 Marie Nagai1
Marie Nagai1 Takahiro Uchida1*
Takahiro Uchida1* Muneharu Yamada1Shuuhei Komatsu1Kohei Ota1Mitsuya Mukae1Hitoshi Iwamoto2Hiroshi Hirano3Miho Karube4Shinya Kaname4
Muneharu Yamada1Shuuhei Komatsu1Kohei Ota1Mitsuya Mukae1Hitoshi Iwamoto2Hiroshi Hirano3Miho Karube4Shinya Kaname4 Takashi Oda1
Takashi Oda1- 1Department of Nephrology and Blood Purification, Kidney Disease Center, Tokyo Medical University Hachioji Medical Center, Hachioji, Japan
- 2Department of Kidney Transplantation Surgery, Kidney Disease Center, Tokyo Medical University Hachioji Medical Center, Hachioji, Japan
- 3Departmet of Diagnostic Pathology, Tokyo Medical University Hachioji Medical Center, Hachioji, Japan
- 4Department of Nephrology and Rheumatology, Kyorin University School of Medicine, Tokyo, Japan
A 57-year-old man who received a kidney transplant 4 years previously owing to unknown underlying disease presented with thrombocytopenia and fever. Hepatosplenomegaly and lymphadenopathy were observed, and development of prominent anasarca and worsening of renal function yielded the diagnosis of TAFRO syndrome. He was treated with high-dose steroids and plasmapheresis, and a thrombopoietin receptor agonist was administered for refractory thrombocytopenia. However, his general condition worsened, and he died on day 92. Histopathological analysis of a kidney autopsy specimen showed thrombotic microangiopathy characterized by glomerular endothelial swelling, mesangiolysis, and double contours of the glomerular capillary walls. His bone marrow showed megakaryocytic hyperplasia with mild reticulin fibrosis. Interestingly, these clinical and pathological features were remarkably similar to those the patient demonstrated before the kidney transplant, suggesting the recurrence of TAFRO syndrome. TAFRO syndrome is a rare systemic disorder whose concept has recently been established, but information on its long-term outcome is scarce. To our knowledge, this is the first case of TAFRO syndrome developing in a kidney transplant recipient, which suggests that disease recurrence occurs many years after the kidney transplant.
Introduction
TAFRO syndrome is a systemic inflammatory disorder characterized by thrombocytopenia, anasarca, fever, renal dysfunction, myelofibrosis, and organomegaly (1). This disorder often has an acute/subacute onset, and the involvement of hypercytokinemia, such as increased levels of interleukin (IL)-6 and vascular endothelial growth factor, which may be triggered by undetected infections (2), is hypothesized in its pathogenesis (3). This is the syndrome of heterogeneous entity; although several diagnostic criteria have been proposed (2, 4), the precise disease mechanism common to this syndrome still remains to be fully understood.
It has been known that this rare clinical entity can occur in association with idiopathic multicentric Castleman disease (iMCD) which presents systemic inflammation and multifocal lymphadenopathy accompanied by characteristic histopathological features. iMCD patients with TAFRO syndrome (iMCD-TAFRO) is now considered to be an aggressive clinical subtype of iMCD (5), and new definition that focuses on histopathological findings of lymph node and categorizes the affected cases into iMCD-TAFRO and TAFRO syndrome without iMCD has been proposed very recently (6).
Many case reports of TAFRO syndrome have been published since it was originally reported in 2010. Renal dysfunction is reportedly observed in more than one-half of patients with this disorder (7) and can be a life-threatening complication, because the degree of renal dysfunction is often very severe and not a few patients require renal replacement therapy (RRT) (8, 9). However, in most cases, discontinuation of RRT is possible after effective treatment, and therefore few patients require maintenance dialysis or a kidney transplant.
We herein report an autopsy case of a kidney transplant recipient with unknown underlying disease, who developed refractory TAFRO syndrome 4 years after the transplant. In the present case, the clinical and pathological features observed before and after the kidney transplant were very similar, suggesting the recurrence of TAFRO syndrome.
Case Presentation
A 57-year-old man who underwent a kidney transplant 4 years previously presented with persistent fever and abdominal back pain, and was admitted to our hospital. Seventeen years previously, he was suspected of having systemic lupus erythematosus (SLE) owing to thrombocytopenia, ascites, and proteinuria, as well as serological positivity for the antinuclear antibody (1:160) and anti-double-stranded (anti-ds) DNA antibody (27.6 IU/mL). However, these autoantibodies became negative soon and were consistently negative thereafter, and hypocomplementemia was never observed. In addition, renal biopsy showed membranoproliferative glomerulonephritis (MPGN) pattern of injury with few immune deposits (Figures 1A–C). Thus, the etiology of his original kidney disease was rather unclear. His bone marrow demonstrated mild myelofibrosis with increased megakaryocytes (data not shown). His renal function gradually deteriorated, and maintenance hemodialysis was initiated 5 years previously. One year after the start of hemodialysis, he received a living-donor kidney transplant. He remained well after the transplant, and was treated with tacrolimus, mycophenolate mofetil, and prednisone at 1 mg daily.
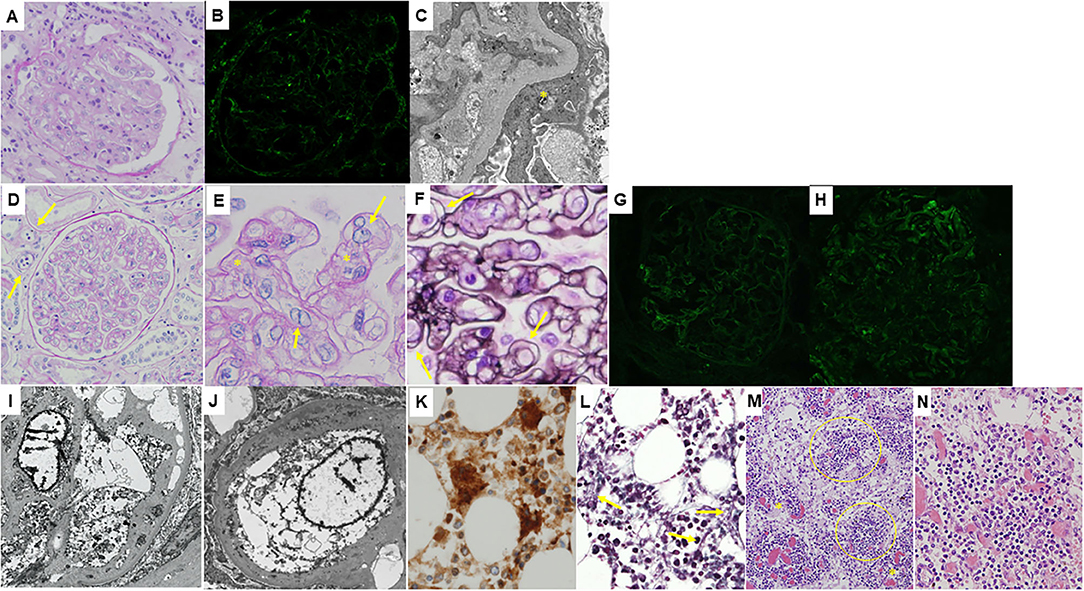
Figure 1. Histological features of the patient. (A–C): Microphotographs of the native kidney. (A) Light microscopy image of a periodic acid-Schiff-stained section showing mesangial proliferation and thickening of the glomerular capillary walls, i.e., membranoproliferative glomerulonephritis pattern. Swelling of glomerular endothelial cells is also seen. (B) Immunofluorescence staining for immunoglobulin G showing only faint deposition in the glomeruli. (C) An electron microscopy section showing no obvious electron-dense deposits. Podocyte foot process effacement is shown (yellow asterisk). (D–N): Microscopic findings of the autopsy samples. (D) Light microscopy image of a periodic acid-Schiff-stained transplanted kidney section showing prominent swelling of glomerular endothelial cells, mesangiolysis, and duplication of the glomerular capillary walls suggesting thrombotic microangiopathy. Peritubular capillaritis is also shown (yellow arrows). A higher magnification image of a periodic acid-Schiff-stained transplanted kidney section demonstrating endothelial swelling (yellow arrows) as well as mesangiolytic lesions (yellow asterisks, E) and a periodic acid-methenamine-silver-stained transplanted kidney section clearly showing duplication of the glomerular capillary walls (yellow arrows, F). Immunofluorescence staining showing weak deposition of immunoglobulin G (G) and complement C3 (H) in the glomeruli. Electron microscopy sections showing swelling of glomerular endothelial cells and double contours of the glomerular basement membrane (I) and widening of the subendothelial space (J). Immunoperoxidase staining for CD42b of the bone marrow tissue showing an increase in megakaryocytes (K). Silver staining of the bone marrow tissue demonstrating mild reticulin myelofibrosis (yellow arrows in L). (M) Atrophic germinal centers (yellow circles) with proliferation of the dilated capillaries in the interfollicular areas (yellow asterisks) were seen in the mildly enlarged lymph nodes (hematoxylin and eosin stain). A higher magnification image of the lymph node showing germinal centers penetrated by hyalinized vessels (hematoxylin and eosin stain, N).
On physical examination, his body temperature was 37.3°C and blood pressure was 132/73 mmHg. Abdominal distension, leg edema, and swelling of the submandibular lymph nodes were observed. His laboratory test results are summarized in Table 1. Urinalysis showed dysmorphic hematuria and nephrotic-range proteinuria. Decoy cells that suggest BK virus infection were never found in the repeated examination of his urine. Blood analysis showed severe thrombocytopenia (platelet count: 2.3 × 104/μL), but hemolytic anemia was absent (the level of lactate dehydrogenase was not elevated and that of haptoglobin was not decreased). Decrease in a disintegrin-like and metalloproteinase with thrombospondin type 1 motifs 13 (ADAMTS13) activity was not seen, and abnormalities of the blood coagulation system to suggest disseminated intravascular coagulation were scarce. Levels of transaminase were normal, but his alkaline phosphatase level was substantially increased. Immunoglobulin (Ig) levels were slightly decreased, but immunoelectrophoresis did not detect any monoclonal proteins. Levels of C-reactive protein and IL-6 were increased, but there were no signs of viral infection, including Epstein-Barr virus or cytomegalovirus, and levels of both complements C3 and C4 were within the normal range. The antinuclear antibody and disease-specific autoantibodies, such as the anti-dsDNA antibody and anti-neutrophil cytoplasmic antibody were negative, and donor-specific antibodies were not detected. Computed tomography displayed pericardial effusion, pleural effusion, massive ascites, retroperitoneal lymphadenopathy, and hepatosplenomegaly, but there was no evidence of malignant solid tumors or signs of infection.
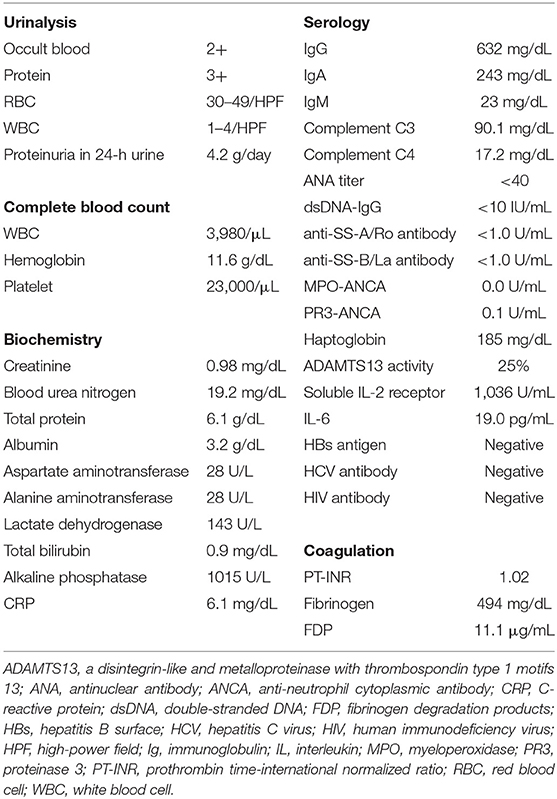
Table 1. Laboratory data of the patient.
After admission, his anasarca worsened, and his renal function rapidly deteriorated. Autoimmune disorders such as Sjogren's syndrome or anti-neutrophil cytoplasmic antibody-associated vasculitis were unlikely because antinuclear antibody and disease-specific autoantibodies were negative. There was no hemolytic anemia or hypocomplementemia, and atypical hemolytic uremic syndrome was also unlikely. Decrease in ADAMTS13 activity was absent, and the possibility of thrombotic thrombocytopenic purpura was excluded. Computed tomography scan did not show evidence of malignancies or infectious disorders. Thus, typical clinical features (thrombocytopenia, anasarca, systemic inflammation, organomegaly, and progressive renal dysfunction) and the exclusion of differential diagnoses led to the diagnosis of TAFRO syndrome. In addition, the clinical course was very similar to that prior to the kidney transplant. Two courses of intravenous methylprednisolone pulse therapy were performed, and the daily dose of steroids was increased, which temporarily resulted in the improvement of renal function and a decrease in C-reactive protein level. However, the patient remained in a condition of transfusion-dependent severe thrombocytopenia, and therefore the administration of romiplostim (a thrombopoietin receptor agonist) was initiated. In addition, five sessions of plasmapheresis using fresh frozen plasma were performed. Although the patient's platelet count increased to some extent by these treatments, it robustly decreased soon after the termination of plasmapheresis, followed by the deterioration of renal function and an increase in C-reactive protein level again. Although a third course of intravenous methylprednisolone pulse therapy was performed, the patient died on day 92 of hospitalization after complaining of chest discomfort.
Consent for autopsy was obtained from the patient's family. The transplanted kidney weighed 420 g, and microscopically showed thrombotic microangiopathy (TMA): there were prominent swelling of glomerular endothelial cells, mesangiolysis, and duplication of the glomerular capillary walls (Figures 1D–F). Thrombi were not found in the glomeruli or peritubular capillaries. Immunofluorescence staining showed focal deposition of Igs and complements (Figures 1G,H). Electron microscopy sections demonstrated glomerular endothelial swelling and widening of the subendothelial space, as well as double contours of the glomerular basement membrane (Figures 1I,J). The bone marrow showed mild hyperplasia with mild reticulin fibrosis, and there was an increase in megakaryocytes (Figures 1K,L). Several lymph nodes were mildly enlarged, and atrophic germinal centers with proliferation of the dilated capillaries in the interfollicular areas were observed (Figure 1M). Germinal centers were penetrated by hyalinized vessels, but neither atypical lymphocytes nor plasma cells were found (Figure 1N). These features of lymph nodes were compatible with hypervascular type of iMCD (10).
Other notable findings were as follows. There was massive ascites and pleural effusion. The spleen weighed 410 g, and showed features of congestion. There were no signs suggesting viral infection or acute cardiovascular events, such as myocardial infarction or aortic dissection. From these autopsy findings, a final diagnosis of TAFRO syndrome was made.
Discussion
To our knowledge, this is the first reported case of TAFRO syndrome developing in a kidney transplant recipient. Based on the recently published definition (6), the present case might be categorized into iMCD-TAFRO; however, the definition had not been clearly proposed when we treated the patient. We therefore diagnosed him simply as TAFRO syndrome according to the diagnostic criteria of the day (4). His clinical course is shown in Figure 2. Because the patient fulfilled the classification criteria of SLE (11, 12) at the time of disease onset, and because the disease concept of TAFRO syndrome had not yet been established, the exclusion of SLE was difficult at that time. However, we considered that SLE was unlikely because of the following reasons: (1) the titers of serum antinuclear antibody and anti-dsDNA antibody were 1:160 and 27.6 IU/mL, respectively, which were not so high, and both titers became negative in the next month and were consistently negative thereafter, (2) hypocomplementemia was never observed, and (3) renal biopsy did not show full-house immune deposits, but only few immune deposits. On the other hand, as summarized in Table 2, the clinical and pathological features observed both before and after the kidney transplant were similar, which suggested recurrent TAFRO syndrome. We performed an English literature search but could not find any previously reported cases of recurrent TAFRO syndrome. Thus, this case is highly suggestive, in which the patient's long-term clinical course and histological features, including changes in both the native and transplanted kidney were observed.
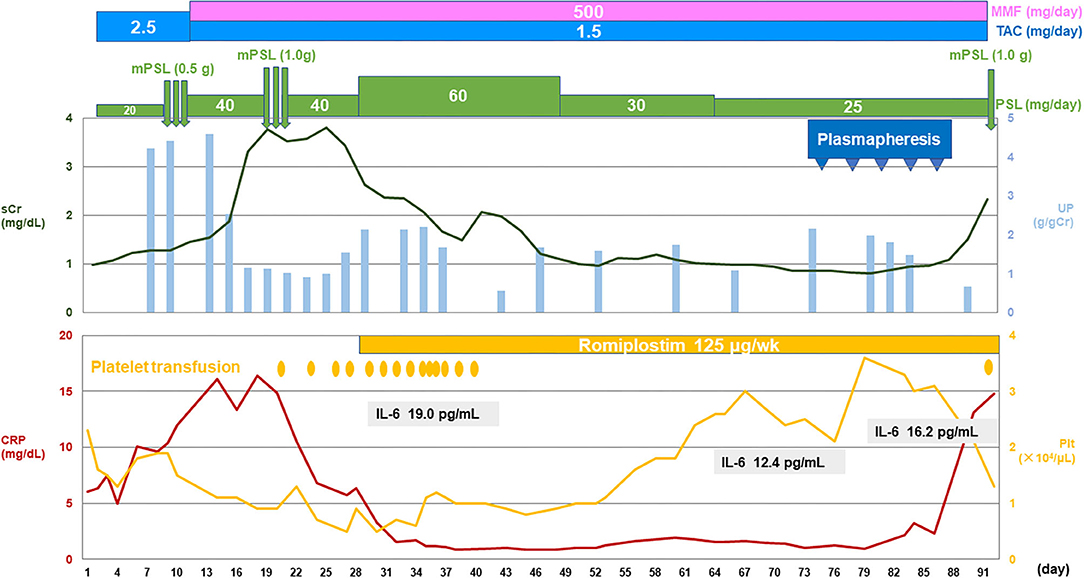
Figure 2. The patient's clinical course.CRP, C-reactive protein; IL, interleukin; MMF, mycophenolate mofetil; mPSL, methylprednisolone; Plt, platelet; sCr, serum creatinine; TAC, tacrolimus; UP, urinary protein.
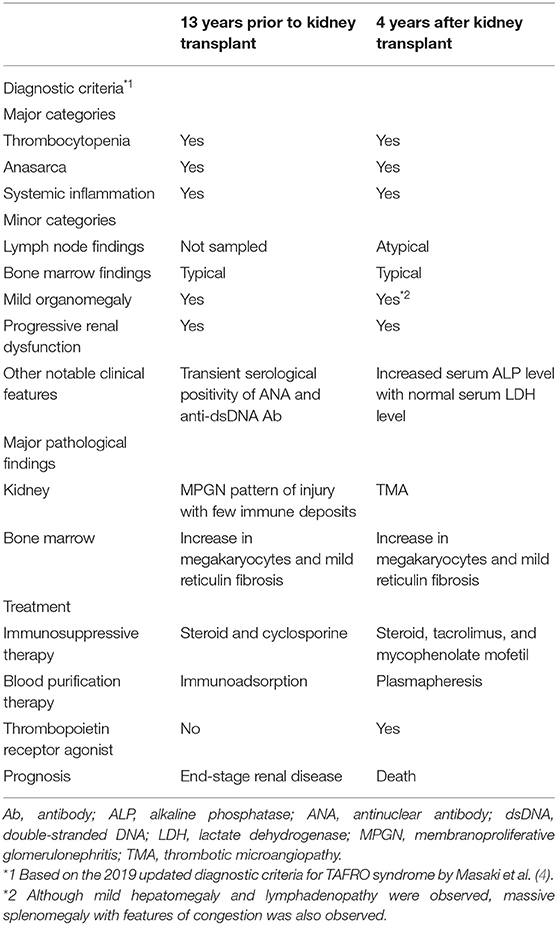
Table 2. Summary of clinical and pathological features of the patient.
Severe renal dysfunction is frequently observed in patients with TAFRO syndrome, and not a few patients require RRT. However, the discontinuation of RRT is reportedly possible in the majority of patients; Mizuno et al. (9) reported that hemodialysis was initiated in 4 out of 7 patients, but was discontinued in all of them. Leurs et al. (8) reported that nine out of 19 patients (including two of the patients reported by Mizuno et al.) required hemodialysis, but only one patient needed maintenance hemodialysis (14). Some types of bacterial infection are supposed to be involved in the pathogenesis of TAFRO syndrome (2, 13), and the recurrence of disease in this patient may also be triggered by infections. However, detailed examination of our patient did not show any evidence of infection. Another possible explanation for the recurrence is that the maintenance immunosuppressive therapy was too mild for the suppression of disease activity.
Light microscopic renal histology findings in patients with TAFRO syndrome have been mainly limited to two patterns: TMA-like glomerulopathy and MPGN-like glomerulopathy (3, 8). In this regard, it has been suggested that TMA-like lesions are seen in the acute phase and MPGN-like lesions are seen in the chronic phase as a result of the TMA-like lesions, and that both lesions are continuous renal pathological conditions in TAFRO syndrome, which may involve glomerular endothelial cell injury and macrophage infiltration (8, 15). In accordance with these previous reports, native kidney biopsy of the present patient showed MPGN pattern of injury with few immune deposits, whereas an autopsy specimen of the transplanted kidney, which was obtained in the acute phase of the disease, showed TMA (Table 2).
Most patients with TAFRO syndrome receive steroid therapy as the first-line treatment. Rituximab (an anti-CD20 antibody), tocilizumab (an anti-IL-6 receptor antibody), and cyclosporine are commonly used in steroid-resistant patients, and a recent study in Japan proposed that rituximab was a promising agent (16). In this regard, it would be reasonable to modify the therapeutic strategy according to the primary disease process, because the process is T cell-dominant in some patients but is possibly plasma cell-dominant in others. However, our patient had been treated with tacrolimus and mycophenolate mofetil in addition to steroids for many years after the kidney transplant, and hence the excessive immunosuppressed state that would be caused by the additional administration of rituximab or tocilizumab was of great concern. Plasmapheresis was performed for the rapid removal of humoral factors that are involved in disease pathogenesis (17). Indeed, the platelet count of our patient was increased to a certain extent, but it robustly decreased soon after termination of plasmapheresis.
In conclusion, we reported the first case of TAFRO syndrome in a kidney transplant recipient. This case suggests that its recurrence occurs many years after the kidney transplant. The further accumulation of cases is needed to investigate the appropriate treatment strategies, including RRT, long-term outcomes, and recurrence of this disorder.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the patient's family for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MN and TU: writing the manuscript draft. TO: critical manuscript revision. MN, TU, SK, KO, MM, HI, and MK: clinical care of the patient. TU, MY, HH, SK, and TO: histological evaluation. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Professor Yasuharu Sato at the Division of Pathophysiology, Okayama University Graduate School of Health Sciences, and Dr. Midori Wakiya at the Department of Diagnostic Pathology of Tokyo Medical University Hachioji Medical Center, for valuable advice on the pathological diagnosis of TAFRO syndrome, and Ms. Yoshie Wakatsuki at the Department of Pathology of Tokyo Medical University Hachioji Medical Center, for expert technical assistance with the histological analyses.
References
1. Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. (2013) 53:57–61. doi: 10.3960/jslrt.53.57
2. Iwaki N, Gion Y, Kondo E, Kawano M, Masunari T, Moro H, et al. Elevated serum interferon gamma-induced protein 10 kDa is associated with TAFRO syndrome. Sci Rep. (2017) 7:42316. doi: 10.1038/srep42316
3. Nagayama Y, Yamano M, Yagame M, Nariyama T, Takahashi M, Kawamoto M, et al. TAFRO syndrome as a cause of glomerular microangiopathy: a case report and literature review. BMC Nephrol. (2019) 20:375. doi: 10.1186/s12882-019-1574-9
4. Masaki Y, Kawabata H, Takai K, Tsukamoto N, Fujimoto S, Ishigaki Y, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol. (2020) 111:155–8. doi: 10.1007/s12185-019-02780-1
5. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood. (2020) 135:1353–64. doi: 10.1182/blood.2019000931
6. Nishimura Y, Fajgenbaum DC, Pierson SK, Iwaki N, Nishikori A, Kawano M, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. (2021) 96:1241–52. doi: 10.1002/ajh.26292
7. Masaki Y, Kawabata H, Takai K, Kojima M, Tsukamoto N, Ishigaki Y, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. (2016) 103:686–92. doi: 10.1007/s12185-016-1979-1
8. Leurs A, Gnemmi V, Lionet A, Renaud L, Gibier JB, Copin MC, et al. Renal pathologic findings in TAFRO syndrome: is there a continuum between thrombotic microangiopathy and membranoproliferative glomerulonephritis? Front Immunol. (2019) 10:1489. doi: 10.3389/fimmu.2019.01489
9. Mizuno H, Sawa N, Watanabe S, Ikuma D, Sekine A, Kawada M, et al. the clinical and histopathological feature of renal manifestation of TAFRO syndrome. Kidney Int Rep. (2020) 5:1172–9. doi: 10.1016/j.ekir.2020.05.004
10. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negatie/idiopathic multicentric Castleman disease. Blood. (2017) 129:1646–57. doi: 10.1182/blood-2016-10-746933
11. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1982) 25:1271–7. doi: 10.1002/art.1780251101
12. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1997) 40:1725. doi: 10.1002/art.1780400928
13. Kageyama C, Igawa T, Gion Y, Iwaki N, Tabata T, Tanaka T, et al. Hepatic campylobacter jejuni infection in patients with castleman-kojima disease (idiopathic multicentric castleman disease with thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly (TAFRO) syndrome). Pathol Int. (2019) 69:572–9. doi: 10.1111/pin.12856
14. Louis C, Vijgen S, Samii K, Chalandon Y, Terriou L, Launay D, et al. TAFRO Syndrome in caucasians: a case report and review of the literature. Front Med. (2017) 4:149. doi: 10.3389/fmed.2017.00149
15. Ito S, Uchida T, Itai H, Yamashiro A, Yamagata A, Matsubara H, et al. Serial manifestation of acute kidney injury and nephrotic syndrome in a patient with TAFRO syndrome. Intern Med. (2018) 57:3129–33. doi: 10.2169/internalmedicine.0806-18
16. Fujimoto S, Kawabata H, Sakai T, Yanagisawa H, Nishikori M, Nara K, et al. Optimal treatments for TAFRO syndrome: a retrospective surveillance study in Japan. Int J Hematol. (2020) 113:73–80. doi: 10.1007/s12185-020-03008-3
Keywords: autopsy, kidney transplant, membranoproliferative glomerulonephritis, TAFRO syndrome, thrombotic microangiopathy
Citation: Nagai M, Uchida T, Yamada M, Komatsu S, Ota K, Mukae M, Iwamoto H, Hirano H, Karube M, Kaname S and Oda T (2021) TAFRO Syndrome in a Kidney Transplant Recipient That Was Diagnosed on Autopsy: A Case Report. Front. Med. 8:747678. doi: 10.3389/fmed.2021.747678
Received: 26 July 2021; Accepted: 06 September 2021;
Published: 04 October 2021.
Edited by:
Sergey Brodsky, Ohio State University Hospital, United StatesReviewed by:
Yoshito Nishimura, Okayama University, JapanBeom Jin Lim, Yonsei University College of Medicine, South Korea
Copyright © 2021 Nagai, Uchida, Yamada, Komatsu, Ota, Mukae, Iwamoto, Hirano, Karube, Kaname and Oda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takahiro Uchida, dHUwNTA5MDIyNEBnbWFpbC5jb20=