 Daniel L. Galvan
Daniel L. Galvan Koki Mise
Koki Mise Farhad R. Danesh
Farhad R. Danesh- 1Section of Nephrology, The University of Texas at MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Nephrology, Rheumatology, Endocrinology and Metabolism, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan
- 3Department of Pharmacology and Chemical Biology, Baylor College of Medicine, Houston, TX, United States
The role and nature of mitochondrial dysfunction in diabetic kidney disease (DKD) has been extensively studied. Yet, the molecular drivers of mitochondrial remodeling in DKD are poorly understood. Diabetic kidney cells exhibit a cascade of mitochondrial dysfunction ranging from changes in mitochondrial morphology to significant alterations in mitochondrial biogenesis, biosynthetic, bioenergetics and production of reactive oxygen species (ROS). How these changes individually or in aggregate contribute to progression of DKD remain to be fully elucidated. Nevertheless, because of the remarkable progress in our basic understanding of the role of mitochondrial biology and its dysfunction in DKD, there is great excitement on future targeted therapies based on improving mitochondrial function in DKD. This review will highlight the latest advances in understanding the nature of mitochondria dysfunction and its role in progression of DKD, and the development of mitochondrial targets that could be potentially used to prevent its progression.
Introduction
The kidney contains a great diversity of cell types in order to perform all of its endocrine and exocrine functions. Importantly, several different cell types in the kidney must act harmoniously in diverse microenvironments for the kidneys to function properly. An early indication as to the importance of mitochondria to the kidney function derives not only from their relative abundance in the kidney, but also the relative distribution of mitochondria specific to the needs and function of the cell type of the kidney with mitochondria-rich cells predominantly distributed in highly metabolically active proximal tubular cells, while podocytes and tubular epithelial cells of thin limb of Henle and collecting ducts exhibit comparatively a lower number of mitochondria (1–5).
Mitochondria are organelles with an endosymbiotic origin critical to proper function of eukaryotic cells. Central to the diverse functions of mitochondria are their bioenergetics properties serving as “powerhouses” of the cell generating adenosine triphosphate (ATP), as well as playing key roles in producing intermediates metabolites, reactive oxygen species (ROS) production, calcium homeostasis and apoptosis (Figure 1). As the most important physiological system for producing chemical energy stored as ATP from glucose, it is not surprising that mitochondria gained early attention as a possible target of diabetes and its micro/macrovascular complications.
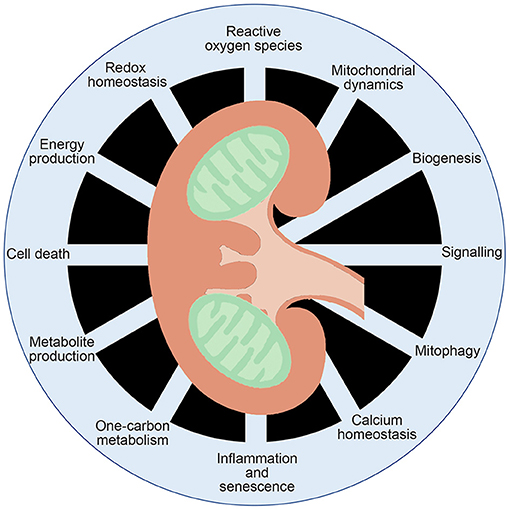
Figure 1. Multifaceted functions of mitochondrial function. Mitochondria are known to generate ATP and metabolites critical for signal transduction, as well as playing key roles in inflammation, calcium homeostasis, redox homeostasis and cell death.
The time course of mitochondrial dysfunction in the kidney has been documented in several experimental models of diabetic kidney disease (DKD) (6, 7). For instance, it was found that mitochondrial changes in size and function preceded histological and biochemical changes associated with kidney damage and these mitochondrial changes evolved with DKD progression (6). Indeed, altered mitochondrial morphology, bioenergetics and increased mitochondrial transition pore opening and ROS were all apparent prior to the presence of albuminuria (6, 8–12). These results suggest that mitochondrial dysfunction could be contributing to diabetic associated kidney damage.
Direct evidence that mitochondrial dysfunction can be a cause of chronic kidney disease (CKD) and DKD can also be gleaned by evaluating renal function in the presence of known mutations of mitochondrial associated proteins. The evidence is strengthened by several studies evaluating mutations in mitochondrial associated proteins that led to kidney dysfunction (13–21). Several independent mutations, relevant to mitochondrial function, result in kidney dysfunction, including prenyl diphosphate synthase subunit 2 (PDSS2) (22–24), mitochondrial inner membrane protein (Mpv17) (25), required for meiotic nuclear division 1 homology (RMND1) (26–30), ATP-binding cassette A1 (ABCA1) (12), apoptosis-inducing factor 1 (AIF1) (31), and several mitochondrial tRNAs (32–36). Podocyte-specific knockout of pdss2 further suggested the possible cell type specific consequences of some of these genes since it resulted in podocyte-associate renal disease. However, kidney damage was not apparent with conditional knockout of pdss2 in tubules, monocytes, or hepatocytes (22, 23). Podocyte knockout of ABCA1 was also shown to predispose the mice to DKD (12). Altogether, the evidence suggests that mitochondrial dysfunction can be a driving and primary cause of CKD and DKD, potentially playing an intrinsic and early role in disease progression. However, despite much interest, the precise nature of the changes to mitochondria and its physiological or pathophysiological significance remains elusive in DKD.
Mitochondrial Function and DKD Progression
Due to the diverse pathways ascribed to mitochondria, there is not a single means to determine their function nor single biochemical assay to define their “health.” However, due to their classically assigned and pivotal role in energy production, many investigators have evaluated alterations in mitochondrial respiratory complexes, oxygen consumption rates, and/or ATP production as “proxy” for mitochondrial dysfunction with DKD progression. The oxygen consumption rate (OCR) measurements in the early phase of DKD (1–4 weeks after diabetic induction) in animal models indicated that metabolic activity was increased in renal cortex and proximal tubular cells (37–40), but subsequently declined with progression of albuminuria in experimental models of DKD (41). This would seem to be consistent with reports of increased respiratory complex activities in early phases of DKD (42, 43). However, other studies report contrasting results indicating decreased mitochondrial respiratory complex activities likely representing later stages of DKD (44–48). Similarly, while ATP levels within the kidney cortex have frequently been found unchanged during progression of DKD (49, 50), significantly lower levels of ATP have also been reported in other studies (6, 51). Our interpretation of these studies is that these results may indicate a compensatory increase in mitochondrial respiration early in DKD which is lost during progression of DKD.
The observations on mitochondrial function during DKD progression focusing mainly on tubular cells seem to be in contrast to the glomerular region of kidney cortex. Since glomerular cells are not as mitochondrial rich as tubules, the reduction in mitochondrial densities may allow for enhanced metabolic plasticity in these cells. Indeed, a number of studies seem to indicate that glomerulus and specifically podocytes have decreased OCR or metabolic activity from early onset of DKD which persist with progression of DKD (9, 52–54). In support of these observations, other reports suggest that mitochondrial respiratory complex activity is also decreased early on with DKD (45, 55, 56). The effects on ATP levels, however, are less clear. While several reports seem to indicate that ATP in podocytes is decreased (6, 9, 52), others have reported no major or little change (49, 50). These paradoxical results are not unexpected since podocytes have been reported to readily utilize glycolysis, possibly exhibiting a more flexible approach to ATP production (3). The inherent tissue differences in mitochondrial number and function highlights some of the limitations in our ability to complete a wholistic picture.
While mitochondria have been clearly demonstrated to be important players in the development and progression of DKD, the intricacies and nature of their dysfunction is not fully understood (57, 58). We and others have reported enhanced mitochondrial fission, increased mitochondrial ROS, and decreased oxidative phosphorylation (OXPHOS) in mouse models of DKD, whereas others have reported conflicting results. It is unclear if differing reports are due to different means of diabetic induction in animal models, the renal cell type examined, species-specific differences, or time of observation in the disease process. It will be an important future goal to reach a consensus on these questions. We will further highlight some of the current knowledge and possible gaps in defining the nature of mitochondrial dysfunction in DKD.
Role of Mitochondrial Dynamics
Mitochondrial dynamics are the processes by which mitochondrial length, shape, and size are determined (59–61). Mitochondria have variable morphologies, even within the same cell, depending on the cell type, cellular needs and signaling cues. In its most basic and rudimentary understanding, mitochondrial morphology appears to be regulated by an ever changing and antagonistic intracellular balance between mitochondrial fission factors and mitochondrial fusion factors (62).
During mitochondrial fission, a mitochondrion is constricted to effectively divide a larger parent mitochondrion into smaller daughter organelles (59, 60). Mitochondrial fusion is the opposite process whereby smaller mitochondria have the outer and inner membranes joined to create a larger mitochondrion (59, 60). The balance between these factors of opposing action ultimately imparts characteristic size and shape of the mitochondria in a tissue-specific manner (Figure 2). Metabolic demands and signaling cues in a cell's microenvironment can push the balance toward mitochondrial fission to generate more fragmented and spherical mitochondria, or conversely toward mitochondrial fusion generating a more tubular and elongated morphology. Since this fluid process provides cells with rapidly responding metabolic flexibility, it is not surprising to realize that mitochondrial dynamics is highly regulated through a spatio-temporally precise cooperation among mitochondria, cytoskeleton, endoplasmic reticulum, and resident and recruited mitochondrial-associated proteins (11, 63–67).
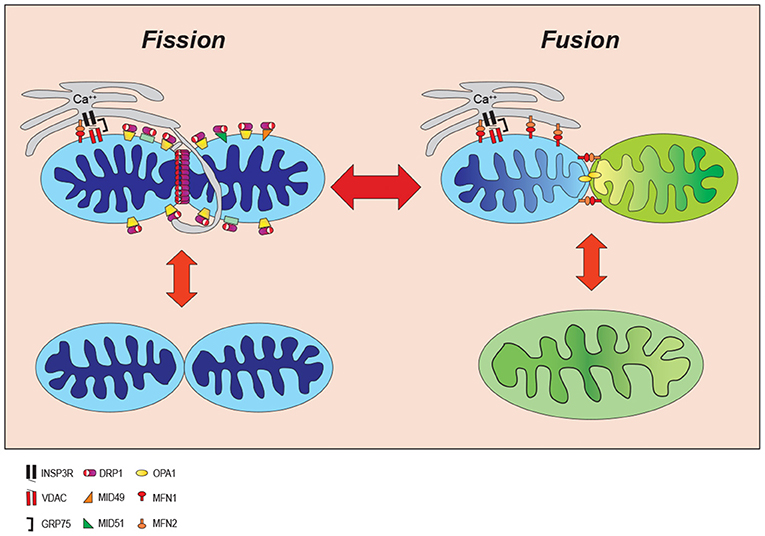
Figure 2. Mitochondrial dynamic. Mitochondria continuously change their size and shape by two opposing processes: mitochondrial fission and fusion. During the mitochondrial fission, mitochondria become fragmented in response to cell stress whereas they form an elongated shape increasing ATP production to adjust to cellular stresses. INSP3R, Inositol trisphosphate receptor; DRP1, dynamin-related protein 1; OPA1, optic atrophy 1; VDAC, voltage-dependent anion channel; MID49/51, mitochondrial dynamics proteins of 49 and 51kD; MFN1/2, mitofusin proteins 1 and 2; GRP75, glucose-regulated protein 75.
While mitochondrial fission can be viewed as a process of sequential discrete steps, the order and independence of each step remains to be fully understood. An early step is marking of the site where the mitochondria will divide. The current model suggests that the endoplasmic reticulum (ER) initially marks fission furrows in the mitochondria where mitochondrial fission will ultimately occur (65, 66, 68). Increases in cytoplasmic calcium drive actin assembly around the ER protein inverted formin 2 (INF2) and the actin polymerization is believed to provide some force for constriction (64, 67). The association of INF2 with mitochondrial localized Spire 1C, links the mitochondrial, actin polymerization event between the two organelles (mitochondria and ER) (69) and will enhance calcium transfer from ER to mitochondria via mitochondrial calcium uniporter 1 (MCU1) initiating constriction of the mitochondrial inner membrane prior to outer membrane constriction in a process which requires activation of the electron transport complexes and the mitochondrial metalloendopeptidase, OMA1 (70). Mitochondrial fission will further proceed by recruitment of the cytoplasmic fission factor, dynamin-related protein 1 (DRP1) to the outer mitochondrial membrane (71–74). DRP1 is recruited to the mitochondrial outer membrane where it oligomerizes to form a ring around the mitochondria at the fission furrow. DRP1 is anchored to the mitochondria by interactions with its mitochondrial receptors including mitochondrial fission 1 (FIS1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins of 49 and 51 kD (MiD49/MiD51). Constriction of the mitochondrial membrane utilizes DRP1-driven GTP-hydrolysis for energy to drive mitochondrial fission.
DRP1 translocation to the mitochondria is further regulated by several posttranslational modifications including phosphorylation (52, 75–80), O-GlcNAcetylation (81), sumoylation (82–84), and S-nitrosylation (85–87). DRP1 activation is also enhanced by binding with actin (88), actin-related proteins (11, 89), AKAP1 (80), cardiolipin, and palmitic acid (90–92).
Mitochondrial fusion, on the other hand, is mediated by another dynamin related protein, optic atrophy 1 (OPA1), at the mitochondrial inner membrane and mitofusin proteins 1 and 2 (MFN1 and 2) at the outer mitochondrial membrane. MFN1/2 can interact both as homo- and hetero-dimers to mediate fusion of the outer mitochondrial membrane. OPA1 appears to be regulated in part by post-translational changes driven by the mitochondrial membrane potential and interactions with the mitochondrial OMA1 zinc metallopeptidase. In addition, to its function in mitochondrial fusion, OPA1 also plays a key role in maintaining mitochondrial cristae morphology and respiratory ETC function by sequestering cytochrome c within the cristae. The importance of these mitochondrial dynamic protein factors to life is evidenced by findings that knockout of several member proteins is embryologically lethal (93–96).
Enhanced mitochondrial fission is reported in multiple cell types of the kidney including tubules and podocytes in animal models of DKD (1, 11, 52, 97–103). In support of these preclinical studies, clinical evidence have revealed increased fragmented mitochondria in several cell types within the kidney cortex of diabetic patients as well (99, 104, 105). Our studies in the db/db model of DKD identified enhanced mitochondrial fission and increased expression of DRP1 in both glomerular endothelial cells and podocytes (8). Importantly, while podocyte-specific depletion of DRP1 had no effect on mitochondrial function, DRP1 deficiency specifically in podocytes in diabetic db/db mice improved DKD progression by improving mitochondrial function suggesting a role for cellular stress to unravel the effect of DRP1 on mitochondrial function (52). The tendency toward mitochondrially fragmented morphology has been tied most strongly to several proteins regulating mitochondrial fission (11, 52, 74, 80, 100, 106, 107). Other studies have confirmed these initial observations in other models of DKD. For example, Myo-inositol oxygenase (MIOX) expression was shown to be increased in kidneys of db/db mice and streptozotocin (STZ)-treated diabetic mice contributing to progression of DKD, and linked to enhanced DRP1 and FIS1 expression with decreased MFN2 expression (98, 108). The Src homologous-collagen homolog adaptor protein, p66Shc, expression and phosphorylation were also increased in kidneys of both db/db mice and STZ-treated diabetic mice, and were found to correlate with increased DRP1 and FIS1 expression and decreased MFN1 expression (99, 109). Knockdown of Fis1 prevented mitochondrial fragmentation, restored MFN1 expression, and reduced p66Src binding to FIS1 under high glucose conditions (99). Dual-specificity protein phosphatase−1 (DUSP1) was shown to be decreased and JNK pathway activation increased in the kidneys of STZ diabetic mice and linked to increased DRP1 and MFF expression with decreased MFN1 and OPA1 expression (101). Finally, the expression of hypoxia inducible factor 1 (HIF1) was conditionally deleted in proximal tubular cells of STZ treated diabetic mice showed enhanced DKD progression with increased expression of DRP1 and FIS1 with decreased MFN1 expression. In vitro it was suggested that HIF1 modulates these changes by its target heme oxygenase-1 (HO-1) (110).
Post-translational modifications of DRP1 and specifically its phosphorylation also seem to play a critical role in pathogenesis of DKD. We and others have found that DRP1 phosphorylation at the human residue S637 and equivalent mouse residue serine 600 (S600 in mouse DRP1 isoform b), hereafter referred to as S600, enhances DRP1 activity and translocation to the mitochondria to mediate enhanced mitochondrial fragmentation (8). We have shown that Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) activation in the diabetic kidney phosphorylates DRP1 at S600 both in vivo and in vitro triggering mitochondrial fragmentation (8). Recently, it was shown that S600 of DRP1 in renal tubules maybe phosphorylated by the compartment directing, A kinase (PRKA) anchor protein 1 (AKAP1), localizing protein kinase A (PKA) to the outer mitochondrial membrane and triggering mitochondrial fragmentation in a STZ model of type-1 diabetes (80). We also provided in vivo evidence indicating that knock-in diabetic db/db mice mutating S600 in DRP1 to the non-phosphorylable alanine at position 600 (S600A) exhibited marked improvement in DKD progression and protected mitochondrial morphology and bioenergetics of podocytes. Mechanistically, it was shown that phosphorylation of DRP1 at S600 enhanced its interaction with both MFF and the actin related protein 2/3 complex (ARP2/3) enhancing mitochondrial localization of DRP1 and triggering mitochondrial fission (11). Similarly, it has been reported that phosphorylated DRP1 was increased and the expression of MFN1 markedly decreased in proximal tubular cells isolated from db/db mice, while treatment of diabetic mice with a β2-agonist, formoterol, decreased phosphorylated DRP1 levels and restores MFN1 levels (51).
While there is a growing body of evidence indicating that mitochondrial fission is a key morphological indicator of kidney damage in DKD, hyperfused and large mitochondria may also have a role in DKD progression (111). Hyper-elongated mitochondria may be an indicator of cellular senescence and associated with mitochondrial DNA damage, loss of mitochondrial membrane potential, and enhanced ROS as well (112–115).
Overall, the evidence seems to indicate that renal damage in DKD is associated with a shift in mitochondrial dynamics toward enhanced fission. The evidence is clear that DRP1 lies at the center of this dynamic and has been found to be increased and/or modified in multiple kidney cell types. These changes are frequently found in conjunction with increased expression of fission proteins such as FIS1 and MFF and decreased expression of MFN1. The functional consequences of tipping the mitochondrial dynamic balance toward fission seem to share deleterious end points such as enhanced ROS contributing to DKD progression.
Mitochondrial Bioenergetics and Oxidative Stress
Cellular ATP is maintained through two interconnected metabolic pathways, glycolysis and oxidative phosphorylation (OXPHOS). During glycolysis, glucose is transported into the cell cytoplasm and converted into 2 molecules of pyruvate to generate 2 ATP molecules. In the absence of oxygen, glycolysis will anaerobically ferment the pyruvate to lactate generating 2 NADH in the cytoplasm. However, in the presence of oxygen, pyruvate will be decarboxylated into acetyl coenzyme A (AcCoA) inside mitochondrial matrix and enter the tricarboxylic acid (TCA) cycle. The TCA cycle is an enzymatically controlled series of oxidation steps culminating in production of CO2 and 8 NADH, 2 FADH2, and 2 ATP molecules. Ultimately OXPHOS can harvest 30–36 ATP from the entry of the NADH and FADH2 per glucose depending upon the amount of proton leak.
OXPHOS is comprised of 4 respiratory complexes (I-IV) within the inner mitochondrial membrane which are collectively referred to as the electron transport chain (ETC) in which a series of redox reactions are converted into a proton motive force by pumping protons into the mitochondrial intermembrane space (Figure 3). Complex I accepts electrons from NADH while complex II accepts electrons from FADH2 and both transfer the electron to coenzyme Q (CoQ). Complex III in conjunction with cytochrome c can accept these electrons and pass them to complex IV with oxygen as the terminal electron acceptor. Complex V or ATP synthase allows passage of protons back to the matrix linked to generation of ATP. Electron escape during ETC reactions is capable of generating ROS which under physiological conditions are both quenched by endogenous antioxidant mechanisms and utilized as important cellular signaling molecules. It has been suggested that increased ROS generation and decreased ROS quenching result in oxidative damage to cellular components and mitochondria capable of resulting in cell death. This apparent paradox may exist with low levels of ROS serving as survival signaling during conditions of stress while once reaching a threshold become damaging to the cell and synergistically contribute to enhanced mitochondrial dysfunction.
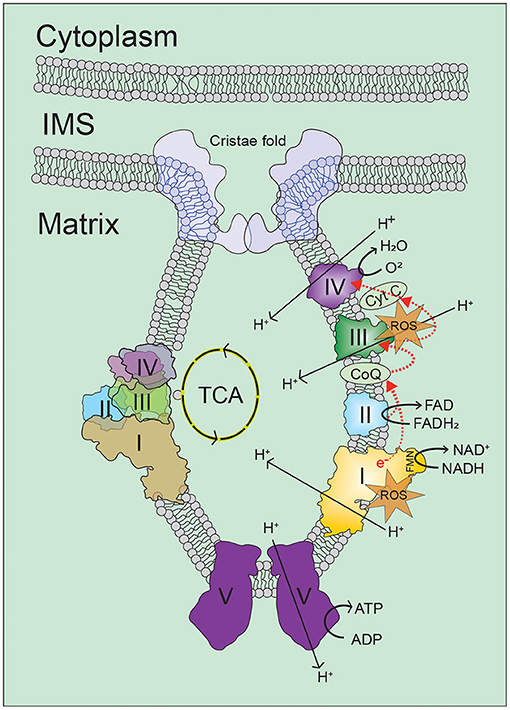
Figure 3. Oxidative phosphorylation. Cellular energy in the form of ATP is mainly generated in mitochondria by the oxidative phosphorylation (OXPHOS) process, in which electrons on the inner-membrane of the mitochondria are passed through a series of mitochondrial complexes (Complexes I-V) in redox reactions. Energy released in these reactions is then coupled to ATP generation. Increase in intracellular levels of NADH and FADH2 drives oxidative phosphorylation, which leads to increase of oxygen consumption and ATP production by ATP synthesis. For more information, refer to the main text. ROS, reactive oxygen species; TCA, tricarboxylic acid; I/II/III/IV/V, mitochondrial respiratory complex I/II/III/IV/V; NADH, reduced nicotinamide adenine dinucleotide; NAD, nicotinamide adenine dinucleotide; FADH2, reduced flavin adenine dinucleotide; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; CoQ, coenzyme Q; Cyt C, cytochrome C.
Superoxide production during ETC transport was first reported in 1966 (116), and has been an area of interest ever since. Complex I (117–120) and complex III (118, 121–124) are believed to be the principle sites of mitochondrial ROS generation during ETC transport, of which complex I is believed to produce the majority of mitochondrial ROS (122, 125–127). Complex III can produce both intermembranous and matrix superoxide during transport of electrons through the quinol (Q)-cycle depending on the mitochondrial membrane potential and oxidation state of cytochrome c (122, 123). Complex I can produce superoxide by two distinguishable mechanisms. When the NADH/NAD+ ratio is high and respiratory chain activity is inhibited, the matrix facing flavin mononucleotide (FMN) site can produce superoxide (119, 128–130). Alternatively, superoxide can be generated when mitochondrial potential drives reverse electron transport at complex I. Reverse transport occurs when the mitochondrial potential is high and CoQ is reduced forcing the reduction of NAD to NADH at the flavin mononucleotide (FMN) site (121, 131–133). In DKD, it has been demonstrated that both complex I (134) and complex III (135) can generate superoxide and increased mitochondrial ROS in the kidney (136–138). Transgenic expression of superoxide dismutase or thioredoxin protected the kidney in mouse models of DKD (139, 140). However, not all antioxidants were equally effective as transgenic glutathione peroxidase-1 expression in STZ-treated mice did not have renal protection (141).
Substantial evidence has accumulated in patients and animal models of DKD indicating that mitochondrial ROS is significantly increased in the kidney and generated the free radical theory of diabetic microvascular complications (142–144). The “Unifying Hypothesis” suggests that chronically driven glucose over production of mitochondrial ROS at the mitochondria leads to cellular and eventual end kidney failure. Increased mitochondrial ROS production has been demonstrated both in vitro and in vivo in multiple mouse models of DKD (39, 41, 56, 100, 109, 145–148). However, an important gap in our current understanding of the role of mitochondrial ROS in DKD pathogenesis is to identify the source(s) of enhanced mitochondrial ROS in DKD. The increased mitochondrial ROS production was initially proposed has been proposed to be linked to mitochondrial dynamics remodeling and biogenesis (149). This suggestion was supported experimentally by some recent studies indicating that overexpression of DRP1 or MFF, as well as knockdown of MFN1/2 together or alone in cultured cells, lead to mitochondrial fragmentation and increased mitochondrial ROS (149–154). Increased expression of p66Shc, NR4A1, ROCK1/DRP1, and HIF1 (hypoxia inducible factor 1) in DKD also caused fragmented mitochondria and increased mitochondrial ROS and apoptosis (8, 98, 99, 101, 110, 155). Decreased expression of DUSP1, MIOX, or PGC1α in the DKD were similarly reported to increase mitochondrial ROS and apoptosis (98, 101, 145). Increased production of mitochondrial ROS appears to be a central effector of cellular damage, but is inherently difficult to measure mitochondrial ROS in vivo due to their multiple species and frequently very short biological half-lives. Indeed, a central challenge in addressing the role of redox biology in DKD progression is to accurately measure mitochondrial ROS. Importantly, studies addressing mitochondrial ROS have resulted in conflicting interpretations mainly because of variations in the detection methods employed with a wide range of experimental approaches, including the use of fluorescent indicators of ROS, electron paramagnetic resonance (EPR), spectrophotometry, and high-performance liquid chromatography (HPLC); each method with its own limitations and advantages and generally specific to the ROS molecule attempting to be measured, cross reactivity, cellular permeability and localization.
We have recently used a transgenic, redox-sensitive GFP based biosensor specifically expressed in the mitochondrial matrix to determine mitochondrial generated ROS in a db/db mouse model in vivo (56). Kidney from transgenic control and diabetic mice were examined by 2-photon microscopy followed by ratio-metric determination of the redox state of the biosensor. Increased mitochondrial ROS in the diabetic kidneys was found which strongly implicated complex I as a key generator as the biosensor was matrix localized and the increase in ROS was prevented by a genetic bypass of complex I. This report and others have utilized mitochondrial targeted antioxidants such as mitoTEMPO, elamipretide, and others to demonstrated reduced mitochondrial ROS correlating with improved histological features of DKD in mouse models (12, 56, 156, 157).
Evidence determining mitochondrial ROS in the kidney of diabetic mice has also been obtained using dihydroethidium (DHE) as the redox sensor (45). Results in these mice were in contrast to the previous studies describing decreased mitochondrial ROS in the diabetic kidney. However, both studies were in agreement in regards to decreased activity of mitochondrial respiratory chain activity and found evidence of oxidative stress in the kidneys of diabetic animals (45, 56). These contrasting findings might be indicative of the difficulty in interpretating the cross-talk among different sources of ROS production (45, 56, 100). One such point of cross-talk in DKD could be derived from NADPH oxidases pathway. The NADPH oxidases of the NOX family are important enzymatic sources of ROS whose main biological function is electron transport across the plasma membrane and generate ROS by reducing oxygen to superoxide and/or hydrogen peroxide (158). At least seven homologs of NOX are present in the human genome: NOX1 to NOX5, DUOX1, and DUOX2. These mainly differ in their activation mechanisms, tissue distribution, and type of ROS production (157). Among different members of NOX family, NOX4 expression has been shown to be increased in the kidneys of diabetic mouse models, and capable of producing different types of ROS, mainly hydrogen peroxide (45, 159–163). However, under stress conditions, NOX4 might be translocated to mitochondria contributing to enhanced mitochondrial ROS by regulating mitochondrial respiratory complex I activity (164, 165). Consistent with this notion, deletion and pharmacological inhibition of NOX4 have been demonstrated to attenuate progression of DKD (161). NOX5 is also increased in the human diabetic kidney but not encoded by the mouse genome. Nevertheless, it has been shown that forced ectopic expression of NOX5 in mouse models leads to accelerated progression of DKD which could be ameliorated by pan-NOX inhibitors (161, 166–168).
While the specific contribution of mitochondrial generated superoxide remains an open question, it is clear that there is enhanced ROS in the kidneys of diabetic mouse models probably arising from multiple sources. The generated ROS is usually carefully balanced to stimulate stress abrogation responses, while not exceeding the cell ability to protect itself from damage through anti-oxidant enzymes. Once the balance is shifted such that the production of ROS exceeds the cells inherent antioxidant protections, an increasing cycle of cell damage is elicited resulting in compromised mitochondrial function, damaged mitochondrial DNA and proteins (169). If the cell cannot re-establish its balance, the end result is cellular death and kidney dysfunction.
Mitochondrial Biogenesis and Mitophagy
Mitochondrial biogenesis and degradation are highly regulated in order to maintain a healthy pool of mitochondria in the cell. However, both of the processes are dysregulated in DKD. Mitochondrial biogenesis refers to the cellular regulation of mitochondrial abundance titrated through an interconnected set of transcription factors. Central among these transcription factors is the peroxisome proliferator-activated receptor gamma (PPAR) coactivator-1 family of transcriptional coactivators (PGC1α/β) and PGC-1-related coactivator (PRC), coined as “master regulators” of mitochondrial biogenesis.
PGC1α was initially identified by the Spiegelman group (170) as a binding partner of PPAR that is highly expressed in tissues with high energy demand such as the kidney. As a coactivator, PGC1 does not bind to DNA promoters directly, but in dimerization with a variety of transcription factors to modulate a series of mitochondrial active gene products (171, 172). A few of the better understood partners of PGC1α include nuclear respiratory factor 1 (NRF1), NRF2, and the estrogen-related receptors (ERR). These heteromeric dimers likely, at least in part, could explain why experimental results with modulating PGC1α appear so highly tissue-specific since the possible dimeric combinations and relative amounts could depend on a specific tissue's expression levels of PGC1α, its various binding partners, and posttranslational modifications. Regardless, the system allows for a high degree of specialization in the regulation of gene products impinging upon mitochondrial biogenesis, mitochondrial gene transcription, fatty acid oxidation, TCA cycle, and OXPHOS. The role of PGC1α as a transcriptional rheostat tuning metabolic cellular function to physiological energy demands has been experimentally demonstrated in a myriad of tissues.
A number of studies have provided strong evidence that decreased PGC1α and reduced mitochondrial biogenesis are key features in the development of DKD. PGC1α has been demonstrated to be significantly decreased in the diabetic kidneys (9, 45, 145, 173–175). STZ treated rats have decreased PGC1α in renal tubules. This is evident in several mouse models of DKD as well. Diabetic OVE26, AKT2, and db/db mouse models have all been illustrated to have decreased PGC1α in the kidneys (176, 177). PGC1α was demonstrated to play a key role in another study examining an enzyme believed to couple glycolysis to mitochondria bioenergetics, pyruvate kinase M2 (PKM2) (10). In this study, podocyte-specific depletion of PKM2 in diabetic mice exacerbated diabetic renal injury, while pharmacological activation of PKM2 protected diabetic mice from kidney damage. Importantly, increased levels of PKM2 were correlated to protection from DKD in diabetic patients. The underlying mechanism proposed was that the protection was due in part to PKM2 linked activation of PGC1α and improved mitochondrial function (10, 178).
Our group has demonstrated that PGC1α could also be regulated by a long non-coding RNA, Tug1 (taurine upregulated 1). We found that Tug1 overexpression protects db/db mice from DKD (9). The protection was linked in vitro to Tug1 binding to PGC1α and improved mitochondrial function. However, another report found that podocyte-specific inducible overexpression of PGC1α in mouse models of DKD failed to offer renal protection (179). High expression levels of PGC1a could potentially drive a mitochondrial substrate preference toward b-oxidation of lipids contributing to worsening phenotype of DKD in experimental models. These results may indicate that PGC1α levels must be regulated and maintained within a very limited range to be beneficial.
PGC1α offers renal protection, at least in part, by driving oxidized nicotinamide adenine dinucleotide (NAD+) biosynthesis (180, 181). The redox imbalance of NADH/NAD+ (reduced/oxidized) is high in the diabetic kidney as electrons from the breakdown of nutrients become stored as NADH and metabolic pathways such as sirtuins consume NAD+. Complex I or lactate dehydrogenase can then regenerate NAD+ through oxidation of NADH (182–185). Modulation and the end balance of these processes determine the NADH/NAD+ ratio and represents one intersection point of PGC1α with sirtuins in mitochondrial biogenesis and bioenergetics.
The family of NAD-dependent deacetylases known as Sirtuins (SIRT1-7) regulate mitochondrial biogenesis and function as a nutritional rheostat which effects mitochondrial function via protein acetylation and have been implicated in several pathologies, including DKD (186–189). Proximal tubular overexpression of SIRT1 protected diabetic mice from DKD. Knockout of SIRT1 exacerbated renal injury in two separate diabetic mouse models and induced albuminuria in non-diabetic animals (190). The SIRT1 agonist, resveratrol, reduced podocyte damage in diabetic mice by activating PGC1α as well as its targets NRF1 and mitochondrial transcription factor 1 (TFAM) to improve mitochondrial function and reduce oxidative stress. SIRT1 has been shown to play a protective role in both tubules and podocytes of diabetic mouse models. The renoprotection stems in part through deacetylation of transcription factors, including PGC1α and PPARy (181, 191, 192). Podocyte-specific overexpression of SIRT1, and several non-specific agonists of SIRT1 such puerarin have been shown to attenuate DKD in animal models (193, 194). A more specific agonist, BF175, was tested and was shown to protect the kidney in type 1 diabetic OVE26 mice (195).
Consistent with the interplay between PGC1α and SIRT1, it has been shown that PGC1α can also increase levels of the mitochondrially-localized, SIRT3 (175, 196, 197). SIRT3 has been demonstrated to regulate mitochondrial function through direct binding to ETC proteins, mitochondrial dynamics, redox protection, and TCA cycle modulation and is the main mitochondrial deacetylase regulating cellular ROS. The SIRT3 agonist, honokol, was tested in BTBR ob/ob mice with type 2 diabetes and determined to be protective in DKD (186). SIRT3 was determined to be significantly decreased in the kidney of BTBR ob/ob mice in conjunction with increased ROS levels. Treatment with Honokol, a Magnolia tree bark extract and SIRT3 activator, reduced albuminuria and podocyte injury in the diabetic mice and was found to restore PGC1α levels in glomerular cells. The protective role of SIRT3 on glomeruli was mediated in part through increased SIRT3 tubular expression and upregulation of tubular nicotinamide phosphoribosyl transferase (Nampt), suggesting a possible tubule-glomerulus retrograde signaling mechanism. The lack of regulation of SIRT3 in glomeruli and postulated tubular-glomerular signaling was also a finding of a study examining SIRT1 in the diabetic kidney (190) where diabetic glomerular damage was improved by selective upregulation of tubular SIRT1.
In contrast to mitochondrial biogenesis, the process of mitophagy is the physiological clearance mechanism for removal of damaged mitochondria from the cell which appears to become overwhelmed in DKD (198–200). Mitophagy appears to have both a ubiquitin-dependent and -independent pathway (201, 202). The ubiquitin-dependent pathway is dependent upon mitochondrial dynamics, energetics, transport, and autophagic factors. The phosphatase and tensin homolog (PTEN) induced putative kinase 1 (PINK1) and Parkin (PRKN) are key mediators of the pathway. Physiologically PINK1 is transported to the inner mitochondrial membrane and proteolytically degraded in a ubiquitin dependent manner. When mitochondria become damaged and depolarized PINK1 is autophosphorylated and stabilized on the outer mitochondrial membrane to recruit PRKN and its E3 ligase activity. Mitochondrial fate is determined by the balance of the ubiquitination/deubiquitination process whereby increased poly-Ub targets the mitochondrion for proteasome destruction. PINK1 can increase mitochondrial fission by indirectly increasing DRP1 activity while the PINK1/PRKN interaction enhances Mfn2 degradation (203–210). Ubiquitin-independent pathway involves several ubiquitin E3 ligases which can localize to mitochondria and recruit autophagic factors.
The kidney has been shown to have a high rate of mitophagy relative to other organs, as well as cell type dependent regulation where podocytes have greater levels of mitophagy relative to tubules (211, 212). Increased mitophagy has been shown to be protective in models of CKD, DKD, and AKI (213–220). The PINK1/PRKN pathway is activated by oxidative stress established in DKD whereas treatment with the mitochondrial antioxidant, MitoQ, has been suggested to protect from DKD by increasing mitophagy levels (218). Tissue-specific knockout of ATG5 in a STZ model of DKD revealed that podocyte deletion induced podocytopathy and glomerulosclerosis while endothelial-specific knockout accelerated progression of DKD and when deleted in both tissues together increased DKD (221). These observations in aggregate suggest a critical role of mitophagy in DKD progression.
Conclusion and Future Perspectives
In this review, we touched the surface of several possibilities by which mitochondrial dysfunction could contribute to the development and progression of DKD, but we recognize that there still much remains to be uncovered. We would like to underscore a few gaps in knowledge for future discoveries.
As of yet, it is difficult to reach a clear consensus on the time course of mitochondrial respiratory activity and OXPHOS changes during progression of DKD. We await the arrival of more specific bioreporters to evaluate specific sources of enhanced ROS in real time in living animals, which ideally could link ROS to their enzymatic source in mitochondria. Similarly, a complete understanding of how mitochondrial dynamics fidelity is regulated and an evaluation of the “coincident detection” to fully integrate multiple organelles and biological factors into a single framework remains to be fully accomplished. The mitochondrial biogenesis pathway, and PGC1α in particular, are attractive therapeutic targets for DKD, but likely await the ability of targeting this pathway selectively in the kidney within a narrow therapeutic window. Finally, mitophagy, the crossroads of diverse signaling pathways, has shown a great promise as a therapeutic target, but the molecular mechanisms by which mitochondrial packaging for mitophagy becomes uncoupled during DKD progression remains unclear.
In conclusion, there have been increasing efforts to better define the nature of mitochondrial dysfunction in DKD over the past two decades. Initial studies utilizing metabolic screening approaches to identify the best possible biomarkers for predicting DKD susceptibility and progression are currently on-going (222, 223). While these and other studies have identified several mitochondrially-derived molecules such as mitochondrial DNA in serum and/or urine as potentially useful markers for DKD progression, none has exceeded expectations and are not currently available for patients care. Looking forward, opportunities in mitochondrial medicine involve the use of “multi-omics” and proteogenomics to provide further insights into the role of mitochondrial biomarkers in predicting DKD progression. A quantitative assessment of mitochondrial dysfunction in patients with DKD could accelerate the identification and development of novel biomarkers and treatments, and improve the ability to assess the efficiency of new drugs by measuring mitochondrial function pre- and post-therapies. Finally, the genetic and hormonal environment of the male and female kidney is significantly different, and these differences have been implicated on the onset and progression of DKD in both Type1 and 2 diabetes (224–227). The impact of gender on mitochondrial bioenergetics and function in kidney diseases has recently been reported (228). While many questions still remain to be carefully addressed, it seems clear that sexually determined differences in mitochondrial biogenesis, bioenergetics, and ROS generation exist, and these differences may also contribute to differences in long-term prognosis in patients with DKD (229, 230). Further research is needed to conclude a causal association between differences in gender, mitochondrial dysfunction and progression of kidney disease in large diabetic population.
Author Contributions
DG is responsible for writing the manuscript and literature research. KM and FD reviewed the manuscript and made significant revisions on the drafts. All authors have read and agreed to the final version of the manuscript.
Funding
This study was supported by grants from the National Institutes of Health RO1DK078900 (FD) and RO1DK091310 (FD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gilbert RE. Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes. (2017) 66:791–800. doi: 10.2337/db16-0796
2. Pfaller W, Rittinger M. Quantitative morphology of the rat kidney. Int J Biochem. (1980) 12:17–22. doi: 10.1016/0020-711X(80)90035-X
3. Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Ozel C, et al. Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep. (2019) 27:1551–66.e5. doi: 10.1016/j.celrep.2019.04.012
4. Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol. (2010) 299:C464–76. doi: 10.1152/ajpcell.00563.2009
5. Muller-Deile J, Schiffer M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front Endocrinol. (2014) 5:209. doi: 10.3389/fendo.2014.00209
6. Coughlan MT, Nguyen TV, Penfold SA, Higgins GC, Thallas-Bonke V, Tan SM, et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin Sci. (2016) 130:711–20. doi: 10.1042/CS20150838
7. Mise K, Galvan DL, Danesh FR. Shaping up mitochondria in diabetic nephropathy. Kidney360. (2020) 1:982–92. doi: 10.34067/KID.0002352020
8. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. (2012) 15:186–200. doi: 10.1016/j.cmet.2012.01.009
9. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. (2016) 126:4205–18. doi: 10.1172/JCI87927
10. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. (2017) 23:753–62. doi: 10.1038/nm.4328
11. Galvan DL, Long J, Green N, Chang BH, Lin JS, Schumacker P, et al. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J Clin Invest. (2019) 129:2807–23. doi: 10.1172/JCI127277
12. Ducasa GM, Mitrofanova A, Mallela SK, Liu X, Molina J, Sloan A, et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J Clin Invest. (2019) 129:3387–400. doi: 10.1172/JCI125316
13. Martin-Hernandez E, Garcia-Silva MT, Vara J, Campos Y, Cabello A, Muley R, et al. Renal pathology in children with mitochondrial diseases. Pediatr Nephrol. (2005) 20:1299–305. doi: 10.1007/s00467-005-1948-z
14. Rotig A, Munnich A. Genetic features of mitochondrial respiratory chain disorders. J Am Soc Nephrol. (2003) 14:2995–3007. doi: 10.1097/01.ASN.0000095481.24091.C9
15. Emma F, Bertini E, Salviati L, Montini G. Renal involvement in mitochondrial cytopathies. Pediatr Nephrol. (2012) 27:539–50. doi: 10.1007/s00467-011-1926-6
16. Emma F, Montini G, Parikh SM, Salviati L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. (2016) 12:267–80. doi: 10.1038/nrneph.2015.214
17. Niaudet P. Renal involvement in mitochondrial cytopathies. Nephrol Ther. (2013) 9:116–24. doi: 10.1016/j.nephro.2012.10.004
18. Seidowsky A, Hoffmann M, Glowacki F, Dhaenens CM, Devaux JP, de Sainte Foy CL, et al. Renal involvement in MELAS syndrome - a series of 5 cases and review of the literature. Clin Nephrol. (2013) 80:456–63. doi: 10.5414/CN107063
19. Finsterer J, Frank M. Prevalence of neoplasms in definite and probable mitochondrial disorders. Mitochondrion. (2016) 29:31–4. doi: 10.1016/j.mito.2016.05.002
20. Sangkhathat S, Kusafuka T, Yoneda A, Kuroda S, Tanaka Y, Sakai N, et al. Renal cell carcinoma in a pediatric patient with an inherited mitochondrial mutation. Pediatr Surg Int. (2005) 21:745–8. doi: 10.1007/s00383-005-1471-0
21. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. (2016) 2:16080. doi: 10.1038/nrdp.2016.80
22. Saiki R, Lunceford AL, Shi Y, Marbois B, King R, Pachuski J, et al. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am J Physiol Renal Physiol. (2008) 295:F1535–44. doi: 10.1152/ajprenal.90445.2008
23. Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. (2008) 4:e1000061. doi: 10.1371/journal.pgen.1000061
24. Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. (2006) 79:1125–9. doi: 10.1086/510023
25. Viscomi C, Spinazzola A, Maggioni M, Fernandez-Vizarra E, Massa V, Pagano C, et al. Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum Mol Genet. (2009) 18:12–26. doi: 10.1093/hmg/ddn309
26. Scialla JJ, Asplin J, Dobre M, Chang AR, Lash J, Hsu CY, et al. Higher net acid excretion is associated with a lower risk of kidney disease progression in patients with diabetes. Kidney Int. (2016) 91:204–215. doi: 10.1016/j.kint.2016.09.012
27. Garcia-Diaz B, Barros MH, Sanna-Cherchi S, Emmanuele V, Akman HO, Ferreiro-Barros CC, et al. Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am J Hum Genet. (2012) 91:729–36. doi: 10.1016/j.ajhg.2012.08.019
28. Janer A, Antonicka H, Lalonde E, Nishimura T, Sasarman F, Brown GK, et al. An RMND1 mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet. (2012) 91:737–43. doi: 10.1016/j.ajhg.2012.08.020
29. Gupta A, Colmenero I, Ragge NK, Blakely EL, He L, McFarland R, et al. Compound heterozygous RMND1 gene variants associated with chronic kidney disease, dilated cardiomyopathy and neurological involvement: a case report. BMC Res Notes. (2016) 9:325. doi: 10.1186/s13104-016-2131-2
30. Shayota BJ, Le NT, Bekheirnia N, Rosenfeld JA, Goldstein AC, Moritz M, et al. Characterization of the renal phenotype in RMND1-related mitochondrial disease. Mol Genet Genomic Med. (2019) 7:e973. doi: 10.1002/mgg3.973
31. Coughlan MT, Higgins GC, Nguyen TV, Penfold SA, Thallas-Bonke V, Tan SM, et al. Deficiency in Apoptosis-Inducing Factor Recapitulates Chronic Kidney Disease via Aberrant Mitochondrial Homeostasis. Diabetes. (2016) 65:1085–98. doi: 10.2337/db15-0864
32. Jansen JJ, Maassen JA, van der Woude FJ, Lemmink HA, van den Ouweland JM, t' Hart LM, et al. Mutation in mitochondrial tRNA(Leu(UUR)) gene associated with progressive kidney disease. J Am Soc Nephrol. (1997) 8:1118–24. doi: 10.1681/ASN.V871118
33. Nakamura S, Yoshinari M, Doi Y, Yoshizumi H, Katafuchi R, Yokomizo Y, et al. Renal complications in patients with diabetes mellitus associated with an A to G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Diabetes Res Clin Pract. (1999) 44:183–9. doi: 10.1016/S0168-8227(99)00051-0
34. Connor TM, Hoer S, Mallett A, Gale DP, Gomez-Duran A, Posse V, et al. Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genet. (2017) 13:e1006620. doi: 10.1371/journal.pgen.1006620
35. D'Aco KE, Manno M, Clarke C, Ganesh J, Meyers KE, Sondheimer N. Mitochondrial tRNA(Phe) mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol. (2013) 28:515–9. doi: 10.1007/s00467-012-2354-y
36. Scaglia F, Vogel H, Hawkins EP, Vladutiu GD, Liu LL, Wong LJ. Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am J Med Genet A. (2003) 123A:172–8. doi: 10.1002/ajmg.a.20315
37. Friederich M, Fasching A, Hansell P, Nordquist L, Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. (2008) 1777:935–40. doi: 10.1016/j.bbabio.2008.03.030
38. Friederich-Persson M, Persson P. Mitochondrial angiotensin II receptors regulate oxygen consumption in kidney mitochondria from healthy and type 1 diabetic rats. Am J Physiol Renal Physiol. (2020) 318:F683–f8. doi: 10.1152/ajprenal.00417.2019
39. Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. (2003) 46:1153–60. doi: 10.1007/s00125-003-1155-z
40. Christensen M, Schiffer TA, Gustafsson H, Krag SP, Norregaard R, Palm F. Metformin attenuates renal medullary hypoxia in diabetic nephropathy through inhibition uncoupling protein-2. Diabetes Metab Res Rev. (2019) 35:e3091. doi: 10.1002/dmrr.3091
41. Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. (2012) 61:2074–83. doi: 10.2337/db11-1437
42. Wu J, Luo X, Yan LJ. Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE). Front Physiol. (2015) 6:98. doi: 10.3389/fphys.2015.00098
43. Wu M, Li S, Yu X, Chen W, Ma H, Shao C, et al. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in STZ-induced diabetic mice. Am J Physiol Renal Physiol. (2019) 317:F593–f605. doi: 10.1152/ajprenal.00076.2019
44. Liu HW, Kao HH, Wu CH. Exercise training upregulates SIRT1 to attenuate inflammation and metabolic dysfunction in kidney and liver of diabetic db/db mice. Nutr Metab. (2019) 16:22. doi: 10.1186/s12986-019-0349-4
45. Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. (2013) 123:4888–99. doi: 10.1172/JCI66218
46. Hunter CA, Kartal F, Koc ZC, Murphy T, Kim JH, Denvir J, et al. Mitochondrial oxidative phosphorylation is impaired in TALLYHO mice, a new obesity and type 2 diabetes animal model. Int J Biochem Cell Biol. (2019) 116:105616. doi: 10.1016/j.biocel.2019.105616
47. Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. (2016) 1:e86976. doi: 10.1172/jci.insight.86976
48. Mustata GT, Rosca M, Biemel KM, Reihl O, Smith MA, Viswanathan A, et al. Paradoxical effects of green tea (Camellia sinensis) and antioxidant vitamins in diabetic rats: improved retinopathy and renal mitochondrial defects but deterioration of collagen matrix glycoxidation and cross-linking. Diabetes. (2005) 54:517–26. doi: 10.2337/diabetes.54.2.517
49. Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, et al. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. (2009) 58:1986–97. doi: 10.2337/db09-0259
50. Sourris KC, Harcourt BE, Tang PH, Morley AL, Huynh K, Penfold SA, et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic Biol Med. (2012) 52:716–23. doi: 10.1016/j.freeradbiomed.2011.11.017
51. Cleveland KH, Brosius FC 3rd, Schnellmann RG. Regulation of mitochondrial dynamics and energetics in the diabetic renal proximal tubule by the β(2)-adrenergic receptor agonist formoterol. Am J Physiol Renal Physiol. (2020) 319:F773–f9. doi: 10.1152/ajprenal.00427.2020
52. Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT, et al. Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J Am Soc Nephrol. (2016) 27:2733–47. doi: 10.1681/ASN.2015101096
53. Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes. (2017) 66:763–78. doi: 10.2337/db16-0695
54. Gujarati NA, Vasquez JM, Bogenhagen DF, Mallipattu SK. The complicated role of mitochondria in the podocyte. Am J Physiol Renal Physiol. (2020) 319:F955–f65. doi: 10.1152/ajprenal.00393.2020
55. Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. (2009) 20:742–52. doi: 10.1681/ASN.2008050514
56. Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. (2017) 92:1051–7. doi: 10.1016/j.kint.2017.05.034
57. Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nat Rev Nephrol. (2018) 14:291–312. doi: 10.1038/nrneph.2018.9
58. Wei PZ, Szeto CC. Mitochondrial dysfunction in diabetic kidney disease. Clin Chim Acta. (2019) 496:108–16. doi: 10.1016/j.cca.2019.07.005
59. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
60. Dai W, Jiang L. Dysregulated mitochondrial dynamics and metabolism in obesity, diabetes, and cancer. Front Endocrinol. (2019) 10:570. doi: 10.3389/fendo.2019.00570
61. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
62. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15:634–46. doi: 10.1038/nrm3877
63. Hatch AL, Ji WK, Merrill RA, Strack S, Higgs HN. Actin filaments as dynamic reservoirs for Drp1 recruitment. Mol Biol Cell. (2016) 27:3109–21. doi: 10.1091/mbc.e16-03-0193
64. Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol. (2018) 217:251–68. doi: 10.1083/jcb.201709111
65. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
66. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. (2016) 17:69–82. doi: 10.1038/nrm.2015.8
67. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. (2013) 339:464–7. doi: 10.1126/science.1228360
68. Tubbs E, Rieusset J. Metabolic signaling functions of ER-mitochondria contact sites: role in metabolic diseases. J Mol Endocrinol. (2017) 58:R87–R106. doi: 10.1530/JME-16-0189
69. Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. (2015) 4:e08828. doi: 10.7554/eLife.08828.023
70. Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun. (2017) 8:15754. doi: 10.1038/ncomms15754
71. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. (2021) 593:435–9. doi: 10.1038/s41586-021-03510-6
72. Chakrabarti R, Higgs HN. Revolutionary view of two ways to split a mitochondrion. Nature. (2021) 593:346–7. doi: 10.1038/d41586-021-01173-x
73. Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun. (2019) 10:2576. doi: 10.1038/s41467-019-10226-9
74. Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. (2016) 540:139–43. doi: 10.1038/nature20555
75. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
76. Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. (2008) 182:573–85. doi: 10.1083/jcb.200802164
77. Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. (2007) 282:21583–7. doi: 10.1074/jbc.C700083200
78. Edwards G, Perkins GA, Kim KY, Kong Y, Lee Y, Choi SH, et al. Loss of AKAP1 triggers Drp1 dephosphorylation-mediated mitochondrial fission and loss in retinal ganglion cells. Cell Death Dis. (2020) 11:254. doi: 10.1038/s41419-020-2456-6
79. Ko HJ, Tsai CY, Chiou SJ, Lai YL, Wang CH, Cheng JT, et al. The phosphorylation status of Drp1-Ser637 by PKA in mitochondrial fission modulates mitophagy via PINK1/parkin to exert multipolar spindles assembly during mitosis. Biomolecules. (2021) 11:424. doi: 10.3390/biom11030424
80. Chen Z, Ma Y, Yang Q, Hu J, Feng J, Liang W, et al. AKAP1 mediates high glucose-induced mitochondrial fission through the phosphorylation of Drp1 in podocytes. J Cell Physiol. (2020) 235:7433–48. doi: 10.1002/jcp.29646
81. Park SJ, Bae JE, Jo DS, Kim JB, Park NY, Fang J, et al. Increased O-GlcNAcylation of Drp1 by amyloid-beta promotes mitochondrial fission and dysfunction in neuronal cells. Mol Brain. (2021) 14:6. doi: 10.1186/s13041-020-00727-w
82. Huang J, Xie P, Dong Y, An W. Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ. (2021) 28:1174–92. doi: 10.1038/s41418-020-00641-7
83. Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. (2004) 14:340–5. doi: 10.1016/j.cub.2004.02.004
84. Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. (2007) 177:439–50. doi: 10.1083/jcb.200610042
85. Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. (2009) 324:102–5. doi: 10.1126/science.1171091
86. Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, et al. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington's disease. Antioxid Redox Signal. (2013) 19:1173–84. doi: 10.1089/ars.2012.4928
87. Lee DS, Kim JE. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. (2018) 9:869. doi: 10.1038/s41419-018-0910-5
88. Fung TS, Ji WK, Higgs HN, Chakrabarti R. Two distinct actin filament populations have effects on mitochondria, with differences in stimuli and assembly factors. J Cell Sci. (2019) 132:jcs234435. doi: 10.1242/jcs.234435
89. Hu J, Zhang H, Li J, Jiang X, Zhang Y, Wu Q, et al. ROCK1 activation-mediated mitochondrial translocation of Drp1 and cofilin are required for arnidiol-induced mitochondrial fission and apoptosis. J Exp Clin Cancer Res. (2020) 39:37. doi: 10.1186/s13046-020-01545-7
90. Kameoka S, Adachi Y, Okamoto K, Iijima M, Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. (2018) 28:67–76. doi: 10.1016/j.tcb.2017.08.011
91. Francy CA, Clinton RW, Frohlich C, Murphy C, Mears JA. Cryo-EM studies of Drp1 reveal cardiolipin interactions that activate the helical oligomer. Sci Rep. (2017) 7:10744. doi: 10.1038/s41598-017-11008-3
92. Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, et al. A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell. (2014) 25:1905–15. doi: 10.1091/mbc.e14-02-0728
93. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. (2003) 160:189–200. doi: 10.1083/jcb.200211046
94. Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. (2007) 16:1307–18. doi: 10.1093/hmg/ddm079
95. Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. (2007) 130:1029–42. doi: 10.1093/brain/awm005
96. Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. (2009) 186:805–16. doi: 10.1083/jcb.200903065
97. Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, et al. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. (2012) 61:2093–104. doi: 10.2337/db11-1640
98. Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. (2015) 26:1304–21. doi: 10.1681/ASN.2014050457
99. Zhan M, Usman I, Yu J, Ruan L, Bian X, Yang J, et al. Perturbations in mitochondrial dynamics by p66Shc lead to renal tubular oxidative injury in human diabetic nephropathy. Clin Sci. (2018) 132:1297–314. doi: 10.1042/CS20180005
100. Gao P, Yang M, Chen X, Xiong S, Liu J, Sun L. DsbA-L deficiency exacerbates mitochondrial dysfunction of tubular cells in diabetic kidney disease. Clin Sci. (2020) 134:677–94. doi: 10.1042/CS20200005
101. Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, et al. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J Cell Physiol. (2019) 234:3043–57. doi: 10.1002/jcp.27124
102. Thomas MC, Burns WC, Cooper ME. Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis. (2005) 12:177–86. doi: 10.1053/j.ackd.2005.01.008
103. Tang SC, Lai KN. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol Dial Transplant. (2012) 27:3049–56. doi: 10.1093/ndt/gfs260
104. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. (2011) 124:444–53. doi: 10.1161/CIRCULATIONAHA.110.014506
105. Ma Y, Chen Z, Tao Y, Zhu J, Yang H, Liang W, et al. Increased mitochondrial fission of glomerular podocytes in diabetic nephropathy. Endocr Connect. (2019) 8:1206–12. doi: 10.1530/EC-19-0234
106. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
107. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. (2013) 83:568–81. doi: 10.1038/ki.2012.441
108. Nayak B, Xie P, Akagi S, Yang Q, Sun L, Wada J, et al. Modulation of renal-specific oxidoreductase/myo-inositol oxygenase by high-glucose ambience. Proc Natl Acad Sci USA. (2005) 102:17952–7. doi: 10.1073/pnas.0509089102
109. Sun L, Xiao L, Nie J, Liu FY, Ling GH, Zhu XJ, et al. p66Shc mediates high-glucose and angiotensin II-induced oxidative stress renal tubular injury via mitochondrial-dependent apoptotic pathway. Am J Physiol Renal Physiol. (2010) 299:F1014–25. doi: 10.1152/ajprenal.00414.2010
110. Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. HIF-1alpha ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. (2020) 53:e12909. doi: 10.1111/cpr.12909
111. Kim K, Lee EY. Excessively enlarged mitochondria in the kidneys of diabetic nephropathy. Antioxidants. (2021) 10:741. doi: 10.3390/antiox10050741
112. Woo CY, Kc R, Kim M, Kim HS, Baek JY, Koh EH. Autophagic flux defect in diabetic kidney disease results in megamitochondria formation in podocytes. Biochem Biophys Res Commun. (2020) 521:660–7. doi: 10.1016/j.bbrc.2019.10.132
113. Kim K, Cha SJ, Choi HJ, Kang JS, Lee EY. Dysfunction of mitochondrial dynamics in drosophila model of diabetic nephropathy. Life. (2021) 11:67. doi: 10.3390/life11010067
114. Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, et al. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. (2007) 282:22977–83. doi: 10.1074/jbc.M700679200
115. Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. (2006) 209:468–80. doi: 10.1002/jcp.20753
116. Jensen PK. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim Biophys Acta. (1966) 122:157–66. doi: 10.1016/0926-6593(66)90057-9
117. Hinkle PC, Butow RA, Racker E, Chance B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J Biol Chem. (1967) 242:5169–73. doi: 10.1016/S0021-9258(18)99410-X
118. Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. (1977) 180:248–57. doi: 10.1016/0003-9861(77)90035-2
119. Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. (2008) 36:976–80. doi: 10.1042/BST0360976
120. Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA. (2006) 103:7607–12. doi: 10.1073/pnas.0510977103
121. Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. (2002) 80:780–7. doi: 10.1046/j.0022-3042.2002.00744.x
122. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. (1985) 237:408–14. doi: 10.1016/0003-9861(85)90293-0
123. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. (2004) 279:49064–73. doi: 10.1074/jbc.M407715200
124. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. (2003) 278:36027–31. doi: 10.1074/jbc.M304854200
125. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. (2010) 45:466–72. doi: 10.1016/j.exger.2010.01.003
126. Forman HJ, Azzi A. On the virtual existence of superoxide anions in mitochondria: thoughts regarding its role in pathophysiology. FASEB J. (1997) 11:374–5. doi: 10.1096/fasebj.11.5.9141504
127. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int J Mol Med. (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
128. Takeshige K, Minakami S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem J. (1979) 180:129–35. doi: 10.1042/bj1800129
129. Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. (2001) 79:266–77. doi: 10.1046/j.1471-4159.2001.00548.x
130. Seo BB, Marella M, Yagi T, Matsuno-Yagi A. The single subunit NADH dehydrogenase reduces generation of reactive oxygen species from complex I. FEBS Lett. (2006) 580:6105–8. doi: 10.1016/j.febslet.2006.10.008
131. Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J Biol Chem. (2007) 282:22040–51. doi: 10.1074/jbc.M703591200
132. Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J Biol Chem. (2004) 279:39414–20. doi: 10.1074/jbc.M406576200
133. Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. (2004) 382:511–7. doi: 10.1042/BJ20040485
134. Forbes JM, Ke BX, Nguyen TV, Henstridge DC, Penfold SA, Laskowski A, et al. Deficiency in mitochondrial complex I activity due to Ndufs6 gene trap insertion induces renal disease. Antioxid Redox Signal. (2013) 19:331–43. doi: 10.1089/ars.2012.4719
135. Zhang H, Zhang HM, Wu LP, Tan DX, Kamat A, Li YQ, et al. Impaired mitochondrial complex III and melatonin responsive reactive oxygen species generation in kidney mitochondria of db/db mice. J Pineal Res. (2011) 51:338–44. doi: 10.1111/j.1600-079X.2011.00894.x
136. Badal SS, Danesh FR. Reactive Oxygen Species (ROS) and Diabetic Nephropathy. In: Laher I, editor. Systems Biology of Free Radicals and Antioxidants. Berlin; Heidelberg: Springer (2014). doi: 10.1007/978-3-642-30018-9_186
137. Lindblom R, Higgins G, Coughlan M, de Haan JB. Targeting mitochondria and reactive oxygen species-driven pathogenesis in diabetic nephropathy. Rev Diabet Stud. (2015) 12:134–56. doi: 10.1900/RDS.2015.12.134
138. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. (2018) 9:119. doi: 10.1038/s41419-017-0135-z
139. DeRubertis FR, Craven PA, Melhem MF, Salah EM. Attenuation of renal injury in db/db mice overexpressing superoxide dismutase: evidence for reduced superoxide-nitric oxide interaction. Diabetes. (2004) 53:762–8. doi: 10.2337/diabetes.53.3.762
140. Hamada Y, Miyata S, Nii-Kono T, Kitazawa R, Kitazawa S, Higo S, et al. Overexpression of thioredoxin1 in transgenic mice suppresses development of diabetic nephropathy. Nephrol Dial Transplant. (2007) 22:1547–57. doi: 10.1093/ndt/gfm099
141. de Haan JB, Stefanovic N, Nikolic-Paterson D, Scurr LL, Croft KD, Mori TA, et al. Kidney expression of glutathione peroxidase-1 is not protective against streptozotocin-induced diabetic nephropathy. Am J Physiol Renal Physiol. (2005) 289:F544–51. doi: 10.1152/ajprenal.00088.2005
142. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. (2001) 414:813–20. doi: 10.1038/414813a
143. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. (2005) 54:1615–25. doi: 10.2337/diabetes.54.6.1615
144. Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl. (2000) 77:S26–30. doi: 10.1046/j.1523-1755.2000.07705.x
145. Guo K, Lu J, Huang Y, Wu M, Zhang L, Yu H, et al. Protective role of PGC-1alpha in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS ONE. (2015) 10:e0125176. doi: 10.1371/journal.pone.0125176
146. Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. (2005) 289:F420–30. doi: 10.1152/ajprenal.00415.2004
147. Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. (2008) 57:1446–54. doi: 10.2337/db08-0057
148. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. (2006) 55:225–33. doi: 10.2337/diabetes.55.01.06.db05-0894
149. Jezek J, Cooper KF, Strich R. Reactive oxygen species and mitochondrial dynamics: the Yin and Yang of mitochondrial dysfunction and cancer progression. Antioxidants. (2018) 7:13. doi: 10.3390/antiox7010013
150. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. (2016) 12:999–1014. doi: 10.1080/15548627.2016.1166318
151. Sanchez-Alvarez R, De Francesco EM, Fiorillo M, Sotgia F, Lisanti MP. Mitochondrial fission factor (MFF) inhibits mitochondrial metabolism and reduces breast cancer stem cell (CSC) activity. Front Oncol. (2020) 10:1776. doi: 10.3389/fonc.2020.01776
152. Tur J, Pereira-Lopes S, Vico T, Marin EA, Munoz JP, Hernandez-Alvarez M, et al. Mitofusin 2 in macrophages links mitochondrial ROS production, cytokine release, phagocytosis, autophagy, and bactericidal activity. Cell Rep. (2020) 32:108079. doi: 10.1016/j.celrep.2020.108079
153. Kulkarni SS, Joffraud M, Boutant M, Ratajczak J, Gao AW, Maclachlan C, et al. Mfn1 deficiency in the liver protects against diet-induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes. (2016) 65:3552–60. doi: 10.1093/med/9780198729426.003.0005
154. Sebastian D, Sorianello E, Segales J, Irazoki A, Ruiz-Bonilla V, Sala D, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. (2016) 35:1677–93. doi: 10.15252/embj.201593084
155. Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, et al. NR4A1 promotes diabetic nephropathy by activating Mff-mediated mitochondrial fission and suppressing Parkin-mediated mitophagy. Cell Physiol Biochem. (2018) 48:1675–93. doi: 10.1159/000492292
156. Chen J, Chen JK, Harris RC. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J Am Soc Nephrol. (2015) 26:1115–25. doi: 10.1681/ASN.2014020192
157. Yang SK, Li AM, Han YC, Peng CH, Song N, Yang M, et al. Mitochondria-targeted peptide SS31 attenuates renal tubulointerstitial injury via inhibiting mitochondrial fission in diabetic mice. Oxid Med Cell Longev. (2019) 2019:2346580. doi: 10.1155/2019/2346580
158. Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. (2014) 76:208–26. doi: 10.1016/j.freeradbiomed.2014.07.046
159. Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, et al. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia. (2003) 46:1428–37. doi: 10.1007/s00125-003-1205-6
160. Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. (2010) 299:F1348–58. doi: 10.1152/ajprenal.00028.2010
161. Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol. (2014) 25:1237–54. doi: 10.1681/ASN.2013070810
162. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA. (2009) 106:14385–90. doi: 10.1073/pnas.0906805106
163. Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, et al. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. (2005) 280:39616–26. doi: 10.1074/jbc.M502412200
164. Hirschhäuser C, Bornbaum J, Reis A, Böhme S, Kaludercic N, Menabò R, et al. NOX4 in mitochondria: yeast two-hybrid-based interaction with complex i without relevance for basal reactive oxygen species? Antioxid Redox Signal. (2015) 23:1106–12. doi: 10.1089/ars.2014.6238
165. Yang Q, Wu FR, Wang JN, Gao L, Jiang L, Li HD, et al. Nox4 in renal diseases: an update. Free Radic Biol Med. (2018) 124:466–72. doi: 10.1016/j.freeradbiomed.2018.06.042
166. Dorotea D, Kwon G, Lee JH, Saunders E, Bae YS, Moon SH, et al. A pan-NADPH oxidase inhibitor ameliorates kidney injury in type 1 diabetic rats. Pharmacology. (2018) 102:180–9. doi: 10.1159/000491398
167. Holterman CE, Thibodeau JF, Towaij C, Gutsol A, Montezano AC, Parks RJ, et al. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J Am Soc Nephrol. (2014) 25:784–97. doi: 10.1681/ASN.2013040371
168. Lee ES, Kim HM, Lee SH, Ha KB, Bae YS, Lee SJ, et al. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic Biol Med. (2020) 161:92–101. doi: 10.1016/j.freeradbiomed.2020.09.024
169. Sakashita M, Tanaka T, Inagi R. Metabolic changes and oxidative stress in diabetic kidney disease. Antioxidants. (2021) 10:1143. doi: 10.3390/antiox10071143
170. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. (1998) 92:829–39. doi: 10.1016/S0092-8674(00)81410-5
171. Baldelli S, Aquilano K, Ciriolo MR. Punctum on two different transcription factors regulated by PGC-1alpha: nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim Biophys Acta. (2013) 1830:4137–46. doi: 10.1016/j.bbagen.2013.04.006
172. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. (2008) 88:611–38. doi: 10.1152/physrev.00025.2007
173. Lee SY, Kang JM, Kim DJ, Park SH, Jeong HY, Lee YH, et al. PGC1alpha activators mitigate diabetic tubulopathy by improving mitochondrial dynamics and quality control. J Diabetes Res. (2017) 2017:6483572. doi: 10.1155/2017/6483572
174. Lee J, Tsogbadrakh B, Yang S, Ryu H, Kang E, Kang M, et al. Klotho ameliorates diabetic nephropathy via LKB1-AMPK-PGC1alpha-mediated renal mitochondrial protection. Biochem Biophys Res Commun. (2021) 534:1040–6. doi: 10.1016/j.bbrc.2020.10.040
175. Lynch MR, Tran MT, Parikh SM. PGC1alpha in the kidney. Am J Physiol Renal Physiol. (2018) 314:F1–F8. doi: 10.1152/ajprenal.00263.2017
176. Ruegsegger GN, Creo AL, Cortes TM, Dasari S, Nair KS. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J Clin Invest. (2018) 128:3671–81. doi: 10.1172/JCI120843
177. Stadler K, Goldberg IJ, Susztak K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr Diab Rep. (2015) 15:40. doi: 10.1007/s11892-015-0611-8
178. Gordin D, Shah H, Shinjo T, St-Louis R, Qi W, Park K, et al. Characterization of glycolytic enzymes and pyruvate kinase M2 in type 1 and 2 diabetic nephropathy. Diabetes Care. (2019) 42:1263–73. doi: 10.2337/dc18-2585
179. Li SY, Park J, Qiu C, Han SH, Palmer MB, Arany Z, et al. Increasing the level of peroxisome proliferator-activated receptor gamma coactivator-1alpha in podocytes results in collapsing glomerulopathy. JCI Insight. (2017) 2:e92930. doi: 10.1172/jci.insight.92930
180. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. (2016) 531:528–32. doi: 10.1038/nature17184
181. Koh JH, Kim JY. Role of PGC-1alpha in the mitochondrial NAD(+) pool in metabolic diseases. Int J Mol Sci. (2021) 22:4558. doi: 10.3390/ijms22094558
182. Hayden MR, Sowers JR. Redox imbalance in diabetes. Antioxid Redox Signal. (2007) 9:865–7. doi: 10.1089/ars.2007.1640
183. Yan LJ. Redox imbalance stress in diabetes mellitus: role of the polyol pathway. Animal Model Exp Med. (2018) 1:7–13. doi: 10.1002/ame2.12001
184. Yan LJ. NADH/NAD(+) redox imbalance and diabetic kidney disease. Biomolecules. (2021) 11:730. doi: 10.3390/biom11050730
185. Wu J, Jin Z, Zheng H, Yan LJ. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. (2016) 9:145–53. doi: 10.2147/DMSO.S106087
186. Locatelli M, Zoja C, Zanchi C, Corna D, Villa S, Bolognini S, et al. Manipulating Sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci Rep. (2020) 10:8418. doi: 10.1038/s41598-020-65423-0
187. Hershberger KA, Martin AS, Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. (2017) 13:213–25. doi: 10.1038/nrneph.2017.5
188. Morigi M, Perico L, Benigni A. Sirtuins in renal health and disease. J Am Soc Nephrol. (2018) 29:1799–809. doi: 10.1681/ASN.2017111218
189. Perico L, Benigni A. The iNADequacy of renal cell metabolism: modulating NAD(+) biosynthetic pathways to forestall kidney diseases. Kidney Int. (2019) 96:264–7. doi: 10.1016/j.kint.2019.03.012
190. Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. (2013) 19:1496–504. doi: 10.1038/nm.3363
191. Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. (2009) 20:98–105. doi: 10.1097/MOL.0b013e328328d0a4
192. Tang BL. Sirt1 and the mitochondria. Mol Cells. (2016) 39:87–95. doi: 10.14348/molcells.2016.2318
193. Zhong Y, Lee K, He JC. SIRT1 is a potential drug target for treatment of diabetic kidney disease. Front Endocrinol. (2018) 9:624. doi: 10.3389/fendo.2018.00624
194. Yacoub R, Lee K, He JC. The role of SIRT1 in diabetic kidney disease. Front Endocrinol. (2014) 5:166. doi: 10.3389/fendo.2014.00166
195. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. (2018) 93:1330–43. doi: 10.1016/j.kint.2017.12.008
196. Brenmoehl J, Hoeflich A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion. (2013) 13:755–61. doi: 10.1016/j.mito.2013.04.002
197. Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, Monsalve M, Ramos AM, Sanchez-Nino MD, et al. The role of PGC-1alpha and mitochondrial biogenesis in kidney diseases. Biomolecules. (2020) 10:347. doi: 10.3390/biom10020347
198. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front Cell Dev Biol. (2020) 8:467. doi: 10.3389/fcell.2020.00467
199. Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol. (2014) 171:1917–42. doi: 10.1111/bph.12503
200. Kume S, Koya D. Autophagy: a novel therapeutic target for diabetic nephropathy. Diabetes Metab J. (2015) 39:451–60. doi: 10.4093/dmj.2015.39.6.451
201. Bhatia D, Choi ME. The emerging role of mitophagy in kidney diseases. J Life Sci. (2019) 1:13–22. doi: 10.36069/JoLS/20191203
202. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
203. Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. (2013) 26:9–18. doi: 10.1016/j.devcel.2013.05.024
204. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. (2015) 6:7527. doi: 10.1038/ncomms8527
205. Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. (2011) 301:H1924–31. doi: 10.1152/ajpheart.00368.2011
206. Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, et al. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol. (2010) 48:1146–56. doi: 10.1016/j.yjmcc.2009.12.004
207. Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. (2010) 6:855–62. doi: 10.4161/auto.6.7.13005
208. McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife. (2018) 7:e32866. doi: 10.7554/eLife.32866.032
209. Basso V, Marchesan E, Peggion C, Chakraborty J, von Stockum S, Giacomello M, et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol Res. (2018) 138:43–56. doi: 10.1016/j.phrs.2018.09.006
210. Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. (2009) 284:22938–51. doi: 10.1074/jbc.M109.035774
211. Kitada M, Ogura Y, Suzuki T, Sen S, Lee SM, Kanasaki K, et al. A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia. (2016) 59:1307–17. doi: 10.1007/s00125-016-3925-4
212. Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. (2010) 120:1084–96. doi: 10.1172/JCI39492
213. Li N, Wang H, Jiang C, Zhang M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp Cell Res. (2018) 369:27–33. doi: 10.1016/j.yexcr.2018.04.025
214. Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. (2018) 14:880–97. doi: 10.1080/15548627.2017.1405880
215. Wang Y, Tang C, Cai J, Chen G, Zhang D, Zhang Z, et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. (2018) 9:1113. doi: 10.1038/s41419-018-1152-2
216. Zhao C, Chen Z, Xu X, An X, Duan S, Huang Z, et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp Cell Res. (2017) 350:390–7. doi: 10.1016/j.yexcr.2016.12.015
217. Zhao C, Chen Z, Qi J, Duan S, Huang Z, Zhang C, et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget. (2017) 8:20988–1000. doi: 10.18632/oncotarget.15470
218. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. (2017) 11:297–311. doi: 10.1016/j.redox.2016.12.022
219. Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes. (2016) 65:755–67. doi: 10.2337/db15-0473
220. Yang YY, Gong DJ, Zhang JJ, Liu XH, Wang L. Diabetes aggravates renal ischemia-reperfusion injury by repressing mitochondrial function and PINK1/Parkin-mediated mitophagy. Am J Physiol Renal Physiol. (2019) 317:F852–f64. doi: 10.1152/ajprenal.00181.2019
221. Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T, et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. (2015) 11:1130–45. doi: 10.1080/15548627.2015.1049799
222. Kwan B, Fuhrer T, Zhang J, Darshi M, Van Espen B, Montemayor D, et al. Metabolomic markers of kidney function decline in patients with diabetes: evidence from the chronic renal insufficiency cohort (CRIC) study. Am J Kidney Dis. (2020) 76:511–20. doi: 10.1053/j.ajkd.2020.01.019
223. Krochmal M, Schanstra JP, Mischak H. Urinary peptidomics in kidney disease and drug research. Expert Opin Drug Discov. (2018) 13:259–68. doi: 10.1080/17460441.2018.1418320
224. Kautzky-Willer A, Harreiter J, Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. (2016) 37:278–316. doi: 10.1210/er.2015-1137
225. Shepard BD. Sex differences in diabetes and kidney disease: mechanisms and consequences. Am J Physiol Renal Physiol. (2019) 317:F456–62. doi: 10.1152/ajprenal.00249.2019
226. Dorman JS, Steenkiste AR, Foley TP, Strotmeyer ES, Burke JP, Kuller LH, et al. Menopause in type 1 diabetic women: is it premature? Diabetes. (2001) 50:1857–62. doi: 10.2337/diabetes.50.8.1857
227. Maric C, Sullivan S. Estrogens and the diabetic kidney. Gend Med. (2008) 5 (Suppl. A):S103–13. doi: 10.1016/j.genm.2008.03.010
228. Sultanova RF, Schibalski R, Yankelevich IA, Stadler K, Ilatovskaya DV. Sex differences in renal mitochondrial function: a hormone-gous opportunity for research. Am J Physiol Renal Physiol. (2020) 319:F1117–24. doi: 10.1152/ajprenal.00320.2020
229. Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R1220–6. doi: 10.1152/ajpregu.00864.2007
Keywords: diabetic kidney disease, mitochondria, mitochondrial dynamics, oxidative phosphorylation, mitochondrial respiratory complexes, bioenergetics
Citation: Galvan DL, Mise K and Danesh FR (2021) Mitochondrial Regulation of Diabetic Kidney Disease. Front. Med. 8:745279. doi: 10.3389/fmed.2021.745279
Received: 21 July 2021; Accepted: 30 August 2021;
Published: 27 September 2021.
Edited by:
Ilse Sofia Daehn, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Alexander Staruschenko, Medical College of Wisconsin, United StatesKrisztian Stadler, Pennington Biomedical Research Center, United States
Copyright © 2021 Galvan, Mise and Danesh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farhad R. Danesh, ZmRhbmVzaEBtZGFuZGVyc29uLm9yZw==