 Silvia Stefania Longoni
Silvia Stefania Longoni Natalia Tiberti
Natalia Tiberti Zeno Bisoffi
Zeno Bisoffi Chiara Piubelli
Chiara Piubelli
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 12 October 2021
Sec. Infectious Diseases – Surveillance, Prevention and Treatment
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.745665
This article is part of the Research Topic Recent Advances in Recombinant Antibody Therapeutics and Diagnostics For Infectious Diseases View all 7 articles
Following the SARS-CoV-2 pandemic, several clinical trials have been approved for the investigation of the possible use of mAbs, supporting the potential of this technology as a therapeutic approach for infectious diseases. The first monoclonal antibody (mAb), Muromonab CD3, was introduced for the prevention of kidney transplant rejection more than 30 years ago; since then more than 100 mAbs have been approved for therapeutic purposes. Nonetheless, only four mAbs are currently employed for infectious diseases: Palivizumab, for the prevention of respiratory syncytial virus (RSV) infections, Raxibacumab and Obiltoxaximab, for the prophylaxis and treatment against anthrax toxin and Bezlotoxumab, for the prevention of Clostridium difficile recurrence. Protozoan infections are often neglected diseases for which effective and safe chemotherapies are generally missing. In this context, drug resistance and drug toxicity are two crucial problems. The recent advances in bioinformatics, parasite genomics, and biochemistry methodologies are contributing to better understand parasite biology, which is essential to guide the development of new therapies. In this review, we present the efforts that are being made in the evaluation of mAbs for the prevention or treatment of leishmaniasis, Chagas disease, malaria, and toxoplasmosis. Particular emphasis will be placed on the potential strengths and weaknesses of biological treatments in the control of these protozoan diseases that are still affecting hundreds of thousands of people worldwide.
The in vitro production of murine monoclonal antibodies (mAbs) was first described in 1975 by Kohler and Milstein, a discovery that earned them the Nobel Prize in 1985 and that revolutionized the clinical practice and biomedical research (1–3). Since then, mAbs have been engineered and stable cell lines able to secrete specific immunoglobulins against the target antigen of interest have been obtained (4). Nowadays there are more than 100 mAbs approved by the US Food and Drug Administration (FDA) (5, 6) and/or by the European Medicines Agency (EMA) (7), and they are classified into four types: murine (–omab), chimeric (–ximab), humanized (~95% human, –zumab), and human (–umab) (3), with the latter being the most successful in terms of tolerability and efficacy. Most of the approved mAbs are used in the field of oncology and immunology, while only a few are directed against infectious diseases, in particular against the respiratory syncytial virus (RSV) (Palivizumab), the anthrax toxin (Raxibacumab and Obiltoxaximab) and the bacterium Clostridium difficile (Bezlotoxumab), for which they are used either for prophylaxis or treatment (6, 7). A therapy using mAbs against protozoan infections is completely missing.
Eleven out of the 20 priority neglected tropical diseases (NTDs) included in the World Health Organization (WHO) portfolio are parasitosis (8). The drugs currently employed to treat these diseases are at least 50 years old, present several side effects and are not 100% efficient partly due to recurrent drug resistance (9–15).
The lack of mAb therapies for parasitosis is to a certain extent due to the neglected status of these diseases, lashing mainly low resource countries, and to high commercial costs of this technology.
In the context of protozoan diseases, two strategies can be followed for the development and use of mAbs. The first consists in the use of antibodies that target host antigens, mostly immune factors. Such a strategy allows modulating host immunity to achieve a more effective response for parasite elimination or at limiting damages due to hyper-inflammation. The main advantages of this type of approach are (i) the possibility of exploiting drug repurposing, thus using drugs already developed, tested in clinical trials, and approved; (ii) the therapeutic efficacy is not undermined by the development of resistance or by antigenic variability; (iii) they might be found particularly useful during chronic infections in which the host response contributes to the pathology. Nonetheless, this strategy requires an in-depth knowledge of the mechanisms of host-pathogen interaction and of immunomodulation, which in the vast majority of the cases are far from being deciphered.
Alternatively, mAbs targeting directly parasitic antigens can be employed to induce parasite elimination through different mechanisms including antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis, and complement-dependent cytotoxicity (16). The identification of the appropriate highly conserved targets for the development of such mAbs can however be cumbersome due to both the phenomenon of antigenic variation that characterizes most protozoa and variability between strains. Moreover, this strategy depends upon a wide knowledge of the parasite life cycle, biochemical processes, and adaptation mechanisms, which unfortunately is often limited.
With this review we intend to do revisit the state-of-the-art of mAb research for protozoan infections, summarizing the most relevant candidate therapeutics proposed and the different strategies. We will present how far research on this field has progressed, from in vitro and animal studies to clinical trials, and which are the main obstacles that have been encountered. In particular, we will deal with mAbs for leishmaniasis, Chagas disease, malaria, and toxoplasmosis, for which important experimental studies or clinical trials are ongoing, as summarized in Tables 1, 2. Possible strategies to overcome the current limits of this technology for the control of parasitic diseases in the context of human public health will also be discussed.
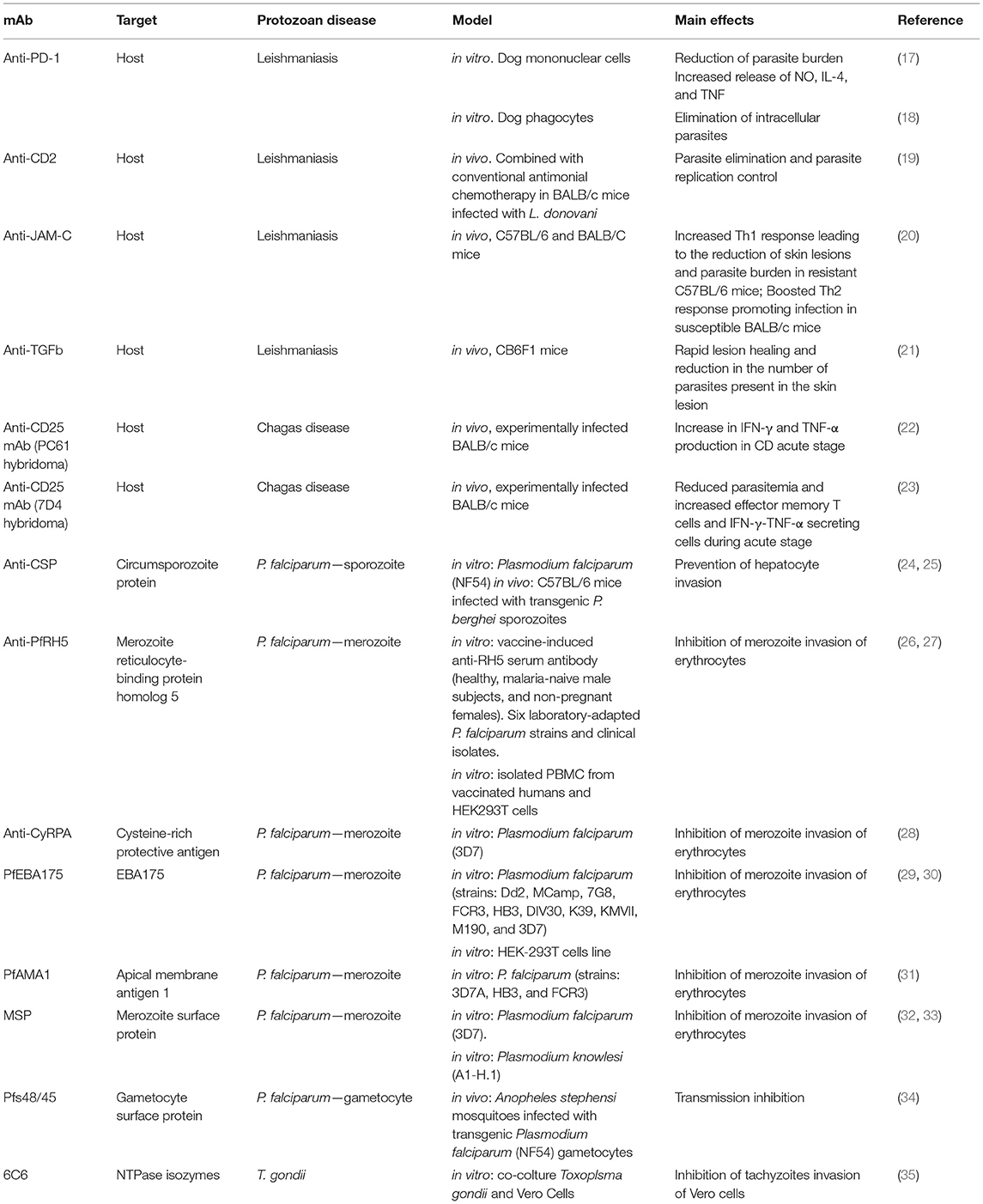
Table 1. mAb evaluated in in vitro and animal models for protozoan diseases treatment.

Table 2. mAb evaluated in clinical trials for protozoan diseases treatment.
The NTD leishmaniasis is endemic in 98 countries in the tropics, subtropics, and southern Europe areas (36), with more than 1 billion people being at risk of infection. Twelve million individuals are currently estimated to be infected and every year 2 million new cases are reported, of which 30,000 of visceral leishmaniasis (VL) and more than 1 million of cutaneous leishmaniasis (CL) (37–40). These data are however underestimated as case reporting is mandatory in only 32 countries (41). Leishmaniasis is caused by Leishmania spp., an obligate intracellular protozoan that specifically infects macrophages. More than 20 species of Leishmania are pathogenic for humans and are transmitted by the bite of a female sandfly. Depending on the species, the parasite can migrate to different organs: to the oropharyngeal cavity-causing mucocutaneous leishmaniasis (MCL), to the viscera causing VL also called Kala-Azar, or it can remain at the skin level causing CL. Leishmaniasis represents a serious public health problem particularly associated with poverty and is responsible for a significant socio-economic burden. VL is the most severe form of the disease since it is lethal if untreated. MCL is a very painful and disfiguring pathology, while CL can cause life-long scars and serious disability or stigma (38–40, 42).
The treatment of leishmaniasis depends on several factors including parasite species, type of disease, co-morbidities, and geographic location (40). Currently available drugs present, however, important drawbacks. Pentavalent antimonials (SbV), meglumine antimoniate (Glucantime®), and sodium stibogluconate (Pentostam®), have been the first-line treatment for leishmaniasis for over 100 years but they have been dismissed for the appearance of resistance and toxicity (43–49). The second-line drugs are pentamidine and amphotericin-B, which however are not optimal for their acute toxicity, high costs, and complex administration requiring hospitalization. Hopes were placed in Miltefosine, a third-line drug against Leishmania spp., for which resistance was however detected after only a decade of use (44, 46, 48, 49). Finally, Paromomycin, despite its low cost, requires parenteral administration and resistance has been reported as well (48, 49). Additional information about leishmaniasis treatment is available in the WHO technical report series 949, “Control of leishmaniasis” (50).
Leishmaniasis cure requires an immunocompetent system because available treatments are not able to eliminate the parasite from the body (40). Thus, drugs able to modulate the host immune response might be useful and, over the past years, attempts to find mAb therapies have been made, although they were mostly limited to in vitro experiments or animal models. An example is the mAb targeting PD-1 (programmed cell death protein 1), which has been approved by the EMA and FDA for different cancer types (51, 52). PD-1 expressed on T cells mediates macrophages differentiation into classically (M1) or alternatively (M2) activated, through its binding to PD-L1 receptor expressed on macrophages surface (53). When associated with its ligand PD-L1, PD-1 induces the deactivation or apoptosis of antigen-specific T lymphocytes, leading to the suppression of tumor necrosis factor α (TNF-α) secretion (17). Despite the role of the axis PD-1/PD-L1during Leishmania spp. infection is still unclear (53), it is known that the differentiation into M2 generates a favorable environment for amastigote survival while the differentiation into M1 leads to parasite death due to the activation of inducible nitric oxide synthase (iNOS) (54, 55).
Although based on pieces of evidence collected through preclinical experimental studies, anti-PD-1/PD-L1 mAbs have been suggested as a potential treatment for leishmaniasis (56). Indeed, PD-1 blocking through mAbs was shown, in vitro, to eliminate the intracellular parasites from phagocytes (18) or to reduce the parasite burden via the increase of NO, IL-4 and TNF-α in mononuclear cells (17), both isolated from naturally infected dogs. Recently, the first in vivo experiment performed in BALB/c mice infected subcutaneously with L. amazonensis showed a reduction in the parasite load in mice treated with anti-PD-1 and anti-PD-L1 mAb compared to untreated animals, while the development of cutaneous lesions seemed not to be affected by the biological treatment (56). The therapeutic effects of anti-PD1 or anti-PD-L1 mAb were also suggested to be potentiated by their combination with traditional drugs (56), although this hypothesis has yet to be confirmed.
Anti-CD2 mAb is another promising example of mAb for the control of VL. CD2 is an immuno-modulator and co-stimulatory molecule that induces the endogenous release of IFN-γ by Th1 cells. The combination of anti-CD2 mAbs with the conventional antimonial chemotherapy resulted in the elimination or the control of parasite replication in BALB/c mice inoculated with sensitive or drug-resistant L. donovani strains (19).
It has previously been demonstrated that TNF-α is necessary for the control of Leishmania spp. infection in a BALB/c mouse model (57). In CL and MCL patients, high levels of TNF-α correlated with major skin lesions (58), while asymptomatic subjects presented moderate levels of TNF-α and INF-γ (59). Following these observations, it was suggested to use anti-TNF-α agents in combination with standard therapy in patients with massive skin sores caused by CL (58). In 2018, Schwartz et al. (60) topically applied anti-TNF-α in combination with paromomycin on CL skin lesions in BALB/c mice infected with L. major. The combination therapy led to smaller size lesions while anti-TNF-α monotherapy did not affect lesion size.
mAb against junctional adhesion molecule C (JAM-C)—involved in leukocyte migration through the endothelium—were reported to increase the Th1 response leading to reduced skin lesions and parasite burden in resistant C57BL/6 mice, while it boosted the Th2 response promoting infection in susceptible BALB/c mice (20). These results agree with previous pieces of evidence on the inhibition of the protective Th1 response in resistant mouse strains following the administration of neutralizing antibodies against pro-inflammatory cytokines, such as IL-12 or IFN-γ, and the induction of resistance to L. major in susceptible mice following inhibition of Th2 cytokines (21). Anti-IL-4 antibody therapy in BALB/c mice was shown effective against chronic borderline leishmaniasis, leading to lesion reduction and a shift toward a Th1 immune response with anti-parasitic effects (61). A mAb against the regulatory cytokine transforming growth factor β (TGF-β) has been studied in intermediate susceptible CB6F1 mice infected with L. major and was shown to induce a rapid lesion healing and a reduction in the number of parasites present in the skin lesion (21).
While promising, to the best of our knowledge these studies have not gone further and the potential use of immunomodulatory mAbs to elicit a Th1 response in the human host has yet to be explored.
Despite several in vitro and in vivo studies, there is currently only one registered clinical trial (ClinicalTrials.gov Identifier: NCT01437020) (62) for the assessment of the safety and efficacy of anti-IL-10 mAb in combination with amphotericin-B for the treatment of visceral leishmaniasis. Unfortunately, the study was withdrawn due to the unavailability of the drug and no additional information is available.
Chagas disease (CD), or American Trypanosomiasis, is an NTD caused by Trypanosoma cruzi and widespread in several countries of Central and South America and in part of Mexico. About 7 million people are estimated to be affected by CD worldwide (63, 64). T. cruzi is transmitted by an invertebrate triatomine bug although outbreaks of orally-transmitted infections via the consumption of contaminated food have also been reported. Congenital transmission can also occur, while blood transfusions or organ transplants are less common transmission routes. After an initial acute stage with mild and unspecific symptoms, lasting for about 2 months, CD remains silent for several years and becomes chronic. In this stage, the protozoa reach the target organs and in 10–40% of infected subjects can generate cardiomyopathy or mega viscera such as hepatomegaly or splenomegaly (63–65). Despite the efforts to discover novel and safe drugs, anti-T. cruzi chemotherapy for both newborns and adults still relies on nifurtimox (NFX) and benznidazole (BZN), two drugs discovered in the '60s with known adverse side effects (64, 66). To improve CD treatment, strategies based on combinations of existing drugs or re-dosing regimens are currently being evaluated (67). Even though the excellent efficacy in reducing the parasitaemia when administered during the acute phase, CD treatment is less effective in preventing the clinical progression when given long time after the initial infection, suggesting a role for the host immune response (68). The mechanisms of interaction between T. cruzi, an obligate intracellular protozoa, and the host's immune system are however complex and far from being completely understood, and how the parasite resists, escapes or subverts the host's immune response to establish a chronic infection is still unclear (69).
As for Leishmania spp. first-line host defenses against T. cruzi depend on the Th1 or Th2 macrophage activation (70). Several studies suggest that the progression to the chronic stage, or its aggravation, might be associated with an immune dysregulation characterized by significantly increased tissue-infiltrating Th1 cells, raised IFN-γ/IL-10 ratio and diminished Treg (71, 72). In children successfully treated with BNZ or NFX it was observed a decline in pro-inflammatory cytokines and chemokines and in T cells expressing IFN-γ and IL-2 along with an increase in IL-7-expressing T cells (73).
The use of mAb to modulate the host immune response might thus have a huge impact on the treatment of chronic CD, although only a few preliminary studies using the murine model are available in the literature.
CD25+ Treg, characterized by the production of IL-10 and TGF-β, were reported to be involved in T. cruzi infection control, even though their specific role has yet to be completely defined (74). The depletion of CD25+ T cells in experimentally infected BALB/c mice using an anti-CD25 mAb, led to a slight increase in IFN-γ and TNF-α production in CD acute stage but not in the chronic stage (22). A different mAb against CD25 was shown to reduce parasitemia and to increase effector memory T cells and IFN-γ/TNF-α-secreting cells in mice when administered during the acute phase, while when administered at the beginning of the chronic phase it was shown to reduce the local inflammatory process in the heart. These results indicate a potential for anti-CD25 mAbs for the treatment of chronic CD and pave the way for more in depth investigations (23). A humanized anti-CD25 mAb, daclizumab, was widely used in the past for the treatment of multiple sclerosis but was withdrawn in 2018 by the EMA because of severe side effects (75). A chimeric anti-CD25 mAb, basiliximab, is currently employed to prevent transplant rejections (76, 77). The use of mAbs against CD25 in mice indicated that Treg inactivation during a T. cruzi infection can reduce the number of inflammatory cells, supporting the possibility of using non-depleting monoclonal antibodies as treatment of chronic CD in humans (22, 23).
The most recent WHO estimates reported 229 million malaria cases worldwide in 2019 and 409,000 deaths, about two thirds of which affecting children under the age of 5 (78). Although both malaria incidence and mortality have declined over the past 20 years, the burden associated with this disease remains high. Five Plasmodium species (P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi) are responsible for human infections and are transmitted by infected female Anopheles mosquitoes. Not surprisingly the majority of the therapeutic and preventive efforts tackle P. falciparum since it is responsible for most malaria cases, even though important issues of drug resistance have developed over the years.
Current recommendations foresee the use of artemisinin combination therapies (ACT) as first-line treatment for uncomplicated malaria infections and artesunate for severe malaria, although other less effective drugs such as quinine, chloroquine or other monotherapies are still available and employed in some circumstances (79). This treatment strategy has substantially contributed in reducing malaria morbidity and mortality over the past 15 years, even though resistance development remains a crucial problem threatening malaria control in both South-East Asia and in the African continent (79, 80).
The introduction of effective anti-malaria vaccines has thus become a priority in the fight against this parasite and numerous studies are ongoing to identify novel candidate antigens since the only vaccine currently available, RTS,S/AS01, has limited efficacy (81). In this context, mAbs are also being evaluated for the prevention or treatment of malaria. Compared to a classical vaccination strategy, mAbs have a relatively short-term effect, since IgG have a plasma half-life of approximately 20–25 days (82); consequently anti-parasitic mAbs might be found particularly useful as a preventive measure among high risk populations of severe and potentially fatal complications. Provided that they are proven to lack off-target reactions that could lead to improper immune activation or infectivity enhancement, mAbs against malaria might be used in children, pregnant women, patients with HIV/AIDS, and migrants or travelers with no previous exposure (83).
The first hints on the potential utility of antibodies to fight human malaria infection date back to the '60s when the efficacy of the passive transfer of immunoglobulin from hyper-immune sera to infected individuals was first assessed (84). Different aspects of Plasmodium spp. life cycle and malaria pathogenesis need however to be taken into account when considering mAbs as therapeutics or prophylaxis. First of all the mechanisms of host immunity to malaria are yet to be fully deciphered, especially concerning humoral immunity (85, 86). Acquired immunity develops after multiple exposures to the pathogen over the years and, although it mitigates the clinical aspects of the disease, it does not prevent re-infection. This might at least in part explain why, in the African continent, children under the age of 5 are more vulnerable and susceptible to develop clinical and severe forms, which in some cases can be fatal.
The second important aspect to take into account is P. falciparum complex life cycle, involving a vector stage and, within the human host, a liver-stage followed by asexual intra-erythrocytic replication (87). Consequently, different mAbs could potentially be designed in order to prevent (i) the infection, if targeting pre-erythrocytic stage (sporozoite); (ii) the clinical disease if targeting asexual erythrocytic stage; (iii) the transmission, if targeting sexual stage (gametocytes) that will not be able to replicate within the mosquito gut (88, 89). Different antigens and epitopes could thus be selected for mAb development, including proteins expressed by merozoites, on the surface of parasitized red blood cells (RBCs) or even by gametocytes. For the successful development of potent mAbs, it is thus essential to better understand the molecular basis of malaria pathogenesis, including erythrocyte invasion mechanisms as well as host immunity, in order to identify highly conserved antigens.
Circumsporozoite protein (CSP), is by far the most investigated target of mAbs for malaria. CSP is the most abundant antigen expressed on sporozoite surface and is involved in parasite attachment to and invasion of hepatocytes through its binding to heparin sulfate proteoglycans (90). The first mAbs against P. falciparum and P. vivax CSP were developed in the '80s and were shown to reduce parasite infectivity in vitro and in vivo (91, 92). Since then, a lot of research has focused on anti-CSP antibodies in order to highlight those with the highest affinity and best efficacy in preventing hepatocyte invasion, and to assess their protective properties (24, 25, 88). Thanks to the clinical trials for RTS,S vaccine, which is based on different portions of CSP, mAbs specific to CSP have become available relatively easily and this explains why mAbs anti-CSP are largely studied. Among anti-PfCSP mAbs, CIS43LS is currently undergoing a phase 1 (ClinicalTrials.gov Identifier: NCT04206332) (62) and a phase 2 clinical trial (ClinicalTrials.gov Identifier: NCT04329104) (62) to determine its safety, tolerability, and efficacy.
mAbs against other parasite stages, especially merozoites, have also been evaluated, although their investigation was more in the context of identifying novel vaccine candidates rather than on their direct use as therapeutics. These include mAbs against the merozoite reticulocyte-binding protein homolog 5 (PfRH5), for which a first vaccine has already been developed (26, 27), cysteine-rich protective antigen (PfCyRPA) (28), PfEBA175 (29, 30), apical membrane antigen 1 (PfAMA1) (31), merozoite surface proteins (MSP) (32, 33), all evaluated for their ability to block erythrocyte invasion being all these proteins involved in the interaction between merozoite and RBC.
The intra-erythrocytic stage can also be inhibited by targeting RBC antigens, as it occurs with meplazumab, a humanized anti-CD147 mAb that inhibits RBC invasion by blocking CD147 interaction with rhoptry-associated protein 2 (RAP2) (93). Preclinical studies have shown good tolerability and efficacy of this mAb which, thanks to its mode of action, could be used for both therapy and prophylaxis and will be further evaluated in a phase 1 clinical study (ClinicalTrials.gov Identifier: NCT04327310) (62). Targeting gametocytes specifically has also been proposed, since transmission-blocking antibodies are able to prevent Plasmodium spp. transmission from the human host to the vector. TB31F is a humanized version of the rat-derived mAb 85RF45.1 which targets Pfs48/45, a gametocyte surface protein involved in male gamete fertility (34). Indeed, antibodies against Pfs18/45 were reported to inhibit zygote development and thus parasite development within the mosquito (94). A phase 1 clinical trial has recently been completed, evaluating TB31F safety and tolerability in malaria naïve subjects. To the best of our knowledge, however, the results are yet to be published (ClinicalTrials.gov Identifier: NCT04238689) (62).
Toxoplasma gondii is a single-cell obligate intracellular parasite responsible for human toxoplasmosis. Felids are the definitive host of T. gondii, in which it completes its life cycle by sexual reproduction, while other warm-blooded animals, including humans, can become intermediate hosts (95–98). The most common infection route for humans is direct contact with cat feces or contaminated soil/water. Food-borne infection can however also occur, especially through the consumption of undercooked meat or contaminated raw fruits and vegetables not properly washed.
T. gondii is among the top 10 food-borne pathogens. One-third of the global population is considered to be infected with T. gondii (99), but the highest prevalence is found in South America (96, 99, 100).
In immunocompetent subjects, human toxoplasmosis is usually asymptomatic (96, 98, 101, 102), while in immuno-compromised individuals it can manifest as an acute, subacute or chronic disease. Clinical presentation can vary from flu-like symptoms to severe extensive lesions in vital organs such as lungs, liver, heart, brain or eyes (103). The infection can be particularly problematic when acquired during pregnancy as T. gondii can pass the placenta and infect the fetus resulting in preterm birth, eye and brain damages including visual and hearing loss, hydrocephaly and microcephaly, or even in fetal death (96–98, 101, 104). During the chronic stage, the parasite enters into a “sleeping” form, called bradyzoite, that forms cysts that colonize different tissues preferentially brain, muscles and eyes (96, 98, 103, 105). Chronic toxoplasmosis is resistant to anti-parasitic therapies and consequently can undergo reactivation/recrudescence (101).
Immune competent subjects recover spontaneously from asymptomatic T. gondii infection, while a combination of antimicrobial agents such as pyrimethamine and sulfadiazine can be administered to symptomatic individuals during the acute phase (98, 106). These drugs present very toxic side effects and cannot be used for a prolonged time. As indicated by the CDC, pregnant women, newborns, infants and immunocompromised patients can require discontinuation of therapy and, as a consequence, parasites will not be completely eliminated (98, 107). During the first decade of 2000, a mAb against nucleoside triphosphate hydrolase (NTPase) was investigated. NTPases are unique enzymes produced by T. gondii and released into the parasitophorous vacuoles (35); they are responsible for the hydrolysis of ATP to ADP and ADP to AMP and their activity leads to T. gondii tachyzoite intracellular survival and replication (108). NTPase activity was shown to be proportional to tachyzoite multiplication rate (109) and mAbs against NTPases were reported to inhibit in vitro the enzymatic activity in a dose-dependent manner in different T. gondii strains, limiting T. gondii invasion and replication within host cells (35, 110). The same enzyme was also evaluated as a candidate antigen for the development of a toxoplasmosis vaccine. Indeed, a “suicidal” DNA vaccine, later upgraded in self-amplifying RNA linked to lipid nanoparticle, based on NTPase-II sequence, was synthesized and shown, in mice, to elicit a Th1 immune response associated with a higher percentage of CD8+ T cells, increased levels of INF-γ, IL-2 and IL-10, and decreased IL-4. This vaccine partially reduced the rate of infection and mortality in both acute and chronic infections, however in-depth studies are yet to be performed (111, 112).
A number of different toxoplasma antigens have been proposed in the last years as candidates for the development of monoclonal antibodies or vaccines, although only preliminary studies have been performed so far. These include rhoptry kinase 18 of Toxoplasma gondii (TgROP18), a key virulence factor that promotes parasite proliferation for which species- and strain-specific mAbs have been obtained (113), and a synthetic peptide from surface antigen 1 (SAG1) (114).
To the best of our knowledge, preliminary experimental studies for the use of mAbs as therapeutics have also been performed for human trichomoniasis and human cryptosporidiosis, although the subject has not been addressed in depth. In the case of Trichomonas vaginalis, an extracellular parasite, it was demonstrated that the inhibition of parasite adhesion to epithelial cells through mAbs reduces parasite motility and protects from infection both in vitro and in the mouse model (115–117). The use of bovine hyper-immune colostrum or mAb against different Cryptosporidium spp. antigens, an apicomplexan protozoon like Plasmodium spp., was instead proposed in the '80s and the '90s to reduce disease severity in human cryptosporidiosis as summarized by Mead (118), although investigations were not pursued further.
A particular case is represented by Trypanosoma brucei spp., the causing agent of human African trypanosomiasis (HAT), for which classical mAbs have not been investigated. This extracellular protozoa has in fact evolved important mechanisms in order to survive in the human bloodstream, involving both immune escape and immune-suppression (119, 120), consequently, antibody-mediated responses were shown to have scarce efficacy in parasite elimination. This also explains why all attempts were undertaken so far to identify an anti-HAT vaccine have failed (121, 122). Nonetheless, an interesting alternative to classical mAb is represented by nanobodies (Nbs), small engineered antibody fragments derived from the heavy chain-only antibodies of camelids, characterized by high stability, specificity for their target, and tolerability (123). Their potential for therapeutic applications for HAT has been recently reviewed by Stijlemans et al. (124) and Nbs against variant surface glycoprotein (VSG) epitopes were reported to display trypanolytic effects in vitro and in vivo (125), supporting the potential of this novel technology.
During the COVID-19 pandemic, the anti-IL-6 mAb Tocilizumab (TCZ) has emerged as an effective treatment (126). Subsequently, 47D11, a human mAb that binds to cells expressing the viral spike protein, was shown to neutralize SARS-CoV-2 in vitro (127). At present, we count more than hundred clinical trials for SARS-CoV-2 treatment (62), for which a promising strategy to at least limit the spread of the virus could be represented by the use of cocktails of neutralizing antibodies (128). The great efforts put in place to fight this pandemic have led to rapid and effective results. Unfortunately, only a few clinical studies are currently ongoing to evaluate the use of mAbs for the treatment or prophylaxis for parasitic infections (Table 2). A number of reasons could explain the paucity of these clinical studies or the failure of many candidates at the pre-clinical level. First, most protozoan diseases are NTDs, affecting the poorest populations in low income countries. mAbs, mainly employed for the treatment of disorders affecting industrialized countries, especially cancers and auto-immune diseases, had an estimated market of about 98 billion USD in sales in 2018 (129), which is expected to increase of about 20% per year, reaching 138.6 billion USD in 2024 (129, 130). Although their use is now well-diffused, the costs of production are still very high and so is the price for the final customer, i.e., the patient or the health care system (131). Consequently, at the current state, it is not economically conceivable to employ such an expensive therapeutic strategy for the fight against NTDs. The case of malaria, which is not listed among NTDs, confirms this first point. More financial efforts have been applied to fight this disease (132, 133) and as a result is the only protozoan disease having several clinical trials ongoing for therapeutic use of mAbs.
Another important factor that could explain the difficulties associated with the introduction of mAb for protozoan diseases, is the complexity of the mechanisms of host-pathogen interaction. Despite great efforts to improve our knowledge of parasite biology and of the mechanisms of parasitism, many aspects are yet to be fully deciphered. Consequently, the translation of evidence collected through in vitro and in vivo experimental studies is often hardly translatable to the more complex human condition. Indeed, experimental mouse models can mimic only few aspects of human pathologies and in some cases different parasite strains need to be employed as human-infecting strains might not be pathogenic in mice (134–136). Another important consideration that needs to be done in the context of parasitic diseases, is the indirect effect that mAb employed for the treatment of other disorders might have on host susceptibility to infections or on the reactivation of latent ones as observed, for instance, for latent leishmaniasis (137) or latent Chagas disease (138).
In the future, one possibility would be to increase the scale production and the stability of the mAb products, in order to considerably decrease the costs (83). A promising alternative is represented by nanobodies, as previously mentioned for HAT. Besides their lower costs and smaller size, these tools have the advantage of being more stable, which make them attractive for disease prophylaxis. Their potential has been investigated not only for HAT; for instance, a single-chain fragment variable (scFv) against T. gondii SAG1, able to achieve a more rapid distribution and better tissue penetration compared to the whole antibody, was reported to efficiently reduce the number of oocyst and the mortality burden in a murine model of congenital toxoplasmosis (139). An engineered single-chain variable fragment derived from mAb-10D8 (scFv-10D8), was shown to adhere to T. cruzi surface and to reduce infectivity in pre-treated cultures, suggesting a potential use of such antibodies as preventive biological drugs (140).
Novel technologies and bioinformatics approaches should thus be exploited to achieve a more in depth knowledge of parasite biology and identify novel and highly specific targets to ad-hoc design mAbs. Such a strategy was in fact already found useful for malaria, for which a number of clinical studies evaluating anti-CSP mAb are currently ongoing.
Strategies based on drug repurposing, in order to employ mAb able to modulate host-immunity to limit damages due to the infection and hyper-inflammation, have also failed. Parasites are in fact well-known to develop effective mechanisms to escape host immune response. Nonetheless, a successful approach might consist in the combination of mAbs with classical anti-parasitic drugs as already proposed for leishmaniasis (19, 60).
In conclusion, mAbs are yet to be found useful in clinical practice for the treatment or prevention of protozoan infections. The main obstacles to the development of mAbs or Nbs therapies to parasitic diseases appear to be related to the production costs and the complexity of the host-pathogen interaction. Considering the rapidity of technology improvement in the development of biologicals, we can foresee that in the near future there would be the possibility for the application of these therapies also for parasitic infections, provided that the preclinical and clinical research can better define the host-parasite mechanisms and reveal the key targets for a specific mAbs or Nbs treatment.
SL: conceptualized the manuscript. SL and NT drafted the manuscript. SL, NT, ZB, and CP reviewed and edited the manuscript. All authors have read and agreed to the submitted version of the manuscript.
This work was supported by the Italian Ministry of Health Fondi Ricerca Corrente—L3P2 to IRCCS Sacro Cuore Don Calabria Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Andrea Fittipaldo for assistance in bibliographic search and Dr. Albert Schram for the English editing of the final version of the present manuscript.
2. Buss NA, Henderson SJ, Mcfarlane M, Shenton JM, De Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmocol. (2012) 12:615–22. doi: 10.1016/j.coph.2012.08.001
3. Bayer V. An overview of monoclonal antibodies. Semin Oncol Nurs. (2019) 35:150927. doi: 10.1016/j.soncn.2019.08.006
4. Casanova Estruch B. Safety profile and practical considerations of monoclonal antibody treatment. Neurol (English Ed). (2013) 28:169–78. doi: 10.1016/j.nrleng.2010.06.004
5. Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. (2021) 20:491–5. doi: 10.1038/d41573-021-00079-7
6. FDA. Available online at: https://purplebooksearch.fda.gov/ (accessed July 9, 2021).
7. EMA. No Title. Available at: https://www.ema.europa.eu/en/medicines [Accessed July 8, 2021]
8. WHO. WHO - NTDs. Available online at: https://www.who.int/teams/control-of-neglected-tropical-diseases (accessed July 7, 2021).
9. Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. (2019) 19:e338–e51. doi: 10.1016/S1473-3099(19)30261-0
10. Capela R, Moreira R, Lopes F. An overview of drug resistance in protozoal diseases. Int J Mol Sci. (2019) 20:5748. doi: 10.3390/ijms20225748
11. Aboelhadid SM, Arafa WM, El-Ashram S, Noaman AF, Shokier KA, Darwish AB, et al. Haemonchus contortus susceptibility and resistance to anthelmintics in naturally infected Egyptian sheep. Acta Parasitol. (2020) 66:329–35. doi: 10.1007/s11686-020-00284-1
12. Zheng Z, Chen J, Ma G, Satoskar AR, Li J. Integrative genomic, proteomic and phenotypic studies of Leishmania donovani strains revealed genetic features associated with virulence and antimony resistance. Parasit Vectors. (2020) 13:510. doi: 10.1186/s13071-020-04397-4
13. Emeto UE, Okolo CC, Nweze NE. Occurrence of Trypanosoma spp. and piroplasm infections of horses at Obollo-Afor southeastern Nigeria and resistance profiles of trypanosomes to isometamidium and diminazene salts. Trop Anim Health Prod. (2020) 52:3745–53. doi: 10.1007/s11250-020-02412-5
14. Fesseha H, Mathewos M, Kidanemariam F. Anthelmintic efficacy of strongyle nematodes to ivermectin and fenbendazole on working donkeys (Equus asinus) in and around Hosaena Town, Southern Ethiopia. Vet Med Int. (2020) 2020:1–7. doi: 10.1155/2020/4868797
15. Crespillo-Andújar C López-Vélez R Trigo E Norman F Díaz-Menéndez M Monge-Maillo B . Comparison of the toxicity of two treatment schemes with benznidazole for chronic Chagas disease: a prospective cohort study in two Spanish referral centres. Clin Microbiol Infect. (2020) 26:384.e1–e4. doi: 10.1016/j.cmi.2019.10.030
16. Monoclonal antibody targets and mechanisms of action. In: Strohl WR, LM. Strohl editors. Therapeutic Antibody Engineering. Elsevier (2012). p. 163–96, 459–595.
17. Chiku VM, Silva KLO, de Almeida BFM, Venturin GL, Leal AAC, de Martini CC, et al. PD-1 function in apoptosis of T lymphocytes in canine visceral leishmaniasis. Immunobiology. (2016) 221:879–88. doi: 10.1016/j.imbio.2016.03.007
18. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol. (2013) 191:5542–50. doi: 10.4049/jimmunol.1301810
19. Bimal S, Sinha S, Singh SK, Narayan S, Kumar V, Verma N, et al. Leishmania donovani: CD2 biased immune response skews the SAG mediated therapy for a predominant Th1 response in experimental infection. Exp Parasitol. (2012) 131:274–82. doi: 10.1016/j.exppara.2012.04.007
20. Ballet R, Emre Y, Jemelin S, Charmoy M, Tacchini-Cottier F, Imhof BA. Blocking junctional adhesion molecule C enhances dendritic cell migration and boosts the immune responses against Leishmania major. PLoS Pathog. (2014) 10:e1004550. doi: 10.1371/journal.ppat.1004550
21. Li J, Hunter CA, Farrell JP. Anti-TGF-beta treatment promotes rapid healing of Leishmania major infection in mice by enhancing in vivo nitric oxide production. J Immunol. (1999) 162:974–9.
22. Sales-Junior PA, Golgher D, Oliveira RV, Vieira V, Arantes RME, Lannes-Vieira J, et al. The regulatory CD4+CD25+ T cells have a limited role on pathogenesis of infection with Trypanosoma cruzi. Microbes Infect. (2008) 10:680–8. doi: 10.1016/j.micinf.2008.03.008
23. Nihei J, Cardillo F, dos Santos WLC, Pontes-de-Carvalho L, Mengel J. Administration of a nondepleting anti-CD25 monoclonal antibody reduces disease severity in mice infected with Trypanosoma cruzi. Eur J Microbiol Immunol. (2014) 4:128–37. doi: 10.1556/EuJMI.4.2014.2.6
24. Murugan R, Scally SW, Costa G, Mustafa G, Thai E, Decker T, et al. Evolution of protective human antibodies against Plasmodium falciparum circumsporozoite protein repeat motifs. Nat Med. (2020) 26:1135–45. doi: 10.1038/s41591-020-0881-9
25. Livingstone MC, Bitzer AA, Giri A, Luo K, Sankhala RS, Choe M, et al. in vitro and in vivo inhibition of malaria parasite infection by monoclonal antibodies against Plasmodium falciparum circumsporozoite protein (CSP). Sci Rep. (2021) 11:5318. doi: 10.1038/s41598-021-84622-x
26. Payne RO, Silk SE, Elias SC, Miura K, Diouf A, Galaway F, et al. Human vaccination against RH5 induces neutralizing antimalarial antibodies that inhibit RH5 invasion complex interactions. JCI Insight. (2017) 2:e96381. doi: 10.1172/jci.insight.96381
27. Alanine DGW, Quinkert D, Kumarasingha R, Mehmood S, Donnellan FR, Minkah NK, et al. Human antibodies that slow erythrocyte invasion potentiate malaria-neutralizing antibodies. Cell. (2019) 178:216–28.e21. doi: 10.1016/j.cell.2019.05.025
28. Chen L, Xu Y, Wong W, Thompson JK, Healer J, Goddard-Borger ED, et al. Structural basis for inhibition of erythrocyte invasion by antibodies to Plasmodium falciparum protein CyRPA. Elife. (2017) 6:e21347. doi: 10.7554/eLife.21347
29. Jiang L, Gaur D, Mu J, Zhou H, Long CA, Miller LH. Evidence for erythrocyte-binding antigen 175 as a component of a ligand-blocking blood-stage malaria vaccine. Proc Natl Acad Sci U S A. (2011) 108:7553–8. doi: 10.1073/pnas.1104050108
30. Chen E, Paing MM, Salinas N, Sim BKL, Tolia NH. Structural and functional basis for inhibition of erythrocyte invasion by antibodies that target Plasmodium falciparum EBA-175. PLoS Pathog. (2013) 9:e1003390. doi: 10.1371/journal.ppat.1003390
31. Maskus DJ, Królik M, Bethke S, Spiegel H, Kapelski S, Seidel M, et al. Characterization of a novel inhibitory human monoclonal antibody directed against Plasmodium falciparum apical membrane antigen 1. Sci Rep. (2016) 6:39462. doi: 10.1038/srep39462
32. Woehlbier U, Epp C, Hackett F, Blackman MJ, Bujard H. Antibodies against multiple merozoite surface antigens of the human malaria parasite Plasmodium falciparum inhibit parasite maturation and red blood cell invasion. Malar J. (2010) 9:77. doi: 10.1186/1475-2875-9-77
33. Han J-H, Cheng Y, Muh F, Ahmed MA, Cho J-S, Nyunt MH, et al. Inhibition of parasite invasion by monoclonal antibody against epidermal growth factor-like domain of Plasmodium vivax merozoite surface protein 1 paralog. Sci Rep. (2019) 9:3906. doi: 10.1038/s41598-019-40321-2
34. Kundu P, Semesi A, Jore MM, Morin MJ, Price VL, Liang A, et al. Structural delineation of potent transmission-blocking epitope I on malaria antigen Pfs48/45. Nat Commun. (2018) 9:4458. doi: 10.1038/s41467-018-06742-9
35. Kikuchi T, Furuta T, Kojima S. Membrane localization and demonstration of isoforms of nucleoside triphosphate hydrolase from Toxoplasma gondii. Parasitology. (2001) 122:15–23. doi: 10.1017/S0031182000007101
36. PAHO. Leishmanisis. Available online at: https://www.paho.org/en/topics/leishmaniasis (accessed October 19, 2020).
37. Morel N, Lassabe G, Elola S, Bondad M, Herrera S, Marí C, et al. A monoclonal antibody-based copro-ELISA kit for canine Echinococcosis to support the PAHO effort for hydatid disease control in South America. PLoS Negl Trop Dis. (2013) 7:e1967. doi: 10.1371/journal.pntd.0001967
38. CDC. Leishmanisis. Available online at: https://www.cdc.gov/parasites/leishmaniasis/index.html (accessed July 14, 2021).
39. Maxfield L, Crane JS. Leishmaniasis. Treasure Island, FL (2021). Available online at: http://www.ncbi.nlm.nih.gov/pubmed/30285351 (accessed July 18, 2021).
40. WHO. Leishmanisis. Available online at: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed July 13, 2021).
41. Tabbabi A. Review of Leishmaniasis in the Middle East and North Africa. Afr Health Sci. (2019) 19:1329. doi: 10.4314/ahs.v19i1.4
42. Okwor I, Uzonna J. Social and economic burden of human leishmaniasis. Am J Trop Med Hyg. (2016) 94:489–93. doi: 10.4269/ajtmh.15-0408
43. Nühs A, Schäfer C, Zander D, Trübe L, Tejera Nevado P, Schmidt S, et al. A novel marker, ARM58, confers antimony resistance to Leishmania spp. Int J Parasitol Drugs Drug Resist. (2014) 4:37–47. doi: 10.1016/j.ijpddr.2013.11.004
44. Ghorbani M, Farhoudi R. Leishmaniasis in humans: drug or vaccine therapy? Drug Des Devel Ther. (2018) 12:25–40. doi: 10.2147/DDDT.S146521
45. Carrió J, Portús M. in vitro susceptibility to pentavalent antimony in Leishmania infantum strains is not modified during in vitro or in vivo passages but is modified after host treatment with meglumine antimoniate. BMC Pharmacol. (2002) 2:11. doi: 10.1186/1471-2210-2-11
46. Ponte-Sucre A, Gamarro F, Dujardin JC, Barrett MP, López-Vélez R, García-Hernández R, et al. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl Trop Dis. (2017) 11:e0006052. doi: 10.1371/journal.pntd.0006052
47. Eddaikra N, Ait-Oudhia K, Kherrachi I, Oury B, Moulti-Mati F, Benikhlef R, et al. Antimony susceptibility of Leishmania isolates collected over a 30-year period in Algeria. PLoS Negl Trop Dis. (2018) 12:e0006310. doi: 10.1371/journal.pntd.0006310
48. Taslimi Y, Zahedifard F, Rafati S. Leishmaniasis and various immunotherapeutic approaches. Parasitology. (2018) 145:497–507. doi: 10.1017/S003118201600216X
49. Sundar S, Singh A. Chemotherapeutics of visceral leishmaniasis: present and future developments. Parasitology. (2018) 145:481–9. doi: 10.1017/S0031182017002116
50. Gonçalves LDR, Soares MR, Nogueira FCS, Garcia C, Camisasca DR, Domont G, et al. Comparative proteomic analysis of whole saliva from chronic periodontitis patients. J Proteomics. (2010) 73:1334–41. doi: 10.1016/j.jprot.2010.02.018
51. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, et al. PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy. J Cell Physiol. (2019) 234:16824–37. doi: 10.1002/jcp.28358
52. ACTIP. Monoclonal Antibodies Approved by the EMA and FDA for Therapeutic Use (status 2017). Available online at: https://www.actip.org/products/monoclonal-antibodies-approved-by-the-ema-and-fda-for-therapeutic-use/ (accessed July 8, 2021).
53. Freitas e Silva R de, Gálvez RI, Pereira VRA, de Brito MEF, Choy SL, Lotter H, et al. Programmed cell death ligand (PD-L)-1 contributes to the regulation of CD4+ T effector and regulatory T cells in cutaneous Leishmaniasis. Front Immunol. (2020) 11:574491. doi: 10.3389/fimmu.2020.574491
54. Balaña-Fouce R, Calvo-Álvarez E, Álvarez-Velilla R, Prada CF, Pérez-Pertejo Y, Reguera RM. Role of trypanosomatid's arginase in polyamine biosynthesis and pathogenesis. Mol Biochem Parasitol. (2012) 181:85–93. doi: 10.1016/j.molbiopara.2011.10.007
55. Ferreira C, Estaquier J, Silvestre R. Immune-metabolic interactions between Leishmania and macrophage host. Curr Opin Microbiol. (2021) 63:231–7. doi: 10.1016/j.mib.2021.07.012
56. da Fonseca-Martins AM, Ramos TD, Pratti JES, Firmino-Cruz L, Gomes DCO, Soong L, et al. Immunotherapy using anti-PD-1 and anti-PD-L1 in Leishmania amazonensis-infected BALB/c mice reduce parasite load. Sci Rep. (2019) 9:20275. doi: 10.1038/s41598-019-56336-8
57. Wilhelm P, Ritter U, Labbow S, Donhauser N, Röllinghoff M, Bogdan C, et al. Rapidly fatal Leishmaniasis in resistant C57BL/6 mice lacking TNF. J Immunol. (2001) 166:4012–19. doi: 10.4049/jimmunol.166.6.4012
58. Barral A, Barral-Netto M, Oliveira F, Cafe V, Rosato AB, Costa JM, et al. Lesion size correlates with Leishmania antigen-stimulated TNF-levels in human cutaneous Leishmaniasis. Am J Trop Med Hyg. (2011) 85:70–3. doi: 10.4269/ajtmh.2011.10-0680
59. Bacellar O, Lessa H, Schriefer A, Machado P, Ribeiro de Jesus A, Dutra WO, et al. Up-regulation of Th1-type responses in mucosal Leishmaniasis patients. Infect Immun. (2002) 70:6734–40. doi: 10.1128/IAI.70.12.6734-6740.2002
60. Schwartz J, Moreno E, Calvo A, Blanco L, Fernández-Rubio C, Sanmartín C, et al. Combination of paromomycin plus human anti-TNF-α antibodies to control the local inflammatory response in BALB/ mice with cutaneous leishmaniasis lesions. J Dermatol Sci. (2018) 92:78–88. doi: 10.1016/j.jdermsci.2018.07.005
61. Uzonna JE, Bretscher PA. Anti-IL-4 antibody therapy causes regression of chronic lesions caused by medium-dose Leishmania major infection in BALB/c mice. Eur J Immunol. (2001) 31:3175–84. doi: 10.1002/1521-4141(200111)31:11<3175::AID-IMMU3175>3.0.CO;2-L1
62. ClinicalTrials.gov. Available online at: https://clinicaltrials.gov/ct2/home (accessed July 7, 2021).
63. CDC. American Trypanosomiasis. Available online at: https://www.cdc.gov/parasites/chagas/index.html (accessed June 15, 2021).
64. WHO. Chagas Disease. Available online at: https://www.who.int/health-topics/chagas-disease#tab=tab_1 (accessed June 15, 2021).
65. Urbina JA. The long road towards a safe and effective treatment of chronic Chagas disease. Lancet Infect Dis. (2018) 18:363–5. doi: 10.1016/S1473-3099(17)30535-2
66. Ruiz-Lancheros E, Chatelain E, Ndao M. Chagas disease treatment efficacy biomarkers: myths and realities. In: Altcheh J, Freilij H, editors. Chagas Disease. Birkhäuser Advances in Infectious Diseases. Cham: Springer (2019) 323–49. doi: 10.1007/978-3-030-00054-7_16
67. Mazzeti AL, Capelari-Oliveira P, Bahia MT, Mosqueira VCF. Review on experimental treatment strategies against trypanosoma cruzi. J Exp Pharmacol. (2021) 13:409–32. doi: 10.2147/JEP.S267378
68. Cunha-Neto E, Chevillard C. Chagas disease cardiomyopathy: immunopathology and genetics. Mediators Inflamm. (2014) 2014:1–11. doi: 10.1155/2014/683230
69. Acevedo GR, Girard MC, Gómez KA. The unsolved jigsaw puzzle of the immune response in chagas disease. Front Immunol. (2018) 9:1929. doi: 10.3389/fimmu.2018.01929
70. Cerbán FM, Stempin CC, Volpini X, Carrera Silva EA, Gea S, Motran CC. Signaling pathways that regulate Trypanosoma cruzi infection and immune response. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165707. doi: 10.1016/j.bbadis.2020.165707
71. Llaguno M, da Silva MV, Batista LR, da Silva DAA, de Sousa RC, de Resende LAPR, et al. T-cell immunophenotyping and cytokine production analysis in patients with Chagas disease 4 years after benznidazole treatment. Infect Immun. (2019) 87:e00103–19. doi: 10.1128/IAI.00103-19
72. de Araújo FF, Vitelli-Avelar DM, Teixeira-Carvalho A, Renato Zuquim Antas P, Assis Silva Gomes J, Sathler-Avelar R, et al. Regulatory T cells phenotype in different clinical forms of Chagas' disease. PLoS Negl Trop Dis. (2011) 5:e992. doi: 10.1371/journal.pntd.0000992
73. Albareda MC, Natale MA, De Rissio AM, Fernandez M, Serjan A, Alvarez MG, et al. Distinct treatment outcomes of antiparasitic therapy in trypanosoma cruzi-infected children is associated with early changes in Cytokines, Chemokines, and T-Cell Phenotypes. Front Immunol. (2018) 9:1958. doi: 10.3389/fimmu.2018.01958
74. Fresno M, Gironès N. Regulatory lymphoid and myeloid cells determine the cardiac immunopathogenesis of Trypanosoma cruzi infection. Front Microbiol. (2018) 9:351. doi: 10.3389/fmicb.2018.00351
75. AIFA(2018). Available online at: https://www.aifa.gov.it/en/-/comunicazione-ema-su-zinbryta-daclizumab-beta-18-05-2018- (accessed June 21, 2021).
76. Ramirez CB, Marino IR. The role of basiliximab induction therapy in organ transplantation. Expert Opin Biol Ther. (2007) 7:137–48. doi: 10.1517/14712598.7.1.137
77. Kim HE, Paik HC, Jeong SJ, Park MS, Kim SY, Lee JG. Basiliximab induction with delayed calcineurin inhibitors for high-risk lung transplant candidates. Yonsei Med J. (2021) 62:164. doi: 10.3349/ymj.2021.62.2.164
78. WHO. World Malaria Report 2020:20 Years of Global Progress and Challenges. (2020). Available online at: https://www.who.int/teams/global-malaria-programme (accessed September 23, 2021).
79. Hanboonkunupakarn B, White NJ. Advances and roadblocks in the treatment of malaria. Br J Clin Pharmacol. (2020). doi: 10.1111/bcp.14474. [Epub ahead of print].
80. Rosenthal PJ. Has artemisinin resistance emerged in Africa? Lancet Infect Dis. (2021) 21:1056–7. doi: 10.1016/S1473-3099(21)00168-7
81. Laurens MB. RTS,S/AS01 vaccine (MosquirixTM): an overview. Hum Vaccin Immunother. (2020) 16:480–9. doi: 10.1080/21645515.2019.1669415
82. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. (2014) 11:20–33. doi: 10.7497/j.issn.2095-3941.2014.01.002
83. Macintyre F, Ramachandruni H, Burrows JN, Holm R, Thomas A, Möhrle JJ, et al. Injectable anti-malarials revisited: discovery and development of new agents to protect against malaria. Malar J. (2018) 17:402. doi: 10.1186/s12936-018-2549-1
84. Cohen S, McGregor IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. (1961) 192:733–7. doi: 10.1038/192733a0
85. Langhorne J, Ndungu FM, Sponaas A-M, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. (2008) 9:725–32. doi: 10.1038/ni.f.205
86. Rogers KJ, Vijay R, Butler NS. Anti-malarial humoral immunity: the long and short of it. Microbes Infect. (2021) 23:104807. doi: 10.1016/j.micinf.2021.104807
87. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. (2005) 5:722–35. doi: 10.1038/nri1686
88. Julien J-P, Wardemann H. Antibodies against Plasmodium falciparum malaria at the molecular level. Nat Rev Immunol. (2019) 19:761–75. doi: 10.1038/s41577-019-0209-5
89. Coelho CH, Jore MM, Canepa GE, Barillas-Mury C, Bousema T, Duffy PE. Antibody therapy goes to insects: monoclonal antibodies can block plasmodium transmission to mosquitoes. Trends Parasitol. (2020) 36:880–3. doi: 10.1016/j.pt.2020.08.009
90. Zhao J, Bhanot P, Hu J, Wang Q. A comprehensive analysis of Plasmodium circumsporozoite protein binding to hepatocytes. PLoS One. (2016) 11:e0161607. doi: 10.1371/journal.pone.0161607
91. Nardin EH, Nussenzweig V, Nussenzweig RS, Collins WE, Harinasuta KT, Tapchaisri P, et al. Circumsporozoite proteins of human malaria parasites Plasmodium falciparum and Plasmodium vivax. J Exp Med. (1982) 156:20–30. doi: 10.1084/jem.156.1.20
92. Zavala F, Cochrane AH, Nardin EH, Nussenzweig RS, Nussenzweig V. Circumsporozoite proteins of malaria parasites contain a single immunodominant region with two or more identical epitopes. J Exp Med. (1983) 157:1947–57. doi: 10.1084/jem.157.6.1947
93. Zhang M-Y, Zhang Y, Wu X-D, Zhang K, Lin P, Bian H-J, et al. Disrupting CD147-RAP2 interaction abrogates erythrocyte invasion by Plasmodium falciparum. Blood. (2018) 131:1111–21. doi: 10.1182/blood-2017-08-802918
94. van Dijk MR, Janse CJ, Thompson J, Waters AP, Braks JAM, Dodemont HJ, et al. A central role for P48/45 in malaria parasite male gamete fertility. Cell. (2001) 104:153–64. doi: 10.1016/S0092-8674(01)00199-4
95. Barrabes A, Duong TH, Reynouard F, Combescot C. [Experimental Schistosoma mansoni parasitosis in golden hamsters. Effects on the intensity of parasitism, and on the rate of antibodies' circulation, after administration of estrogen, testosterone, or progesterone (author's transl)]. Ann Parasitol Hum Comp. (1980) 55:671–7. doi: 10.1051/parasite/1980556671
96. Aguirre AA, Longcore T, Barbieri M, Dabritz H, Hill D, Klein PN, et al. The one health approach to toxoplasmosis: epidemiology, control, and prevention strategies. Ecohealth. (2019) 16:378–90. doi: 10.1007/s10393-019-01405-7
97. Kochanowsky JA, Koshy AA. Toxoplasma gondii. Curr Biol. (2018) 28:R770–R1. doi: 10.1016/j.cub.2018.05.035
98. CDC. Parasites - Toxoplasmosis (Toxoplasma infection). Available online at: https://www.cdc.gov/parasites/toxoplasmosis/index.html (accessed March 29, 2021).
99. Rostami A, Riahi SM, Gamble HR, Fakhri Y, Nourollahpour Shiadeh M, Danesh M, et al. Global prevalence of latent toxoplasmosis in pregnant women: a systematic review and meta-analysis. Clin Microbiol Infect. (2020) 26:673–83. doi: 10.1016/j.cmi.2020.01.008
100. Almeria S, Dubey JP. Foodborne transmission of Toxoplasma gondii infection in the last decade. An overview. Res Vet Sci. (2021) 135:371–85. doi: 10.1016/j.rvsc.2020.10.019
102. FAO. Parasites in Foods: An Invisible Threat. Bangkok: Food Safety Technical Toolkit for Asia and the Pacific (2021).
103. PARA-SITE. Available online at: https://parasite.org.au/para-site/introduction/index.html (accessed July 14, 2021).
104. Montoya JG, Remington JS. Management of Toxoplasma gondii infection during pregnancy. Clin Infect Dis. (2008) 47:554–66. doi: 10.1086/590149
105. Długońska H, Grzybowski MM. Functional exhaustion of T lymphocytes in chronic toxoplasmosis. Ann Parasitol. (2013) 59:109–12.
106. Dunay IR, Gajurel K, Dhakal R, Liesenfeld O, Montoya JG. Treatment of toxoplasmosis: historical perspective, animal models, and current clinical practice. Clin Microbiol Rev. (2018) 31:e00057–17. doi: 10.1128/CMR.00057-17
107. Konstantinovic N, Guegan H, Stäjner T, Belaz S, Robert-Gangneux F. Treatment of toxoplasmosis: current options and future perspectives. Food Waterborne Parasitol. (2019) 15:e00036. doi: 10.1016/j.fawpar.2019.e00036
108. Asai T, Miura S, Sibley LD, Okabayashi H, Takeuchi T. Biochemical and molecular characterization of nucleoside triphosphate hydrolase isozymes from the parasitic protozoan Toxoplasma gondii. J Biol Chem. (1995) 270:11391–7. doi: 10.1074/jbc.270.19.11391
109. Olias P, Sibley LD. Functional analysis of the role of Toxoplasma gondii nucleoside triphosphate hydrolases I and II in acute mouse virulence and immune suppression. Infect Immun. (2016) 84:1994–2001. doi: 10.1128/IAI.00077-16
110. Tan F, Hu X, Pan C-W, Ding J-Q, Chen X-G. Monoclonal antibodies against nucleoside triphosphate hydrolase-II can reduce the replication of Toxoplasma gondii. Parasitol Int. (2010) 59:141–6. doi: 10.1016/j.parint.2009.12.007
111. Zheng L, Hu Y, Hua Q, Luo F, Xie G, Li X, et al. Protective immune response in mice induced by a suicidal DNA vaccine encoding NTPase-II gene of Toxoplasma gondii. Acta Trop. (2017) 166:336–42. doi: 10.1016/j.actatropica.2016.12.004
112. Luo F, Zheng L, Hu Y, Liu S, Wang Y, Xiong Z, et al. Induction of protective immunity against Toxoplasma gondii in mice by nucleoside triphosphate hydrolase-II (NTPase-II) self-amplifying RNA vaccine encapsulated in lipid nanoparticle (LNP). Front Microbiol. (2017) 8:605. doi: 10.3389/fmicb.2017.00605
113. Zhang F, Su R, Han C, Wang Y, Li J, Zhu J, et al. Production and characterization of monoclonal antibodies against Toxoplasma gondii ROP18 with strain-specific reactivity. Parasitology. (2020) 147:940–8. doi: 10.1017/S0031182020000177
114. Naghi Vishteh M, Javadi Mamaghani A, Rashidi S, Seyyed Tabaei SJ, Jeddi-Tehrani M. Peptide-based monoclonal antibody production against SAG1 (P30) protein of Toxoplasma gondii. Monoclon Antib Immunodiagn Immunother. (2020) 39:51–6. doi: 10.1089/mab.2019.0041
115. Krieger JN, Torian BE, Hom J, Tam MR. Inhibition of Trichomonas vaginalis motility by monoclonal antibodies is associated with reduced adherence to HeLa cell monolayers. Infect Immun. (1990) 58:1634–9. doi: 10.1128/iai.58.6.1634-1639.1990
116. Hernández H, Marcet R, Figueredo M, Garrido N, Sarracent J. Comparative study of epitopes recognized by two monoclonal antibodies that protects mice against Trichomonas vaginalis challenge. Exp Parasitol. (2008) 118:583–6. doi: 10.1016/j.exppara.2007.10.005
117. Hernandez H, Sariego I, Garber G, Delgado R, Lopez O, Sarracent J. Monoclonal antibodies against a 62 kDa proteinase of Trichomonas vaginalis decrease parasite cytoadherence to epithelial cells and confer protection in mice. Parasite Immunol. (2004) 26:119–25. doi: 10.1111/j.0141-9838.2004.00690.x
118. Mead JR. Prospects for immunotherapy and vaccines against Cryptosporidium. Hum Vaccin Immunother. (2014) 10:1505–13. doi: 10.4161/hv.28485
119. Stijlemans B, Caljon G, Van Den Abbeele J, Van Ginderachter JA, Magez S, De Trez C. Immune evasion strategies of Trypanosoma brucei within the mammalian host: progression to pathogenicity. Front Immunol. (2016) 7:233. doi: 10.3389/fimmu.2016.00233
120. Onyilagha C, Uzonna JE. Host immune responses and immune evasion strategies in African Trypanosomiasis. Front Immunol. (2019) 10:2738. doi: 10.3389/fimmu.2019.02738
121. Magez S, Radwanska M. African trypanosomiasis and antibodies: implications for vaccination, therapy and diagnosis. Future Microbiol. (2009) 4:1075–87. doi: 10.2217/fmb.09.65
122. La Greca F, Magez S. Vaccination against trypanosomiasis. Hum Vaccin. (2011) 7:1225–33. doi: 10.4161/hv.7.11.18203
123. Steeland S, Vandenbroucke RE, Libert C. Nanobodies as therapeutics: big opportunities for small antibodies. Drug Discov Today. (2016) 21:1076–113. doi: 10.1016/j.drudis.2016.04.003
124. Stijlemans B, De Baetselier P, Caljon G, Van Den Abbeele J, Van Ginderachter JA, Magez S. Nanobodies as tools to understand, diagnose, and treat African Trypanosomiasis. Front Immunol. (2017) 8:724. doi: 10.3389/fimmu.2017.00724
125. Stijlemans B, Caljon G, Natesan SKA, Saerens D, Conrath K, Pérez-Morga D, et al. High affinity nanobodies against the Trypanosome brucei VSG are potent trypanolytic agents that block endocytosis. PLoS Pathog. (2011) 7:e1002072. doi: 10.1371/journal.ppat.1002072
126. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab treatment in COVID-19: a single center experience. J Med Virol. (2020) 92:814–8. doi: 10.1002/jmv.25801
127. Wang C, Li W, Drabek D, Okba NMA, van Haperen R, Osterhaus ADME, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. (2020) 11:2251. doi: 10.1038/s41467-020-16452-w
128. Chaqroun A, Hartard C, Schvoerer E. Anti-SARS-CoV-2 vaccines and monoclonal antibodies facing viral variants. Viruses. (2021) 13:1171. doi: 10.3390/v13061171
129. Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. (2019) 37:9–16. doi: 10.1016/j.tibtech.2018.05.014
130. Yang O, Qadan M, Ierapetritou M. Economic analysis of batch and continuous biopharmaceutical antibody production: A review. J Pharm Innov. (2020) 15:182–200. doi: 10.1007/s12247-018-09370-4
131. Hernandez I, Bott SW, Patel AS, Wolf CG, Hospodar AR, Sampathkumar S, et al. Pricing of monoclonal antibody therapies: higher if used for cancer? Am J Manag Care. (2018) 24:109–12.
132. Bill & Melinda Gates foundation. Malaria. Available online at: https://www.gatesfoundation.org/our-work/programs/global-health/malaria (accessed September 10, 2021).
133. RBM Partnership to End Malaria. Coordinated Action Towards Ending Malaria. Available online at: https://endmalaria.org/ (accessed September 10, 2021).
134. Mukhopadhyay D, Arranz-Solís D, Saeij JPJ. Influence of the host and parasite strain on the immune response during toxoplasma infection. Front Cell Infect Microbiol. (2020) 10:580425. doi: 10.3389/fcimb.2020.580425
135. Honoré S, Garin YJ-F, Sulahian A, Gangneux J-P, Derouin F. Influence of the host and parasite strain in a mouse model of visceral Leishmania infantum infection. FEMS Immunol Med Microbiol. (1998) 21:231–9. doi: 10.1111/j.1574-695X.1998.tb01170.x
136. Loria-Cervera EN, Andrade-Narvaez FJ. ANIMAL MODELS FOR THE STUDY OF LEISHMANIASIS IMMUNOLOGY. Rev Inst Med Trop São Paulo. (2014) 56:1–11. doi: 10.1590/S0036-46652014000100001
137. Català A, Roé E, Dalmau J, Pomar V, Muñoz C, Yelamos O, et al. Anti-tumour necrosis factor-induced visceral and cutaneous leishmaniasis: case report and review of the literature. Dermatology. (2015) 230:204–7. doi: 10.1159/000370238
138. Vacas AS, Gomez-Santana LV, Torre AC, Galimberti RL. Reactivation of Chagas-Mazza disease during treatment with infliximab. An Bras Dermatol. (2017) 92:899–900. doi: 10.1590/abd1806-4841.20177346
139. di Tommaso A, Juste MO, Lakhrif Z, Mévélec M-N, Borowczyk C, Hammeni P, et al. Engineering and functional evaluation of neutralizing antibody fragments against congenital toxoplasmosis. J Infect Dis. (2021) 224:705–14. doi: 10.1093/infdis/jiab141
Keywords: monoclonal antibody, toxoplasmosis, Chagas disease, malaria, leishmaniasis, protozoa
Citation: Longoni SS, Tiberti N, Bisoffi Z and Piubelli C (2021) Monoclonal Antibodies for Protozoan Infections: A Future Reality or a Utopic Idea? Front. Med. 8:745665. doi: 10.3389/fmed.2021.745665
Received: 22 July 2021; Accepted: 17 September 2021;
Published: 12 October 2021.
Edited by:
Livia Visai, University of Pavia, ItalyReviewed by:
Xiaojiong Jia, Harvard Medical School, United StatesCopyright © 2021 Longoni, Tiberti, Bisoffi and Piubelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Stefania Longoni, c2lsdmlhLmxvbmdvbmlAc2Fjcm9jdW9yZS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.