type and hepatocellular specific TGF-β receptor type II deficiency mice demonstrated that TGF-β signaling in hepatocytes promotes lipid accumulation by regulating lipid metabolism and enhancing cell death in hepatocytes that accumulate lipid, leading to the development of hepatic steatosis, inflammation, and fibrosis (183). Previous studies have shown a significant increase in TGF-β expression in the liver of patients with NASH fibrosis (184). Preclinical results showed that TGF-β inhibitor Galunisertib affected parenchymal cell fate by regulating the biochemical composition of deposited extracellular matrix and inhibited the progression of liver fibrosis, but did not significantly improve the pathological grading of fibrosis in Abcb4ko mice (185). At present, there have been clinical studies on TGF-β inhibitors restraining fibrosis in other organs (idiopathic pulmonary fibrosis and myelofibrosis), but there are no clinical studies related to liver fibrosis. It is believed that researchers will conduct many clinical studies on TGF-β inhibitors in the fibrosis process of MAFLD in the future.
The burden of MAFLD/NAFLD is increasing rapidly with the ongoing metabolic disease epidemic (6, 7). MAFLD/NAFLD have shared and predominate causes from nutrition overload to persistent liver damage and eventually lead to the development of liver fibrosis and cirrhosis (10, 186). Discoveries have revealed that the pathogenesis of fibrosis in NAFLD involves multiple mechanisms and factors, such as lipid metabolism, inflammation, cell apoptosis, oxidative stress, extracellular matrix formation, and intestinal flora, as well as genetic and epigenetic regulation (17, 134). Reagents specifically targeting these pathways and receptor/ligand interactions have been developed, including agents acting on lipid synthesis, i.e., GLP-1 agonists, ACC inhibitors, FXR agonists, PPAR-α/δ agonists, and THR-β agonists, agents acting on cell stress and apoptosis, i.e., vitamin E and caspase inhibitors, agents acting on the innate immune system and inflammation, i.e., ASK1 inhibitors, TLR4 inhibitors, VAP-1 inhibitors, and CCR2/5 antagonists, and agents acting on other mechanisms, i.e., LOXL2 monoclonal antibodies and galectin-3 inhibitors (Supplementary Table 1). Although substantial advances have been made in the development of novel antifibrotic targets and therapeutic compounds, very few have reached clinical primary endpoints without significant side effects in large clinical studies. Therefore, it is important to recognize the boundaries and drawbacks of the traditional paths of drug discovery. First, since the majority of mechanistic investigations are based on rodent models, the application of models in large animals, such as non-human primates, with a closer resemblance to humans in the preclinical phase would likely allow for a higher chance of translating basic discoveries to clinical practice. Second, as the pathways involved in fibrosis are complex and targeting one mechanism may trigger alternative compensatory mechanisms, combination therapies targeting multiple profibrotic pathways could be promising in achieving successful antifibrotic interventions in patients with MAFLD/NAFLD. Third, the inconsistent results in previous trials have indicated that there may be large variations in the genetic predisposition and mechanisms involved in the pathogenesis of MAFLD/NAFLD fibrosis among individuals. A one-size-fits-all strategy would not be applicable for the treatment of MAFLD/NAFLD. Fourth, although liver biopsy is the gold standard for diagnosing NASH and assessing the stage of fibrosis in patients with NAFLD, this methodology misses the systemic evaluation of liver pathology and involves interobserver variations and biopsy bias. Liver biopsy cannot be performed for screening and follow-up in large populations due to its well-known limitations. There is an urgent need to improve the methodology in the evaluation of liver fibrosis.
In summary, the high prevalence of MAFLD/NAFLD paired with end-stage complications emphasizes the need for the discovery of effective and safe pharmaceutical treatments. In the current situation, one should keep in mind that appropriate lifestyle interventions with improvements in metabolic risk factors can potentially impede the development of MAFLD/NAFLD (13). In addition to accelerating the discovery of new pharmacotherapeutics, personalized medicine, combination therapies targeting multiple profibrotic pathways, and different methodologies for evaluating fibrosis would be beneficial for the development of new treatment strategies with good tolerability and efficacy.
WQ and TM collected the data and drafted the first edition of the paper. All authors listed have made a substantial, intellectual and direct contributions to this review, and authorized the publication of it.
This work was supported by grants from the National Key R&D Program of China (2016YFF0101504, 2020YFC2004702), the National Science Foundation of China (81630011, 81970364, 81970070, 81770053, 81870171, and 81970011), the Hubei Science and Technology Support Project (2019BFC582, 2018BEC473), and Medical flight plan of Wuhan University (TFJH2018006).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.761538/full#supplementary-material
1. Eslam M, Sanyal AJ, George J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014. doi: 10.1053/j.gastro.2019.11.312
2. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
3. Younossi ZM, Rinella ME, Sanyal AJ, Harrison SA, Brunt EM, Goodman Z, et al. From NAFLD to MAFLD: implications of a premature change in terminology. Hepatology. (2021) 73:1194–8. doi: 10.1002/hep.31420
4. Ciardullo S, Perseghin G. Prevalence of NAFLD, MAFLD and associated advanced fibrosis in the contemporary United States population. Liver Int. (2021) 41:1290–3. doi: 10.1111/liv.14828
5. Huang Q, Zou X, Wen X, Zhou X. Ji L. NAFLD or MAFLD: which has closer association with all-cause and cause-specific mortality?-Results from NHANES III. Front Med. (2021) 8:693507. doi: 10.3389/fmed.2021.693507
6. Lin S, Huang J, Wang M, Kumar R, Liu Y, Liu S, et al. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. (2020) 40:2082–9. doi: 10.1111/liv.14548
7. Wai-Sun WV, Lai-Hung WG, Woo J, Abrigo JM, Ka-Man CC, She-Ting SS, et al. Impact of the new definition of metabolic associated fatty liver disease on the epidemiology of the disease. Clin Gastroenterol Hepatol. (2020) 19:2161.e5–71.e5. doi: 10.1016/j.cgh.2020.10.046
8. Younossi Z, Tacke F, Arrese M, Chander SB, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. (2019) 69:2672–82. doi: 10.1002/hep.30251
9. Zhou J, Zhou F, Wang W, Zhang XJ Ji YX, Zhang P, et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. (2020) 71:1851–64. doi: 10.1002/hep.31150
10. Zhou JH, Cai JJ, She ZG Li HL. Noninvasive evaluation of nonalcoholic fatty liver disease: current evidence and practice. World J Gastroenterol. (2019) 25:1307–26. doi: 10.3748/wjg.v25.i11.1307
11. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
12. Chen Z, Yu Y, Cai J, Li H. Emerging molecular targets for treatment of nonalcoholic fatty liver disease. Trends Endocrinol Metab. (2019) 30:903–14. doi: 10.1016/j.tem.2019.08.006
13. Cai J, Zhang XJ Li H. Progress and challenges in the prevention and control of nonalcoholic fatty liver disease. Med Res Rev. (2019) 39:328–48. doi: 10.1002/med.21515
14. Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun WV, Castellanos M, Aller-de LFR, Metwally M, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology. (2018) 155:443–57. doi: 10.1053/j.gastro.2018.04.034
15. Heyens L, Busschots D, Koek GH, Robaeys G, Francque S. Liver fibrosis in non-alcoholic fatty liver disease: from liver biopsy to non-invasive biomarkers in diagnosis and treatment. Front Med. (2021) 8:615978. doi: 10.3389/fmed.2021.615978
16. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. (2018) 68–69:435–51. doi: 10.1016/j.matbio.2018.04.006
17. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. (2021) 184:2537–64. doi: 10.1016/j.cell.2021.04.015
18. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. (2019) 76:99–128. doi: 10.1007/s00018-018-2947-0
19. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol. (2020) 73:505–15. doi: 10.1016/j.jhep.2020.04.003
20. Mann JP, Pietzner M, Wittemans LB, Rolfe EL, Kerrison ND, Imamura F, et al. Insights into genetic variants associated with NASH-fibrosis from metabolite profiling. Hum Mol Genet. (2020) 29:3451–63. doi: 10.1093/hmg/ddaa162
21. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. (2020) 158:1913–28. doi: 10.1053/j.gastro.2019.11.311
22. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
23. Delli BA, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative stress in non-alcoholic fatty liver disease. an updated mini review. Front Med (Lausanne). (2021) 8:595371. doi: 10.3389/fmed.2021.595371
24. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
25. Cai J, Zhang XJ Li H. Role of innate immune signaling in non-alcoholic fatty liver disease. Trends Endocrinol Metab. (2018) 29:712–22. doi: 10.1016/j.tem.2018.08.003
26. Cai J, Zhang XJ Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. (2019) 70:1026–37. doi: 10.1002/hep.30506
27. Roh YS, Seki E. Chemokines and chemokine receptors in the development of NAFLD. Adv Exp Med Biol. (2018) 1061:45–53. doi: 10.1007/978-981-10-8684-7_4
28. Xu M, Liu PP Li H. Innate immune signaling and its role in metabolic and cardiovascular diseases. Physiol Rev. (2019) 99:893–948. doi: 10.1152/physrev.00065.2017
29. Bai L, Li H. Innate immune regulatory networks in hepatic lipid metabolism. J Mol Med (Berl). (2019) 97:593–604. doi: 10.1007/s00109-019-01765-1
30. Kazankov K, Jorgensen S, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
31. Breuer DA, Pacheco MC, Washington MK, Montgomery SA, Hasty AH, Kennedy AJ. CD8(+) T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. (2020) 318:G211–24. doi: 10.1152/ajpgi.00040.2019
32. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. (2021) 12:4474. doi: 10.1038/s41467-021-24734-0
33. Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han JH, et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology. (2015) 61:2067–79. doi: 10.1002/hep.27761
34. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-Driven activation of intrahepatic b cells aggravates nonalcoholic steatohepatitis through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
35. Dewidar B, Kahl S, Pafili K, Roden M. Metabolic liver disease in diabetes - from mechanisms to clinical trials. Metabolism. (2020) 111S:154299. doi: 10.1016/j.metabol.2020.154299
36. Barcena-Varela M, Colyn L, Fernandez-Barrena MG. Epigenetic mechanisms in hepatic stellate cell activation during liver fibrosis and carcinogenesis. Int J Mol Sci. (2019) 20:2507. doi: 10.3390/ijms20102507
37. Campisano S, La Colla A, Echarte SM, Chisari AN. Interplay between early-life malnutrition, epigenetic modulation of the immune function and liver diseases. Nutr Res Rev. (2019) 32:128–45. doi: 10.1017/S0954422418000239
38. Muller TD, Finan B, Bloom SR, D'Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
39. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
40. Liu L, Yan H, Xia M, Zhao L, Lv M, Zhao N, et al. Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab Res Rev. (2020) 36:e3292. doi: 10.1002/dmrr.3292
41. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A Placebo-Controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. (2021) 384:1113–24. doi: 10.1056/NEJMoa2028395
42. Boland ML, Laker RC, Mather K, Nawrocki A, Oldham S, Boland BB, et al. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab. (2020) 2:413–31. doi: 10.1038/s42255-020-0209-6
43. Hartman ML, Sanyal AJ, Loomba R, Wilson JM, Nikooienejad A, Bray R, et al. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care. (2020) 43:1352–5. doi: 10.2337/dc19-1892
44. Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. (2020) 29:135–41. doi: 10.1080/13543784.2020.1668374
45. Bates J, Vijayakumar A, Ghoshal S, Marchand B, Yi S, Kornyeyev D, et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J Hepatol. (2020) 73:896–905. doi: 10.1016/j.jhep.2020.04.037
46. Stiede K, Miao W, Blanchette HS, Beysen C, Harriman G, Harwood HJ, et al. Acetyl-coenzyme a carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: a randomized, double-blind, crossover study. Hepatology. (2017) 66:324–34. doi: 10.1002/hep.29246
47. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. (2018) 155:1463–73. doi: 10.1053/j.gastro.2018.07.027
48. Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. (2021) 73:625–43. doi: 10.1002/hep.31622
49. Ross TT, Crowley C, Kelly KL, Rinaldi A, Beebe DA, Lech MP, et al. Acetyl-CoA carboxylase inhibition improves multiple dimensions of NASH pathogenesis in model systems. Cell Mol Gastroenterol Hepatol. (2020) 10:829–51. doi: 10.1016/j.jcmgh.2020.06.001
50. Baardman J, Verberk S, van der Velden S, Gijbels M, van Roomen C, Sluimer JC, et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat Commun. (2020) 11:6296. doi: 10.1038/s41467-020-20141-z
51. Guo L, Guo YY Li BY, Peng WQ, Chang XX, Gao X, et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J Biol Chem. (2019) 294:11805–16. doi: 10.1074/jbc.RA119.008708
52. Yang X, Sun D, Xiang H, Wang S, Huang Y, Li L, et al. Hepatocyte SH3RF2 deficiency is a key aggravator for nonalcoholic fatty liver disease. Hepatology. (2021) 74:1319–38. doi: 10.1002/hep.31863
53. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. (2016) 5:e11612. doi: 10.7554/eLife.11612
54. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. (2013) 440:105–11. doi: 10.1016/j.bbrc.2013.09.037
55. Feng X, Zhang L, Xu S, Shen AZ. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: an updated review. Prog Lipid Res. (2020) 77:101006. doi: 10.1016/j.plipres.2019.101006
56. Pinkosky SL, Filippov S, Srivastava RA, Hanselman JC, Bradshaw CD, Hurley TR, et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. (2013) 54:134–51. doi: 10.1194/jlr.M030528
57. Cicero A, Fogacci F, Hernandez AV, Banach M. Efficacy and safety of bempedoic acid for the treatment of hypercholesterolemia: a systematic review and meta-analysis. PLoS Med. (2020) 17:e1003121. doi: 10.1371/journal.pmed.1003121
58. Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. (2021) 50:101238. doi: 10.1016/j.molmet.2021.101238
59. Hu Y, He W, Huang Y, Xiang H, Guo J, Che Y, et al. FASN-suppressor screening identifies SNX8 as a novel therapeutic target for NAFLD. Hepatology. (2021). doi: 10.1002/hep.32045. [Epub ahead of print].
60. Beysen C, Schroeder P, Wu E, Brevard J, Ribadeneira M, Lu W, et al. Inhibition of fatty acid synthase with FT-4101 safely reduces hepatic de novo lipogenesis and steatosis in obese subjects with non-alcoholic fatty liver disease: results from two early-phase randomized trials. Diabetes Obes Metab. (2021) 23:700–10. doi: 10.1111/dom.14272
61. Syed-Abdul MM, Parks EJ, Gaballah AH, Bingham K, Hammoud GM, Kemble G, et al. Fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology. (2020) 72:103–18. doi: 10.1002/hep.31000
62. Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled Ph2a trial. Gastroenterology. (2021) 2021: S0016-5085(21)03276-5. doi: 10.1053/j.gastro.2021.07.025
63. Rau M, Geier A. An update on drug development for the treatment of nonalcoholic fatty liver disease - from ongoing clinical trials to future therapy. Expert Rev Clin Pharmacol. (2021) 14:333–40. doi: 10.1080/17512433.2021.1884068
64. Lai K, Kweon SM, Chi F, Hwang E, Kabe Y, Higashiyama R, et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology. (2017) 152:1477–91. doi: 10.1053/j.gastro.2017.01.021
65. Gu X, Sun R, Chen L, Chu S, Doll MA Li X, et al. Neutral ceramidase mediates nonalcoholic steatohepatitis by regulating monounsaturated fatty acids and gut IgA(+) b cells. Hepatology. (2021) 73:901–19. doi: 10.1002/hep.31628
66. Iruarrizaga-Lejarreta M, Varela-Rey M, Fernandez-Ramos D, Martinez-Arranz I, Delgado TC, Simon J, et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun. (2017) 1:911–27. doi: 10.1002/hep4.1107
67. Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2014) 12:2085–91. doi: 10.1016/j.cgh.2014.04.038
68. Guirguis E, Grace Y, Bolson A, DellaVecchia MJ, Ruble M. Emerging therapies for the treatment of nonalcoholic steatohepatitis: a systematic review. Pharmacotherapy. (2021) 41:315–28. doi: 10.1002/phar.2489
69. Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. (2021) 33:1671–84. doi: 10.1016/j.cmet.2021.06.012
70. Schumacher JD, Kong B, Wu J, Rizzolo D, Armstrong LE, Chow MD, et al. Direct and indirect effects of fibroblast growth factor (FGF) 15 and FGF19 on liver fibrosis development. Hepatology. (2020) 71:670–85. doi: 10.1002/hep.30810
71. van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J, et al. A translational mouse model for NASH with advanced fibrosis and atherosclerosis expressing key pathways of human pathology. Cells-Basel. (2020) 9:2014. doi: 10.3390/cells9092014
72. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. (2015) 385:956–65. doi: 10.1016/S0140-6736(14)61933-4
73. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. (2019) 394:2184–96. doi: 10.1016/S0140-6736(19)33041-7
74. Polyzos SA, Kountouras J, Mantzoros CS. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: expectations and concerns. Metabolism. (2020) 104:154144. doi: 10.1016/j.metabol.2020.154144
75. Pockros PJ, Fuchs M, Freilich B, Schiff E, Kohli A, Lawitz EJ, et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. (2019) 39:2082–93. doi: 10.1111/liv.14209
76. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. (2020) 72:58–71. doi: 10.1002/hep.31205
77. Erstad DJ, Farrar CT, Ghoshal S, Masia R, Ferreira DS, Chen YI, et al. Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP-305, a novel farnesoid X receptor agonist. Hepatol Commun. (2018) 2:821–35. doi: 10.1002/hep4.1193
78. An P, Wei G, Huang P, Li W, Qi X, Lin Y, et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. (2020) 40:1655–69. doi: 10.1111/liv.14490
79. Fiorucci S, Biagioli M, Sepe V, Zampella A, Distrutti E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. (2020) 29:623–32. doi: 10.1080/13543784.2020.1763302
80. Henriksson E, Andersen B. FGF19 and FGF21 for the treatment of NASH-Two sides of the same coin? Differential and overlapping effects of FGF19 and FGF21 from mice to human. Front Endocrinol. (2020) 11:601349. doi: 10.3389/fendo.2020.601349
81. Talukdar S, Kharitonenkov A. FGF19 and FGF21: In NASH we trust. Mol Metab. (2021) 46:101152. doi: 10.1016/j.molmet.2020.101152
82. Zhou M, Wang X, Phung V, Lindhout DA, Mondal K, Hsu JY, et al. Separating tumorigenicity from bile acid regulatory activity for endocrine hormone FGF19. Cancer Res. (2014) 74:3306–16. doi: 10.1158/0008-5472.CAN-14-0208
83. Zhou M, Yang H, Learned RM, Tian H, Ling L. Non-cell-autonomous activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat Commun. (2017) 8:15433. doi: 10.1038/ncomms15433
84. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2018) 391:1174–85. doi: 10.1016/S0140-6736(18)30474-4
85. Harrison SA, Rossi SJ, Paredes AH, Trotter JF, Bashir MR, Guy CD, et al. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology. (2020) 71:1198–212. doi: 10.1002/hep.30590
86. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. (2021) 160:219–31. doi: 10.1053/j.gastro.2020.08.004
87. Rinella ME, Trotter JF, Abdelmalek MF, Paredes AH, Connelly MA, Jaros MJ, et al. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol. (2019) 70:735–44. doi: 10.1016/j.jhep.2018.11.032
88. Geng L, Lam K, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. (2020) 16:654–67. doi: 10.1038/s41574-020-0386-0
89. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. (2019) 392:2705–17. doi: 10.1016/S0140-6736(18)31785-9
90. Abdelmalek MF, Charles ED, Sanyal AJ, Harrison SA, Neuschwander-Tetri BA, Goodman Z, et al. The FALCON program: Two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemp Clin Trials. (2021) 104:106335. doi: 10.1016/j.cct.2021.106335
91. Stanislaus S, Hecht R, Yie J, Hager T, Hall M, Spahr C, et al. A novel Fc-FGF21 with improved resistance to proteolysis, increased affinity toward beta-Klotho, and enhanced efficacy in mice and cynomolgus monkeys. Endocrinology. (2017) 158:1314–27. doi: 10.1210/en.2016-1917
92. Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. (2021) 27:1262–71. doi: 10.1038/s41591-021-01425-3
93. Han X, Wu Y, Yang Q, Cao G. Peroxisome proliferator-activated receptors in the pathogenesis and therapies of liver fibrosis. Pharmacol Ther. (2021) 222:107791. doi: 10.1016/j.pharmthera.2020.107791
94. Tong J, Han CJ, Zhang JZ, He WZ, Zhao GJ, Cheng X, et al. Hepatic interferon regulatory factor 6 alleviates liver steatosis and metabolic disorder by transcriptionally suppressing peroxisome proliferator-activated receptor gamma in mice. Hepatology. (2019) 69:2471–88. doi: 10.1002/hep.30559
95. He J, Hong B, Bian M, Jin H, Chen J, Shao J, et al. Docosahexaenoic acid inhibits hepatic stellate cell activation to attenuate liver fibrosis in a PPARgamma-dependent manner. Int Immunopharmacol. (2019) 75:105816. doi: 10.1016/j.intimp.2019.105816
96. Sandoval-Rodriguez A, Monroy-Ramirez HC, Meza-Rios A, Garcia-Banuelos J, Vera-Cruz J, Gutierrez-Cuevas J, et al. Pirfenidone is an agonistic ligand for PPARalpha and improves NASH by activation of SIRT1/LKB1/pAMPK. Hepatol Commun. (2020) 4:434–49. doi: 10.1002/hep4.1474
97. Poo JL, Torre A, Aguilar-Ramirez JR, Cruz M, Mejia-Cuan L, Cerda E, et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol Int. (2020) 14:817–27. doi: 10.1007/s12072-020-10069-3
98. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. (2016) 165:305–15. doi: 10.7326/M15-1774
99. Bril F, Biernacki DM, Kalavalapalli S, Lomonaco R, Subbarayan SK, Lai J, et al. Role of vitamin e for nonalcoholic steatohepatitis in patients with type 2 diabetes: a randomized controlled trial. Diabetes Care. (2019) 42:1481–8. doi: 10.2337/dc19-0167
100. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. (2020) 72:613–26. doi: 10.1016/j.jhep.2019.10.023
101. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. (2016) 150:1147–59. doi: 10.1053/j.gastro.2016.01.038
102. Siddiqui MS, Idowu MO, Parmar D, Borg BB, Denham D, Loo NM, et al. A phase 2 double blinded, randomized controlled trial of saroglitazar in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. (2020) 2020:S1542-3565(20)31509-3. doi: 10.1016/j.cgh.2020.10.051
103. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-alpha/gamma agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology. (2021) 74:1809–24. doi: 10.1002/hep.31843
104. Goyal O, Nohria S, Goyal P, Kaur J, Sharma S, Sood A, et al. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: a prospective, observational, real world study. Sci Rep. (2020) 10:21117. doi: 10.1038/s41598-020-78342-x
105. Boyer-Diaz Z, Aristu-Zabalza P, Andres-Rozas M, Robert C, Ortega-Ribera M, Fernandez-Iglesias A, et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J Hepatol. (2021) 74:1188–99. doi: 10.1016/j.jhep.2020.11.045
106. Kannt A, Wohlfart P, Madsen AN, Veidal SS, Feigh M, Schmoll D. Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br J Pharmacol. (2021) 178:2412–23. doi: 10.1111/bph.15427
107. Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2019) 394:2012–24. doi: 10.1016/S0140-6736(19)32517-6
108. Harrison SA, Bashir M, Moussa SE, McCarty K, Pablo FJ, Taub R, et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol Commun. (2021) 5:573–88. doi: 10.1002/hep4.1657
109. Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. (2018) 2018:9547613. doi: 10.1155/2018/9547613
110. Alberti G, Gana JC, Santos JL. Fructose, omega 3 fatty acids, and vitamin E: involvement in pediatric Non-Alcoholic fatty liver disease. Nutrients. (2020) 12:3531. doi: 10.3390/nu12113531
111. Sodum N, Kumar G, Bojja SL, Kumar N, Rao CM. Epigenetics in NAFLD/NASH: targets and therapy. Pharmacol Res. (2021) 167:105484. doi: 10.1016/j.phrs.2021.105484
112. Nagashimada M, Ota T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life. (2019) 71:516–22. doi: 10.1002/iub.1991
113. Vilar-Gomez E, Vuppalanchi R, Gawrieh S, Ghabril M, Saxena R, Cummings OW, et al. Vitamin E improves transplant-free survival and hepatic decompensation among patients with nonalcoholic steatohepatitis and advanced fibrosis. Hepatology. (2020) 71:495–509. doi: 10.1002/hep.30368
114. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. (2010) 362:1675–85. doi: 10.1056/NEJMoa0907929
115. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. (2011) 305:1659–68. doi: 10.1001/jama.2011.520
116. Hoofnagle JH, Van Natta ML, Kleiner DE, Clark JM, Kowdley KV, Loomba R, et al. Vitamin E and changes in serum alanine aminotransferase levels in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2013) 38:134–43. doi: 10.1111/apt.12352
117. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. (2007) 297:842–57. doi: 10.1001/jama.297.8.842
118. Klein EA, Thompson IJ, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT). JAMA. (2011) 306:1549–56. doi: 10.1001/jama.2011.1437
119. Curtis AJ, Bullen M, Piccenna L, McNeil JJ. Vitamin E supplementation and mortality in healthy people: a meta-analysis of randomised controlled trials. Cardiovasc Drugs Ther. (2014) 28:563–73. doi: 10.1007/s10557-014-6560-7
120. Aune D, Keum N, Giovannucci E, Fadnes LT, Boffetta P, Greenwood DC, et al. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: a systematic review and dose-response meta-analysis of prospective studies. Am J Clin Nutr. (2018) 108:1069–91. doi: 10.1093/ajcn/nqy097
121. Akazawa Y, Nakao K. To die or not to die: death signaling in nonalcoholic fatty liver disease. J Gastroenterol. (2018) 53:893–906. doi: 10.1007/s00535-018-1451-5
122. Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. (2018) 24:2661–72. doi: 10.3748/wjg.v24.i25.2661
123. Li X, Dong G, Xiong H, Diao H, A. narrative review of the role of necroptosis in liver disease: a double-edged sword. Ann Transl Med. (2021) 9:422. doi: 10.21037/atm-20-5162
124. Shojaie L, Iorga A, Dara L. Cell death in liver diseases: a review. Int J Mol Sci. (2020) 21:9682. doi: 10.3390/ijms21249682
125. Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, et al. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. (2015) 35:953–66. doi: 10.1111/liv.12570
126. Shiffman M, Freilich B, Vuppalanchi R, Watt K, Chan JL, Spada A, et al. Randomised clinical trial: emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. (2019) 49:64–73. doi: 10.1111/apt.15030
127. Frenette CT, Morelli G, Shiffman ML, Frederick RT, Rubin RA, Fallon MB, et al. Emricasan improves liver function in patients with cirrhosis and high model for end-stage liver disease scores compared with placebo. Clin Gastroenterol Hepatol. (2019) 17:774–83. doi: 10.1016/j.cgh.2018.06.012
128. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol. (2020) 72:816–27. doi: 10.1016/j.jhep.2019.11.024
129. Garcia-Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, Lawitz E, et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J Hepatol. (2020) 72:885–95. doi: 10.1016/j.jhep.2019.12.010
130. Frenette C, Kayali Z, Mena E, Mantry PS, Lucas KJ, Neff G, et al. Emricasan to prevent new decompensation in patients with NASH-related decompensated cirrhosis. J Hepatol. (2021) 74:274–82. doi: 10.1016/j.jhep.2020.09.029
131. Weinberg EM, Curry MP, Frenette CT, Regenstein FG, Schiff ER, Goodman ZD, et al. Multicenter, Double-Blind, randomized trial of emricasan in hepatitis C-Treated liver transplant recipients with residual fibrosis or cirrhosis. Liver Transpl. (2021) 27:568–79. doi: 10.1002/lt.25934
132. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. (2013) 14:996–1006. doi: 10.1038/ni.2691
133. Wang XA, Deng S, Jiang D, Zhang R, Zhang S, Zhong J, et al. CARD3 deficiency exacerbates diet-induced obesity, hepatosteatosis, and insulin resistance in male mice. Endocrinology. (2013) 154:685–97. doi: 10.1210/en.2012-1911
134. Yu Y, Cai J, She Z, Li H. Insights into the epidemiology, pathogenesis, and therapeutics of nonalcoholic fatty liver diseases. Adv Sci (Weinh). (2019) 6:1801585. doi: 10.1002/advs.201801585
135. Kovalic AJ, Satapathy SK, Chalasani N. Targeting incretin hormones and the ASK-1 pathway as therapeutic options in the treatment of non-alcoholic steatohepatitis. Hepatol Int. (2018) 12:97–106. doi: 10.1007/s12072-018-9854-1
136. Wang PX Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, et al. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med. (2017) 23:439–49. doi: 10.1038/nm.4290
137. Younossi ZM, Stepanova M, Anstee QM, Lawitz EJ, Wai-Sun WV, Romero-Gomez M, et al. Reduced Patient-Reported outcome scores associate with level of fibrosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. (2019) 17:2552–60. doi: 10.1016/j.cgh.2019.02.024
138. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatol. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
139. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. (2018) 67:549–59. doi: 10.1002/hep.29514
140. Zhang L, Tian R, Yao X, Zhang XJ, Zhang P, Huang Y, et al. Milk fat Globule-Epidermal growth Factor-Factor 8 improves hepatic steatosis and inflammation. Hepatology. (2021) 73:586–605. doi: 10.1002/hep.31277
141. An SY, Jang YJ, Lim HJ, Han J, Lee J, Lee G, et al. Milk fat Globule-EGF factor 8, secreted by mesenchymal stem cells, protects against liver fibrosis in mice. Gastroenterology. (2017) 152:1174–86. doi: 10.1053/j.gastro.2016.12.003
142. Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med. (2018) 24:84–94. doi: 10.1038/nm.4453
143. Wang Y, Wen H, Fu J, Cai L, Li PL, Zhao CL, et al. Hepatocyte TNF receptor-associated factor 6 aggravates hepatic inflammation and fibrosis by promoting lysine 6-linked polyubiquitination of apoptosis signal-regulating kinase 1. Hepatology. (2020) 71:93–111. doi: 10.1002/hep.30822
144. Bai L, Chen MM, Chen ZD, Zhang P, Tian S, Zhang Y, et al. F-box/WD repeat-containing protein 5 mediates the ubiquitination of apoptosis signal-regulating kinase 1 and exacerbates nonalcoholic steatohepatitis in mice. Hepatology. (2019) 70:1942–57. doi: 10.1002/hep.30537
145. Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. (1995) 270:2008–11. doi: 10.1126/science.270.5244.2008
146. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. (2005) 6:1087–95. doi: 10.1038/ni1255
147. Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. (2014) 49:185–94. doi: 10.1007/s00535-013-0931-x
148. Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci USA. (2010) 107:844–9. doi: 10.1073/pnas.0909781107
149. Ji YX, Huang Z, Yang X, Wang X, Zhao LP, Wang PX, et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat Med. (2018) 24:213–23. doi: 10.1038/nm.4461
150. Liu D, Zhang P, Zhou J, Liao R, Che Y, Gao MM, et al. TNFAIP3 interacting protein 3 overexpression suppresses nonalcoholic steatohepatitis by blocking TAK1 activation. Cell Metab. (2020) 31:726–40. doi: 10.1016/j.cmet.2020.03.007
151. Zhao Y, Wang F, Gao L, Xu L, Tong R, Lin N, et al. Ubiquitin-Specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology. (2018) 68:897–917. doi: 10.1002/hep.29889
152. Zhang Y, Wan J, Xu Z, Hua T, Sun Q. Exercise ameliorates insulin resistance via regulating TGFbeta-activated kinase 1 (TAK1)-mediated insulin signaling in liver of high-fat diet-induced obese rats. J Cell Physiol. (2019) 234:7467–74. doi: 10.1002/jcp.27508
153. An S, Zhao LP, Shen LJ, Wang S, Zhang K, Qi Y, et al. USP18 protects against hepatic steatosis and insulin resistance through its deubiquitinating activity. Hepatology. (2017) 66:1866–84. doi: 10.1002/hep.29375
154. Wang S, Yan ZZ, Yang X, An S, Zhang K, Qi Y, et al. Hepatocyte DUSP14 maintains metabolic homeostasis and suppresses inflammation in the liver. Hepatology. (2018) 67:1320–38. doi: 10.1002/hep.29616
155. Wang J, Ma J, Nie H, Zhang XJ, Zhang P, She ZG, et al. Hepatic regulator of g protein signaling 5 ameliorates nonalcoholic fatty liver disease by suppressing transforming growth factor beta-activated kinase 1-c-Jun-N-Terminal kinase/p38 signaling. Hepatology. (2021) 73:104–25. doi: 10.1002/hep.31242
156. Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. (2008) 47:729–36. doi: 10.1002/hep.22034
157. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. (2013) 57:577–89. doi: 10.1002/hep.26081
158. Zhao GN, Zhang P, Gong J, Zhang XJ, Wang PX, Yin M, et al. Tmbim1 is a multivesicular body regulator that protects against non-alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4. Nat Med. (2017) 23:742–52. doi: 10.1038/nm.4334
159. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. (2016) 126:859–64. doi: 10.1172/JCI83885
160. Shu X, Wang M, Xu H, Liu Y, Huang J, Yao Z, et al. Extracts of Salvia-Nelumbinis naturalis ameliorate nonalcoholic steatohepatitis via inhibiting Gut-Derived endotoxin mediated TLR4/NF-kappaB activation. Evid Based Complement Alternat Med. (2017) 2017:9208314. doi: 10.1155/2017/9208314
161. Hsu MC, Liu SH, Wang CW, Hu NY, Wu E, Shih YC, et al. JKB-122 is effective, alone or in combination with prednisolone in Con A-induced hepatitis. Eur J Pharmacol. (2017) 812:113–20. doi: 10.1016/j.ejphar.2017.07.012
162. McNab G, Reeves JL, Salmi M, Hubscher S, Jalkanen S, Adams DH. Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterology. (1996) 110:522–8. doi: 10.1053/gast.1996.v110.pm8566600
163. Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. (2015) 125:501–20. doi: 10.1172/JCI73722
164. Kurkijarvi R, Adams DH, Leino R, Mottonen T, Jalkanen S, Salmi M. Circulating form of human vascular adhesion protein-1 (VAP-1): increased serum levels in inflammatory liver diseases. J Immunol. (1998) 161:1549–57.
165. Oksuz Z, Ucbilek E, Serin MS, Yaras S, Temel GO, Sezgin O. Circulating vascular adhesion protein-1(VAP-1): a possible biomarker for liver fibrosis associated with chronic hepatitis B and C. Braz J Microbiol. (2020) 51:1757–63. doi: 10.1007/s42770-020-00379-x
166. Kubota R, Reid MJ, Lieu KL, Orme M, Diamond C, Tulberg N, et al. Comparison of inhibitor and substrate selectivity between rodent and human vascular adhesion protein-1. Mediators Inflamm. (2020) 2020:3270513. doi: 10.1155/2020/3270513
167. Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. (2009) 174:1766–75. doi: 10.2353/ajpath.2009.080632
168. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. (2009) 50:185–97. doi: 10.1002/hep.22952
169. Li BH, He FP, Yang X, Chen YW, Fan JG. Steatosis induced CCL5 contributes to early-stage liver fibrosis in nonalcoholic fatty liver disease progress. Transl Res. (2017) 180:103–17. doi: 10.1016/j.trsl.2016.08.006
170. Sun D, Yang X, Wu B, Zhang XJ Li H, She ZG. Therapeutic potential of G protein-coupled receptors against nonalcoholic steatohepatitis. Hepatology. (2021). doi: 10.1002/hep.31852. [Epub ahead of print].
171. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE. (2016) 11:e158156. doi: 10.1371/journal.pone.0158156
172. Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
173. Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl oxidase (LOX) family members: rationale and their potential as therapeutic targets for liver fibrosis. Hepatology. (2020) 72:729–41. doi: 10.1002/hep.31236
174. Murawaki Y, Kusakabe Y, Hirayama C. Serum lysyl oxidase activity in chronic liver disease in comparison with serum levels of prolyl hydroxylase and laminin. Hepatology. (1991) 14:1167–73.
175. Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. (2017) 66:1697–708. doi: 10.1136/gutjnl-2016-312473
176. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. (2018) 155:1140–53. doi: 10.1053/j.gastro.2018.07.006
177. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A. (2006) 103:5060–5. doi: 10.1073/pnas.0511167103
178. Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE. (2013) 8:e83481. doi: 10.1371/journal.pone.0083481
179. Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. Placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther. (2016) 44:1183–98. doi: 10.1111/apt.13816
180. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. (2020) 158:1334–45. doi: 10.1053/j.gastro.2019.11.296
181. Dooley S, Ten DP. TGF-beta in progression of liver disease. Cell Tissue Res. (2012) 347:245–56. doi: 10.1007/s00441-011-1246-y
182. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. (2005) 115:209–18. doi: 10.1172/JCI24282
183. Yang L, Roh YS, Song J, Zhang B, Liu C, Loomba R, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology. (2014) 59:483–95. doi: 10.1002/hep.26698
184. Cayon A, Crespo J, Mayorga M, Guerra A, Pons-Romero F. Increased expression of Ob-Rb and its relationship with the overexpression of TGF-beta1 and the stage of fibrosis in patients with nonalcoholic steatohepatitis. Liver Int. (2006) 26:1065–71. doi: 10.1111/j.1478-3231.2006.01337.x
185. Hammad S, Cavalcanti E, Werle J, Caruso ML, Dropmann A, Ignazzi A, et al. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch Toxicol. (2018) 92:2297–309. doi: 10.1007/s00204-018-2231-y
Keywords: liver fibrosis, metabolic associated fatty liver disease, drug target, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, cirrhosis
Citation: Qu W, Ma T, Cai J, Zhang X, Zhang P, She Z, Wan F and Li H (2021) Liver Fibrosis and MAFLD: From Molecular Aspects to Novel Pharmacological Strategies. Front. Med. 8:761538. doi: 10.3389/fmed.2021.761538
Received: 20 August 2021; Accepted: 27 September 2021;
Published: 22 October 2021.
Edited by:
Aldo Torre, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoReviewed by:
Ling Yang, Huazhong University of Science and Technology, ChinaCopyright © 2021 Qu, Ma, Cai, Zhang, Zhang, She, Wan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhigang She, emdzaGVAd2h1LmVkdS5jbg==; Feng Wan, d2FuZmVuZ0BoZ3l5Lm9yZy5jbg==; Hongliang Li, bGlobEB3aHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.